

Abstract
A wide variety of temperate animals rely on length of day (photoperiodism) to anticipate and prepare for changing seasons by regulating the timing of development, reproduction, dormancy, and migration. Although the molecular basis of circadian rhythms regulating daily activities is well defined, the molecular basis for the photoperiodic regulation of seasonal activities is largely unknown. We use geographic variation in the photoperiodic control of diapause in the pitcher-plant mosquito Wyeomyia smithii to create the first QTL map of photoperiodism in any animal. For critical photoperiod (CPP), we detect QTL that are unique, a QTL that is sex linked, QTL that overlap with QTL for stage of diapause (SOD), and a QTL that interacts epistatically with the circadian rhythm gene, timeless. Results presented here confirm earlier studies concluding that CPP is under directional selection over the climatic gradient of North America and that the evolution of CPP is genetically correlated with SOD. Despite epistasis between timeless and a QTL for CPP, timeless is not located within any detectable QTL, indicating that it plays an ancillary role in the evolution of photoperiodism in W. smithii. Finally, we highlight one region of the genome that includes loci contributing to CPP, SOD, and hormonal regulation of development.
THE annual change in day length at temperate latitudes provides a highly reliable indicator of future seasonal events and a wide variety of organisms use day length (photoperiod) to time their development, reproduction, dormancy, and migration. Concordance between individual photoperiodic response and local climate is an essential component of fitness for animals living in the temperate zone (Bradshaw et al. 2004), and modification of photoperiodic response is an important adaptation of animals during range expansion (Danilevskii 1965; Cooke 1977; Tauber et al. 1986; Danks 1987; Lounibos et al. 2003) or when confronted with rapid climate change (Bradshaw and Holzapfel 2001a, 2006). Photoperiodism provides an ecologically relevant, highly heritable trait whose adaptive significance in a temperate seasonal environment is not questioned. Length of the favorable or growing season in North America decreases with increasing latitude (Bradshaw 1976). Consequently, the optimal time to enter diapause advances to an earlier day in the year and the day length used to switch from active development to diapause [hereafter, the critical photoperiod (CPP)] is positively correlated with latitude and altitude among a wide variety of temperate arthropods (Danilevskii 1965; Taylor and Spalding 1986; Danks 1987). The consistent genetic change in photoperiodic response over geographic and climatic gradients provides one of the most robust examples of repeated adaptive evolution in nature.
Although much progress has been made in identifying the genetic components of the circadian clock regulating daily activities, the molecular basis of the photoperiodic timer regulating seasonal activities is conspicuously absent from the literature. For insects, the model system for circadian rhythmicity has been Drosophila melanogaster; the Canton-S strain of D. melanogaster is photoperiodic for ovarian diapause but only over a very narrow range of temperatures, and its diapausing status must be determined destructively from dissection of the ovaries. Hence, while “D. melanogaster, with its unrivalled genetic background has provided a foundation of uncovering the molecular basis of the circadian mechanism, … it is probably less useful as a model for photoperiodism. Further studies should examine species with a much more robust [photoperiodic] response” (Saunders 2002, p. 481). Toward the goal of understanding the molecular basis of photoperiodism and its adaptive evolution, we developed QTL maps for CPP and stage of diapause in the pitcher-plant mosquito Wyeomyia smithii, using a southern (31°N) and a northern (57°N) population recently collected from nature.
W. smithii lays its eggs and completes its preadult development only within the water-filled leaves of the carnivorous purple pitcher plant Sarracenia purpurea, where it undergoes a larval diapause that is initiated, maintained, and terminated by photoperiod over a broad range of temperatures. Southern populations (≤36°N) diapause primarily in the fourth larval instar, while northern populations (≥40°N) diapause primarily in the third larval instar (Bradshaw and Lounibos 1977). The earlier stage of diapause in northern populations provides them with a fail-safe opportunity to enter an additional fourth instar diapause in response to uncertain vernal environments (Lounibos and Bradshaw 1975). In W. smithii, the heritability of CPP increases from 0.15 in southern populations to 0.70 in northern populations (Bradshaw and Holzapfel 2001b) and CPP increases with latitude and altitude with R2 repeatedly >90% (Bradshaw and Holzapfel 2001a). Within populations polymorphic for stage of larval diapause (SOD), SOD is negatively genetically correlated with CPP (W. Bradshaw and C. Holzapfel, unpublished results) and the daily expression of the circadian rhythm gene, timeless, differs between diapausing instars (Mathias et al. 2005). Within northern populations of W. smithii, the expression of timeless is inversely correlated with critical photoperiod (Mathias et al. 2005). Nonetheless, critical photoperiod is not correlated with either the period or the amplitude of response to the Nanda–Hamner protocol, the most frequently used experiment to infer a causal connection between the circadian clock and photoperiodism (Bradshaw et al. 2006). Estimates from line crosses for the minimum number of effective factors underlying genetic differences in critical photoperiod between populations range from 5 to 20 and involve additive and nonadditive genetic effects, including both dominance and epistasis (Hard et al. 1992, 1993; Lair et al. 1997). The above observations suggest that CPP is a complex polygenic trait, that expression of CPP and SOD are related through pleiotropy, and that there are correlated evolutionary trends in CPP, SOD, and timeless expression. Consequently, we developed QTL maps of critical photoperiod and stage of diapause from F2 hybrids between populations exhibiting extreme differences in both characters, using timeless as well as other genetic markers to construct the underlying linkage map.
We sought to address four main questions: First, are there QTL for CPP that do not overlap with SOD and are therefore potentially capable of independent evolution? Second, are there QTL for CPP that do overlap with QTL for SOD and may therefore include pleiotropic genes responsible for their genetic covariation within and between populations? Third, are there QTL for CPP that involve dominance and epistasis that may account for these nonadditive genetic differences in CPP between southern and northern populations? Fourth, is timeless included within or does it interact epistatically with QTL for CPP?
MATERIALS AND METHODS
Generation of the F2 mapping population:
To derive an F2 mapping population, a single pair of adults was mated in the parental generation and their F1 offspring were mass swarmed. The parental pair was descended from wild-caught individuals collected from Florida (FL) and Alberta (AL), Canada (Table 1). A full-sib family from FL was used to generate a partially inbred line with reduced heterozygosity. The AB population was forced through a bottleneck of 30 individuals. Because the AB locality is at the extreme edge of W. smithii's range where heterozygosity is low (Armbruster et al. 1998), we did not further inbreed this population. A single FL♀ × AB♂ cross produced 19 F1 offspring that were allowed to mate en masse to generate the F2 mapping population. Both parents were frozen at −70° following reproduction. Consistently with long-established geographic variation in W. smithii (Bradshaw and Lounibos 1977), the FL female diapaused in the fourth instar and had a short CPP of 13.4 hr; the AB male diapaused in the third instar and had a long critical photoperiod of 17.4 hr (Table 1A).
TABLE 1.
Geographic and phenotypic data for parental and F2 generations
| Population | Location | Latitude, longitude | CPP (hr)a | SODb |
|---|---|---|---|---|
| A. Parental generation | ||||
| FL | NW Florida | 31.0°N, 86.5°W | 13.4 | Fourth instar |
| AB | NE Alberta | 57.5°N, 111.3°W | 17.4 | Third instar |
| Sex | Third instar | Fourth instar | Combined |
|---|---|---|---|
| B. F2 generation: mean CPP ± 2 SE (n) according to sex and instar of diapause | |||
| Female | 15.63 ± 0.17 (52) | 14.81 ± 0.12 (85) | 15.12 ± 0.12 (137) |
| Male | 15.79 ± 0.14 (86) | 15.26 ± 0.15 (42) | 15.62 ± 0.11 (127) |
| Combined | 15.73 ± 0.11 (137) | 14.96 ± 0.10 (127) | 15.36 ± 0.08 (264) |
CPP, critical photoperiod determined with increasing day length as in Lair et al. (1997).
