

Abstract
The proportion of human genetic variation due to differences between populations is modest, and individuals from different populations can be genetically more similar than individuals from the same population. Yet sufficient genetic data can permit accurate classification of individuals into populations. Both findings can be obtained from the same data set, using the same number of polymorphic loci. This article explains why. Our analysis focuses on the frequency, ω, with which a pair of random individuals from two different populations is genetically more similar than a pair of individuals randomly selected from any single population. We compare ω to the error rates of several classification methods, using data sets that vary in number of loci, average allele frequency, populations sampled, and polymorphism ascertainment strategy. We demonstrate that classification methods achieve higher discriminatory power than ω because of their use of aggregate properties of populations. The number of loci analyzed is the most critical variable: with 100 polymorphisms, accurate classification is possible, but ω remains sizable, even when using populations as distinct as sub-Saharan Africans and Europeans. Phenotypes controlled by a dozen or fewer loci can therefore be expected to show substantial overlap between human populations. This provides empirical justification for caution when using population labels in biomedical settings, with broad implications for personalized medicine, pharmacogenetics, and the meaning of race.
DISCUSSIONS of genetic differences between major human populations have long been dominated by two facts: (a) Such differences account for only a small fraction of variance in allele frequencies, but nonetheless (b) multilocus statistics assign most individuals to the correct population. This is widely understood to reflect the increased discriminatory power of multilocus statistics. Yet Bamshad et al. (2004) showed, using multilocus statistics and nearly 400 polymorphic loci, that (c) pairs of individuals from different populations are often more similar than pairs from the same population. If multilocus statistics are so powerful, then how are we to understand this finding?
All three of the claims listed above appear in disputes over the significance of human population variation and “race.” In particular, the American Anthropological Association (1997, p. 1) stated that “data also show that any two individuals within a particular population are as different genetically as any two people selected from any two populations in the world” (subsequently amended to “about as different”). Similarly, educational material distributed by the Human Genome Project (2001, p. 812) states that “two random individuals from any one group are almost as different [genetically] as any two random individuals from the entire world.” Previously, one might have judged these statements to be essentially correct for single-locus characters, but not for multilocus ones. However, the finding of Bamshad et al. (2004) suggests that an empirical investigation of these claims is warranted.
In what follows, we use several collections of loci genotyped in various human populations to examine the relationship between claims a, b, and c above. These data sets vary in the numbers of polymorphic loci genotyped, population sampling strategies, polymorphism ascertainment methods, and average allele frequencies. To assess claim c, we define ω as the frequency with which a pair of individuals from different populations is genetically more similar than a pair from the same population. We show that claim c, the observation of high ω, holds with small collections of loci. It holds even with hundreds of loci, especially if the populations sampled have not been isolated from each other for long. It breaks down, however, with data sets comprising thousands of loci genotyped in geographically distinct populations: In such cases, ω becomes zero. Classification methods similarly yield high error rates with few loci and almost no errors with thousands of loci. Unlike ω, however, classification statistics make use of aggregate properties of populations, so they can approach 100% accuracy with as few as 100 loci.
MATERIALS AND METHODS
Data sets:
Three data sets were used. Loci or individuals with >10% missing data were not included in any data set (loci were pruned first and then individuals). The first data set (“insertions”) consists of 175 polymorphic transposable element insertion loci (100 Alu and 75 L1) previously genotyped in 259 individuals. The population sample consists of 104 individuals from sub-Saharan Africa, 54 East Asians, 61 individuals of northern European ancestry, and 40 individuals from Andhra Pradesh, India (Watkins et al. 2005; Witherspoon et al. 2006). The second data set (“microarray”) consists of 9922 biallelic single-nucleotide polymorphism (SNP) loci genotyped in 278 individuals (55 Africans, 42 African Americans, 40 Native Americans, 22 Indians, 20 East Asians, 62 Europeans, 18 Hispano–Latinos from Puerto Rico, and 19 individuals from New Guinea). This data set is derived from that of Shriver et al. (2005). The third data set (“resequenced”) is derived from the 10 ENCODE regions of the HapMap project, release 16c.1 of phase I, June 2005 (International HapMap Consortium 2005). These regions were resequenced in 48 individuals to identify SNPs without ascertainment bias in favor of loci with common polymorphisms. These SNPs were then genotyped in 209 unrelated individuals: 60 Yoruba in Ibadan, Nigeria (YRI); 60 Utah residents with ancestry from northern and western Europe (CEU, from the CEPH diversity panel); and 89 Japanese in Tokyo, Japan, plus Han Chinese in Beijing, China (CHB + JPT). Our subset consists of 14,258 SNPs. All markers in all three data sets are biallelic. The proportions of missing genotypes are 2.4, 2.1, and 0.36%, respectively.
Data subsampling:
To examine the effect of population sampling (i.e., the effects of comparing relatively isolated populations vs. more closely related or admixed ones), two subsets were constructed from each of the insertions and microarray main data sets: one consisting of the entire data set, with all its labeled populations, and another consisting of East Asian, European, and sub-Saharan African population groups only. The resequenced data set consists only of the latter three population groups.
To investigate the effect of allele frequency, these five data subsets were subdivided according to three further treatments: loci with common polymorphisms (with minor allele frequency, MAF, > 0.1); loci with rare polymorphisms (MAF < 0.1); and all polymorphic loci, regardless of frequency. Henceforth we refer to these classes of loci as rare polymorphisms, common polymorphisms, or all polymorphisms. For this classification, allele frequencies were computed across the entire sample in the parent data set. To investigate the effect of incrementally increasing the number of loci used, loci from each of these 15 data subsets were sampled (without replacement) to produce 200 independent data sets with numbers of loci varying in 21 steps on a logarithmic scale from 10 to the maximum.
Pairwise genetic distance:
We use the “shared alleles” genetic distance (Chakraborty and Jin 1993; Bowcock et al. 1994; Mountain and Cavalli-Sforza 1997), which defines the distance between two individuals at a locus as one minus half the number of alleles they share. The genetic distance between individuals is the average of their per-locus distances. Pairs of individuals are classified as “within population” or “between population” according to whether the individuals were sampled from the same or different groups of populations as defined above.
