

Abstract
Unigene alignments obtained from cDNA libraries made using multiple individuals are not currently used to estimate population heterozygosity, as they are known to harbor mutations created during library construction. We describe an estimator of population heterozygosity that utilizes only SNPs unlikely to be library construction artifacts.
EXPRESSED sequence tag (EST) projects have become a popular and cost-effective means of initially cataloging a large number of genes in biological systems without genome projects (reviewed in Rudd 2003). DNA sequencing of several thousand randomly chosen clones from a cDNA library allows thousands of different transcripts to be identified. However, since the likelihood of observing a given transcript is proportional to the expression level of that transcript in the tissue from which the library is derived, often transcripts are represented by several EST sequences. In a typical EST project, using an inbred line as the starting material to construct the cDNA library, ESTs associated with the same transcript can be assembled into a Unigene cluster and the consensus sequence associated with that assembly is referred to as a Unigene. On the other hand, if the ESTs contributing to a Unigene cluster are associated with cDNA libraries obtained from different individuals, or different inbred lines, then single nucleotide polymorphisms (SNPs) can be identified from the resulting alignments (Picoult-Newberg et al. 1999). Although SNPs obtained from haphazard collections of ESTs may have utility as markers, it would be difficult to estimate per-site heterozygosity from such a resource, since the unknown ascertainment scheme could bias any estimates.
On the other hand, if a cDNA library were constructed from an equimolar collection of RNAs from an infinite number of outbred individuals, the alignments associated with different Unigene clusters could be used to estimate per-site heterozygosity using standard population genetics methods for estimating diversity (e.g., Hartl and Clark 1997). Standard methods could also be applied to a Unigene cluster obtained from a library derived from a finite number of individuals provided the alignment depth of that cluster is much less than twice the number of individuals used to create the cDNA library to ensure that alleles sampled in the Unigene cluster are likely to be independent of one another. However, the application of standard methods for estimating per-site heterozygosity to collections of ESTs has generally been avoided, as the DNA sequences obtained from EST projects are often believed to be associated with a relatively high rate of point mutation arising from replication errors. These replication errors are introduced into the cDNA by the reverse transcriptase enzyme during the first-strand synthesis required to convert RNA to DNA, and to a lesser extent during other steps in which the library is manipulated.
Despite the potential high rate of point mutations in EST collections, it is likely that polymorphic sites with a minor allele count of ≥2 in an alignment of at least four ESTs (the minor allele cannot have a frequency >1 in shallower alignments) are true SNPs. Unlike true SNPs, the point mutations associated with library creation are unlikely to result in the exact same mutation event and will thus almost always be singletons regardless of alignment depth. Here we describe a method that provides an unbiased estimate of the per-site heterozygosity that relies only on information obtained from those SNPs with a minor allele count of ≥2 in alignments of depth ≥4 and use the method to estimate per-site heterozygosity from an EST project. With an estimate of per-site heterozygosity in hand, we are able to additionally estimate the fraction of singleton polymorphic sites that are actual SNPs as opposed to errors introduced during cDNA library construction.