SOD, stage of diapause determined at 30 days post-eclosion from larvae reared at 21° on diapause-inducing short days (light:dark = 8:16).
CPP and SOD in the F2 generation:
F2 larvae were reared from day of hatch under diapause-inducing, short-day conditions (light:dark = 8:16 at 21°) and scored for stage of diapause at 30 days post hatch. W. smithii are photoperiodic for the termination as well as the initiation of larval diapause so we were able to synchronize the F2 in diapause and then place the synchronized F2 on increasing day lengths to stimulate sequential development according to their individual critical photoperiods. Diapausing larvae were exposed to an initial day length of 13.25 hr that increased 3 min/day. Upon pupation, the length of the previous day was recorded as an individual's critical photoperiod, along with its sex and stage of diapause (Table 1B). Each pupa was transferred to a 1.5-ml centrifuge tube and stored at −70°.
DNA extraction:
Genomic DNA from the experimental animals, as well as that used to develop molecular markers from stock populations, was extracted with a DNeasy tissue kit (QIAGEN, Chatsworth, CA) following the protocol for insects in Appendix G of the QIAGEN manual. DNA was eluted in 30 μl of buffer AE, of which 5 μl was used for amplified fragment length polymorphisms (AFLPs) and 2 μl for amplification with the Genomiphi genomic DNA amplification kit (GE Healthcare). The Genomiphi kit uses random hexamers and a strand-displacing DNA polymerase from bacteriophage Phi29 to amplify small quantities of genomic DNA, which can then be used as a template for polymerase chain reaction (PCR) amplification using gene-specific primers. Following the manufacturer's protocol, 2 μl of genomic DNA per individual was amplified in a reaction volume of 20 μl and then diluted with sterile water to a total volume of 100 μl. The remaining DNA from the DNeasy extraction was stored at −70°, while the amplified DNA and the DNA set aside for AFLPs was stored at −20°.
Gene-based markers:
Partial sequences of 23 genes previously isolated in W. smithii were screened for polymorphisms in the FL and AB parents. Fragments were amplified via PCR, cloned using a TA Topo cloning kit (Invitrogen, San Diego), and sequenced on a capillary sequencer. Sequences were aligned using the web-based program MultAlin (Corpet 1988; http://prodes.toulouse.inra.fr/multalin/multalin.html) and searched by eye for polymorphisms. Once found, sequence differences among the parents were confirmed by restriction endonuclease digestion where possible (i.e., a restriction site found in one sequence was absent in the other due to one or more base-pair differences within the restriction site).
Once a polymorphism was confirmed, restriction digests were also used to verify homozygosity of alternate alleles in the parents and to genotype all F2 individuals. PCR primers were designed around polymorphisms so that, following a digest, the three genotypes could be easily scored on an agarose gel: homozygotes for one allele show a single uncut band, homozygotes for the other allele show two shorter bands, and heterozygotes show all three bands. Prior to restriction endonuclease digestion, polymorphic regions were amplified via PCR using 2.0 μl 10× Taq polymerase buffer, 0.32 μl 10 mm dNTPs, 0.4 μl 10 μm forward primer, 0.4 μl 10 μm reverse primer, 0.8 μl DNA template, 0.08 μl Taq DNA polymerase (5 units/μl), and 16.0 μl sterile H2O for a final reaction volume of 20 μl. Reaction conditions were 95° for 5 min plus 35 cycles of 95° for 30 sec, 60° for 30 sec, and 72° for 30 sec, plus a final 72° extension for 5 min. To confirm a positive reaction, 5 μl of the PCR product was electrophoresed on a 1% agarose gel. The remaining product served as template for the restriction digest, all of which was performed in a final volume of 40 μl with 1 unit of enzyme. Each digest proceeded for 8 hr at 37° with the exception of those using BsmBI, which were performed at 55°.
An initial screen of parental genotypes for fragments of 23 genes revealed fixed single nucleotide polymorphisms at eight loci that could be easily scored via restriction endonuclease digestion (appendix). In addition, a 96-bp insertion/deletion was found in an intron near the 3′-end of locus Ws13043, which provided a ninth codominant marker. Of the remaining genes, numerous polymorphisms were found but were unusable due to heterozygosity in one of the two parents. All F2 individuals were genotyped for the nine loci with fixed polymorphisms, and only one marker departed from the expected 1:2:1 ratio for a codominant marker in an F2 intercross (cutoff of χ2 = 9.21 for α = 0.01, 2 d.f.). However, transmission distortion was only minor at this locus, as the genotypic ratio fits expected values for α = 0.001 (cutoff of χ2= 13.82, 2 d.f.). Furthermore, transmission distortion for codominant markers is less of a concern compared to dominant markers since confirmation of homozygosity is possible in each parent.
APPENDIX.
Polymorphic markers used to distinguish FL and AB alleles in the F2 generation
| Genea | Primers (5′ → 3′) | Amplicon length | Restrictionb enzyme | Allele cut | Linkage group | Marker no. | Position (cM) |
|---|---|---|---|---|---|---|---|
| A. Gene-based codominant markers | |||||||
| Ws5096 | GATGATCGATCTGGAGCACATGGCTAAAACAGGCTACTTCA | 331 | HinfI | AB | 2 | 8 | 50.0 |
| Ws13043 | CGGCCGTGACTTATGCTAAAGCAGGAACTCCGCCATAATA | 182 | NA | NA | 2 | 17 | 123.8 |
| 278c | |||||||
| UbcD4 | CCAGCAGTGGATATGGGTGTGCGGCTCAAATCCCTAAAA | 398 | EcoRV | FL | 2 | 18 | 124.6 |
| timeless | GGTGGTGATCTGGAGCAAATCCACGCTTGGGTTGAGCAGAT | 624 | BsmBI | AB | 3 | 1 | 0.0 |
| Ws10014 | CCCCAATGGTGATGTTTCTCAGCCCGATTTTTGGAATGAT | 503 | EcoRI | AB | 3 | 2 | 4.1 |
| WsRpL17 | TCCGCAAAGCAATATTACCGGCTTGTGTCGAGCACACATT | 499 | SfaNI | FL | 3 | 4 | 18.0 |
| Ws31075 | AAGGTTGGCTGTCCATTCACGAAACGGTTTTCGTTTCGAG | 488 | BccI | AB | 3 | 5 | 22.6 |
| l(1)G0156 | GAGGCGGTTGATGTAACTCCTGGATCCTGCACCATATTCA | 540 | AciI | AB | 3 | 12 | 53.1 |
| Ws13994 | CGGAAAAAGTCCAAATGTTGTTTTTGGTTCACCTGGATCGT | 258 | HindIII | FL | 3 | 13 | 54.3 |
| Marker name/selective primersd | Dominant population | Linkage group | Marker no. | Position (cM) |
|---|---|---|---|---|
