

Abstract
We tested the hypothesis that the histamine H3-receptor (H3R)-mediated attenuation of norepinephrine (NE) exocytosis from cardiac sympathetic nerves results not only from a Gαi-mediated inhibition of the adenylyl cyclase-cAMP-PKA pathway, but also from a Gβγi-mediated activation of the MAPK-PLA2 cascade, culminating in formation of an arachidonate metabolite with anti-exocytotic characteristics (e.g., PGE2). We report in Langendorff-perfused guinea-pig hearts and isolated sympathetic nerve endings (cardiac synaptosomes), H3R-mediated attenuation of K+-induced NE exocytosis was prevented by MAPK and PLA2 inhibitors, and by cyclooxygenase and EP3-receptor (EP3R) antagonists. Moreover, H3R activation resulted in MAPK phosphorylation in H3R-transfected SH-SY5Y neuroblastoma cells, and in PLA2 activation and PGE2 production in cardiac synaptosomes; H3R-induced MAPK phosphorylation was prevented by an anti-βγ peptide. Synergism between H3R and EP3R agonists (i.e., imetit and sulprostone, respectively) suggested PGE2 may be a downstream effector of the anti-exocytotic effect of H3R activation. Furthermore, the anti-exocytotic effect of imetit and sulprostone was potentiated by the N-type Ca2+-channel antagonist ω-conotoxin GVIA, and prevented by an anti-Gβγ peptide. Our findings suggest an EP3R Gβγi-induced decrease in Ca2+ influx through N-type Ca2+-channels is involved in PGE2/EP3R-mediated attenuation of NE exocytosis elicited by H3R activation. Conceivably, activation of the Gβγi subunit of H3R and EP3R may also inhibit Ca2+ entry directly, independent of MAPK intervention. As heart failure, myocardial ischemia and arrhythmic dysfunction are associated with excessive local NE release, attenuation of NE release by H3R activation is cardioprotective. Thus, the uncovering of a novel H3R signaling pathway may ultimately bear therapeutic significance in hyper-adrenergic states.
1. Introduction
Sympathetic nerve terminals in the guinea pig [1;2] and human [3] heart express histamine H3-receptors (H3R). H3R activation reduces norepinephrine (NE) exocytosis and is associated with a marked decrease in the peak intraneuronal Ca2+ ([Ca2+]i) response [4]. We recently reported that the H3R-mediated attenuation of NE exocytosis involves an H3R-Gi/Go coupling, adenylyl cyclase inhibition by Gαi, decreased cAMP formation and diminished PKA activity [5]. Diminished PKA activity is likely to result in reduced phosphorylation of voltage-operated Ca2+-channels (VOCC), which would be reflected in a decrease in Ca2+ current (ICa). Thus, it is plausible that the H3R-mediated attenuation of NE exocytosis, and the associated reduction in [Ca2+]i, results from a decreased Ca2+ influx via VOCC, due to diminished activity of the adenylyl cyclase-cAMP-PKA pathway.
In addition to adenylyl cyclase inhibition, receptors coupled to pertussis toxin-sensitive heterotrimeric G proteins (e.g., H3R) are known to stimulate phospholipase A2 (PLA2) via the Gαi subunit [6–8]. Furthermore, H3R couple to the MAPK cascade [9] which contributes to PLA2 phosphorylation and stimulation of its catalytic activity [10]. PLA2 activation initiates the arachidonic acid cascade with the ultimate formation of various eicosanoids, including PGE2. PGE2 has been shown to inhibit NE release from sympathetic nerves by activating presynaptic EP3-receptors (EP3R) [11;12]. Accordingly, we hypothesized that the H3R-mediated attenuation of NE exocytosis results not only from a decreased adenylyl cyclase-cAMP-PKA function, but also involves another signaling pathway entailing the activation of MAPK and PLA2, and the eventual formation of an arachidonate metabolite with anti-exocytotic characteristics, most likely PGE2. We tested this hypothesis both at the subcellular (i.e., cardiac synaptosomes) and whole organ level (i.e., Langendorff-perfused heart).
2. Methods and Materials
2.1 Isolated Heart
All experiments were approved by the IACUC of Weill Cornell Medical College. Male adult Hartley guinea pigs (350 to 500 g; Charles River Labs., Wilmington, MA) were anesthetized with CO2 and rapidly exsanguinated. Hearts were excised and immediately immersed in ice-cold Krebs-Henseleit solution (mM: NaCl, 118; KCl, 4.7; MgSO4·7H2O, 1.2; NaHCO3, 24; KH2PO4, 1.1; glucose 10 and CaCl2·2H2O, 2.5) equilibrated with 95% O2/5% CO2. Hearts were perfused at constant pressure (40 cm H2O) in a Langendorff apparatus with warmed Krebs-Henseleit solution (37°C), containing desipramine (0.1 μM) and atropine (1 μM). ECG was recorded on-line using needle electrodes (400 Hz recording frequency) and analyzed with Powerlab/8SP (AD Instruments, Colorado Springs, CO). Only hearts with a stable sinus rhythm were included in the study. To elicit NE release, two custom-made stainless steel paddles were gently attached to the heart and kept parallel to the intraventricular septum. After 20 min of stabilization, two sequential field stimulations (5 Hz, 2 msec, 5 V, 60 sec) using PowerLab/8SP were applied 15 min apart from each other. Coronary effluent was collected before and during stimulation for 2 min. NE overflow into the coronary effluent (i.e., NE exocytosis) was measured by high pressure liquid chromatography with electrochemical detection (HPLC-EC)[2] and expressed as the ratio between the second and first stimulation (S2/S1). The amount of released NE was very similar during two consecutive stimulations (S2/S1 = 0.992 ± 0.012; n = 7). Subsequently, a concentration-response curve (0.03–3 μM, n = 3–6) for the imetit-induced attenuation of NE exocytosis was constructed and the IC50 was found to be ~0.3 μM. In subsequent experiments the anti-exocytotic effect of imetit (at its IC50) was re-assessed in hearts perfused with the H3R antagonist clobenpropit (50 nM), the PLA2 inhibitor methyl arachidonyl fluorophosphonate (MAFP; 10 μM) or the EP3R antagonist ONO-AE3-240 (10 nM).
2.2 Isolation of Cardiac Synaptosomes
Guinea-pig hearts were isolated as described above and perfused for 15 min in the Langendorff apparatus to ensure that no blood traces remained in the coronary circulation. Hearts were then minced in ice-cold 0.32 M sucrose containing 1 mM EGTA, pH 7.4. Minced tissue was digested with 40 mg collagenase (Type II, Worthington Biochemicals, Lakewood, NJ) per 10 ml HEPES-buffered saline solution (HBS) per gram of wet heart weight for 1 hour at 37°C. HBS contained 1 mM pargyline to prevent enzymatic destruction of synaptosomal NE. After low-speed centrifugation (10 minutes at 120 g at 4°C), the resulting pellet was suspended in 10 volumes of 0.32 M sucrose and homogenized with a Teflon/glass homogenizer. The homogenate was spun at 650 g for 10 min at 4°C and the pellet rehomogenized and respun. The pellet containing cellular debris was discarded, and the supernatants from the last two spins were combined and equally subdivided into 10 to 12 tubes. Each tube was centrifuged for 20 min at 20,000 g at 4°C. This pellet, which contained cardiac synaptosomes, was resuspended in HBS to a final volume of 500 μL in the presence or absence of pharmacological agents in a water bath at 37°C. Each suspension functioned as an independent sample and was used only once. In every experiment, one sample was untreated (control, basal NE release), and others were incubated with drugs for 10 min. When antagonists were used, samples were incubated with the antagonist for 10 min before incubation with the agonist. Controls were incubated for an equivalent length of time without drugs. NE exocytosis was elicited by incubating samples for 5 minutes with K+ 100 mM (osmolarity was maintained constant by adjusting the NaCl concentration). At the end of the incubation period, each sample was centrifuged for 20 min (20,000 g at 4°C). The supernatant was assayed for NE content by high pressure liquid chromatography (HPLC) with electrochemical detection [2]. The pellet was assayed for protein content by a modified Lowry procedure [2].