Dissimilarity fraction ω̂:
Let ω be the probability that a pair of individuals randomly chosen from different populations is genetically more similar than an independent pair chosen from any single population. We compute all possible pairwise genetic distances, classify them as within- or between-population distances (the sets dW or dB, respectively), and then calculate the frequency with which dW > dB (that is, a within-population pair is more dissimilar than a between-population pair). This fraction, 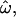 is an estimator of ω. The expected value of
is an estimator of ω. The expected value of 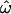 ranges from 0 to 0.5 (regardless of the number of populations). At
ranges from 0 to 0.5 (regardless of the number of populations). At 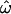 = 0, individuals are always more similar to members of their own population than to members of other populations; at
= 0, individuals are always more similar to members of their own population than to members of other populations; at  = 0.5, individuals are as likely to be more similar to members of other populations as to members of their own. The distributions of pairwise genetic distances implied here resemble the common ancestry profiles proposed by Mountain and Ramakrishnan (2005), who use a different measure of genetic distance. The shared-alleles distance used here generally yields slightly lower values of
= 0.5, individuals are as likely to be more similar to members of other populations as to members of their own. The distributions of pairwise genetic distances implied here resemble the common ancestry profiles proposed by Mountain and Ramakrishnan (2005), who use a different measure of genetic distance. The shared-alleles distance used here generally yields slightly lower values of 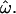
Centroid misclassification rate CC:
The centroid classification method is also based on pairwise genetic distances, with one critical difference: Every individual is compared to the centroid of each population, rather than to every other individual. The centroid is the genetic average of a population, an individual whose pseudogenotypes at each locus are the frequencies of the genotypes in that population (not including the individual being compared to the centroid). This genetic distance is equivalent to the average of the genetic distances from an individual to all other individuals in the target population. Each individual is then assigned to the population with the closest centroid, as in Cornuet et al. (1999). These assignments are compared to the known populations of origin, and the proportion of individuals misclassified is reported as CC. The expected classification error for random assignment of individuals to populations is 1 − 1/n, where n is the number of populations.
Population trait value misclassification rate CT:
Our definition of CT is implicit in the theoretical illustrations of Risch et al. (2002) and Edwards (2003). These authors used simplified models to show how modest differences between populations can nonetheless enable accurate classification. In both cases, population membership is treated as an additive quantitative genetic trait controlled by many loci of equal effect, and individuals are divided into populations on the basis of their trait values.
This method is inherently limited to dividing individuals into just two clusters using only biallelic loci, so we limit our definitions to that situation. Consider individuals sampled from two populations, A and B, and genotyped at many biallelic loci. At each locus, we identify the allele whose frequency is higher in population A and assign it a value of 0. The other allele (more frequent in B than in A) is assigned a value of 1. Let qij represent the genotype of individual i at locus j, defined as the average of the assigned values of the two alleles carried by that individual at that locus. Now define qi as the average of qij over all loci j (so qi is a polygenic quantitative genetic trait). Given these definitions, if populations A and B are typified by even slightly different allele frequencies at many loci, then qi will usually be smaller for a member of population A than for a member of population B. Thus the value of the trait qi indicates membership in one population or the other, so we call qi the “population trait” value of individual i.
Individuals are assigned to population A or B depending on whether their population trait value qi falls below or above some dividing criterion qC, respectively. In the case of just two populations, these assignments are compared to the known origins of the individuals, and the proportion misclassified is reported as CT. The classification criterion qC is chosen as follows. Let 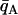 be the mean of qi taken over all individuals in population A, and define
be the mean of qi taken over all individuals in population A, and define 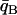 similarly for population B. If the distributions of qi for individuals from the two populations are symmetric with equal variance, then letting qC = (
similarly for population B. If the distributions of qi for individuals from the two populations are symmetric with equal variance, then letting qC = (  +
+ 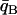 )/2 minimizes misclassification (cf. Risch et al. 2002; Edwards 2003). To better account for unequal variances, we generalize slightly and solve for a criterion qC such that r(qC) = s(qC) and
)/2 minimizes misclassification (cf. Risch et al. 2002; Edwards 2003). To better account for unequal variances, we generalize slightly and solve for a criterion qC such that r(qC) = s(qC) and 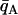 < qC <
< qC < 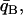 where r and s are normal probability density functions with means and variances estimated from the distributions of qi for populations A and B, respectively.
where r and s are normal probability density functions with means and variances estimated from the distributions of qi for populations A and B, respectively.
To extend this inherently pairwise approach to more than two populations, assignments for each individual are initially computed with reference to each possible pair of populations. The values (0 or 1) assigned to particular alleles, the criterion qC, and all qi are calculated anew for each pair of populations. Individuals are finally assigned to a population only if they were assigned to it in all pairwise comparisons involving that population. The proportion of individuals misclassified (or not classified, since this method can fail to classify individuals) is reported as CT. For comparison, a “single-locus” classification error rate is computed by using this method to classify individuals using each locus singly and then averaging the results over all loci.
RESULTS
Distributions of distances:
The statistics 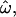 CC, and CT are closely related by design. To illustrate the relationships between them, the distributions of the genetic measures that underlie them are shown in Figure 1. For simplicity, only two populations (Europeans and sub-Saharan Africans) and 50 typical loci randomly chosen from the insertions data set are used. The distributions of pairwise genetic distances for within- and between-population pairs of individuals (Figure 1A) overlap considerably even for these geographically isolated populations. The dissimilarity fraction,
CC, and CT are closely related by design. To illustrate the relationships between them, the distributions of the genetic measures that underlie them are shown in Figure 1. For simplicity, only two populations (Europeans and sub-Saharan Africans) and 50 typical loci randomly chosen from the insertions data set are used. The distributions of pairwise genetic distances for within- and between-population pairs of individuals (Figure 1A) overlap considerably even for these geographically isolated populations. The dissimilarity fraction, 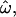 is 20%, indicating that between-population pairs are more similar than within-population pairs one-fifth of the time. In contrast, the distributions of individuals' distances to the centroids of their own or different populations (Figure 1B) show much less overlap, resulting in CC = 4.2%. The population trait value distributions for Africans and Europeans overlap for just three individuals, yielding CT = 1.8%. Classifications using model-based methods such as Structure (Pritchard et al. 2000) achieve 90% accuracy or better using the same data (Bamshad et al. 2003; Witherspoon et al. 2006).
is 20%, indicating that between-population pairs are more similar than within-population pairs one-fifth of the time. In contrast, the distributions of individuals' distances to the centroids of their own or different populations (Figure 1B) show much less overlap, resulting in CC = 4.2%. The population trait value distributions for Africans and Europeans overlap for just three individuals, yielding CT = 1.8%. Classifications using model-based methods such as Structure (Pritchard et al. 2000) achieve 90% accuracy or better using the same data (Bamshad et al. 2003; Witherspoon et al. 2006).