| B. Dominant AFLP markers | ||||
| EAGCMCTA.o | AB | 1 | 1 | 0.0 |
| EAGCMCTA.c | AB | 1 | 2 | 13.4 |
| EAGCMCTT.s | AB | 1 | 3 | 17.1 |
| EACCMCTT.i | AB | 1 | 4 | 25.9 |
| EAACMCTA.l | FL | 1 | 5 | 33.7 |
| EAGCMCTA.h | FL | 2 | 1 | 0.0 |
| EAGCMCTA.i | AB | 2 | 2 | 5.1 |
| EAACMCTA.f | AB | 2 | 3 | 13.9 |
| EAGCMCTA.g | FL | 2 | 4 | 16.2 |
| EAGCMCTT.k | AB | 2 | 5 | 21.8 |
| EACCMCTT.c | FL | 2 | 6 | 27.1 |
| EAGCMCTA.m | FL | 2 | 7 | 47.3 |
| EAACMCTA.e | FL | 2 | 9 | 66.0 |
| EACCMCTT.o | FL | 2 | 10 | 84.6 |
| EACCMCTT.q | FL | 2 | 11 | 88.7 |
| EAGCMCTT.t | FL | 2 | 12 | 92.8 |
| EAGCMCTT.o | AB | 2 | 13 | 95.8 |
| EAGCMCTA.d | FL | 2 | 14 | 101.9 |
| EACCMCTT.a | FL | 2 | 15 | 108.0 |
| EAGCMCTT.n | AB | 2 | 16 | 117.0 |
| EAGCMCTT.h | AB | 2 | 19 | 134.1 |
| EAACMCTA.d | AB | 2 | 20 | 151.2 |
| EAGCMCTA.b | FL | 3 | 3 | 14.7 |
| EAGCMCTT.q | AB | 3 | 6 | 31.1 |
| EAGCMCTT.i | FL | 3 | 7 | 31.7 |
| EACCMCTT.l | FL | 3 | 8 | 38.9 |
| EAACMCTA.h | FL | 3 | 9 | 42.5 |
| EAGCMCTT.r | AB | 3 | 10 | 42.9 |
| EACCMCTT.m | AB | 3 | 11 | 48.6 |
| EAACMCTA.m | AB | 3 | 14 | 56.7 |
| EAGCMCTA.a | AB | 3 | 15 | 58.7 |
| EAGCMCTT.v | FL | 3 | 16 | 61.0 |
| EAGCMCTT.y | AB | 3 | 17 | 65.0 |
| EAGCMCTA.n | FL | 3 | 18 | 67.4 |
| EAGCMCTT.j | AB | 3 | 19 | 85 |
| EAACMCTA.c | AB | 3 | 20 | 102.0 |
The genes are named according to the putative ortholog from TBLASTX results of the W. smithii sequence against the genome of D. melanogaster. The numbered genes are unannotated on FlyBase and the prefix Cg- has been replaced by Ws- for W. smithii. The GenBank accession nos. for the nine genes, in descending order, are EF094841–EF094856 and AY943312.
The restriction endonuclease used in genotyping (see materials and methods for details).
The polymorphism for Ws13043 is a 96-bp insertion/deletion. Genotyping in the F2 was performed by scoring length differences of PCR amplicons instead of by restriction endonuclease digestion.
The EcoRI and MseI core primer sequences for both the pre- and selective amplification steps of the AFLP protocol are 5′-GACTGCGTACCAATTC-3′ and 5′-GATGAGTCCTGAGTAA-3′, respectively. The three bases after the letters E and M in each marker name denote the three arbitrary nucleotides added to the 3′-end of the core sequence for selective amplification. The lowercase letter at the end of the name distinguishes between multiple markers generated by the same set of selective primers.
AFLPs:
The AFLP technique generates numerous polymorphic markers in four steps: (1) digestion of genomic DNA with two restriction endonucleases, one rare and one common cutter; (2) ligation of oligonucleotide adapters to the DNA fragments; (3) selective PCR amplification of fragment sets using primers with a core sequence plus one to three arbitrary nucleotides at the 3′-end; and (4) gel electrophoresis to separate the amplified fragments. The major advantages of this technique are that it does not require a priori knowledge about DNA sequence and that it generates multiple polymorphic markers with a single PCR reaction.
To find AFLPs in W. smithii, the protocol of Vos et al. (1995) for “complex genomes” was followed with minor changes and the substitution of fluorescently labeled primers for [γ-33P]. Briefly, 5 μl of genomic DNA was digested for 3 hr at 37° with 1 unit MseI and 2.5 units EcoRI followed by a 20-min incubation at 65°. Adaptor ligation was performed by incubating the digested DNA for 3 hr at 37° with 12.5 pmol MseI adaptor, 1.25 pmol EcoRI adaptor, 1.25 μm ATP, and 0.5 unit T4, followed by a 20-min incubation at 65° (for adaptor sequences, see Vos et al. 1995). The restriction-ligation product was then diluted 5:1 with low TE.
Following ligation, the protocol required two PCR amplifications: the first (preamplification) used primers with a single selective nucleotide at the 3′-end, while the second (selective amplification) used primers with three selective primers at the 3′-end. Each preamplification reaction was performed using 5 μl of the diluted restriction-ligation product as template plus 2.5 μl 10× Taq polymerase buffer, 1.5 μl 25 mm MgCl2, 0.5 μl 10 mm dNTPs (2.5 mm each), 1.0 μl 10 μm EcoRI + A primer, 1.0 μl 10 μm MseI + C primer, 0.1 μl Taq polymerase (5 units/μl), and 13.4 μl sterile H2O for a final volume of 25 μl (see Table 2 for the EcoRI and MseI core primer sequences). PCR reaction conditions were those given in Vos et al. (1995) for primers with a single selective nucleotide. The preamplification product was diluted 5:1 with low TE, and 5 μl of the dilution was used as template for the selective amplification step. The PCR reaction mix was the same as for the previous step with the exception of the primers and their concentrations. For selective amplification, 0.5 pmol of a fluorescently labeled EcoRI + 3 nucleotide primer and 8 pMol of an unlabeled MseI + 3 nucleotide primer were used (see Table 2 for the EcoRI and MseI core primer sequences and the selective nucleotides used for each marker). PCR reaction conditions were those given in Vos et al. (1995) for primers with three selective nucleotides.
TABLE 2.
Locations and effects of QTL for critical photoperiod and stage of diapause
| QTL | Linkage group | Positiona (cM) | LRb | ac | dd | |d/a|e | R2f |
|---|---|---|---|---|---|---|---|
| A. CPP | |||||||
| 1 | 1 | 11.0 | 137.32*** | 0.467 | 0.235 | 0.504 | 0.207 |
| 2 | 1 | 14.4 | 48.99*** | 0.381 | 0.210 | 0.550 | 0.065 |
| 3 | 2 | 13.9 | 14.81* | 0.158 | −0.058 | 0.367 | 0.016 |
| 4 | 2 | 45.1 | 16.96* | −0.528 | −0.110 | 0.208 | 0.048 |
| 5 | 2 | 50.0 | 14.95* | 0.450 | −0.069 | 0.153 | 0.015 |
| 6 | 2 | 93.8 | 29.92** | 0.187 | −0.200 | 1.070 | 0.034 |
| 7 | 2 | 122.0 | 49.96*** | 0.500 | −0.187 | 0.374 | 0.069 |
| 8 | 2 | 125.6 | 91.84*** | 0.438 | −0.182 | 0.416 | 0.115 |
| 9 | 3 | 54.3 | 46.34*** | 0.289 | −0.075 | 0.261 | 0.048 |
| B. SOD | |||||||
| 1 | 1 | 21.1 | 46.91** | −0.294 | −0.020 | 0.068 | 0.164 |
| 2 | 2 | 88.6 | 17.33* | 0.205 | 0.244 | 1.190 | 0.079 |
| 3 | 2 | 108.0 | 28.01* | 0.223 | 0.291 | 1.304 | 0.105 |
| 4 | 2 | 124.6 | 29.84* | −0.378 | 0.198 | 0.525 | 0.075 |
Each QTL position is the point on a linkage group where the likelihood ratio reaches the highest value after crossing the significance threshold (α = 0.05).
