

Abstract
A maximum-likelihood estimator for pairwise relatedness is presented for the situation in which the individuals under consideration come from a large outbred subpopulation of the population for which allele frequencies are known. We demonstrate via simulations that a variety of commonly used estimators that do not take this kind of misspecification of allele frequencies into account will systematically overestimate the degree of relatedness between two individuals from a subpopulation. A maximum-likelihood estimator that includes FST as a parameter is introduced with the goal of producing the relatedness estimates that would have been obtained if the subpopulation allele frequencies had been known. This estimator is shown to work quite well, even when the value of FST is misspecified. Bootstrap confidence intervals are also examined and shown to exhibit close to nominal coverage when FST is correctly specified.
THE use of molecular marker data to infer the degree of relatedness between two individuals is of interest in a variety of contexts (see Weir et al. 2006 for a review). Several estimators have been developed for the case in which the loci are unlinked and the individuals are not inbred. These include Thompson's maximum-likelihood estimator (Thompson 1975; Milligan 2003) and a variety of other estimators (e.g., Queller and Goodnight 1989; Li et al. 1993; Ritland 1996; Lynch and Ritland 1999; Wang 2002).
The above methods generally assume that allele frequencies are known without error, but Wang (2002) also considered the case in which allele frequencies were estimated from a sample of size N from the population in which relatedness is to be estimated. In this work, we consider a different situation: the case in which the individuals examined belong to a subpopulation of the population to which known allele frequencies apply. An example of this situation would be when “European” allele frequencies are used in estimating the relatedness between two individuals who happen to be Italian.
The effect of using allele frequencies from the overall population is to shift the reference from which we measure relatedness back in time. As an illustration, consider the case in which two individuals share copies of an allele that is common in their subpopulation, but very rare in the population as a whole. Using the subpopulation allele frequencies, the fact that both individuals have this allele provides little evidence for relatedness. When the population allele frequencies are used, however, the evidence for relatedness becomes strong. The difference is that, when the population allele frequencies are used, relatedness is implicitly measured with respect to the time when the population allele frequencies held in the subpopulation, that is, before the subpopulation split from the ancestral population (which may be assumed to have allele frequencies similar to that of the current overall population—there is an implicit assumption here that the overall population is so large that its allele frequencies remain roughly unchanged over time). In this scenario, the relatedness estimate using the population allele frequencies is affected by the generations during which the allele frequencies in the subpopulation were diverging via drift from that in the overall population. From that perspective, the abundant copies of the allele in the subpopulation may all be copies of one allele in the ancestral population and these two individuals both have copies because they share common ancestry. Hence, even though the individuals may not be closely related with respect to recent generations, the fact that they share alleles that are rare in the overall population provides evidence that they may be closely related with respect to their more distant ancestry. The difference in allele frequencies between the subpopulation and the overall population is itself suggestive of relatedness between the individuals: Both are consequences of finite population size.
In contrast to the estimate of relatedness found by applying population allele frequencies, the estimate using the subpopulation allele frequencies ignores the evolutionary history during which the allele frequencies in the subpopulation drifted to their current states. Hence, this estimate measures relatedness relative to the period of time when the current subpopulation allele frequencies began to (approximately) hold.
Depending on a researcher's particular interests, he or she may prefer the estimate from using the overall population frequencies or that obtained from the subpopulation frequencies. The choice would depend upon the timescale of the researcher's scientific question.
As an example, suppose a researcher was interested in estimating rates of extrapair paternity in some species of birds. This is inherently a question of relatedness in just the preceding generation—whether a mother bird's social mate is in fact the father of her offspring or, if the mother's social mate is unavailable for testing, a question of whether her chicks are full or half siblings. From the perspective of this researcher, blindly using the population allele frequencies would inflate the degree of relatedness between the individuals (by including evolutionary relatedness that is irrelevant to this study).
On the other hand, if a species is in danger of extinction, a researcher might be interested in determining the relatedness between individuals to determine which individuals might be bred with each other to maximize the genetic diversity maintained in the population. In this case, if the researcher is interested in creating a population with maximum heterozygosity, he or she is concerned with relatedness going back for many generations and so might prefer to use the population allele frequencies and an estimation procedure that takes inbreeding into account.