2.3 cPLA2 Assay
Guinea-pig heart synaptosomes were incubated with the H3R agonist imetit (100 nM)[13] or the Ca2+-ionophore A23187 (10 μM) for 10 min, either alone or in the presence of the H3R antagonist clobenpropit [14]. When clobenpropit (50 nM) was used, samples were incubated with it for 10 min before incubation with imetit or A23187. Controls were incubated for an equivalent length of time without drugs. Following low-speed centrifugation the synaptosomal pellet was isolated and lysed in 10x lysis buffer (Cell Signaling Technology, Danvers, MA). Samples were then centrifuged at 12,000 g at 4°C and the resulting supernatant was assayed for cPLA2 activity using a cPLA2 assay kit (Cayman Chemical, Ann Arbor, MI), following the manufacturers protocol.
2.4 PGE2 Assay
Following low-speed centrifugation, supernatants from guinea-pig heart synaptosomal preparations were assayed for PGE2 release using a PGE2 EIA kit (Cayman Chemical, Ann Arbor, MI), following the manufacturers protocol.
2.5 Detection of p38 and JNK MAPK activation
A human neuroblastoma cell line stably transfected with the H3R (SH-SY5Y-H3) was kindly supplied by Dr T. W. Lovenberg, Johnson & Johnson Pharmaceutical R&D, LLC (San Diego, CA) cells were maintained in a 1:1 ratio of Eagle’s and Ham’s F-12 [4]. SH-SY5Y-H3 minimal essential medium mixture, supplemented with 10% fetal bovine serum, 2 mM L-glutamine, 450 μg/ml geneticin, 50 units/ml penicillin, and 50 μg/ml streptomycin at 37°C, 5% CO2. Cell culture media and supplements were purchased from Mediatech, Inc. (Herndon, VA). Cells were grown to confluence in 6-well plates. MAPK phosphorylation (i.e., an indication of MAPK activation) cells as elicited by incubating SH-SY5Y-H3 with the H3R agonist imetit (100 nM), clobenpropit (CBP; 50 nM) for in the absence or presence of the H3R antagonist10 min at 37°C, or with anti-βγ peptide (1μM) in the presence or absence of imetit. cells were then lysed (Lysis buffer; Cell Signaling Technology Inc., SH-SY5Y-H3 Beverly, MA). Samples of lysate (15 μg/lane) were prepared with 5x Tris-glycine SDS sample buffer and boiled for 5 min before separation on 10–20% gradient Tris-glycine SDS-polyacrylamide minigels (Invitrogen, Carlsbad, CA). Electrophoresis was carried out at 200 V, 40 mA/gel for 1 h. Gels were then electrotransferred to polyvinylidine difluoride (PVDF) membranes (Immobilon-P; Millipore, Billerica, MA) for 90 min at 200 V, 300 mA, 4ºC. Membranes were blocked for 2 h in blocking buffer [Tris-buffered saline (TBS) containing 0.1% Tween 20, 5% (w/v) non-fat dry milk]. Phospho-p38 antibody (Biosource; Camarillo, CA) (1:1,000) and phospho-JNK antibody (Cell Signaling Technology Inc., Beverly, MA) (1:1,000) diluted in primary antibody dilution buffer (TBS containing 0.1% Tween 20, 5% bovine serum albumin) were incubated with the PVDF membrane overnight at 4°C. The PVDF membrane was washed three times with TBST and then horseradish peroxidase-coupled anti-rabbit IgG (Cell Signaling Technology Inc., Beverly, MA) was added at a 1:3,000 dilution in blocking buffer for 1 h. The PVDF membrane was then washed three times with TBST and the protein of interest was detected using enhanced chemiluminescence (Millipore, Billerica, MA) followed by exposure to X-ray film (Biomax MR; Eastman Kodak, Rochester, NY). Bands were analyzed by densitometry using Fluorchem™ 8800 (Alpha Innotech, San Leandro, CA).
2.6 Drugs and Chemicals
Indomethacin, imetit, ω-conotoxin GVIA, clobenpropit, A23187 and nifedipine were purchased from Sigma-Aldrich Chemical Co (St Louis, MO). MAFP was purchased from Biomol Research Laboratories (Plymouth Meeting, PA). PD 98059, SB 202190, SB 202474, SP600125 and N1-Methyl-1,9-pyrazoloanthrone were purchased from CalbioChem (La Jolla, CA). Sulprostone was purchased from Cayman Chemical (Ann Arbor, MI). The anti-βγ peptide was purchased from AnaSpec, Inc. (San Jose, CA). L-798,106 was a gift from Merck Frosst Canada & Co. ONO-AE3-240 was a gift from ONO Pharmaceutical Co., LTD., Japan. ONO-AE3-240 was dissolved in 100% ethanol and the anti-Gβγ peptide in 10% NH4OH. All other drugs were dissolved in dimethyl sulfoxide (DMSO). Further dilutions were made with HEPES buffer; at the concentration used, DMSO, ethanol and NH4OH did not affect NE release.
3. Results
3.1 MAPK activation plays a role in the H3R-induced inhibition of NE exocytosis
We first determined whether H3R stimulation with the selective agonist imetit [13] results in MAPK activation. For this, we used the H3R-transfected neuroblastoma cell line SH-SY5Y (SH-SY5Y-H3) [4]. As illustrated in Figure 1, treatment of SH-SY5Y-H3 cells with imetit (100 nM) resulted in a marked increase in the phosphorylation of p38 (panel A) and JNK (panel B). The selective H3R antagonist clobenpropit (50 nM) prevented this effect (Fig. 1A and B). Furthermore, the imetit-induced activation of MAPK was prevented by incubation with the anti-βγ peptide (Fig. 1B). This suggested that the H3R-coupled Gβγ subunit is involved in the activation of MAPK by imetit. Since these findings suggested that H3R stimulation results in MAPK activation, we next investigated whether the H3R-induced inhibition of NE exocytosis is dependent upon MAPK activation. For this, we first assessed whether MAPK inhibition modified the imetit-induced attenuation of NE exocytosis in cardiac synaptosomes. Depolarization of synaptosomes with K+ (100 mM) resulted in a ~25% increase in endogenous NE release above basal level (Fig. 2A-C). In the presence of imetit (100 nM), K+-induced NE release was inhibited by ~40–60% (Fig. 2A-C), an effect which we have previously shown to be prevented by clobenpropit [5], indicating that the inhibition of the K+-induced NE exocytosis is mediated by H3R activation. As shown in Figure 2A, the MEK/ERK inhibitor PD98059 [15] diminished the anti-exocytotic effect of imetit by ~30% at 3 μM and abolished it at 10 μM. Similarly, the p38 inhibitor SB202190 [16] diminished the effect of imetit by ~60% at 30 nM and abolished it at 100 nM (Fig. 2B), while the JNK inhibitor SP600125 [17] diminished the effect of imetit by ~30% at 150 nM and abolished it at 200 nM (Fig. 2C). In contrast, no antagonism of imetit occurred with compound SB202474, a pyridinyl imidazole analog of SB202190 that does not bind p38 [18], nor with N1-methyl-1,9-pyrazoloanthrone, an analog of SP600125 that is over 100-fold less potent than SP600125 at inhibiting JNK [17]. We found that in the presence of SB202474 (10 μM) and N1-methyl-1,9-pyrazoloanthrone (10 μM), imetit inhibited NE exocytosis by 54.0 ± 4.7 (± SEM; n = 12) and 53.6 ± 2.3 % (± SEM; n = 4; NS), respectively, as compared with a 59.8 ± 7.7 % inhibition with imetit alone (100 nM). Notably, none of the MAPK inhibitors affected basal NE release or K+-induced NE exocytosis in the absence of imetit (see legend to Fig. 2).
Figure 1. H3R activation increases the phosphorylation of p38 and JNK MAPK.