The likelihood-ratio test statistic for H0/HA: *P < 0.05, **P < 0.01, and ***P < 0.001.
Additive effect of the QTL in hours for CPP and for proportion of one larval instar for SOD. The sign indicates whether the QTL effect is toward the northern (positive) or southern (negative) parent.
Dominance effect of the QTL: the sign indicates whether the QTL effect is toward the northern (positive) or southern (negative) parent. The scale is in hours for CPP and instar stages for SOD.
Absolute value of the ratio of dominance to additive effects; a QTL is considered completely dominant when |d/a| = 1, partially dominant when |d/a| is between 0 and 1, additive when|d/a| = 0.
Proportion of the phenotypic variance explained by the QTL.
After selective amplification, 10 μl of loading dye was added to all samples, which were then denatured for 3 min at 95°. A total of 1.5 μl of each denatured sample was loaded on a 5.7% denaturing polyacrylamide gel (25 cm plates) and run at 1500 V, 20 mA, 40 W on a Li-cor 4200 sequencer. Gel images were collected by the Li-cor software for 10 frames at a scan speed of 4 and saved as TIFF files. Polymorphic AFLPs were scored by eye and verified independently by at least two of the authors using the program RFLPscan 3.0 (Scanalytics).
Initially, 16 primer combinations were screened with the above protocol using DNA from the FL and AB stock populations to identify the most promising primer sets based on clarity, repeatability, and number of polymorphic bands. Four combinations were chosen and used to genotype the two parents and all F2 individuals for 77 polymorphic markers. Once scored, the genotypes for each marker were entered into a spreadsheet as 0's or 1's for absent or present, respectively, and then converted into Mapmaker 3.0 format for dominant markers. A segregation ratio was then calculated for each AFLP and a chi-square test (cutoff of χ2 = 6.64 for α = 0.01, 1 d.f.) was used to test goodness of fit to the 3:1 Mendelian expectation. Only the 36 AFLPs showing the expected 3:1 ratio were included in the linkage map (appendix).
Linkage map construction:
The F2 mapping population consisted of 264 individuals genotyped for 45 markers. Initially, the markers were separated into two groups of overlapping data sets, one with the 36 dominant AFLP markers, the other with the 9 codominant markers. Each set was then used to produce two separate linkage maps using Mapmaker 3.0 (Lander et al. 1987). The codominant markers on both maps provided landmarks so that the two could be merged into one map. This method was chosen to avoid long stretches of AFLPs with positions biased by linkage phase, which in turn can lead to the misordering of closely linked markers of the opposite phase.
To generate the two linkage-phase maps, the first step was to sort each subset of AFLP plus codominant markers into likely linkage groups (LGs) using the “group” command with the Kosambi mapping function (Kosambi 1944) (two-point linkage criteria: minimum LOD 6.0, maximum distance between markers of 30 cM). Marker order and position was then estimated using the “compare” command and then refined with the “ripple” command. The complete set of markers was then remapped using the “try,” “compare,” and “ripple” commands. The final process was expedited by using the two linkage-phase maps as guides.
Homology with other mosquitoes:
To find orthologs of W. smithii genes in other mosquitoes, we used the TBLASTX algorithm to search the Anopheles gambiae and Aedes aegypti genomes in the Ensembl database (Birney et al. 2004; http://www.ensembl.org/index.html). We used the default parameters for the program, which compares a translated DNA query with a translated DNA database. In cases with more than one match, the best TBLASTX match was assumed to represent the corresponding ortholog. For all nine genes, there were substantial decreases in both E-values and BLAST scores between the first and second matches, suggesting that each is a single-copy gene in both An. gambiae and Ae. aegypti.
Mapping the sex locus:
In mosquitoes of the subfamily Culicinae (which includes W. smithii), males are heterozygous (Mm) and females are homozygous recessive (mm) (Gilchrist and Haldane 1947). The sex-determining locus in W. smithii was mapped by performing a chi-square test for sex ratio and marker genotype. The expectation is that the three possible genotypes of each gene-based marker and the two possible genotypes of each AFLP will have a 1:1 sex ratio. The point of maximal departure from this expectation approximates the position of the sex locus (Wilcox 1995).
Composite interval mapping:
QTL underlying geographic variation in both CPP and SOD were mapped in the F2 generation using composite interval mapping (CIM) (Zeng 1994) via Windows QTL Cartographer version 2.5 (Wang et al. 2006). Unlike critical photoperiod, which is a continuous trait, stage of diapause is categorical with two possible states in W. smithii. Although CIM was developed to analyze continuous characters, it also works for categorical traits that have an underlying polygenic basis (McIntyre et al. 2001). In essence, CIM combines interval mapping (Lander et al. 1987) with multiple regression to test for the presence of QTL within each marker interval, while using specific markers as cofactors to account statistically for QTL outside the test interval. The likelihood-ratio (LR) test statistic is −2 ln(L0/L1), where L0/L1 is the ratio of the likelihood of the null hypothesis (i.e., no QTL in the test interval) to the likelihood of the alternative hypothesis (i.e., a QTL present in the test interval) (Basten et al. 2004). Two parameters affecting QTL detection with CIM are (1) number of marker cofactors in the multiple regression and (2) size of the exclusion window flanking the test interval. The number of marker cofactors is left to the user's discretion with a default value of 5. Increasing cofactor number may improve the resolution of linked QTL (Basten et al. 2004), but also increases the risk of type 2 error and an overly conservative map. The other key parameter, window size, is also left to the user's discretion (default value of 10 cM). Essentially, this parameter removes from the analysis any marker cofactor located within the specified window flanking the test interval. Thus, if the window size is broad, closely linked markers with large effects are not taken into account and may therefore inflate the likelihood ratio of a given interval. Conversely, making the window size too narrow may eliminate or diminish a true QTL signal.
Both linkage and genetic background are factors that must be considered, given that W. smithii has only three pairs of chromosomes and that epistasis contributes to geographic variation in CPP in this species (Hard et al. 1992; Lair et al. 1997). We varied the number of conditioning markers from 5 to 20 and the exclusion window from 2.5 to 20 cM. We sought a compromise between the marker number and window size that minimized the effects of linkage, while not eliminating the background effect of non-QTL regions. Ultimately, we used the 10 markers with the greatest effect (based on stepwise forward regression) as cofactors with an exclusion window of 2.5 cM.
Under these parameters, the likelihood-ratio test statistic was computed at every centimorgan across all marker intervals. QTL significance thresholds for all parameter sets were estimated by permutation tests. Briefly, trait data and marker genotypes were permuted 1000 times and the maximum-likelihood ratio statistic across all intervals was recorded for each permutation. Likelihood statistics computed from the original data that exceeded the 50th greatest likelihood-ratio statistic from the permuted data were significant at the level of α = 0.05 under the null hypothesis (Churchill and Doerge 1994).
Estimation of QTL effects:
Both additive (a) and dominance (d) effects and the proportion of phenotypic variance explained were estimated for each QTL using Windows QTL Cartographer version 2.5 (Wang et al. 2006). Briefly, estimates of a and d were obtained by maximum likelihood through an expectation/conditional maximization algorithm (Meng and Rubin 1993). The proportion of the variance explained by a QTL was estimated by the equation 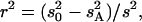 where s2 is the trait variance,
where s2 is the trait variance, 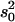 is the sample variance of the residuals under the null model, and
is the sample variance of the residuals under the null model, and 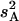 is the variance of the residuals under the alternative model (Basten et al. 2004).
is the variance of the residuals under the alternative model (Basten et al. 2004).