Figure 1.—

Frequency distributions of the underlying genetic measures used to compute 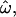 CC, and CT, for a subset of 50 loci genotyped in 104 sub-Saharan African and 61 European individuals of the insertions data set. The measures shown are (A) 13,530 pairwise genetic distances for within- and between-population pairs of individuals (in blue and red, respectively); (B) 330 genetic distances between each individual and the centroid of each population, for an individual's known population of origin (blue) or other population (red); and (C) 165 population trait values qi for individuals computed relative to the African vs. European population pair. Alleles more common in Africa than in Europe are assigned a value of 0; those more common in Europe are assigned a value of 1. The classification criterion qC is marked. The qi distributions for Africans and Europeans are green and yellow, respectively. The areas of overlap between the distributions do not correspond directly to the dissimilarity fraction or misclassification rates. Distributions that do not overlap imply that
CC, and CT, for a subset of 50 loci genotyped in 104 sub-Saharan African and 61 European individuals of the insertions data set. The measures shown are (A) 13,530 pairwise genetic distances for within- and between-population pairs of individuals (in blue and red, respectively); (B) 330 genetic distances between each individual and the centroid of each population, for an individual's known population of origin (blue) or other population (red); and (C) 165 population trait values qi for individuals computed relative to the African vs. European population pair. Alleles more common in Africa than in Europe are assigned a value of 0; those more common in Europe are assigned a value of 1. The classification criterion qC is marked. The qi distributions for Africans and Europeans are green and yellow, respectively. The areas of overlap between the distributions do not correspond directly to the dissimilarity fraction or misclassification rates. Distributions that do not overlap imply that 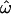 = 0 and all individuals can be correctly classified. In C, only three individuals are misclassified. Means and standard deviations are indicated above each distribution by vertical ticks and horizontal bars. The horizontal axes share the same scale. As the number of polymorphic loci used increases, the variances of these distributions decrease while their means remain roughly constant. As a result, the statistics
= 0 and all individuals can be correctly classified. In C, only three individuals are misclassified. Means and standard deviations are indicated above each distribution by vertical ticks and horizontal bars. The horizontal axes share the same scale. As the number of polymorphic loci used increases, the variances of these distributions decrease while their means remain roughly constant. As a result, the statistics 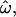 CC, and CT decrease as more loci are used.
CC, and CT decrease as more loci are used.
The variances of the distributions are much greater for the individual-to-individual comparisons (Figure 1A) than for the centroid-to-individual comparisons (Figure 1B). The distribution means are nearly identical, however, so the distributions overlap more in Figure 1A than in 1B, and thus 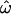 > CC. The difference in variances is due to the fact that each genetic distance to a centroid (each datum in Figure 1B) is equivalent to the average of a sizable subset of pairwise genetic distances represented in Figure 1A (see materials and methods). That averaging step eliminates considerable variation and produces the narrower distributions of Figure 1B.
> CC. The difference in variances is due to the fact that each genetic distance to a centroid (each datum in Figure 1B) is equivalent to the average of a sizable subset of pairwise genetic distances represented in Figure 1A (see materials and methods). That averaging step eliminates considerable variation and produces the narrower distributions of Figure 1B.
The simplifications introduced by Risch et al. (2002) and Edwards (2003) allow an alternative view, represented in Figure 1C. Here, each individual i is assigned a unidimensional genetic location qi (the individual's population trait value; see materials and methods). The trait distance between any two individuals x and y is now just the horizontal distance between them, |qx–qy|. This simplification is possible only in the two-population case and requires a population-specific coding of allele states, so the trait distance is not equivalent to the genetic distances represented in Figure 1, A and B. Nonetheless, it is instructive to consider the analogy using Figure 1C as a guide. For example, an African individual x with qx = 0.52 will be more similar to a European y with qy = 0.60 than to another African z with qz = 0.4. Yet that individual x will still be closer to the population mean trait value for Africans (qA ≅ 0.48, the African centroid) than to the mean value of Europeans (qB ≅ 0.68). It follows that many individuals like this one will be correctly classified (yielding low CC and CT) even though they are often more similar to individuals of the other population than to members of their own population (yielding high 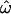 ).
).
To empirically and quantitatively understand the relationships and contrasts between 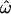 and the misclassification rates CC and CT, we examine three primary factors that influence them: the number of polymorphic loci used, the allele frequencies at those loci, and the degree of differentiation between the populations examined.
and the misclassification rates CC and CT, we examine three primary factors that influence them: the number of polymorphic loci used, the allele frequencies at those loci, and the degree of differentiation between the populations examined.
Data subset statistics:
Three data sets, labeled insertions, microarray, and resequenced, were used, and 15 subsets were constructed from these to examine the effects of different data collection strategies (see materials and methods). Table 1 lists the 15 data subsets and reports 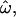 CC, and CT (each computed over all loci in each data subset) as well as the expected value of CT when only a single locus is used. Table 1 also gives values of five descriptive statistics for each data subset: the proportion of genetic variance explained by interpopulation differences (FST); the observed proportions of heterozygotes (% het); the absolute differences in allele frequencies between population pairs (averaged),
CC, and CT (each computed over all loci in each data subset) as well as the expected value of CT when only a single locus is used. Table 1 also gives values of five descriptive statistics for each data subset: the proportion of genetic variance explained by interpopulation differences (FST); the observed proportions of heterozygotes (% het); the absolute differences in allele frequencies between population pairs (averaged), 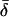 ; the fraction of polymorphisms that are rare (MAF < 0.1) in at least one population and at the same time common (MAF > 0.1) in another population (% rare and common); and the fraction of loci that are monomorphic in at least one population and common polymorphisms in another (% fixed and common). The values observed are typical of human population genetic data sets (Nei 1973; Dean et al. 1994; International HapMap Consortium 2005; Shriver et al. 2005; Witherspoon et al. 2006).
; the fraction of polymorphisms that are rare (MAF < 0.1) in at least one population and at the same time common (MAF > 0.1) in another population (% rare and common); and the fraction of loci that are monomorphic in at least one population and common polymorphisms in another (% fixed and common). The values observed are typical of human population genetic data sets (Nei 1973; Dean et al. 1994; International HapMap Consortium 2005; Shriver et al. 2005; Witherspoon et al. 2006).
TABLE 1.