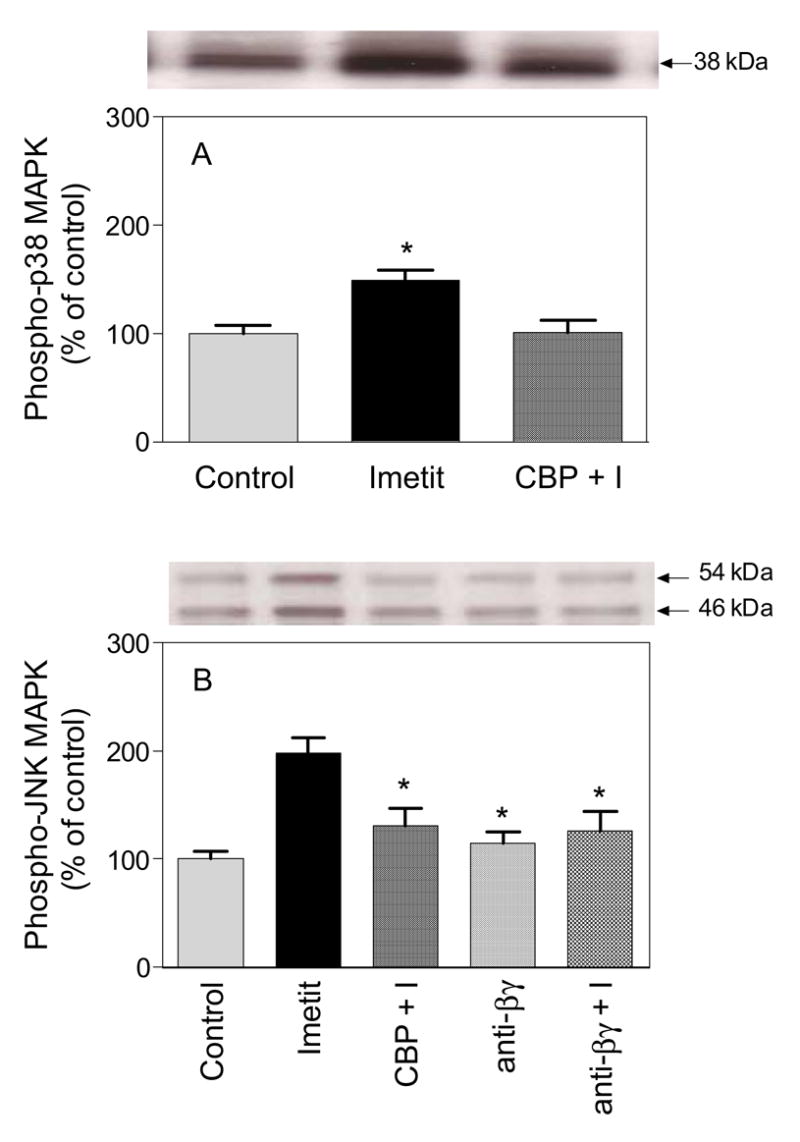
SH-SY5Y-H3 neuroblastoma cells were incubated in the absence (control) or presence of imetit (100 nM), and clobenpropit (CPB; 50 nM) + imetit (100 nM)(CBP + I) or the anti-βγ peptide (1 μM) + imetit for 10 min at 37ºC. Cells were then lysed and equal amounts of protein (15 μg/lane) were run on SDS-PAGE gel followed by Western blot analysis using antibodies against phospho-p38 MAPK (panel A) or against phospho-JNK MAPK (panel B). As expected, a single band at 38 kDa was visualized for p38 MAPK and double bands at 46 kDa and 54 kDa were detected for JNK MAPK (representing JNK1 and JNK2, respectively). Bars are means (± SEM; n = 4–5. *P<0.05 from control (by ANOVA followed by post-hoc Dunnett’s test).
Figure 2. MAPK inhibition prevents the H3R-mediated attenuation of NE exocytosis from guinea-pig heart synaptosomes.
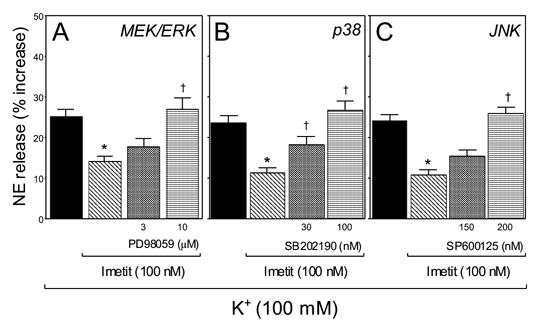
The histamine H3R agonist imetit (100 nM) attenuates the release of endogenous NE elicited by K+ depolarization (100 mM) of cardiac sympathetic nerve endings in vitro. Pretreatment of synaptosomes with the MEK/ERK inhibitor PD 98059 (3 and 10 μM; panel A), the p38 inhibitor SB 202190 (30 and 100 nM; panel B) and the JNK inhibitor SP600125 (150 and 200 nM; panel C) markedly inhibited the anti-exocytotic effect of imetit. Bars are mean increases in NE release above own basal level (± SEM; n = 8–17). * And †, significantly different from K+ alone and imetit, respectively (P<0.01 by ANOVA followed by post-hoc Dunnett’s test). Basal NE release was 1.27 ± 0.06 pmol/mg protein; it increased to 1.57 ± 0.04 pmol/mg with K+ 100 mM (n = 41). In the presence of PD 98059 (10 μM), SB202190 (100 nM) or SP600125 (200 nM), basal NE level was 1.27 ± 0.04, 1.29 ± 0.02 and 1.28 ± 0.03 pmol/mg (n = 4, 4, 4), respectively, while K+-induced NE exocytosis was 1.32 ± 0.06, 1.39 ± 0.03 and 1.37 ± 0.03 (n = 4, 4, 4), respectively.
3.2 PLA2 and cyclooxygenase activation plays a role in the H3R-induced inhibition of NE exocytosis
Since cPLA2 is a substrate for MAPK, and phosphorylation by MAPK increases the enzymatic activity of cPLA2 [10], we next assessed whether the H3R-mediated inhibition of NE exocytosis via MAPK entails the activation of cPLA2 in sympathetic nerve endings. Incubation of guinea-pig heart synaptosomes with imetit (100 nM) significantly enhanced the level of cPLA2 activity (~25%; Fig. 3A). Pretreatment with the selective H3R antagonist clobenpropit (50 nM) [5] prevented the imetit-induced increase of cPLA2 activity, indicating that this increase was dependent on H3R activation. Similar to imetit, the Ca2+-ionophore A23187 (10 μM) also enhanced cPLA2 activity (~20%), however this effect was not inhibited by clobenpropit (50 nM; Fig. 3A).
Figure 3. Stimulation of H3R enhances cPLA2 activity and PGE2 production in guinea-pig heart synaptosomes.
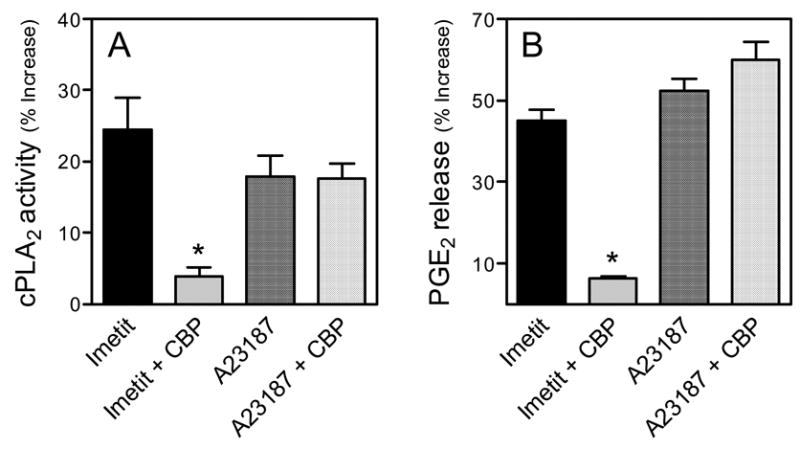
The H3R agonist imetit (100 nM) and the Ca2+ ionophore A23187 (10 μM) enhance cPLA2 activity (Panel A) and PGE2 production (Panel B) in cardiac sympathetic nerve endings. The H3R antagonist clobenpropit (CBP; 50 nM) antagonizes the effects of imetit, but not those of the ionophore. Bars are mean increases above control (± SEM; n= 3 and 4, for A and B respectively). *, Significantly different from imetit alone (P<0.01 by one-way ANOVA with Bonferroni post-hoc test). Basal cPLA2 activity was 20.48 ± 4.4 nmol/min/μg protein. Basal PGE2 level was 154.95 ± 14.64 pg/mg protein.
Given that the anti-exocytotic effect of H3R activation was associated with an increase in cPLA2 activity, we questioned whether cPLA2 activation leads to the downstream production of arachidonate metabolites capable of inhibiting NE exocytosis. For this, we determined whether cPLA2 inhibition reduced the anti-exocytotic effect of imetit in guinea-pig heart synaptosomes. Depolarization of synaptosomes with K+ (100 mM) resulted in a ~30% increase in endogenous NE release above basal level (Fig. 4A-D). In the presence of imetit (100 nM), K+-induced NE release was inhibited by ~40–60% (Fig. 4A-D), an effect due to H3R activation since, as mentioned above for Figure 2, it was prevented by clobenpropit. As shown in Figure 4A, the PLA2 inhibitor MAFP (10 μM) prevented the anti-exocytotic effect of imetit in cardiac synaptosomes. Similarly, the cyclooxygenase inhibitor indomethacin (10 μM) also prevented the effect of imetit (Fig. 4B). These results suggested that a cyclooxygenase product of the arachidonic acid cascade may mediate the anti-exocytotic effect of imetit. Notably, neither the PLA2 inhibitor MAFP nor the cyclooxygenase inhibitor indomethacin affected basal NE release or K+-induced NE exocytosis in the absence of imetit (see legend to Fig. 4).