The methodology in this article is relevant to the first of these researchers: We present a methodology for estimating the degree of relatedness between individuals that would have been obtained had the researcher been able to use current subpopulation allele frequencies instead of the overall population frequencies.
To create such an estimator, we need to be able to characterize the variation between the allele frequencies in a subpopulation and those in the overall population. This variation between allele frequencies between two or more populations can be summarized by the population structure parameter, θ (also known as FST). Balding and Nichols (1997) estimated θ using data from studies performed by Krane et al. (1992) and Budowle and Monson (1994) in which mixed Caucasian allele frequencies at variable number tandem repeat (VNTR) loci were compared to allele frequencies in various European subpopulations (e.g., Norway, Spain, Turkey). They examined three loci in each data set and found that, for the six loci examined, θ was generally <0.01, although at one locus larger values of θ could not be ruled out. Weir (1994) estimated a common θ-value for Apache, Navajo, and Pima populations using allele frequencies calculated from a pool of the three populations and obtained values of 0.02, 0.041, 0.097, 0.032, and 0.111 at the five VNTR loci considered. In a more recent study, Weir et al. (2005) estimated θ using two large SNP data sets. The HapMap data set (International HapMap Consortium 2005) contained data on four human subpopulations: Caucasians of European descent, Yoruba from Ibadan, Nigeria, Han Chinese from Beijing, and Japanese from Tokyo. The genomewide estimate of θ using all four of these populations was 0.13. The Perlegen (Hinds et al. 2005) data set, which contained European Americans, African Americans, and Han Chinese from the Los Angeles area, yielded 0.10 as an estimate of θ.
Values of θ estimated for animal populations are often even higher. Kretzmann et al. (2003) considered samples from five subpopulations of the Egyptian vulture (Neophron percnopterus) from the Iberian peninsula, Canary Islands, and Balearic Islands. They used genotypes at nine microsatellite loci to estimate θ-values between pairs of the populations (or, in the language of this work, they estimated a common θ for each pair of subpopulations, using the allele frequencies estimated from a pool of the two samples). Of the 10 pairwise θ-estimates, 3 were <0.015, 3 were between 0.05 and 0.1, 3 were between 0.1 and 0.15, and 1 was 0.295. In a similar study, Marshall and Ritland (2002) used 10 microsatellite loci to examine the genetic differentiation among 11 subpopulations of black bear (Ursus americanus) in the Pacific Northwest. Of the 55 pairwise θ-estimates from this study, 5 were <0.05, 28 were between 0.05 and 0.10, 18 were between 0.10 and 0.15, and 4 were ≥0.15.
In this article, we apply a maximum-likelihood approach to relationship estimation, based upon a generalization of Thompson's (1976) likelihood in which we account for population structure by including θ in our model. This model is the same as that given in Ayres (2000), but, whereas Ayres used the model to present formulas for some specific likelihood ratios, we present the likelihood equations in their general form and use them to find maximum-likelihood estimators.
THEORY AND METHODS
In this section, we begin by outlining the likelihood method developed by Thompson (1975). In notation we follow the treatment given in Milligan (2003): Our Table 1 and Figure 1 are essentially identical to those in Milligan's article. We then proceed to explain the model we use to describe the relationship between allele frequencies in the subpopulation and those in the ancestral population and, using this model, derive the likelihood analogous to that of Thompson.
TABLE 1.