On the basis of a simple extension of Fu (1995; Equation 7), and assuming samples from a single large random mating population are used to construct the cDNA library, we estimate heterozygosity per base pair conditional on the number of alleles sampled (n) and the number of copies of the minor allele observed (i) as 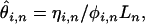 where ηi,n is the number of SNPs with a minor allele frequency of i over all aligned positions of depth n in the EST collection, Ln is the total number of bases aligned to depth n, and
where ηi,n is the number of SNPs with a minor allele frequency of i over all aligned positions of depth n in the EST collection, Ln is the total number of bases aligned to depth n, and
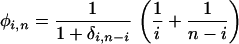 |
Here, δi,n−i is Kroneker's delta, which is equal to one if i = n − i and zero otherwise. Each 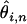 is an unbiased estimator of θ, as the
is an unbiased estimator of θ, as the 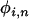 term effectively “corrects” for the fact the SNP is ascertained to be at a frequency i in an alignment of depth n. To obtain an overall estimate of θ we must calculate a weighted average over the estimates of
term effectively “corrects” for the fact the SNP is ascertained to be at a frequency i in an alignment of depth n. To obtain an overall estimate of θ we must calculate a weighted average over the estimates of 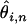 obtained for different values of i and n. The weight we propose using is
obtained for different values of i and n. The weight we propose using is
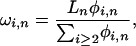 |
the product of the total length of the alignment at depth n and the proportion of that length expected to be segregating a SNP having a minor allele count ≥2 (thus ωi,n is defined only for i ≥ 2 and n ≥ 4). If, for example, some set of aligned sequences has a majority of the alignment at depth d and a small fraction of the alignment at a depth less than d, our scheme weights more highly those estimates of 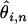 from the more deeply aligned majority of the sequence. The above estimates of
from the more deeply aligned majority of the sequence. The above estimates of 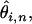 and their associated weights (ωi,n), suggest a least-squares function that can be minimized to obtain a global estimate of θ from a set of aligned ESTs:
and their associated weights (ωi,n), suggest a least-squares function that can be minimized to obtain a global estimate of θ from a set of aligned ESTs:
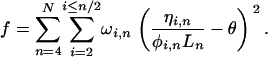 |
This function is simply the squared difference between the estimate of 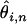 for each possible minor allele count and alignment depth consistent with SNPs seen at least twice, and the unknown true value of θ, with each term weighted in an appropriate manner. N should be chosen to be much less than twice the total number of individuals contributing to the cDNA library to avoid considering deep alignments where a single naturally occurring allele may be sampled multiple times. Differentiating f with respect to θ we obtain an estimator of
for each possible minor allele count and alignment depth consistent with SNPs seen at least twice, and the unknown true value of θ, with each term weighted in an appropriate manner. N should be chosen to be much less than twice the total number of individuals contributing to the cDNA library to avoid considering deep alignments where a single naturally occurring allele may be sampled multiple times. Differentiating f with respect to θ we obtain an estimator of
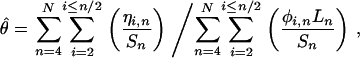 |
where
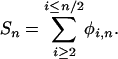 |
Finally, we can separately estimate heterozygosity from singleton SNPs by conditioning on i = 1 and summing over all alignments regardless of depth. This results in an estimate of
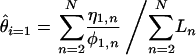 |
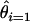 is then used to estimate the per-site per-EST probability of observing an error as
is then used to estimate the per-site per-EST probability of observing an error as 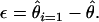 Similarly the fraction of all observed singleton SNPs that are likely to be mutations resulting from library construction (as opposed to actual SNPs) is
Similarly the fraction of all observed singleton SNPs that are likely to be mutations resulting from library construction (as opposed to actual SNPs) is 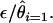
Beldade et al. (2006) carried out an EST project in Bicyclus anynana, a butterfly model for the study of evolution and development. The ESTs were generated from 3′ reads (in an attempt to maximize alignment depths within Unigene clusters) from five cDNA developmental stage-specific normalized libraries sequenced in roughly equal proportions that were each derived from between 20 and 66 diploid individuals. We initially examined all Unigene clusters visually and split or removed alignments for which extremely high diversity in the aligned region suggested mis-assembly. This resulted in a collection of 1257 Unigene clusters consisting of at least two ESTs. Aligned bases with PHRED quality scores of 20 or lower were converted to missing, polymorphic IN/DELs scored as missing, and the first 10 bases of each trimmed read scored as missing regardless of PHRED score (the first 10 bases of a read are generally unreliable). On the basis of these potentially ragged alignments we tabulated the total number of positions observed as a function of minor allele count and alignment depth (Table 1). At positions where the total nonmissing alignment depth was greater than N = 15 alleles, we randomly chose 15 alleles to contribute to the estimate of heterozygosity, which we estimate to be 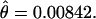 This estimate of
This estimate of 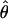 is consistent with data obtained from sequencing a very limited number of 3′-UTRs at the Distal-less locus in B. anynana (Beldade et al. 2002) and similar to a widely cited number of 0.6% for Drosophila melanogaster.
is consistent with data obtained from sequencing a very limited number of 3′-UTRs at the Distal-less locus in B. anynana (Beldade et al. 2002) and similar to a widely cited number of 0.6% for Drosophila melanogaster.