Data set descriptions and summary results
| Data set | MAF classa | Populationsb | Individuals | Loci |
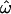 %c %c
|
CC%d | CT%e | CT%, single locusf |
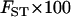 g g
|
% rare and commonh | % fixed and commoni |
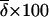 j j
|
% het.k |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Insertions | All | Distinct (3) | 219 | 175 | 10 | 0.46 | 0 | 56 | 14 | 21 | 10 | 18 | 32 |
| Insertions | Rare | Distinct (3) | 219 | 21 | 47 | 58 | 39 | 66 | 12 | 57 | 33 | 5.7 | 7.0 |
| Insertions | Common | Distinct (3) | 219 | 154 | 9.1 | 0.46 | 0 | 55 | 14 | 16 | 7.1 | 20 | 36 |
| Insertions | All | All (4) | 259 | 175 | 15 | 3.1 | 1.2 | 65 | 12 | 23 | 11 | 15 | 32 |
| Insertions | Rare | All (4) | 259 | 21 | 50 | 76 | 46 | 73 | 8.3 | 62 | 43 | 4.3 | 7.5 |
| Insertions | Common | All (4) | 259 | 154 | 13 | 3.1 | 0.77 | 64 | 12 | 18 | 6.5 | 17 | 36 |
| Microarray | All | Distinct (3) | 137 | 9,922 | 0 | 0 | 0 | 56 | 15 | 26 | 12 | 20 | 32 |
| Microarray | Rare | Distinct (3) | 137 | 920 | 17 | 7.3 | 0 | 65 | 10 | 87 | 56 | 9.5 | 10 |
| Microarray | Common | Distinct (3) | 137 | 9,002 | 0 | 0 | 0 | 55 | 15 | 19 | 7.8 | 21 | 34 |
| Microarray | All | All (8) | 278 | 9,922 | 3.1 | 10 | 1.1 | 82 | 13 | 44 | 27 | 18 | 32 |
| Microarray | Rare | All (8) | 278 | 851 | 36 | 72 | 1.4 | 84 | 8.9 | 98 | 88 | 7.9 | 11 |
| Microarray | Common | All (8) | 278 | 9,071 | 3.3 | 8.3 | 1.1 | 81 | 13 | 39 | 21 | 19 | 34 |
| Resequenced | All | All (3) | 209 | 14,233 | 8.2 | 0 | 0 | 57 | 13 | 40 | 29 | 11 | 17 |
| Resequenced | Rare | All (3) | 209 | 8,389 | 29 | 45 | 0.96 | 59 | 9.6 | 51 | 43 | 4.6 | 4.7 |
| Resequenced | Common | All (3) | 209 | 5,844 | 5.9 | 0.96 | 0 | 53 | 14 | 23 | 8.4 | 19 | 34 |
Sets of loci with MAF < 0.1 (rare), MAF > 0.1 (common), or any MAF (all).
Sets of individuals representing all populations in a data set or only the more distinct populations.
Dissimilarity fraction.
Centroid misclassification rate.
Population trait value misclassification rate.
Trait value misclassification rate based on a single locus, averaged over all loci.
The proportion of variance in allelic frequencies attributable to population differences.
Percentage of loci with MAF < 0.1 in one population and MAF > 0.1 in another.
Percentage of loci that are monomorphic in one population and polymorphic with MAF > 0.1 in another.
Average difference in allele frequency between populations, averaged across alleles, loci, and population pairs.
Percentage of individuals heterozygous at each locus, averaged across all loci.
Dependency of ω̂ on number of loci:
Figure 2 shows the dependency of 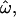 CC, and CT on the number of loci for each of the 15 data subsets listed in Table 1. As the number of included loci increases,
CC, and CT on the number of loci for each of the 15 data subsets listed in Table 1. As the number of included loci increases, 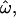 CC and CT decrease. This is the expected behavior for CC (Smouse and Chakraborty 1986; Manel et al. 2002; Campbell et al. 2003) and CT (Risch et al. 2002; Edwards 2003). However,
CC and CT decrease. This is the expected behavior for CC (Smouse and Chakraborty 1986; Manel et al. 2002; Campbell et al. 2003) and CT (Risch et al. 2002; Edwards 2003). However, 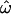 does not decrease nearly as rapidly. Figure 2A shows the results for a diverse sample of individuals genotyped at 175 insertion loci, a number that is typical of many studies of human genetic diversity published during the last decade. The downward trend in
does not decrease nearly as rapidly. Figure 2A shows the results for a diverse sample of individuals genotyped at 175 insertion loci, a number that is typical of many studies of human genetic diversity published during the last decade. The downward trend in 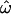 is apparent, but even with the full data set it remains at 15% (with all four population groups; Table 1). Across all data sets and using <100 polymorphisms,
is apparent, but even with the full data set it remains at 15% (with all four population groups; Table 1). Across all data sets and using <100 polymorphisms, 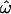 generally exceeds 10% (Figure 2). With <100 loci, then, it will often be the case that two individuals from different populations are more similar to one another than are two individuals from the same population.
generally exceeds 10% (Figure 2). With <100 loci, then, it will often be the case that two individuals from different populations are more similar to one another than are two individuals from the same population.
Figure 2.—

Behavior of the dissimilarity fraction ( 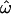 ) and error rates of the “centroid” (CC) and “population trait” (CT) classification methods (red, blue, and green lines, respectively) for each of 15 data subsets (see Table 1 and materials and methods). The number of loci subsampled varies in 21 logarithmic steps from 10 to the maximum for each data subset. At each step, all three statistics were computed for 200 subsampled data sets. Lines indicate the medians of the resulting distributions. Within each section, separate series represent three polymorphism frequency subsets: rare (MAF < 10%, blue contours), common (MAF > 10%, green), and all (all polymorphisms, black; see key). Results computed from the data subsets derived from the insertion, microarray, and resequenced data sets are shown in A and B, C and D, and E, respectively. A and C show results from analyses that use only the three most distinct population groups (Europeans, East Asians, and sub-Saharan Africans, abbreviated Eu, EA, and Af), while B and D show results based on all populations in the insertion and microarray data sets, respectively (Indian, Native American, New Guinean, African American, and Hispano–Latino, abbreviated In, NA, NG, AfAm, and HL). E uses all three population groups in the resequenced data set.