QTL sign test:
To test for evidence that the parental phenotypes diverged through natural selection, a QTL sign test (Orr 1998) was performed using the additive effects. Under the null model of neutral evolution, the expectation is that an equal number of antagonistic (i.e., plus and minus) alleles are responsible for the phenotypic difference. In contrast, directional selection should favor the accumulation of consistently signed alleles, which in this case are plus alleles toward the northern parent. To perform the test, we determined the conditional probability of observing by chance n “plus” QTL of m total, given the phenotypic difference (R) between parents. Prior to calculating this probability, the additive effects were fitted to a gamma distribution to approximate shape and scale parameters required by the test. The threshold for heterozygous QTL effect was set at 0.1 and the test was performed using program code downloaded from the website cited in Orr (1998).
Epistasis:
For CPP, digenic epistasis was assessed using ANOVA models to evaluate interactions between all possible marker pairs. For each ANOVA, marker genotypes were used as factors and markers were considered epistatic if there was a significant interaction term. For SOD, digenic epistasis was assessed using log-linear models with frequency as the dependent variable and the trait and marker genotypes as factors. Two models were then computed: (1) a model including all factors, all two-way interactions, and the three-way interaction and (2) a model including all factors and all two-way interactions (i.e., the three-way interaction was excluded). A likelihood-ratio test was then used to determine the significance of the three-way interaction term. Two markers were considered epistatic if the log-linear model including the three-way interaction had a significantly higher likelihood than the model excluding the term.
To account for multiple testing in the epistasis analysis, we performed the Benjamini–Hochberg test (Benjamini and Hochberg 1995; Pavlidis 2003), a post-hoc false discovery rate with the expected value of Q, defined in the expression Q = V/(V + S), where V is the number of false rejections and S is the number of correct rejections of the null hypothesis (Sabatti et al. 2003). For all three traits, a significance level of α = 0.001 led to at most one false-positive result and was thus set as the level of significance in this study. All ANOVAs and Benjamini–Hochberg tests were performed using the statistical program R (R Development Core Team 2006).
RESULTS
Trait values for parents and F2 generation:
The individuals crossed in the parental generation differed in CPP by 4 hr (Table 1A). In the F2 generation, males and larvae diapausing as third instars, respectively, had longer CPPs than females (F1,262 = 35.42, P < 0.0001) and larvae diapausing as fourth instars (F1,262= 106.77, P < 0.0001) (Table 1B).
Linkage map:
The W. smithii FL × AB linkage map consists of 45 marker loci spanning 286.9 cM on three linkage groups (Figure 1). Average interval length or marker spacing (s) was estimated at s = 6.82 cM by dividing the summed length of all linkage groups by the number of intervals (Fishman et al. 2001). Genome length (L) was estimated using two different methods. For the first method, we assumed a random distribution of markers and added 2s to the length of each linkage group to account for the ends of chromosomes beyond the terminal markers (Fishman et al. 2001). This approach yielded an estimate of L = 327.2 cM. For the second method, we multiplied the length of each linkage group by the factor (m + 1)/(m − 1), where m is the number of the markers for a specific linkage group (Chakravarti et al. 1991). This approach yielded an estimate of L = 330.4 cM. Next, we calculated map coverage from c = 1 − e−2dn/L (Fishman et al. 2001). Using L = 330, we found that 93.5 and 99.6% of the genome lies within 10 and 20 cM of a marker, respectively. Thus, the proportion of the genome that is included in our linkage map is well covered. Finally, we approximated the relationship between linkage map units and units of DNA sequence by dividing W. smithii's estimated genome size of 850 Mb (Rao and Rai 1990) by L = 330 cM, yielding a minimum average of 2.58 Mb/cM.
Figure 1.—

Linkage map for W. smithii showing the linkage groups (Lg) based on the gene-based and AFLP markers in the appendix.
Correspondence to other mosquitoes:
By convention, chromosomes of culicine mosquitoes are named according to length, with the shortest designated as chromosome 1, the longest as chromosome 2, and the intermediate as chromosome 3 (Clements 1992, p. 3). Hence, numbers were assigned to the three linkage groups according to the chromosome that each is most likely to represent (Figure 1).
Position of the sex locus:
Using a chi-square test, we found that the sex locus is on linkage group 1. The sex ratio of the two genotypes for each AFLP marker on this linkage group departed significantly from the 1:1 expectation, while no marker genotypes on linkage groups 2 and 3 departed from a 1:1 ratio. On linkage group 1, the markers EAGCMCTA.c and EAGCMCTT.s have the highest chi-square values, with the former having only 1 male and 49 female recessive homozygotes and the latter having 0 male and 52 female recessive homozygotes. We therefore estimate that the sex locus lies within the 2.3 cM region between these two markers.
QTL for CPP:
CIM detected nine QTL underlying geographic variation in CPP, accounting for 61.7% of the variation in CPP between FL and AB (Figure 2, Table 2). Two QTL each account for >10% of the variance, one located on linkage group 1 (QTL 1, 20.7%) and the other on linkage group 2 (QTL 8, 11.5%). Of the remaining seven QTL, one is located on linkage group 1 (QTL 2, 6.5%), five on linkage group 2 (QTL 3–7, 1.5–6.9%), and one on linkage group 3 (QTL 9, 4.8%). Additive effects were generally positive, as 8 of 9 QTL were toward the northern parent with a longer CPP. Orr's (1998) QTL sign test indicated that significant directional selection has acted on CPP (P = 0.039).
Figure 2.—

Composite interval map for critical photoperiod. (Top) Likelihood ratios. (Bottom) The corresponding R2 for each of the nine QTL. Digenic epistatic interactions between markers are shown as brackets subtended by a dashed line in the top graph. For clarity, individual markers from Figure 1 are shown only if they are mentioned in the results and discussion. For the gene-based markers, both homozygotes and the heterozygote were distinguishable.
Dominance effects were generally negative and toward the southern parent with a shorter CPP (Table 2). Dominance (d/a) was complete for QTL 6, accounting for 3.4% of the variance in CPP, and lowest for QTL 5, accounting for 1.5% of the variance; the two major QTL (1 and 8) had intermediate levels of dominance.
QTL for SOD:
CIM resolved four QTL for SOD, accounting for 42.3% of the variation in SOD between FL and AB (Figure 3, Table 2). Two QTL each account for >10% of the variance, one located on linkage group 1 (QTL 1, 16.4%) and the other on linkage group 2 (QTL 3, 10.5%). Both of the remaining two QTL are located on linkage group 2 (QTL 2, 7.9%; QTL 4, 7.5%) and none is detected on linkage group 3. Additive effects were evenly split between positive toward the northern parent diapausing in the third instar and negative toward the southern parent diapausing in the fourth instar. Orr's (1998) sign test could not be performed on QTL for SOD because a minimum of six QTL are necessary for the test to be valid. Dominance effects were generally positive toward the northern parent (Table 2). Dominance (d/a) was complete for QTL 2 and 3, accounting for 18.4% of the variance in SOD; intermediate for QTL 4, accounting for 7.5% of the variance; and virtually absent for QTL 1, accounting for 16.4% of the variance.
Figure 3.—

Composite interval map for stage of diapause. Top and bottom graphs are as in Figure 2. The dashed red line plots QTL for critical photoperiod as a reference. Note that the QTL for critical photoperiod are scaled down to fit the lower values of the likelihood ratios for SOD and that only the significant QTL for critical photoperiod are shown. The digenic epistatic interaction in LG2 is shown as a bracket subtended by a dashed line in the top graph.