TABLE 1.
Number of SNPs as a function of minor allele count (i, columns) and alignment depth (n, rows) in 1257 Unigene clusters
|
i
|
||||||||
|---|---|---|---|---|---|---|---|---|
| n | 0 | 1 | 2 | 3 | 4 | 5 | 6 | 7 |
| 2 | 265,245 | 4,512 | ||||||
| 3 | 85,559 | 1,874 | ||||||
| 4 | 49,382 | 1,081 | 233 | |||||
| 5 | 31,676 | 727 | 253 | |||||
| 6 | 16,795 | 394 | 114 | 47 | ||||
| 7 | 16,283 | 459 | 68 | 102 | ||||
| 8 | 10,483 | 278 | 69 | 58 | 30 | |||
| 9 | 8,152 | 199 | 46 | 31 | 41 | |||
| 10 | 7,017 | 205 | 21 | 26 | 20 | 14 | ||
| 11 | 5,709 | 137 | 26 | 11 | 33 | 19 | ||
| 12 | 4,905 | 152 | 24 | 15 | 16 | 16 | 4 | |
| 13 | 3,287 | 113 | 15 | 6 | 10 | 8 | 5 | |
| 14 | 2,959 | 124 | 7 | 6 | 7 | 9 | 2 | 1 |
| 15 | 24,386 | 794 | 127 | 71 | 58 | 68 | 32 | 30 |
The column corresponding to i = 0 tabulates monomorphic positions.
In theory, for cases where N is large relative to the number of individuals used to construct the cDNA library, a single allele can be represented multiple times in an alignment, which will result in downward biases in the estimate of 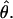 In practice, the particular choice of N we employ does not have a large effect on our estimate of heterozygosity. We estimated
In practice, the particular choice of N we employ does not have a large effect on our estimate of heterozygosity. We estimated 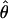 for every value of N between 4 and 15, randomly discarding alleles to achieve the desired N at positions with deeper alignments. Estimates ranged from 0.00836 to 0.00861, and
for every value of N between 4 and 15, randomly discarding alleles to achieve the desired N at positions with deeper alignments. Estimates ranged from 0.00836 to 0.00861, and 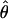 is not a function of N. The vast majority of aligned bases at a depth >3 (87%) and SNPs at frequencies of ≥2 (80%) are associated with alignment depths of ≤10 (Table 1); thus, changing N does not appear to greatly affect our estimate of heterozygosity.
is not a function of N. The vast majority of aligned bases at a depth >3 (87%) and SNPs at frequencies of ≥2 (80%) are associated with alignment depths of ≤10 (Table 1); thus, changing N does not appear to greatly affect our estimate of heterozygosity.
For N = 15, we furthermore estimate 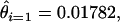 and it follows that ɛ is ∼0.94% and that ∼53% of observed singleton “SNPs” are likely to be mutations generated during library creation and propagation. Our estimate that 0.94% of bases in the EST project are mutations introduced as part of the EST project seems high, but error rates as high as 3% are reported to be associated with EST projects (reviewed in Rudd 2003). Our analysis suggests that a singleton SNP is as likely to be a mutation generated during library construction or propagation as a real segregating SNP. Thus, it would not seem prudent to design SNP genotyping assays for singleton SNPs identified as part of this project, the majority of such assays would be monomorphic when applied to actual butterfly populations.
and it follows that ɛ is ∼0.94% and that ∼53% of observed singleton “SNPs” are likely to be mutations generated during library creation and propagation. Our estimate that 0.94% of bases in the EST project are mutations introduced as part of the EST project seems high, but error rates as high as 3% are reported to be associated with EST projects (reviewed in Rudd 2003). Our analysis suggests that a singleton SNP is as likely to be a mutation generated during library construction or propagation as a real segregating SNP. Thus, it would not seem prudent to design SNP genotyping assays for singleton SNPs identified as part of this project, the majority of such assays would be monomorphic when applied to actual butterfly populations.