Figure 4. Inhibition of PLA2 and cyclooxygenase, and blockade of EP3R prevent the H3R-mediated attenuation of NE exocytosis in guinea-pig heart synaptosomes.
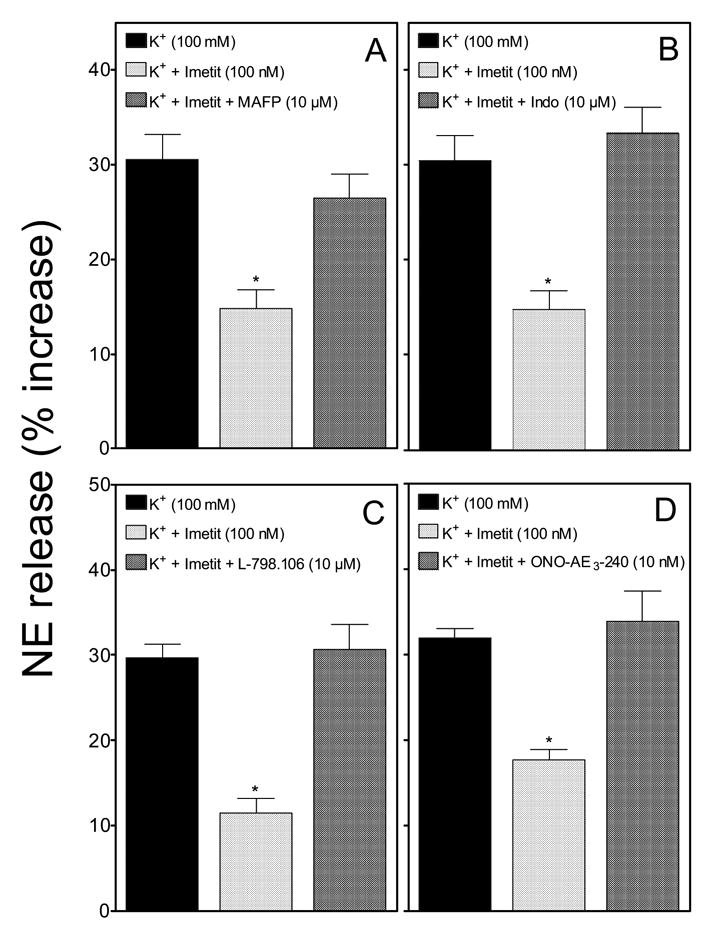
The H3R agonist imetit (100 nM) attenuates the release of endogenous NE from K+-depolarized (100 mM) sympathetic nerve endings. Incubation of synaptosomes with the PLA2 inhibitor MAFP (10 μM; panel A), the cyclooxygenase inhibitor indomethacin (10 μM; panel B), and the EP3R antagonists L-798,106 (10 μM; panel C) and ONO-AE3 -240 (10 nM; panel D), prevented the anti-exocytotic effect of imetit. Synaptosomes treated with MAFP or indomethacin were isolated from hearts previously perfused with these agents. Bars are mean increases in NE release above own basal level (± SEM; n = 12). *, Significantly different from K+ alone (p<0.01 by ANOVA with post-hoc Dunnett’s test). Basal NE release was 1.27 ± 0.04 pmol/mg protein (n = 44); it increased to 1.66 ± 0.04 pmol/mg with K+ 100 mM (n = 36). In the presence of MAFP (10 μM) or indomethacin (10 μM), basal NE release was 1.24 ± 0.04 and 1.24 ± 0.05 pmol/mg (n = 12 and 12), respectively, while K+-induced NE exocytosis was 1.5 ± 0.07 and 1.43 ± 0.07 pmol/mg (n = 12 and 12), respectively. In the presence of the EP3R antagonists L-798,106 (10 μM) or ONO-AE3-240 (10 nM), basal NE level was 1.27 ± 0.05 and 1.47 ± 0.03 pmol/mg (n = 12 and 12), respectively, while K+-induced NE exocytosis was 1.44 ± 0.05 and 1.7 ± 0.04 pmol/mg (n = 12 and 12), respectively..
3.3 Increased PGE2 production is involved in the H3R-induced inhibition of NE exocytosis: role of EP3R at subcellular and whole organ levels
Among the various cyclooxygenase products of the arachidonate cascade, PGE2 is known to inhibit NE exocytosis [12]. Accordingly, we determined whether H3R activation leads to increased PGE2 production. As shown in Figure 3B, incubation of guinea-pig heart synaptosomes with imetit (100 nM) or the Ca2+-ionophore A23187 (10 μM) was associated with a ~50% increase in PGE2 production. The H3R antagonist clobenpropit (50 nM) again prevented the effect of imetit but not that of the ionophore (Fig. 3B). This suggested that the H3R-induced attenuation of NE exocytosis associated with PLA2 activation might be due to the downstream generation of PGE2.
We next investigated whether PGE2 may be signaling via EP3R, which are known to mediate the PGE2-induced inhibition of NE exocytosis [11;12]. To this end, we used two selective EP3R antagonists L-798,106 [19] and ONO-AE3-240 [20]. As shown in Figure 4C and D, both EP3R antagonists (L-798,106 at 10 μM and ONO-AE3-240 at 10 nM) abolished the imetit-induced attenuation of NE exocytosis in cardiac synaptosomes. The EP3R antagonists did not affect basal NE release or K+-induced NE exocytosis in the absence of imetit (see legend to Fig. 4).
Our findings in cardiac synaptosomes and cultured cells suggested that stimulation of H3R results in MAPK and cPLA2 activation, leading to PGE2 formation, activation of EP3R on sympathetic terminals and inhibition of NE exocytosis. We next extended this research to the whole heart to verify that this H3R pathway is operational at the intact organ level. Hearts were isolated from guinea pigs, perfused in a Langendorff apparatus and subjected to electrical field stimulation to elicit NE release from sympathetic nerves. As shown in Figure 5A, activation of H3R with imetit caused a concentration-dependent decline in NE overflow into the coronary effluent. In the 30 nM-3 μM concentration range, imetit inhibited NE exocytosis by ~15–35%. Figure 5B shows that the anti-exocytotic effect of imetit (300 nM) was prevented not only by H3R blockade with clobenpropit (50 nM), but also by PLA2 blockade with MAFP (10 μM) and by EP3R blockade with ONO-AE3-240 (10 nM). This suggests that in the intact organ, as well as in synaptosomes, inhibition of NE exocytosis by H3R activation involves the PLA2-dependent generation of an endogenous EP3R agonist (e.g., PGE2) capable of inhibiting NE release.
Figure 5. Inhibition of PLA2 and blockade of EP3R prevent the H3R-mediated attenuation of NE exocytosis in guinea-pig hearts ex vivo.