Epistasis for CPP:
Fourteen interactions were significant using the false discovery rate procedure, which estimates that no more than 1 of the 14 interactions is a false positive (Table 3A). These 14 interactions fell into three clusters, each associated with a specific marker in a separate linkage group (Figures 4 and 5). In linkage group 1, the marker EAGCMCTA.o at 0 cM interacted with three other markers, two of which spanned CPP QTL 2 (Figure 2) and the sex locus (Figure 1). In linkage group 2, the marker EACCMCTT.a at 108 cM interacted with markers in CPP QTL 5 (Ws5096), CPP QTL 6 (EAGCMCTT.t), SOD QTL 3 (EACCMCTT.a), and with two markers in SOD QTL 4 (Ws13043, UbcD4) (Figures 2 and 3). In linkage group 3, timeless at 0 cM interacted with a marker in CPP QTL 9 [l(1)G0156] (Figure 2).
TABLE 3.
Epistatic interactions between marker pairs affecting variation in critical photoperiod and stage of diapause
| Marker 1 | Linkage group | Position (cM) | Marker 2 | Linkage group | Position (cM) | Interaction P-value |
|---|---|---|---|---|---|---|
| A. Critical photoperiod | ||||||
| EAGCMCTA.o | 1 | 0.0 | EAGCMCTA.c | 1 | 13.4 | 1.32E-07 |
| EAGCMCTA.o | 1 | 0.0 | EAGCMCTT.s | 1 | 17.1 | 3.19E-08 |
| EAGCMCTA.o | 1 | 0.0 | EACCMCTT.i | 3 | 38.9 | 1.01E-06 |
| EACCMCTT.a | 2 | 108.0 | Ws5096 | 2 | 50.0 | 7.83E-04 |
| EACCMCTT.a | 2 | 108.0 | EAACMCTA.e | 2 | 66.0 | 1.76E-04 |
| EACCMCTT.a | 2 | 108.0 | EAGCMCTT.t | 2 | 92.8 | 1.29E-04 |
| EACCMCTT.a | 2 | 108.0 | Ws13043 | 2 | 123.8 | 4.41E-04 |
| EACCMCTT.a | 2 | 108.0 | WsUbcD4 | 2 | 124.6 | 8.25E-04 |
| Wstimeless | 3 | 0.0 | EAGCMCTA.b | 3 | 14.7 | 1.70E-04 |
| Wstimeless | 3 | 0.0 | EACCMCTT.l | 3 | 38.9 | 7.43E-04 |
| Wstimeless | 3 | 0.0 | EAACMCTA.h | 3 | 42.5 | 2.34E-04 |
| Wstimeless | 3 | 0.0 | Wsl(1)G0156 | 3 | 53.1 | 8.26E-04 |
| Wstimeless | 3 | 0.0 | EAGCMCTT.v | 3 | 61.0 | 3.12E-04 |
| Wstimeless | 3 | 0.0 | EAGCMCTA.n | 3 | 67.4 | 6.16E-05 |
| B. Stage of diapause | ||||||
| EACCMCTT.a | 2 | 108.0 | EACCMCTT.q | 2 | 88.7 | 6.72E-04 |
Figure 4.—

Epistatic interactions affecting critical photoperiod (CPP) in LG1 (top row) and LG2 (bottom two rows). Note that the AFLP markers are dominant so that individuals are scored either as homozygous recessive or as combined homozygous dominant plus heterozygote. The first marker is indicated on the horizontal axis, the second marker at the top of each plot. Nonparallel lines indicate epistatic interaction. SoHom and NoHom indicate southern (FL) and northern (AB) homozygotes; Het indicates a north–south heterozygote. The error bars show ± 1 standard error.
Figure 5.—

Epistatic interactions affecting critical photoperiod in LG3. All involve timeless, which is codominant. Abbreviations are as in Figure 4.
Epistasis for SOD:
Only one interaction was significant using the false discovery rate criterion (Table 3B). In linkage group 2, marker EACCMCTT.a at 108 cM within SOD QTL 3 interacted with EACCMCTT.q at 88.7 cM within CPP QTL 2 (Figures 2 and 3).
DISCUSSION
To our knowledge, the QTL map in Figure 2 provides the first such map of CPP in any animal. The underlying linkage map (Figure 1) shows three linkage groups, consistent with the haploid number of chromosomes in W. smithii (Moeur and Istock 1982), and shows the sex locus on the shortest chromosome, as in other mosquitoes (Clements 1992, p. 2). In Figures 2 and 3, we mark nine QTL for CPP and four QTL for SOD. We note first that this number may be an overestimate because CPP QTL 1–2, 4–5, and 7–8 and SOD QTL 2–4 may each constitute a single QTL; conversely, there are undoubtedly many genes embedded within each QTL and, hence, additional QTL below our level of resolution (Erickson et al. 2004; Mackay 2004). Second, because of our sample size of 264 F2 individuals, we are likely overestimating the magnitude of QTL effects, the variation of effects among QTL, and both the additive and dominance effects in Table 2 (Beavis 1998). With these caveats in mind, the QTL map of CPP confirms earlier conclusions that a complex genetic architecture underlies photoperiodism in W. smithii (Bradshaw and Holzapfel 2000, 2001b; Bradshaw et al. 2005). CPP QTL 1 plus QTL 3–5 account for 29% of the phenotypic variation in CPP, have no detectable overlap with QTL for SOD, and are not involved in any of the significant epistatic interactions. The remaining five QTL for CPP (2, 6–9), accounting for 33% of the variation, were sex linked, overlapped with QTL for SOD, or contained markers with significant epistatic interactions.
Females in the F2 hybrids had shorter CPPs than males (Table 1B), likely due to linkage of the sex locus with QTL 2 for CPP (Figure 2) because the female grandparent came from the southern locality with the short CPP (Table 1). In Drosophila littoralis, critical photoperiod is inherited primarily as a single autosomal Mendelian unit, but the X chromosome of a southern population exhibited a recessive factor having some influence on the expression of diapause (Lumme 1981, p. 243). Hybrids between D. lummei females and D. virilis males with a white-eye marker and backcrossed with D. virilis males showed that photoperiodic diapause was controlled by a monofactorial unit on the X chromosome (Lumme and Keränen 1978). In D. triauraria, phenotypic frequencies among recombinant inbred lines suggest that the difference in the photoperiodic response between diapausing and nondiapausing strains is due to genes at three or four loci, at least one of which is located on the X chromosome (Kimura and Yoshida 1995). Photoperiodic control of diapause among species of Drosophila therefore generally involves a less complex genetic architecture than is found in W. smithii but, like W. smithii, often involves sex-linked genes.
The latter half of the second chromosome from ∼66 to 134 cM represents a region of overlap between QTL for CPP and SOD as well as epistatic interactions for CPP between EACCMCTT.a and five other markers within the QTL for CPP and SOD (Figures 2 and 3). The epistatic interaction between EACCMCTT.a and WsUbcD4 in this region is important because in the genome of the mosquito An. gambiae UbcD4 is tightly linked with putative orthologs of the Drosophila genes protein on ecdysone puffs (Pep) and Ecdysone-inducible gene L3 (ImpL3) (Birney et al. 2004; Montgomery et al. 2004). Both of these genes are activated during development by ecdysteroid (Amero et al. 1993; Andres et al. 1993). The implication is that genes in this region of the second chromosome are involved in complex interactions coordinating the external environment (photoperiod) with both active development (ecdysteroid-activated genes) and diapause (SOD).