Probabilities for various identity-by-state modes, given modes of identity-by-descent
| IBS mode | Allelic state | Identity-by-descent mode Sj
|
||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| S1 | S2 | S3 | S4 | S5 | S6 | S7 | S8 | S9 | ||
 |
AiAi, AiAi | pi | 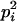 |
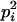 |
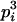 |
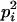 |
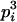 |
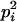 |
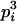 |
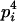 |
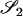 |
AiAi, AjAj | 0 | pipj | 0 | pipj | 0 |
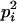 pj pj
|
0 | 0 | 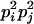 |
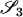 |
AiAi, AiAj | 0 | 0 | pipj | 2 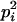 pj pj
|
0 | 0 | 0 |
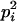 pj pj
|
2 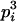 pj pj
|
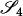 |
AiAi, AjAk | 0 | 0 | 0 | 2pipjpk | 0 | 0 | 0 | 0 | 2 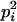 pjpk pjpk
|
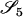 |
AiAj, AiAi | 0 | 0 | 0 | 0 | pipj | 2 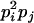
|
0 | 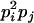 |
2 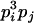
|
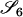 |
AjAk, AiAi | 0 | 0 | 0 | 0 | 0 | 2pipjpk | 0 | 0 | 2 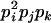
|
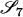 |
AiAj, AiAj | 0 | 0 | 0 | 0 | 0 | 0 | 2pipj | pipj(pi + pj) | 4 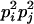
|
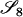 |
AiAj, AiAk | 0 | 0 | 0 | 0 | 0 | 0 | 0 | pipjpk | 4 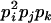
|
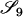 |
AiAj, AkAl | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 4pipjpkpl |
Here, alleles with different subscripts are distinct, and pi is the frequency of allele Ai.
Figure 1.—

Jacquard's identity-by-descent modes. Each group of four dots represents an IBD mode between two individuals. The top pair of dots represents the two alleles in individual 1 and the bottom pair of dots represents the two alleles in individual 2. Lines connect alleles that are IBD.
Identity-by-descent and relationship estimation:
Two alleles are said to be identical by descent (IBD) if they are both copies of an allele present in some previous generation. Individuals that are related are more likely than unrelated individuals to have similar genotypes because they have an increased probability of sharing alleles IBD from a recent common ancestor. When we speak of relationship estimation, we generally refer to the estimation of one or more parameters related to the probability that alleles are shared IBD between two individuals. One such parameter is the coancestry coefficient, θXY, which represents the probability that an allele chosen at random from individual X is IBD to an allele chosen at random from individual Y. An equivalent parameter is the relatedness coefficient, r = 2θXY.
Jacquard (1972) described a set of nine identity-by-descent modes that give a full description of the possible IBD relationships between the set of four alleles possessed by two (possibly inbred) individuals. These are denoted S1, …, S9 and are shown in Figure 1. The probability that a pair of individuals will be in IBD mode Si is denoted Δi.
As an example, if two noninbred individuals are full siblings (that is, they share both a mother and a father and the mother and father are unrelated), then Δ7 = 0.25, Δ8 = 0.5, and Δ9 = 0.25. All other IBD modes are impossible for noninbred full siblings. Indeed, all IBD modes other than S7, S8, and S9 can occur only if one or both of the two individuals are inbred.
If it is assumed that two related individuals are not inbred, then only three IBD modes are possible: S7, S8, and S9. These can be described more simply by noting the number of alleles shared IBD between the two individuals. IBD mode S7 corresponds to the case in which two alleles are shared IBD between the two individuals, whereas S8 and S9 correspond to the sharing of one and zero alleles, respectively. In this case, the relevant probabilities, Δ7, Δ8, and Δ9 correspond to the probabilities that the pair share two, one, and zero alleles, respectively, and are often denoted by k2, k1, and k0. Note that k2 + k1 + k0 = 1.
It is usually not possible to look at the alleles in two individuals and infer their IBD mode. We can, however, tell which alleles are identical by state (IBS), that is, which alleles share the same allelic type. There are 9 IBS modes, denoted 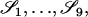 and these are listed in the “Allelic state” column in Table 1.
and these are listed in the “Allelic state” column in Table 1.
The genetic information about the relationship between two individuals that can be found using unlinked loci pertains exclusively to the estimation of the proportion of loci in the genome that are in each IBD state. Hence, with unlinked loci, two relationships with identical single-locus IBD probabilities are indistinguishable. As an example, noninbred half-sibling, grandparent–grandchild, and avuncular relationships all have k0 = 0.5, k1 = 0.5, and k2 = 0.0, so it is impossible to distinguish between these relationship types with unlinked loci.