) and error rates of the “centroid” (CC) and “population trait” (CT) classification methods (red, blue, and green lines, respectively) for each of 15 data subsets (see Table 1 and materials and methods). The number of loci subsampled varies in 21 logarithmic steps from 10 to the maximum for each data subset. At each step, all three statistics were computed for 200 subsampled data sets. Lines indicate the medians of the resulting distributions. Within each section, separate series represent three polymorphism frequency subsets: rare (MAF < 10%, blue contours), common (MAF > 10%, green), and all (all polymorphisms, black; see key). Results computed from the data subsets derived from the insertion, microarray, and resequenced data sets are shown in A and B, C and D, and E, respectively. A and C show results from analyses that use only the three most distinct population groups (Europeans, East Asians, and sub-Saharan Africans, abbreviated Eu, EA, and Af), while B and D show results based on all populations in the insertion and microarray data sets, respectively (Indian, Native American, New Guinean, African American, and Hispano–Latino, abbreviated In, NA, NG, AfAm, and HL). E uses all three population groups in the resequenced data set.
The power of large numbers of common polymorphisms is most apparent in the microarray data set, comparing the European, East Asian, and sub-Saharan African population groups (Figure 2C). 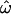 approaches zero (median 0.12%) with 1000 polymorphisms. This implies that, when enough loci are considered, individuals from these population groups will always be genetically most similar to members of their own group. In general, CC and CT decrease more rapidly and to lower values than
approaches zero (median 0.12%) with 1000 polymorphisms. This implies that, when enough loci are considered, individuals from these population groups will always be genetically most similar to members of their own group. In general, CC and CT decrease more rapidly and to lower values than 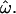
Allele frequency effects:
The “rare” polymorphism subsets defy this trend by converging toward high values of 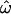 as loci are added. This is largely because the frequencies of rare polymorphisms are necessarily quite similar across populations, whereas higher-frequency polymorphisms have the potential to differ more. For example, the frequency of an allele with an overall MAF of 5% can differ by at most
as loci are added. This is largely because the frequencies of rare polymorphisms are necessarily quite similar across populations, whereas higher-frequency polymorphisms have the potential to differ more. For example, the frequency of an allele with an overall MAF of 5% can differ by at most 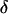 = 10% between two populations (absent in one, at 10% frequency in another). This situation yields
= 10% between two populations (absent in one, at 10% frequency in another). This situation yields 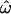 > 0 and very poor classification accuracy, since most between-population pairs are identical but some within-population pairs differ. In contrast, an allele with an overall frequency of 50% across two populations could be fixed in one and absent in the other, resulting in
> 0 and very poor classification accuracy, since most between-population pairs are identical but some within-population pairs differ. In contrast, an allele with an overall frequency of 50% across two populations could be fixed in one and absent in the other, resulting in 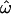 = 0 and allowing perfect classification. It is these frequency differences that allow populations to be distinguished, so the data sets with lower
= 0 and allowing perfect classification. It is these frequency differences that allow populations to be distinguished, so the data sets with lower 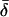 (and thus generally lower FST) have lower classification power.
(and thus generally lower FST) have lower classification power.
The sensitivity of these statistics to allele frequencies explains some differences between the data sets. The microarray data set exhibits strong ascertainment bias for common polymorphisms, and it is with this data set that 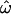 drops most rapidly and to its lowest values (Figure 2, C and D). The insertions data set exhibits a weaker ascertainment bias and includes more rare polymorphisms, so
drops most rapidly and to its lowest values (Figure 2, C and D). The insertions data set exhibits a weaker ascertainment bias and includes more rare polymorphisms, so 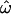 remains higher (Figure 2, A and B). Similarly, CC and CT drop more rapidly for the microarray data set than for the insertion data set. The resequenced data set polymorphisms were ascertained by resequencing a sizable panel of individuals from the genotyped populations and thus include many rare polymorphisms, but this is partially offset by the equally large number of common polymorphisms (Figure 2E). The classification methods are less affected by the inclusion of rare polymorphisms.
remains higher (Figure 2, A and B). Similarly, CC and CT drop more rapidly for the microarray data set than for the insertion data set. The resequenced data set polymorphisms were ascertained by resequencing a sizable panel of individuals from the genotyped populations and thus include many rare polymorphisms, but this is partially offset by the equally large number of common polymorphisms (Figure 2E). The classification methods are less affected by the inclusion of rare polymorphisms.
Population sampling effects:
We contrast two choices: sets of populations that have been relatively isolated from each other by geographic distance and barriers since the earliest migrations of modern humans out of Africa and sets that include populations that were founded more recently, are geographically closer to one another and therefore more likely to exchange migrants, or have recently experienced a large genetic influx from another population in the set. Sampling only from the more distinct populations yields lower 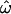 -values, as expected. Figure 2, A, C, and E, shows the results of using only the three most distinct population groups (Europeans, East Asians, and sub-Saharan Africans). Figure 2, B and D, expands the samples used in Figure 2, A and C, to include recently founded and/or geographically intermediate populations (Indians in the insertions data set and New Guineans, South Asians, and Native Americans in the microarray data set) and “admixed” populations (i.e., those that have recently received many migrants from different populations, such as the African American and Hispano–Latino groups in the microarray data set). With just 175 loci, choosing to sample distinct populations vs. more closely related ones makes only a modest difference (insertions data set, compare Figure 2A to 2B; Table 1). The effect of population sampling becomes more pronounced when ≥1000 loci are available. In the microarray data set,
-values, as expected. Figure 2, A, C, and E, shows the results of using only the three most distinct population groups (Europeans, East Asians, and sub-Saharan Africans). Figure 2, B and D, expands the samples used in Figure 2, A and C, to include recently founded and/or geographically intermediate populations (Indians in the insertions data set and New Guineans, South Asians, and Native Americans in the microarray data set) and “admixed” populations (i.e., those that have recently received many migrants from different populations, such as the African American and Hispano–Latino groups in the microarray data set). With just 175 loci, choosing to sample distinct populations vs. more closely related ones makes only a modest difference (insertions data set, compare Figure 2A to 2B; Table 1). The effect of population sampling becomes more pronounced when ≥1000 loci are available. In the microarray data set, 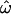 drops to zero at 1000 loci if only distinct populations are sampled. With geographically intermediate and admixed populations added, however,
drops to zero at 1000 loci if only distinct populations are sampled. With geographically intermediate and admixed populations added, however, 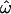 reaches an asymptotic value of 3.1%, CC remains well above zero, and even CT does not reach zero (microarray data, Figure 2, C and D; Table 1).
reaches an asymptotic value of 3.1%, CC remains well above zero, and even CT does not reach zero (microarray data, Figure 2, C and D; Table 1).