It is well known that an exponentially growing or structured population can result in a skew toward rare alleles (Slatkin and Hudson 1991). If our estimators were inappropriately applied to a growing population it would underestimate 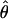 and overestimate ɛ, although the magnitude of allele frequency skew introduced by demography would likely be an order of magnitude smaller than the difference between
and overestimate ɛ, although the magnitude of allele frequency skew introduced by demography would likely be an order of magnitude smaller than the difference between 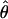 and ɛ observed here. It will be of interest to apply our estimator to other appropriate data sets to obtain additional estimates of ɛ, although we are unaware of other EST collections derived from an equimolar collection of RNAs obtained from a large number of outbred individuals. If this assumption is not satisfied then estimates of
and ɛ observed here. It will be of interest to apply our estimator to other appropriate data sets to obtain additional estimates of ɛ, although we are unaware of other EST collections derived from an equimolar collection of RNAs obtained from a large number of outbred individuals. If this assumption is not satisfied then estimates of 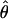 are unlikely to be reliable.
are unlikely to be reliable.
In addition to obtaining a global estimate of 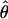 over all clusters, it may be of interest to obtain estimates of
over all clusters, it may be of interest to obtain estimates of 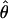 for every cluster that has at least some aligned region(s) deeper than n = 4. Clusters with abnormally high estimates of
for every cluster that has at least some aligned region(s) deeper than n = 4. Clusters with abnormally high estimates of 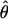 are candidates for experiencing overdominant selection (or a lack of selective constraints), whereas those with abnormally low estimates of
are candidates for experiencing overdominant selection (or a lack of selective constraints), whereas those with abnormally low estimates of 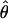 are candidates for having recently experienced selective sweeps (or for being under strong selective constraints). In Figure 1 we plot estimates of
are candidates for having recently experienced selective sweeps (or for being under strong selective constraints). In Figure 1 we plot estimates of 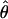 associated with SNPs with a minor allele frequency of at least 2 (and hence n ≥ 4) against a measure of the “total opportunity” to measure heterozygosity in that cluster. We define that opportunity as
associated with SNPs with a minor allele frequency of at least 2 (and hence n ≥ 4) against a measure of the “total opportunity” to measure heterozygosity in that cluster. We define that opportunity as 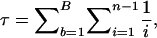 where B is the total number of aligned positions in a cluster and n is the total nonmissing alignment depth (i.e., excluding polymorphic INDELS and low-quality bases) at each position. Since we count only SNPs with a minor allele count of ≥2, τ is defined only at aligned positions having a depth of ≥4. Under Wright–Fisher sampling τθ is the expected number of segregating sites for a given assembly; hence, clusters associated with larger τ should be associated with more accurate measures of θ. Figure 1 shows that the central tendency in the estimation of θ is largely independent of τ (the solid line is a spline through the data), and clusters associated with larger values of τ show less variation about that tendency (which is expected since those estimates of θ are more accurate). If close outgroup sequences were available we could identify clusters experiencing selection or selective constraints as those falling a long way from the spline, with the distance required to achieve statistical significance a decreasing function of τ.
where B is the total number of aligned positions in a cluster and n is the total nonmissing alignment depth (i.e., excluding polymorphic INDELS and low-quality bases) at each position. Since we count only SNPs with a minor allele count of ≥2, τ is defined only at aligned positions having a depth of ≥4. Under Wright–Fisher sampling τθ is the expected number of segregating sites for a given assembly; hence, clusters associated with larger τ should be associated with more accurate measures of θ. Figure 1 shows that the central tendency in the estimation of θ is largely independent of τ (the solid line is a spline through the data), and clusters associated with larger values of τ show less variation about that tendency (which is expected since those estimates of θ are more accurate). If close outgroup sequences were available we could identify clusters experiencing selection or selective constraints as those falling a long way from the spline, with the distance required to achieve statistical significance a decreasing function of τ.