In the drosophilid fly Chymomyza costata, the nonphotoperiodic diapause strain has arrhythmic eclosion, does not enter diapause on short days, and has mutations at the circadian rhythm genes period and timeless. The mutation at the period locus renders flies behaviorally arrhythmic but they remain normally photoperiodic if they possess wild-type timeless. The double mutant is both behaviorally arrhythmic and nonphotoperiodic (Pavelka et al. 2003). Flesh flies (Sarcophaga bullata) that have elevated expression of period and timeless are behaviorally arrhythmic and nonresponsive to short days for the induction of diapause (Goto et al. 2006). Finally, period null mutants in the Canton-S strain of D. melanogaster are behaviorally arrhythmic but have a robust photoperiodic response curve, albeit shifted toward shorter day lengths (Saunders 1990). In combination, these results indicate that even if a functional, period-based circadian clock is not necessary for photoperiodic time measurement in flies, an individual circadian clock gene, such as timeless, may still have an ancillary effect on photoperiodism independently of and incidentally to its role in circadian rhythmicity.
In W. smithii, the expression of timeless covaries with CPP geographically and with SOD within a polymorphic population (Mathias et al. 2005). However, timeless does not overlap with any QTL for SOD (Figures 2 and 3), suggesting that any functional connection between timeless with SOD is due to differences between the Alberta and Florida populations in regulatory regions (e.g., transcription factors) located elsewhere in the genome (Arnosti 2003; Wray et al. 2003) or within a QTL for CPP on the third chromosome below our level of detection. Nevertheless, timeless, or a gene closely linked to it, does have multiple epistatic interactions with other markers on the third chromosome, including markers within QTL 9 for CPP (Figures 2 and 5). These results indicate that timeless itself plays no detectable role in the evolution of SOD, but that timeless may play an ancillary role in the evolution of photoperiodism in W. smithii.
In summary, we use geographic variation between natural populations of W. smithii to illustrate the evolution of two physiological traits essential to fitness in a seasonal environment: the photoperiodic timing of hibernal diapause (CPP) and the developmental stage of diapause itself (SOD). Significant positive, additive effects for CPP substantiate earlier conclusions that, in W. smithii, CPP has undergone directional selection on a latitudinal scale. The main region of overlap in QTL for CPP and SOD also coincides with two developmental genes activated by ecdysteroid, revealing a portion of the W. smithii genome that is involved in active development, diapause, and their photoperiodic regulation. QTL for CPP are involved in a number of epistatic interactions, reflecting genetic differences due to epistasis in CPP between southern and northern populations identified in earlier studies. Finally, the key circadian clock gene, timeless, does not overlap with any detectable QTL for CPP but does interact epistatically with a marker in one of them, suggesting that timeless may play an indirect role in the evolution of the photoperiodic timer. This genetic architecture underlying photoperiodism not only has permitted diversification of W. smithii from the Gulf of Mexico to northern Canada, but also has enabled it to track recent rapid climate change.
Acknowledgments
We thank K. Emerson, M. Haley, P. Phillips, J. Postlethwait, and M. Whitlock for useful discussion and for reading previous versions of the manuscript; B. Kolaczkowski and K. Emerson for computational support, and D. Houle and two anonymous reviewers for helpful comments. We gratefully acknowledge support from National Institutes of Health training grant to G. F. Sprague (23T2GM07413-26) and from the National Science Foundation through a Doctoral Dissertation Improvement Grant for D.M. (IBN-0408154) and research grants DEB-9806278, IBN-9814438, IOB-0415653, and IOB-0445710 to W.E.B.
References
- Amero, S. A., M. J. Matunis, E. L. Matunis, J. W. Hockensmith, G. Raychaudhuri et al., 1993. A unique ribonucleoprotein complex assembles preferentially on ecdysone-responsive sites in Drosophila melanogaster. Mol. Cell. Biol. 13: 5323–5330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andres, A. J., J. C. Fletcher, F. D. Karim and C. S. Thummel, 1993. Molecular analysis of the initiation of insect metamorphosis: a comparative study of Drosophila ecdysteroid-regulated transcription. Dev. Biol. 160: 388–404. [DOI] [PubMed] [Google Scholar]
- Armbruster, P. A., W. E. Bradshaw and C. M. Holzapfel, 1998. Effects of postglacial range expansion on allozyme and quantitative genetic variation in the pitcher-plant mosquito, Wyeomyia smithii. Evolution 52: 1697–1704. [DOI] [PubMed] [Google Scholar]
- Arnosti, D. N., 2003. Analysis and function of transcriptional regulatory elements: insights from Drosophila. Annu. Rev. Entomol. 48: 579–602. [DOI] [PubMed] [Google Scholar]
- Basten, C. J., B. S. Weir and Z. B. Zeng, 2004. QTL Cartographer: A Reference Manual and Tutorial for QTL Mapping. Department of Statistics, North Carolina State University, Raleigh, NC.
- Beavis, W. D., 1998. QTL analysis: power, precision, and accuracy, pp. 145–162 in Molecular Dissection of Complex Traits, edited by A. H. Patterson. CRC Press, Boca Raton, FL.
- Benjamini, Y., and Y. Hochberg, 1995. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Statist. Soc. B 57: 289–300. [Google Scholar]
- Birney, E., T. D. Andrews, P. Bevan, M. Caccamo, Y. Chen et al., 2004. An overview of ensembl. Genet. Res. 14: 925–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradshaw, W. E., 1976. Geography of photoperiodic response in a diapausing mosquito. Nature 262: 384–386. [DOI] [PubMed] [Google Scholar]
- Bradshaw, W. E., and C. M. Holzapfel, 2000. The evolution of genetic architectures and the divergence of natural populations, pp. 245–263 in Epistasis and the Evolutionary Process, edited by J. B. Wolf, E. D. Brodie, III and M. J. Wade. Oxford University Press, New York.
- Bradshaw, W. E., and C. M. Holzapfel, 2001. a Genetic shift in photoperiodic response correlated with global warming. Proc. Natl. Acad. Sci. USA 98: 14509–14511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradshaw, W. E., and C. M. Holzapfel, 2001. b Phenotypic evolution and the genetic architecture underlying photoperiodic time measurement. J. Insect Physiol. 47: 809–820. [Google Scholar]
- Bradshaw, W. E., and C. M. Holzapfel, 2006. Evolutionary response to rapid climate change. Science 312: 1477–1478. [DOI] [PubMed] [Google Scholar]
- Bradshaw, W. E., and L. P. Lounibos, 1977. Evolution of dormancy and its photoperiodic control in pitcher-plant mosquitoes. Evolution 31: 546–567. [DOI] [PubMed] [Google Scholar]
- Bradshaw, W. E., P. A. Zani and C. M. Holzapfel, 2004. Adaptation to temperate climates. Evolution 58: 1748–1762. [DOI] [PubMed] [Google Scholar]
- Bradshaw, W. E., B. P. Haggerty and C. M. Holzapfel, 2005. Epistasis underlying a fitness trait within a natural population of the pitcher-plant mosquito, Wyeomyia smithii. Genetics 169: 485–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradshaw, W. E., C. M. Holzapfel and D. Mathias, 2006. Circadian rhythmicity and photoperiodism in the pitcher-plant mosquito: Can the seasonal timer evolve independently of the circadian clock? Am. Nat. 167: 601–605. [DOI] [PubMed] [Google Scholar]
- Chakravarti, A., L. K. Lasher and J. E. Reefer, 1991. A maximum likelihood method for estimating genome length using genetic linkage data. Genetics 128: 175–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Churchill, G. A., and R. W. Doerge, 1994. Empirical threshold values for quantitative trait mapping. Genetics 138: 963–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clements, A. N., 1992. The Biology of Mosquitoes: Development, Nutrition, and Reproduction. Chapman & Hall, London.
- Cooke, B. D., 1977. Factors limiting the distribution of the wild rabbit in Australia. Proc. Ecol. Soc. Aust. 10: 113–120. [Google Scholar]
- Corpet, F., 1988. Multiple sequence alignment with hierarchical clustering. Nucleic Acids Res. 16: 10881–10890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danilevskii, A. S., 1965. Photoperiodism and Seasonal Development in Insects. Oliver & Boyd, Edinburgh.