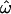 also appears to reach a nonzero asymptotic value in the resequenced data set, instead of continuing to trend downward as would be expected given the distinct populations used. This may be due to the fact that many of the polymorphisms in that data set are physically linked and therefore nonindependent. Overall, the responses of the two classification methods to data set composition variables are qualitatively similar to the behavior of
also appears to reach a nonzero asymptotic value in the resequenced data set, instead of continuing to trend downward as would be expected given the distinct populations used. This may be due to the fact that many of the polymorphisms in that data set are physically linked and therefore nonindependent. Overall, the responses of the two classification methods to data set composition variables are qualitatively similar to the behavior of 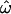 (Figure 2). The most apparent difference is that the misclassification rates (CC and CT) decrease much more rapidly, and to lower values, than
(Figure 2). The most apparent difference is that the misclassification rates (CC and CT) decrease much more rapidly, and to lower values, than  does as the number of loci considered increases.
does as the number of loci considered increases.
DISCUSSION
It has long been appreciated that differences between human populations account for only a small fraction of the total variance in allele frequencies (typically presented as FST values of 10–15%; Lewontin 1972; Nei and Roychoudhury 1972; Latter 1980; Barbujani et al. 1997; Jorde et al. 2000; Watkins et al. 2003; International HapMap Consortium 2005; Rosenberg et al. 2005). Such observations triggered controversy from the outset. Some geneticists concluded the differences were negligible (Lewontin 1972); others disagreed (Mitton 1978). Despite the limited data, it soon became apparent that even a modest number of loci should allow accurate assignment of individuals to populations (Mitton 1978; Smouse et al. 1982).
More recently, the Human Genome Project (2001) (HGP) highlighted the basic genetic similarity of all humans, yet subsequent analyses demonstrated that genetic data can be used to accurately classify humans into populations (Rosenberg et al. 2002, 2005; Bamshad et al. 2003; Turakulov and Easteal 2003; Tang et al. 2005; Lao et al. 2006). Risch et al. (2002) and Edwards (2003) used theoretical illustrations to show why accurate classification is possible despite the slight differences in allele frequencies between populations. These illustrations suggest that, if enough loci are considered, two individuals from the same population may be genetically more similar (i.e., more closely related) to each other than to any individual from another population (as foreshadowed by Powell and Taylor 1978). Accordingly, Risch et al. (2002, p. 2007.5) state that “two Caucasians are more similar to each other genetically than a Caucasian and an Asian.” However, in a reanalysis of data from 377 microsatellite loci typed in 1056 individuals, Europeans proved to be more similar to Asians than to other Europeans 38% of the time (Bamshad et al. 2004; population definitions and data from Rosenberg et al. 2002).
With the large and diverse data sets now available, we have been able to evaluate these contrasts quantitatively. Even the pairwise relatedness measure, 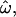 can show clear distinctions between populations if enough polymorphic loci are used. Observations of high
can show clear distinctions between populations if enough polymorphic loci are used. Observations of high 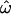 and low classification errors are the norm with intermediate numbers of loci (up to several hundred). These results bear out the observations of Bamshad et al. (2004). The high
and low classification errors are the norm with intermediate numbers of loci (up to several hundred). These results bear out the observations of Bamshad et al. (2004). The high 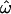 observed there was due primarily to the slow rate of decrease of
observed there was due primarily to the slow rate of decrease of  with increasing numbers of loci. Although Rosenberg et al. (2002) achieved a very low misclassification rate with the same data, far more loci would be needed to reduce
with increasing numbers of loci. Although Rosenberg et al. (2002) achieved a very low misclassification rate with the same data, far more loci would be needed to reduce 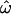 to similarly small values (assuming such values could be reached at all for those populations).
to similarly small values (assuming such values could be reached at all for those populations).
Thus the answer to the question “How often is a pair of individuals from one population genetically more dissimilar than two individuals chosen from two different populations?” depends on the number of polymorphisms used to define that dissimilarity and the populations being compared. The answer,  can be read from Figure 2. Given 10 loci, three distinct populations, and the full spectrum of polymorphisms (Figure 2E), the answer is
can be read from Figure 2. Given 10 loci, three distinct populations, and the full spectrum of polymorphisms (Figure 2E), the answer is 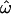 ≅ 0.3, or nearly one-third of the time. With 100 loci, the answer is ∼20% of the time and even using 1000 loci,
≅ 0.3, or nearly one-third of the time. With 100 loci, the answer is ∼20% of the time and even using 1000 loci, 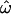 ≅ 10%. However, if genetic similarity is measured over many thousands of loci, the answer becomes “never” when individuals are sampled from geographically separated populations.
≅ 10%. However, if genetic similarity is measured over many thousands of loci, the answer becomes “never” when individuals are sampled from geographically separated populations.
On the other hand, if the entire world population were analyzed, the inclusion of many closely related and admixed populations would increase 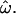 This is illustrated by the fact that
This is illustrated by the fact that 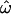 and the classification error rates, CC and CT, all remain greater than zero when such populations are analyzed, despite the use of >10,000 polymorphisms (Table 1, microarray data set; Figure 2D). In a similar vein, Romualdi et al. (2002) and Serre and Pääbo (2004) have suggested that highly accurate classification of individuals from continuously sampled (and therefore closely related) populations may be impossible. However, those studies lacked the statistical power required to answer that question (see Rosenberg et al. 2005).
and the classification error rates, CC and CT, all remain greater than zero when such populations are analyzed, despite the use of >10,000 polymorphisms (Table 1, microarray data set; Figure 2D). In a similar vein, Romualdi et al. (2002) and Serre and Pääbo (2004) have suggested that highly accurate classification of individuals from continuously sampled (and therefore closely related) populations may be impossible. However, those studies lacked the statistical power required to answer that question (see Rosenberg et al. 2005).
How can the observations of accurate classifiability be reconciled with high between-population similarities among individuals? Classification methods typically make use of aggregate properties of populations, not just properties of individuals or even of pairs of individuals. For instance, the centroid classification method computes the distances between individuals and population centroids and then clusters individuals around the nearest centroid. The population trait method relies on information about the frequencies of each allele in each population to compute individual trait values and on the means and variances of the trait distributions to classify individuals. The Structure classification algorithm (Pritchard et al. 2000) also relies on aggregate properties of populations, such as Hardy–Weinberg and linkage equilibrium. In contrast, the pairwise distances used to compute 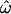 make no use of population-level information and are strongly affected by the high level of within-groups variation typical of human populations. This accounts for the difference in behavior between
make no use of population-level information and are strongly affected by the high level of within-groups variation typical of human populations. This accounts for the difference in behavior between 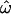 and the classification results.
and the classification results.