Thompson's model:
Thompson (1975) assumed a model in which the individuals under consideration came from a single population in Hardy–Weinberg equilibrium. In such a situation, two random alleles that are not IBD may be considered to be two random draws from the population of alleles. Under this assumption, the probabilities of observing each of the nine possible IBS modes, conditioned on the IBD mode, are shown in Table 1. Using these probabilities, the single-locus likelihood of a relationship specified by Δ, between two individuals whose IBS mode is 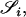 can be found by conditioning on the IBD mode as follows:
can be found by conditioning on the IBD mode as follows:
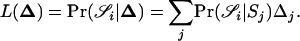 |
(1) |
Multilocus likelihoods for unlinked loci are formed by taking the product of the single-locus likelihoods.
Thompson (1975) used these likelihood equations primarily for the purpose of constructing likelihood ratios to compare the probability of the observed marker data under two competing hypotheses concerning the relationship between the individuals. Milligan (2003) used the likelihood to estimate k2, k1, and k0 and hence obtain an estimate for θXY = 0.5k2 + 0.25k1. He then compared the performance of the maximum-likelihood estimator to the performance of various method-of-moments estimators.
Model with population substructure:
Unlike Thompson's model, in which the individuals come from a single panmictic population with the given allele frequencies, we consider the case in which individuals come from a subpopulation of the population for which allele frequencies are known. For populations in equilibrium and loci for which the postmutation state of an allele is independent of its premutation state, it has been shown (Wright 1951; Griffiths 1979) that allele frequencies among the subpopulations follow a Dirichlet distribution. Under similar assumptions, Balding and Nichols (1994) derived equations for joint allele probabilities within a subpopulation using the population allele frequencies; these results match the moments of a Dirichlet distribution.
The Dirichlet distribution depends upon the population structure parameter described above. This parameter, θ, can be thought of as a correlation among alleles in the subpopulation: The probability that the first allele drawn from the population is A is pA (because, although we do not know the allele frequencies specific to the subpopulation, we do know that the expected allele frequency in the subpopulation is the same as the population allele frequency pA) and, given that the first allele drawn was A, the probability that the second allele drawn will also be A is pA + θ(1 − pA). The equilibrium assumption means that θ is not changing over time.
One can also think of θ as an IBD probability. The current population allele frequencies approximate those of an ancestral population from which the subpopulation descended. When we estimate IBD using the subpopulation allele frequencies, we use Thompson's model in which IBD is measured with respect to some previous generation when the current subpopulation allele frequencies held. Using the population allele frequencies forces our frame of reference back to the ancestral population. Two alleles that are merely IBS with respect to the subpopulation model may be IBD when the longer population history is taken into account. In this context, θ represents the probability that any two alleles in the subpopulation are IBD with respect to the ancestral population.
In Thompson's model, two individuals have a relationship specified by Δ and alleles that are not IBD are considered to be drawn independently at random according to the subpopulation allele frequencies. Using population allele frequencies instead of subpopulation frequencies forces us to consider the subpopulation alleles within a longer evolutionary framework, where our “not IBD” alleles are no longer drawn independently because they may be IBD to previously seen alleles when IBD is measured with respect to the ancestral population.
To calculate the likelihood under a model in which the subpopulations are related to the overall population with population structure parameter θ, we first note that Equation 1 still holds, but the calculation of 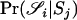 will now be undertaken under the assumption that our individuals belong to a subpopulation of the population from which the allele frequencies apply.
will now be undertaken under the assumption that our individuals belong to a subpopulation of the population from which the allele frequencies apply.
Under the Dirichlet model, joint probabilities for sets of alleles can be calculated as described, for example, in Weir (2003). In particular, if (p1, p2, …, pn) are the population allele frequencies of alleles A1, …, An, at a locus, the probability that a sample of alleles from the subpopulation will contain t1 alleles of type A1, t2 alleles of type A2, and so forth, is given by
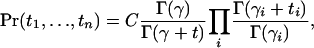 |
(2) |
where Γ indicates the usual gamma function, γi = (1 − θ)pi/θ, 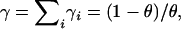 and C is a constant indicating the number of possible orderings in which we could have drawn a sample with these allele counts.
and C is a constant indicating the number of possible orderings in which we could have drawn a sample with these allele counts.