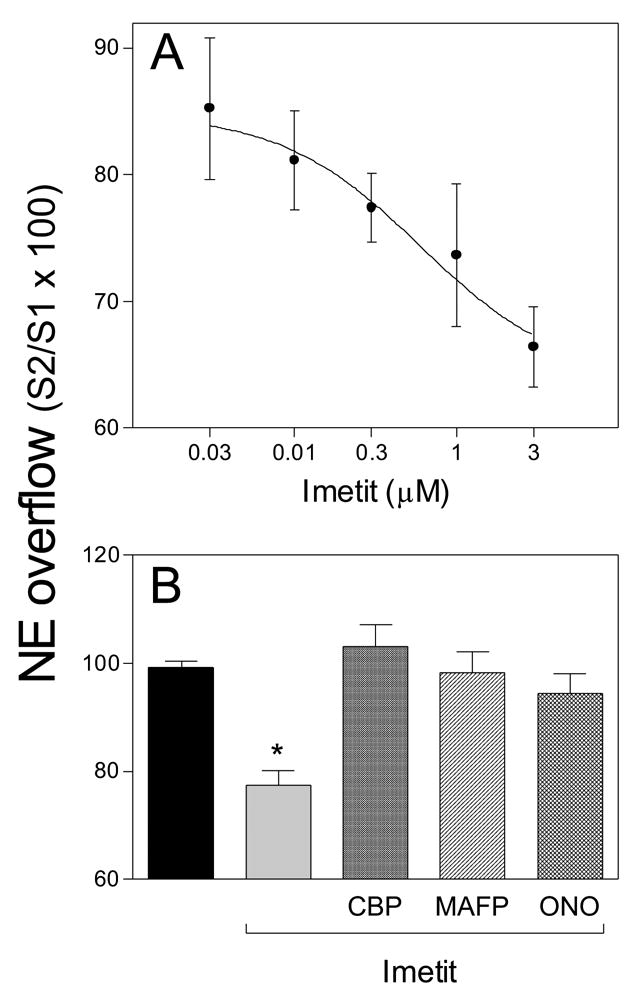
NE was released into the coronary effluent by transmural electrical field stimulation of sympathetic nerve terminals. Panel A: concentration-response curve for the anti-exocytotic effect of imetit (IC50 ~300 nM). Points are mean NE overflow values (i.e., NE exocytosis) expressed as the ratio between the second and first stimulation (S2/S1 × 100; ± SEM; n = 5–7). Panel B: the anti-exocytotic effect of imetit (300 nM) is prevented by perfusion of the hearts with the H3R antagonist clobenpropit (CBP; 50 nM), the PLA2 inhibitor MAFP (10 μM) and the EP3R antagonist ONO-AE3 -240 (10 nM). Bars are mean NE overflow values (± SEM; n = 5–7). *, Significantly different from transmural electrical field stimulation in the absence of drugs (control; p<0.01 by ANOVA with post-hoc Dunnett’s test).
To further strengthen the case for downstream EP3R involvement in the H3R-mediated anti-exocytotic effect of imetit, we next assessed whether the EP3R agonist sulprostone would act synergistically with imetit in attenuating NE exocytosis. Shown in Figure 6A and B are the concentration-response curves for the anti-exocytotic effects of imetit and sulprostone, respectively. The inhibition of K+-induced NE release ranged between ~5 and ~60% with imetit 3–100 nM (Fig. 6A) and between ~3 and ~50% with sulprostone 10–300 nM (Fig. 6B). When imetit and sulprostone were combined, each at the lowest concentration tested (i.e., 3 and 10 nM, causing a ~10 and ~5% inhibition of NE release, respectively), the inhibition of NE release increased to ~45%; i.e., an inhibition 3-fold greater than the arithmetic sum of the two single inhibitions (Fig. 6C). The synergism between imetit and sulprostone provided further support for the notion that a downstream EP3R agonist such as PGE2 is involved in the H3R-mediated anti-exocytotic effect of imetit. As expected, neither PLA2 inhibition with MAFP, nor cyclooxygenase inhibition with indomethacin, significantly modified the sulprostone-induced attenuation of NE exocytosis [Absolute values for NE release (not shown in Fig. 6 or its legend) were (n = 4): Basal, 1.42 ± 0.05 pmol/mg; K+ 100 mM, 1.71 ± 0.03; K+ + sulprostone 300 nM, 1.38 ± 0.05; K+ + MAFP 10 μM + sulprostone, 1.5 ± 0.06; K+ + indomethacin 10 μM + sulprostone, 1.5 ± 0.07].
Figure 6. H3R and EP3R agonists act synergistically to attenuate NE exocytosis from guinea-pig heart synaptosomes.
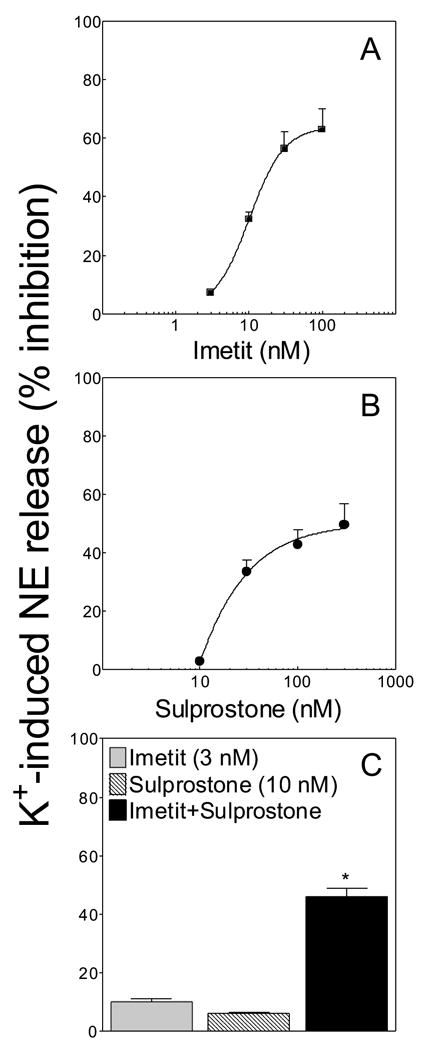
Panels A and B: concentration-response curves for the anti-exocytotic effect of imetit and sulprostone, respectively. Panel C, inhibition of NE exocytosis from cardiac synaptosomes by subthreshold concentrations of imetit (3 nM) and sulprostone (10 nM) administered either alone or in combination; note that a significant attenuation of NE release occurs when imetit is combined with sulprostone (*, Significantly different from the sum of imetit + sulprostone, P<0.001 by unpaired t test). Points and bars represent mean percent inhibition of K+-induced NE exocytosis (± SEM; n = 8–16). Basal NE release was 1.36 ± 0.05 pmol/mg protein (n = 36). In the presence of imetit (100 nM) NE release was 1.32 ± 0.08 pmol/mg protein (n = 12), while in the presence of sulprostone (300 nM) NE release was 1.39 ± 0.05 pmol/mg protein (n = 8).
3.4 Mechanism(s) of PGE2-mediated anti-exocytotic effect of H3R activation
We next questioned whether the PGE2-mediated anti-exocytotic effect of H3R activation may result from an inhibition of Ca2+ influx into sympathetic nerve terminals. Since the synergism between the anti-exocytotic effects of imetit and sulprostone suggested a similar mechanism of action, we compared the effects of imetit and sulprostone, each in combination with the selective N-type and L-type Ca2+-channel inhibitors, ω-conotoxin GVIA and nifedipine, respectively. As shown in Figure 7A and B, when a subthreshold anti-exocytotic concentration of imetit (3 nM) or sulprostone (10 nM) was used in combination with a subthreshold concentration of ω-conotoxin (0.1 nM) or nifedipine (0.1 μM), a marked synergistic anti-exocytotic effect was observed. ω-CTX (0.1 nM) and nifedipine (0.1 μM) did not significantly affect basal NE release (see legend to Fig. 7). These findings suggest that a decrease in Ca2+ influx through both N- and L-type Ca2+-channels is likely to be involved in the PGE2/EP3R-mediated attenuation of NE exocytosis elicited by H3R activation.
Figure 7. N- and L-type Ca2+-channel antagonists act synergistically with H3R and EP3R agonists to attenuate NE exocytosis in guinea-pig heart synaptosomes.
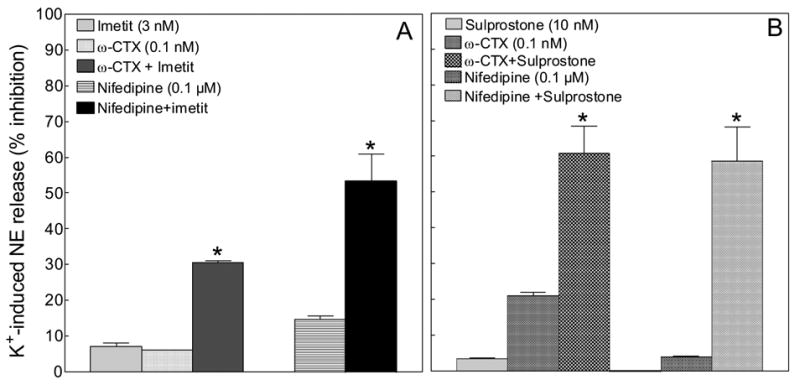
Panel A, inhibition of NE exocytosis from cardiac synaptosomes by subthreshold concentrations of imetit (3 nM), ω-conotoxin GVIA (ω-CTX; 0.1 nM) and nifedipine (0.1 μM) administered either alone or in combination. Panel B, inhibition of NE exocytosis from cardiac synaptosomes by subthreshold concentrations of sulprostone (10 nM), ω-CTX (0.1 nM) and nifedipine (0.1 μM) administered either alone or in combination. Note that a significant attenuation of NE release occurs when imetit or sulprostone is combined with ω-CTX or nifedipine (*, significantly different from the sum of imetit + ω-CTX, imetit + nifedipine, sulprostone + ω-CTX, sulprostone + nifedipine; P<0.01 by unpaired t test). Bars represent mean percent inhibition of K+-induced NE exocytosis (± SEM; n = 12). Basal NE release was 1.23 ± 0.05 pmol/mg protein (n = 24). In the presence of ω-CTX (0.1 nM) or nifedipine (0.1 μM) NE release was 1.32 ± 0.09 and 1.18 ± 0.04 pmol/mg protein, respectively (n = 6 and 8).