Since an individual's geographic ancestry can often be inferred from his or her genetic makeup, knowledge of one's population of origin should allow some inferences about individual genotypes. To the extent that phenotypically important genetic variation resembles the variation studied here, we may extrapolate from genotypic to phenotypic patterns. Resequencing studies of gene-coding regions show patterns similar to those seen here (e.g., Stephens et al. 2001), and many common disease-associated alleles are not unusually differentiated across populations (Lohmueller et al. 2006). Thus it may be possible to infer something about an individual's phenotype from knowledge of his or her ancestry.
However, consider a hypothetical phenotype of biomedical interest that is determined primarily by a dozen additive loci of equal effect whose worldwide distributions resemble those in the insertion data set (e.g., with 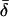 = 0.15; Table 1). Given these assumptions, the genetic distance used in computing
= 0.15; Table 1). Given these assumptions, the genetic distance used in computing 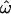 and CC is equivalent to a phenotypic distance, so Figure 2 can be used to analyze this hypothetical trait. Figure 2A shows that a trait determined by 12 such loci will typically yield
and CC is equivalent to a phenotypic distance, so Figure 2 can be used to analyze this hypothetical trait. Figure 2A shows that a trait determined by 12 such loci will typically yield 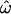 = 0.31 (0.20–0.41) and CC = 0.14 (0.054–0.29; medians and 90% ranges). About one-third of the time (
= 0.31 (0.20–0.41) and CC = 0.14 (0.054–0.29; medians and 90% ranges). About one-third of the time (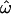 = 0.31) an individual will be phenotypically more similar to someone from another population than to another member of the same population. Similarly, individuals will be more similar to the average or “typical” phenotype of another population than to the average phenotype in their own population with a probability of ∼14% (CC = 0.14). It follows that variation in such a trait will often be discordant with population labels.
= 0.31) an individual will be phenotypically more similar to someone from another population than to another member of the same population. Similarly, individuals will be more similar to the average or “typical” phenotype of another population than to the average phenotype in their own population with a probability of ∼14% (CC = 0.14). It follows that variation in such a trait will often be discordant with population labels.
The population groups in this example are quite distinct from one another: Europeans, sub-Saharan Africans, and East Asians. Many factors will further weaken the correlation between an individual's phenotype and their geographic ancestry. These include considering more closely related or admixed populations, studying phenotypes influenced by fewer loci, unevenly distributed effects across loci, nonadditive effects, developmental and environmental effects, and uncertainties about individuals' ancestry and actual populations of origin. The typical frequencies of alleles that influence a phenotype are also relevant, as our results show that rare polymorphisms yield high values of 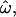 CC, and CT, even when many such polymorphisms are studied. This implies that complex phenotypes influenced primarily by rare alleles may correspond poorly with population labels and other population-typical traits (in contrast to some Mendelian diseases). However, the typical frequencies of alleles responsible for common complex diseases remain unknown. A final complication arises when racial classifications are used as proxies for geographic ancestry. Although many concepts of race are correlated with geographic ancestry, the two are not interchangeable, and relying on racial classifications will reduce predictive power still further.
CC, and CT, even when many such polymorphisms are studied. This implies that complex phenotypes influenced primarily by rare alleles may correspond poorly with population labels and other population-typical traits (in contrast to some Mendelian diseases). However, the typical frequencies of alleles responsible for common complex diseases remain unknown. A final complication arises when racial classifications are used as proxies for geographic ancestry. Although many concepts of race are correlated with geographic ancestry, the two are not interchangeable, and relying on racial classifications will reduce predictive power still further.
The fact that, given enough genetic data, individuals can be correctly assigned to their populations of origin is compatible with the observation that most human genetic variation is found within populations, not between them. It is also compatible with our finding that, even when the most distinct populations are considered and hundreds of loci are used, individuals are frequently more similar to members of other populations than to members of their own population. Thus, caution should be used when using geographic or genetic ancestry to make inferences about individual phenotypes.
Acknowledgments
We thank Jinchuan Xing, Michael Bamshad, Dennis O'Rourke, and Thomas Doak for thoughtful comments. This work was supported by National Science Foundation grants BCS-0218338 (M.A.B.), BCS-0218370 (L.B.J.), and EPS-0346411 (M.A.B.); by National Institutes of Health grant GM-59290 (L.B.J. and M.A.B.); by the Louisiana Board of Regents Millennium Trust Health Excellence Fund HEF (2000-05)-05 (M.A.B.), (2000-05)-01 (M.A.B.), and (2001-06)-02 (M.A.B.); and by the Intramural Research Program of the National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health.
References
- American Anthropological Association, 1997. Response to OMB directive 15: race and ethnic standards for federal statistics and administrative reporting (original statement at http://web.archive.org/web/19990507115624/http://www.ameranthassn.org/ombnews.htm; amended 2000 statement at http://www.aaanet.org/gvt/ombdraft.htm).