- Danks, H. V., 1987. Insect Dormancy: An Ecological Perspective. Biological Survey of Canada (Terrestrial Arthropods), Ottawa.
- Erickson, D. L., C. B. Fenster, H. K. Stenøien and D. Price, 2004. Quantitative trait locus analyses and the study of evolutionary process. Mol. Ecol. 13: 2505–2522. [DOI] [PubMed] [Google Scholar]
- Fishman, L., A. J. Kelly, E. Morgan and J. H. Willis, 2001. A genetic map in the Mimulus guttatus species complex reveals transmission ratio distortion due to heterospecific interactions. Genetics 159: 1701–1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilchrist, B. M., and J. B. S. Haldane, 1947. Sex linkage and sex determination in a mosquito, Culex molestus. Hereditas 33: 175–190. [Google Scholar]
- Goto, S. G., B. Han and D. L. Denlinger, 2006. A nondiapausing variant of the flesh fly, Sarcophaga bullata, that shows arrhythmic adult eclosion and elevated expression of two circadian clock genes, period and timeless. J. Insect Physiol. 52: 1213–1218. [DOI] [PubMed] [Google Scholar]
- Hard, J. J., W. E. Bradshaw and C. M. Holzapfel, 1992. Epistasis and the genetic divergence of photoperiodism between populations of the pitcher-plant mosquito, Wyeomyia smithii. Genetics 131: 389–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hard, J. J., W. E. Bradshaw and C. M. Holzapfel, 1993. The genetic basis of photoperiodism and its evolutionary divergence among populations of the pitcher-plant mosquito, Wyeomyia smithii. Am. Nat. 142: 457–473. [DOI] [PubMed] [Google Scholar]
- Kimura, M. T., and T. Yoshida, 1995. A genetic analysis of photoperiodic reproductive diapause in Drosophila triauraria. Physiol. Entomol. 20: 253–256. [Google Scholar]
- Kosambi, D. D., 1944. The estimation of map distances from recombination values. Ann. Eugen. 12: 172–175. [Google Scholar]
- Lair, K. P., W. E. Bradshaw and C. M. Holzapfel, 1997. Evolutionary divergence of the genetic architecture underlying photoperiodism in the pitcher-plant mosquito, Wyeomyia smithii. Genetics 147: 1873–1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lander, E. S., P. Green, J. Abrahamson, A. Barlow, M. Daley et al., 1987. MAPMAKER: an interactive computer package for constructing primary genetic linkage maps of experimental and natural populations. Genomics 1: 174–181. [DOI] [PubMed] [Google Scholar]
- Lounibos, L. P., and W. E. Bradshaw, 1975. A second diapause in Wyeomyia smithii: seasonal incidence and maintenance by photoperiod. Can. J. Zool. 53: 215–221. [DOI] [PubMed] [Google Scholar]
- Lounibos, L. P., R. L. Escher and R. Lorenço-De-Oliveira, 2003. Asymmetric evolution of photoperiodic diapause in temperate and tropical invasive populations of Aedes albopictus (Diptera: Culicidae). Ann. Entomol. Soc. Am. 96: 512–518. [Google Scholar]
- Lumme, J., 1981. Localization of the genetic unit controlling the photoperiodic adult diapause in Drosophila littoralis. Hereditas 94: 241–244. [Google Scholar]
- Lumme, J., and L. Keränen, 1978. Photoperiodic diapause in Drosophila lummei Hackman is controlled by an X-chromosomal factor. Hereditas 89: 261–262. [Google Scholar]
- Mackay, T. F. C., 2004. The genetic architecture of quantitative traits: lessons from Drosophila. Curr. Opin. Genet. Dev. 14: 253–257. [DOI] [PubMed] [Google Scholar]
- Mathias, D., L. Jacky, W. E. Bradshaw and C. M. Holzapfel, 2005. Geographic and developmental variation in expression of the circadian rhythm gene, timeless, in the pitcher-plant mosquito, Wyeomyia smithii. J. Insect Physiol. 51: 661–667. [DOI] [PubMed] [Google Scholar]
- McIntyre, L. M., C. J. Coffman and R. W. Doerge, 2001. Detection and localization of a single binary trait locus in experimental populations. Genet. Res. 78: 79–92. [DOI] [PubMed] [Google Scholar]
- Meng, X. L., and D. B. Rubin, 1993. Maximum likelihood estimation via the ECM algorithm: a general framework. Biometrika 80: 267–278. [Google Scholar]
- Moeur, J. E., and C. A. Istock, 1982. Chromosomal polymorphisms in the pitcher-plant mosquito, Wyeomyia Smithii. Chromosoma 84: 623–651. [Google Scholar]
- Montgomery, S. B., T. Astakhova, M. Bilenky, E. Birney, T. Fu et al., 2004. Sockeye: a 3D environment for comparative genomics. Genet. Res. 14: 956–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orr, H. A., 1998. Testing natural selection vs. genetic drift in phenotypic evolution using quantitative trait locus data. Genetics 149: 2099–2104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavelka, J., K. Shimada and V. Kostal, 2003. TIMELESS: A link between fly's circadian and photoperiodic clocks? Eur. J. Entomol. 100: 255–266. [Google Scholar]
- Pavlidis, P., 2003. Using ANOVA for gene selection from microarray studies of the nervous system. Methods 31: 282–289. [DOI] [PubMed] [Google Scholar]
- Rao, P. N., and K. S. Rai, 1990. Genome evolution in the mosquitoes and other closely related members of superfamily Culicoidea. Hereditas 113: 139–144. [DOI] [PubMed] [Google Scholar]
- R Development Core Team, 2006. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing, Vienna.
- Sabatti, C., S. Service and N. Freimer, 2003. False discovery rate in linkage and association genome screens for complex disorders. Genetics 164: 829–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunders, D. S., 1990. The circadian basis of ovarian diapause regulation in Drosophila melanogaster: Is the period gene causally involved in photoperiodic time measurement? J. Biol. Rhythms 5: 315–331. [DOI] [PubMed] [Google Scholar]
- Saunders, D. S., 2002. Insect Clocks. Elsevier, Amsterdam.
- Tauber, M. J., C. A. Tauber and S. Masaki, 1986. Seasonal Adaptations of Insects. Oxford University Press, New York.
- Taylor, F., and J. B. Spalding, 1986. Geographical patterns in the photoperiodic induction of hibernal diapause, pp. 66–85 in The Evolution of Insect Life Cycles, edited by F. Taylor and R. Karban. Springer-Verlag, New York.
- Vos, P., R. Hogers, M. Bleeker, M. Reijans, T. van de Lee et al., 1995. AFLP: a new technique for DNA fingerprinting. Nucleic Acids Res. 23: 4407–4414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, S., C. J. Basten and Z. B. Zeng, 2006. Windows QTL Cartographer. Department of Statistics, North Carolina State University, Raleigh, NC.
- Wilcox, P., 1995. Genetic Dissection of Fusiform Rust Resistance in Loblolly Pine. Ph.D. Thesis, North Carolina State University, Raleigh, NC.
- Wray, G. A., M. W. Hahn, E. Abouheif, J. P. Balhoff, M. Pizer et al., 2003. The evolution of transcriptional regulation in eukaryotes. Mol. Biol. Evol. 20: 1377–1419. [DOI] [PubMed] [Google Scholar]
- Zeng, Z. B., 1994. Precision mapping of quantitative trait loci. Genetics 136: 1457–1468. [DOI] [PMC free article] [PubMed] [Google Scholar]