EP3R and H3R are coupled to a Gi protein [12;21]. The Gβγi subunit is known to inhibit Ca2+ entry [22]; thus, the Gβγi subunit could play a role in the H3R-mediated attenuation of NE exocytosis. To test this hypothesis we used a phosducin-like membrane-permeable Gβγ-blocking peptide [23]. As shown in Figure 8, the H3R agonist imetit (100 nM) and the EP3R agonist sulprostone (300 nM) each caused a ~60% attenuation of K+-induced NE exocytosis from guinea-pig heart synaptosomes. The inhibitory effects of imetit and sulprostone were prevented by the anti-βγ peptide (1 μM) (Fig. 8). Notably, the anti-βγ peptide (1 μM) had no effect on basal NE release (see legend to Fig. 8). Thus, Ca2+-channel blockade via the βγ-subunit of the Gi-protein may mediate the H3R- and EP3R-induced inhibition of NE exocytosis in cardiac sympathetic nerve terminals.
Figure 8. A membrane-permeable Gβγ-blocking peptide inhibits the H3R- and EP3R-induced attenuation of NE release from guinea-pig heart synaptosomes.
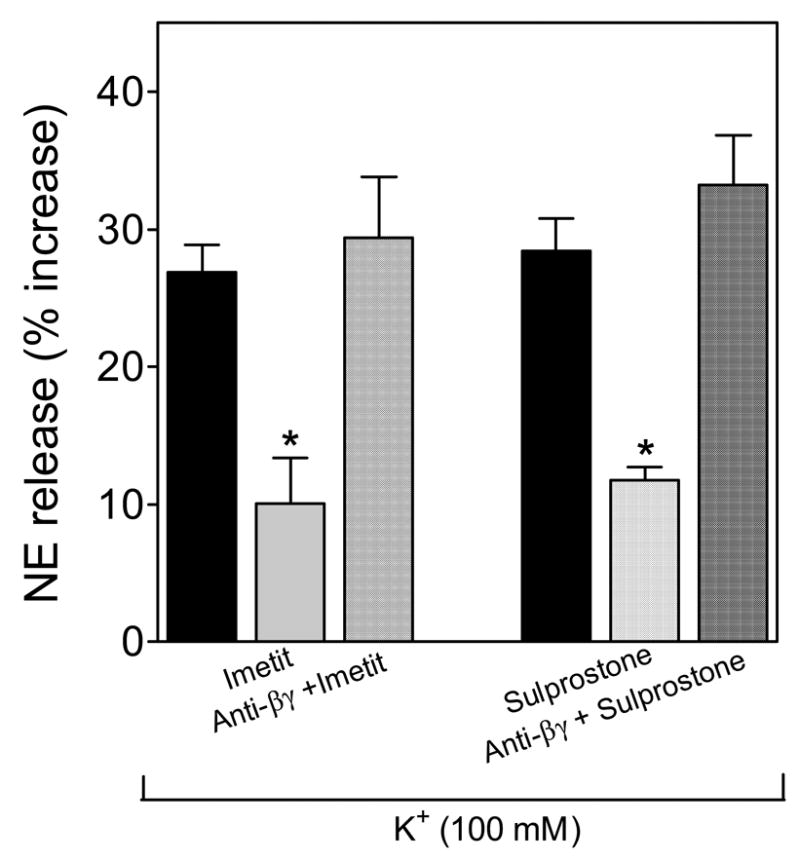
The EP3R and H3R agonists sulprostone (300 nM) and imetit (100 nM) each attenuates the release of endogenous NE from K+-depolarized (100 mM) sympathetic nerve endings. Pretreatment of isolated synaptosomes with the phosducin-like anti-Gβγ peptide (1 μM) prevents the anti-exocytotic effect of each imetit and sulprostone. Bars are mean increases in NE release above own basal level (± SEM; n = 8). *, Significantly different from K+ alone (p<0.01 by ANOVA with post-hoc Dunnett’s test). Basal NE release was 1.73 ± 0.05 pmol/mg protein (n = 8). In the presence of the anti-Gβγ peptide (1 μM) NE release was 1.84 ± 0.08 pmol/mg protein (n = 4).
4. Discussion
In this study, we report on a new signaling pathway involved in the H3R-mediated inhibition of NE exocytosis from cardiac sympathetic nerve terminals. This pathway entails the activation of MAPK and subsequent phosphorylation of cPLA2. The downstream formation of arachidonic acid metabolites inhibits NE exocytosis, most likely by decreasing Ca2+ entry into sympathetic nerve terminals.
Two observations had attracted our attention. Firstly, the finding that H3R are positively coupled to the MAPK cascade in COS-7 cells [9], and secondly, that H3R activation results in PLA2 activation [24]. Given that H3R are Gi-coupled [12;21], and that the βγi subunit is responsible for the activation of MAPK by Gi-coupled receptors [25], we assumed that H3R would activate MAPK via βγi. Indeed, we found that imetit activates MAPK in SH-SY5Y-H3 cells, an optimal model of sympathetic nerve endings and that the anti-βγ peptide prevents this effect of imetit (see Fig. 1). Whether H3R-coupled Gαi contributes to MAPK activation [26] remains to be tested at this point. Since MAPK activation contributes to PLA2 phosphorylation and stimulation of its catalytic activity [10], we hypothesized that PLA2 activation initiates the arachidonate cascade, leading to the downstream formation of PGE2, which is in large part responsible for the H3R-induced attenuation of NE exocytosis. Our findings are consistent with this hypothesis.
Individual pharmacological inhibition of each MAPK pathway (i.e., MEK/ERK, p38 and JNK) significantly attenuated the anti-exocytotic effect of imetit. Therefore, all three MAPK pathways are likely to be involved in the H3R-mediated attenuation of NE exocytosis. Whether these pathways complement and/or supplement each other in contributing to the H3R-induced anti-exocytotic effect is uncertain at this time. Nonetheless, our findings suggest that the H3R-mediated MAPK activation results in PLA2 phosphorylation and formation of arachidonate metabolites that play a major role in the attenuation of NE exocytosis. Indeed, we found that activation of H3R in cardiac sympathetic nerve terminals is associated with an increase in PLA2 activity and PGE2 formation, and that these downstream events are selectively prevented by H3R blockade. Moreover, the H3R-induced attenuation of NE exocytosis in cardiac sympathetic nerve endings was abolished by PLA2 or cyclooxygenase inhibition, as well as by blockade of EP3R. This suggests that PGE2 is the cyclooxygenase metabolite of the arachidonate cascade involved in the anti-exocytotic effect elicited by the H3R-initiated activation of the MAPK-PLA2 pathway.
This conclusion is further supported by our findings in the whole heart ex vivo. In these experiments, the transmural stimulation of sympathetic nerves elicited NE exocytosis, which was attenuated by activation of H3R. The H3R-mediated attenuation of NE exocytosis was prevented by PLA2 inhibition and by EP3R blockade. These results demonstrate that the H3R-initiated activation of the MAPK-PLA2 pathway also functions at the organ level and is not purely characteristic of isolated sympathetic nerve terminals. Interestingly, at concentrations of 100 and 300 nM, imetit was an equieffective inhibitor of NE exocytosis in synaptosomes and isolated heart, respectively. These differences in imetit concentration probably reflect technical differences between the two preparations and the different methods used to elicit NE exocytosis. In any event, the antiexocytotic effect of imetit was prevented in the isolated heart by the same concentrations of CBP, MAFP and ONO-AE3 -240 found to be effective in the synaptosomes.