- Bamshad, M., S. Wooding, B. A. Salisbury and J. C. Stephens, 2004. Deconstructing the relationship between genetics and race. Nat. Rev. Genet. 5: 598–609. [DOI] [PubMed] [Google Scholar]
- Bamshad, M. J., S. Wooding, W. S. Watkins, C. T. Ostler, M. A. Batzer et al., 2003. Human population genetic structure and inference of group membership. Am. J. Hum. Genet. 72: 578–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbujani, G., A. Magagni, E. Minch and L. L. Cavalli-Sforza, 1997. An apportionment of human DNA diversity. Proc. Natl. Acad. Sci. USA 94: 4516–4519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowcock, A. M., A. Ruiz-Linares, J. Tomfohrde, E. Minch, J. R. Kidd et al., 1994. High resolution of human evolutionary trees with polymorphic microsatellites. Nature 368: 455–457. [DOI] [PubMed] [Google Scholar]
- Campbell, D., P. Duchesne and L. Bernatchez, 2003. AFLP utility for population assignment studies: analytical investigation and empirical comparison with microsatellites. Mol. Ecol. 12: 1979–1991. [DOI] [PubMed] [Google Scholar]
- Chakraborty, R., and L. Jin, 1993. A unified approach to study hypervariable polymorphisms: statistical considerations of determining relatedness and population distances. Exper. Suppl. 67: 153–175. [DOI] [PubMed] [Google Scholar]
- Cornuet, J. M., S. Piry, G. Luikart, A. Estoup and M. Solignac, 1999. New methods employing multilocus genotypes to select or exclude populations as origins of individuals. Genetics 153: 1989–2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dean, M., J. C. Stephens, C. Winkler, D. A. Lomb, M. Ramsburg et al., 1994. Polymorphic admixture typing in human ethnic populations. Am. J. Hum. Genet. 55: 788–808. [PMC free article] [PubMed] [Google Scholar]
- Edwards, A. W., 2003. Human genetic diversity: Lewontin's fallacy. BioEssays 25: 798–801. [DOI] [PubMed] [Google Scholar]
- Human Genome Project, 2001. The human genome. Nature 409: 812. [Google Scholar]
- International HapMap Consortium, 2005. A haplotype map of the human genome. Nature 437: 1299–1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorde, L. B., W. S. Watkins, M. J. Bamshad, M. E. Dixon, C. E. Ricker et al., 2000. The distribution of human genetic diversity: a comparison of mitochondrial, autosomal, and Y-chromosome data. Am. J. Hum. Genet. 66: 979–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lao, O., K. van Duijn, P. Kersbergen, P. de Knijff and M. Kayser, 2006. Proportioning whole-genome single-nucleotide-polymorphism diversity for the identification of geographic population structure and genetic ancestry. Am. J. Hum. Genet. 78: 680–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latter, B., 1980. Genetic differences within and between populations of the major human subgroups. Am. Nat. 116: 220–237. [Google Scholar]
- Lewontin, R. C., 1972. The apportionment of human diversity. Evol. Biol. 6: 381–398. [Google Scholar]
- Lohmueller, K. E., M. M. Mauney, D. Reich and J. M. Braverman, 2006. Variants associated with common disease are not unusually differentiated in frequency across populations. Am. J. Hum. Genet. 78: 130–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manel, S., P. Berthier and G. Luikart, 2002. Detecting wildlife poaching: identifying the origin of individuals with Bayesian assignment tests and multilocus genotypes. Conserv. Biol. 16: 650–659. [Google Scholar]
- Mitton, J. B., 1977. Genetic differentiation of races of man as judged by single-locus and multilocus analyses. Am. Nat. 111: 203–212. [Google Scholar]
- Mitton, J. B., 1978. Measurement of differentiation: reply to Lewontin, Powell, and Taylor. Am. Nat. 112: 1142–1144. [Google Scholar]
- Mountain, J. L., and L. L. Cavalli-Sforza, 1997. Multilocus genotypes, a tree of individuals, and human evolutionary history. Am. J. Hum. Genet. 61: 705–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mountain, J. L., and U. Ramakrishnan, 2005. Impact of human population history on distributions of individual-level genetic distance. Hum. Genomics 2: 4–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nei, M., 1973. Analysis of gene diversity in subdivided populations. Proc. Natl. Acad. Sci. USA 70: 3321–3323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nei, M., and A. K. Roychoudhury, 1972. Gene differences between Caucasian, Negro, and Japanese populations. Science 177: 434–436. [DOI] [PubMed] [Google Scholar]
- Powell, J. R., and C. E. Taylor, 1978. Are human races “substantially” different genetically? Am. Nat. 112: 1139–1142. [Google Scholar]
- Pritchard, J. K., M. Stephens and P. Donnelly, 2000. Inference of population structure using multilocus genotype data. Genetics 155: 945–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Risch, N., E. Burchard, E. Ziv and H. Tang, 2002. Categorization of humans in biomedical research: genes, race and disease. Genome Biol. 3: 2007.1–2007.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romualdi, C., D. Balding, I. S. Nasidze, G. Risch, M. Robichaux et al., 2002. Patterns of human diversity, within and among continents, inferred from biallelic DNA polymorphisms. Genome Res. 12: 602–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg, N. A., J. K. Pritchard, J. L. Weber, H. M. Cann, K. K. Kidd et al., 2002. Genetic structure of human populations. Science 298: 2381–2385. [DOI] [PubMed] [Google Scholar]
- Rosenberg, N. A., S. Mahajan, S. Ramachandran, C. Zhao, J. K. Pritchard et al., 2005. Clines, clusters, and the effect of study design on the inference of human population structure. PLoS Genet. 1: e70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serre, D., and S. Pääbo, 2004. Evidence for gradients of human genetic diversity within and among continents. Genome Res. 14: 1679–1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shriver, M. D., R. Mei, E. J. Parra, V. Sonpar, I. Halder et al., 2005. Large-scale SNP analysis reveals clustered and continuous patterns of human genetic variation. Hum. Genomics 2: 81–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smouse, P. E., and R. Chakraborty, 1986. The use of restriction fragment length polymorphisms in paternity analysis. Am. J. Hum. Genet. 38: 918–939. [PMC free article] [PubMed] [Google Scholar]
- Smouse, P. E., R. S. Spielman and M. H. Park, 1982. Multiple-locus allocation of individuals to groups as a function of the genetic variation within and differences among human populations. Am. Nat. 119: 445. [Google Scholar]
- Stephens, J. C., J. A. Schneider, D. A. Tanguay, J. Choi, T. Acharya et al., 2001. Haplotype variation and linkage disequilibrium in 313 human genes. Science 293: 489–493. [DOI] [PubMed] [Google Scholar]
- Tang, H., T. Quertermous, B. Rodriguez, S. L. Kardia, X. Zhu et al., 2005. Genetic structure, self-identified race/ethnicity, and confounding in case-control association studies. Am. J. Hum. Genet. 76: 268–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turakulov, R., and S. Easteal, 2003. Number of SNPs loci needed to detect population structure. Hum. Hered. 55: 37–45. [DOI] [PubMed] [Google Scholar]
- Watkins, W. S., A. R. Rogers, C. T. Ostler, S. Wooding, M. J. Bamshad et al., 2003. Genetic variation among world populations: inferences from 100 Alu insertion polymorphisms. Genome Res. 13: 1607–1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watkins, W. S., B. V. Prasad, J. M. Naidu, B. B. Rao, B. A. Bhanu et al., 2005. Diversity and divergence among the tribal populations of India. Ann. Hum. Genet. 69: 680–692. [DOI] [PubMed] [Google Scholar]
- Witherspoon, D. J., E. E. Marchani, W. S. Watkins, C. T. Ostler, S. P. Wooding et al., 2006. Human population genetic structure and diversity inferred from polymorphic L1(LINE-1) and Alu insertions. Hum. Hered. 62: 30–46. [DOI] [PubMed] [Google Scholar]