Figure 1.—

Plot of heterozygosity, 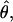 as a function of τ for SNPs with minor allele counts of ≥2 (in alignments of depth of four or greater). Points correspond to 425 Unigene clusters matching these criteria. The black line is a spline, using the “ksmooth” function in R (http://www.r-project.org) with a window size of 500, and gray lines are 500 bootstrap estimates of the same spline.
as a function of τ for SNPs with minor allele counts of ≥2 (in alignments of depth of four or greater). Points correspond to 425 Unigene clusters matching these criteria. The black line is a spline, using the “ksmooth” function in R (http://www.r-project.org) with a window size of 500, and gray lines are 500 bootstrap estimates of the same spline.
In this note we describe an estimator of the per-site heterozygosity (θ) and the per-site error rate (ɛ) that can be applied to DNA sequence data obtained from EST projects. At present, these parameters are not routinely estimated from ESTs collections, because it is generally believed that the error rates associated with individual EST sequences preclude estimating heterozygosity (and vice versa). By assuming that SNPs seen at least twice in an EST collection are true SNPs (and those seen only once are suspect) and that cDNA libraries are constructed in a manner such that different ESTs are likely different sampled alleles from a single large randomly mating population, population genetics theory allows for estimation of both θ and ɛ. It will be of interest to apply these estimators to other appropriate EST collections to determine how variable ɛ is over different EST projects and additionally examine the effect of library normalization on these parameter estimates. In projects where ɛ is large relative to θ, it would make little sense to develop SNP assays for singleton SNPs, whereas in the opposite cases it may be worthwhile to develop such markers.
We also note that the estimation procedure described here can be applied to gene sequence collections other than EST projects. For example, if a DNA pool derived from several dozen individuals is used as a template for PCR, the resulting amplicon cloned, and several clones sequenced (a useful approach when polymorphic IN/DELs prevent direct sequencing), the described method may also be of utility.
Acknowledgments
Comments from B. Gaut, J. Gruber, P. Morrell, and two anonymous reviewers improved this manuscript. This work was supported by National Science Foundation DEB-0235697. A PERL script implementing these estimators is available from the author.
References
- Beldade, P., S. Rudd, J. Gruber and A. D. Long, 2006. A wing expressed sequence tag resource for Bicyclus anynana butterflies, an evo-devo model. BMC Genomics 7: 130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beldade, P., P. Brakefield and A. D. Long, 2002. Contribution of Distal-less to quantitative variation in butterfly eyespots. Nature 415: 315–318. [DOI] [PubMed] [Google Scholar]
- Fu, Y., 1995. Statistical properties of segregating sites. Theor. Pop. Biol. 48: 172–197. [DOI] [PubMed] [Google Scholar]
- Hartl, D. L., and A. G. Clark, 1997. Principles of Population Genetics, Ed. 3. Sinauer Associates, Sunderland, MA.
- Picoult-Newberg, L., T. E. Ideker, M. G. Pohl, S. L. Taylor, M. A. Donaldson et al., 1999. Mining SNPs from EST databases. Genome Res. 9: 167–174. [PMC free article] [PubMed] [Google Scholar]
- Rudd, S., 2003. Expressed sequence tags: alternative or complement to whole genome sequences. Trends Plant Sci. 8: 321–329. [DOI] [PubMed] [Google Scholar]
- Slatkin, M., and R. R. Hudson, 1991. Pairwise comparisons of mitochondrial DNA sequences in stable and exponentially growing populations. Genetics 129: 555–562. [DOI] [PMC free article] [PubMed] [Google Scholar]