Additionally, we discovered that imetit and the EP3R agonist sulprostone act synergistically to attenuate NE exocytosis elicited by K+ depolarization of sympathetic nerve terminals. This suggests that imetit and sulprostone share a common mechanism of action and favors the hypothesis that a downstream EP3R agonist such as PGE2 is involved in the H3R-mediated anti-exocytotic effect of imetit. Furthermore, we found that both imetit and sulprostone acted synergistically with the Ca2+-channel blockers ω-conotoxin GVIA and nifedipine in inhibiting NE exocytosis. This suggests that a decrease in Ca2+ influx through both N- and L-type Ca2+-channels is likely to be involved in the PGE2/EP3R-mediated attenuation of NE exocytosis elicited by H3R activation.
Although the N-type Ca2+-channels are the dominant Ca2+ entry pathway triggering sympathetic transmitter release [27;28], it is possible that L-type Ca2+-channels may also participate in NE exocytosis and be inhibited by H3R and EP3R activation [5]. H3R and EP3R are both coupled to a Gi protein [12;21]. The Gβγi subunit is known to inhibit Ca2+ entry via N-type Ca2+-channels [22;29]. Thus, we hypothesized that signaling via the Gβγi subunit could represent a common mechanism of action in the attenuation of NE exocytosis by H3R and EP3R. Indeed, we found that the membrane-permeable Gβγ-blocking peptide [23], which prevented the imetit-induced MAPK activation in SH-SY5Y-H3 cells, significantly attenuated the inhibition of K+-induced NE exocytosis elicited by imetit and sulprostone in cardiac sympathetic nerve endings. This suggests that neuronal Ca2+-channel blockade via the Gβγi-subunit plays a role in the H3R- and EP3R-mediated inhibition of NE exocytosis and that MAPK is an important upstream step in this pathway.
While it is well established that the Gβγ dimer inhibits N-type Ca2+-channel activity [29], Gβγ may also inhibit presynaptic L-type Ca2+-channels [30;31]. Further, Gβγi is known to decrease adenylyl cyclase activity [32]. Thus, it is possible that in addition to the attenuation of adenylyl cyclase by Gαi [5], Gβγi will also play a role in the H3R-mediated decrease in cAMP. A decrease in intracellular cAMP would diminish PKA activity and phosphorylation of L-type Ca2+ channels resulting in a decreased Ca2+ influx. This could explain mechanistically why nifedipine synergized with both imetit and sulprostone in their anti-exocytotic effect. In addition, the H3R-mediated attenuation of NE exocytosis may also result from an inhibition of Ca2+ influx by direct coupling of the Gβγi subunit to N-type Ca2+-channels [33–35]. At any rate, our findings suggest that the H3R Gβγi-subunit plays a pivotal role in the inhibition of NE exocytosis, most likely via the MAPK-PLA2-PGE2-EP3R pathway, but conceivably also via direct coupling to the N-type Ca2+-channels.
In conclusion, we have identified a novel signaling pathway (see Fig. 9) whereby stimulation of H3R on sympathetic nerve endings results in the intraneuronal activation of the MAPK cascade. Activated MAPK phosphorylates cPLA2 which is then translocated to the cellular membrane, with the consequent formation of arachidonic acid from membrane phospholipids, and the subsequent production of PGE2 via cyclooxygenase. PGE2 then activates EP3R on the neuronal membrane, and the Gβγi subunit of EP3R inhibits Ca2+ entry, thus attenuating NE exocytosis. This novel pathway likely functions in concert with the traditional H3R/Gαi-induced inhibition of adenylyl cyclase [5], which leads to a decreased phosphorylation of Ca2+-channels, diminished Ca2+ entry and thus, attenuation of NE exocytosis. Conceivably, the Gβγi subunit of H3R could also directly inhibit Ca2+ entry without MAPK intervention. Potential H3R/EP3R cross-talk is likely; thus, MAPK could be activated by Gβγi and Gαi from both H3R and EP3R, and the N-type Ca2+-channel could be inhibited by Gβγi from both H3R and EP3R.
Figure 9. Proposed MAPK-PLA2-PGE2-EP3R signaling pathway involved in the H3R-mediated attenuation of NE exocytosis in cardiac sympathetic nerve terminals.
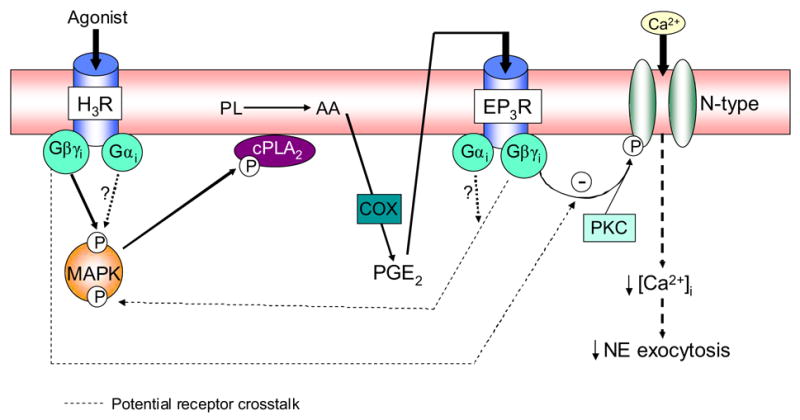
Stimulation of H3R on sympathetic nerve endings results in the Gβγi-mediated intraneuronal activation of the MAPK cascade. MAPK activation phosphorylates cPLA2 which is then translocated to the cellular membrane, with the consequent formation of arachidonic acid (AA) from membrane phospholipids (PL), and the subsequent production of PGE2 via cyclooxygenase (COX). PGE2 activates EP3R on the neuronal membrane, and the Gβγi subunit of EP3R inhibits Ca2+ entry, thus attenuating NE exocytosis. The Gβγi subunit of H3R may also directly inhibit Ca2+ entry without MAPK intervention.
Acknowledgments
This work was supported by NIH grants HL73400, HL34215, HL46403, HL47073 and DK60726. There are no conflicts of interest.
Abbreviations
- [Ca2+]i
intraneuronal Ca2+
- CBP
clobenpropit
- COX
cyclooxygenase
- DMSO
dimethyl sulfoxide
- EP3R
EP3-receptors
- H3R
histamine H3-receptors
- ICa
Ca2+ current
- MAFP
methyl arachidonyl fluorophosphonate
- MAPK
mitogen activated protein kinase
- NE
norepinephrine
- PGE2
prostaglandin E2
- PKA
protein kinase A
- PLA2
phospholipase A2
- VOCC
voltage-operated Ca2+-channels
- ω-CTX
ω-conotoxin GVIA
Footnotes
Conflicts of interest: none
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Imamura M, Poli E, Omoniyi AT, Levi R. Unmasking of activated histamine H3-receptors in myocardial ischemia: their role as regulators of exocytotic norepinephrine release. J Pharmacol Exp Ther. 1994;271:1259–1266. [PubMed] [Google Scholar]
- 2.Seyedi N, Win T, Lander HM, Levi R. Bradykinin B2-receptor activation augments norepinephrine exocytosis from cardiac sympathetic nerve endings. Mediation by autocrine/paracrine mechanisms . Circ Res. 1997;81:774–784. doi: 10.1161/01.res.81.5.774. [DOI] [PubMed] [Google Scholar]
- 3.Imamura M, Seyedi N, Lander HM, Levi R. Functional identification of histamine H3-receptors in the human heart. Circ Res. 1995;77:206–210. doi: 10.1161/01.res.77.1.206. [DOI] [PubMed] [Google Scholar]
- 4.Silver RB, Poonwasi KS, Seyedi N, Wilson SJ, Lovenberg TW, Levi R. Decreased intracellular calcium mediates the histamine H3-receptor-induced attenuation of norepinephrine exocytosis from cardiac sympathetic nerve endings. Proc Natl Acad Sci USA. 2002;99:501–506. doi: 10.1073/pnas.012506099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Seyedi N, Mackins CJ, Machida T, Reid AC, Silver RB, Levi R. Histamine H3-receptor-induced attenuation of norepinephrine exocytosis: A decreased protein kinase A activity mediates a reduction in intracellular calcium. J Pharmacol Exp Ther. 2005;312:1–9. doi: 10.1124/jpet.104.072504. [DOI] [PubMed] [Google Scholar]
- 6.Felder CC, Williams HL, Axelrod J. A transduction pathway associated with receptors coupled to the inhibitory guanine nucleotide binding protein Gi that amplifies ATP-mediated arachidonic acid release. Proc Natl Acad Sci U S A. 1991;88:6477–6480. doi: 10.1073/pnas.88.15.6477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hunt TW, Carroll RC, Peralta EG. Heterotrimeric G proteins containing Gai3 regulate multiple effector enzymes in the same cell. Activation of phospholipases C and A2 and inhibition of adenylyl cyclase. J Biol Chem. 1994;269:29565–29570. [PubMed] [Google Scholar]
- 8.Burke JR, Davern LB, Gregor KR, Todderud G, Alford JG, Tramposch KM. Phosphorylation and calcium influx are not sufficient for the activation of cytosolic phospholipase A2 in U937 cells: requirement for a Gi alpha-type G-protein. Biochim Biophys Acta. 1997;1341:223–237. doi: 10.1016/s0167-4838(97)00085-x. [DOI] [PubMed] [Google Scholar]
- 9.Drutel G, et al. Identification of rat H3 receptor isoforms with different brain expression and signaling properties. Mol Pharmacol. 2001;59:1–8. [PubMed] [Google Scholar]
- 10.Lin LL, Wartmann M, Lin AY, Knopf JL, Seth A, Davis RJ. cPLA2 is phosphorylated and activated by MAP kinase. Cell. 1993;72:269–278. doi: 10.1016/0092-8674(93)90666-e. [DOI] [PubMed] [Google Scholar]
- 11.Molderings GJ, Likungu J, Göthert M. Modulation of noradrenaline release from the sympathetic nerves of human right atrial appendages by presynaptic EP3- and DP-receptors. Naunyn Schmiedebergs Arch Pharmacol. 1998;358:440–444. doi: 10.1007/pl00005276. [DOI] [PubMed] [Google Scholar]
- 12.Boehm S, Kubista H. Fine tuning of sympathetic transmitter release via ionotropic and metabotropic presynaptic receptors. Pharmacol Rev. 2002;54:43–99. doi: 10.1124/pr.54.1.43. [DOI] [PubMed] [Google Scholar]
- 13.Garbarg M, et al. S-[2-(4-imidazolyl)ethyl]isothiourea, a highly specific and potent histamine H3 receptor agonist. J Pharmacol Exp Ther. 1992;263:304–310. [PubMed] [Google Scholar]
- 14.Van der Goot H, Schepers MJP, Sterk GJ, Timmerman H. Isothiourea analogues of histamine as potent agonists or antagonists of the histamine H3-receptor. Eur J Med Chem. 1992;27:511–517. [Google Scholar]
- 15.Alessi DR, Cuenda A, Cohen P, Dudley DT, Saltiel AR. PD 098059 is a specific inhibitor of the activation of mitogen-activated protein kinase kinase in vitro and in vivo. J Biol Chem. 1995;270:27489–27494. doi: 10.1074/jbc.270.46.27489. [DOI] [PubMed] [Google Scholar]
- 16.Wilson KP, et al. The structural basis for the specificity of pyridinylimidazole inhibitors of p38 MAP kinase. Chem Biol. 1997;4:423–431. doi: 10.1016/s1074-5521(97)90194-0. [DOI] [PubMed] [Google Scholar]
- 17.Bennett BL, et al. SP600125, an anthrapyrazolone inhibitor of Jun N-terminal kinase. Proc Natl Acad Sci U S A. 2001;98:13681–13686. doi: 10.1073/pnas.251194298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fabian MA, et al. A small molecule-kinase interaction map for clinical kinase inhibitors. Nat Biotechnol. 2005;23:329–336. doi: 10.1038/nbt1068. [DOI] [PubMed] [Google Scholar]
- 19.Clarke DL, Giembycz MA, Patel HJ, Belvisi MG. E-Ring 8-isoprostanes inhibit ACh release from parasympathetic nerves innervating guinea-pig trachea through agonism of prostanoid receptors of the EP3-subtype. Br J Pharmacol. 2004;141:600–609. doi: 10.1038/sj.bjp.0705648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Amano H, et al. Host Prostaglandin E2-EP3 Signaling Regulates Tumor-Associated Angiogenesis and Tumor Growth. The Journal of Experimental Medicine. 2003;197:221–232. doi: 10.1084/jem.20021408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hill SJ, et al. International Union of Pharmacology. XIII. Classification of histamine receptors. Pharmacol Rev. 1997;49:253–278. [PubMed] [Google Scholar]
- 22.Borgland SL, Connor M, Ryan RM, Ball HJ, Christie MJ. Prostaglandin E2 inhibits calcium current in two sub-populations of acutely isolated mouse trigeminal sensory neurons. J Physiol. 2002;539:433–444. doi: 10.1113/jphysiol.2001.013322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chang M, Zhang L, Tam JP, Sanders-Bush E. Dissecting G Protein-coupled Receptor Signaling Pathways with Membrane-permeable Blocking Peptides. J Biol Chem. 2000;275:7021–7029. doi: 10.1074/jbc.275.10.7021. [DOI] [PubMed] [Google Scholar]
- 24.Morisset S, et al. High constitutive activity of native H3 receptors regulates histamine neurons in brain. Nature. 2000;408:860–864. doi: 10.1038/35048583. [DOI] [PubMed] [Google Scholar]
- 25.Coso OA, Teramoto H, Simonds WF, Gutkind JS. Signaling from G protein-coupled receptors to c-Jun kinase involves beta gamma subunits of heterotrimeric G proteins acting on a Ras and Rac1-dependent pathway. J Biol Chem. 1996;271:3963–3966. doi: 10.1074/jbc.271.8.3963. [DOI] [PubMed] [Google Scholar]
- 26.Mochizuki N, et al. Activation of the ERK/MAPK pathway by an isoform of rap1GAP associated with G alpha(i) Nature. 1999;400:891–894. doi: 10.1038/23738. [DOI] [PubMed] [Google Scholar]
- 27.Lipscombe D, Kongsamut S, Tsien RW. Alpha-adrenergic inhibition of sympathetic neurotransmitter release mediated by modulation of N-type calcium-channel gating. Nature. 1989;340:639–642. doi: 10.1038/340639a0. [DOI] [PubMed] [Google Scholar]
- 28.Zhu Y, Yakel JL. Modulation of Ca2+ currents by various G protein-coupled receptors in sympathetic neurons of male rat pelvic ganglia. J Neurophysiol. 1997;78:780–789. doi: 10.1152/jn.1997.78.2.780. [DOI] [PubMed] [Google Scholar]
- 29.Dolphin AC. G protein modulation of voltage-gated calcium channels. Pharmacol Rev. 2003;55:607–627. doi: 10.1124/pr.55.4.3. [DOI] [PubMed] [Google Scholar]
- 30.Callaghan B, Koh SD, Keef KD. Muscarinic M2 receptor stimulation of Cav1.2b requires phosphatidylinositol 3-kinase, protein kinase C, and c-Src. Circ Res. 2004;94:626–633. doi: 10.1161/01.RES.0000118248.17466.B7. [DOI] [PubMed] [Google Scholar]
- 31.Ivanina T, Blumenstein Y, Shistik E, Barzilai R, Dascal N. Modulation of L-type Ca2+ channels by Gbeta gamma and calmodulin via interactions with N and C termini of alpha 1C. J Biol Chem. 2000;275:39846–39854. doi: 10.1074/jbc.M005881200. [DOI] [PubMed] [Google Scholar]
- 32.Taussig R, Iniguez-Lluhi JA, Gilman AG. Inhibition of adenylyl cyclase by Gi alpha. Science. 1993;261:218–221. doi: 10.1126/science.8327893. [DOI] [PubMed] [Google Scholar]
- 33.Herlitze S, Garcia DE, Mackie K, Hille B, Scheuer T, Catterall WA. Modulation of Ca2+ channels by G-protein beta gamma subunits. Nature. 1996;380:258–262. doi: 10.1038/380258a0. [DOI] [PubMed] [Google Scholar]
- 34.Ikeda SR. Voltage-dependent modulation of N-type calcium channels by G-protein beta gamma subunits. Nature. 1996;380:255–258. doi: 10.1038/380255a0. [DOI] [PubMed] [Google Scholar]
- 35.Catterall WA. Structure and regulation of voltage-gated Ca2+ channels. Annu Rev Cell Dev Biol. 2000;16:521–555. doi: 10.1146/annurev.cellbio.16.1.521. [DOI] [PubMed] [Google Scholar]