Define Mi, j = [(1 − θ)pi + jθ], for i = 1, 2, … and j = 0, 1, … . Suppose the single-locus joint genotype, g, of two individuals contains ti alleles of type Ai (so ti ∈ {0, 1, 2, 3, 4}), and let h denote the number of heterozygous individuals in the pair (so h ∈ {0, 1, 2}). Then
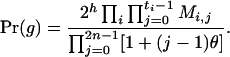 |
(3) |
Table 2 shows the probabilities of the nine possible joint IBS states given the nine possible IBD states. Note that, when θ = 0, these reduce to the probabilities given in Table 1. Using these probabilities, the likelihood can be calculated as in Equation 1. In determining the maximum-likelihood estimator, we consider Δ1, …, Δ9, which refer to IBD probabilities measured with respect to the subpopulation, to be parameters while θ, which measures the background IBD that comes from the relationship between the subpopulation and the ancestral population, is considered to be a known constant. As in Thompson's model, we assume that genotypes at distinct unlinked loci are independent and, hence, the multilocus likelihood is the product of the single-locus likelihoods.
TABLE 2.
Probabilities for various identity-by-state modes, given modes of identity-by-descent when the individuals being compared belong to a subpopulation of the population from which the allele frequencies are estimated
| IBS mode | Allelic state | Identity-by-descent mode
|
||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| S1 | S2 | S3 | S4 | S5 | S6 | S7 | S8 | S9 | ||
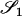 |
AiAi, AiAi | 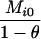 |
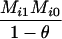 |
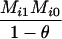 |
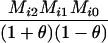 |
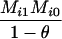 |
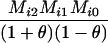 |
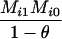 |
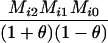 |
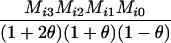 |
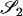 |
AiAi, AjAj | 0 | 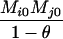 |
0 | 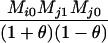 |
0 | 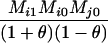 |
0 | 0 | 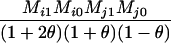 |
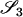 |
AiAi, AiAj | 0 | 0 | 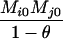 |
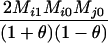 |
0 | 0 | 0 | 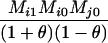 |
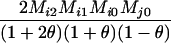 |
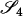 |
AiAi, AjAk | 0 | 0 | 0 | 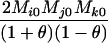 |
0 | 0 | 0 | 0 | 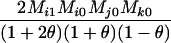 |
 |
AiAj, AiAi | 0 | 0 | 0 | 0 | 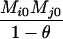 |
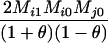 |
0 | 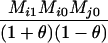 |
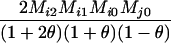 |
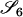 |
AjAk, AiAi | 0 | 0 | 0 | 0 | 0 | 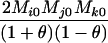 |
0 | 0 | 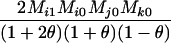 |
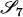 |
AiAj, AiAj | 0 | 0 | 0 | 0 | 0 | 0 | 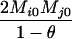 |
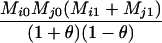 |
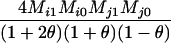 |
 |
AiAj, AiAk | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 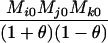 |
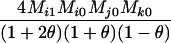 |
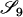 |
AiAj, AkAl | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 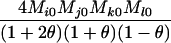 |
As in Table 1, alleles with different subscripts are distinct.
When θ = 0, the likelihood with population structure is identical to that of Thompson. To distinguish between the two maximum-likelihood estimators when θ > 0, we refer to the maximum-likelihood estimator (MLE) calculated with θ = 0 as the reduced model (r)MLE and the general MLE introduced here as the full model (f)MLE.
Parameter space:
In the analyses presented in this article, we consider the case in which the subpopulation is large and outbred. In this case, Δ1 = … = Δ6 = 0 and the likelihood is a function of k0, k1, and k2. It is also true that, in an outbred population, the IBD parameters k2, k1, and k0 are subject to the constraint 4k2k0 < 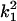 (Thompson 1976), and we have incorporated this constraint into our estimations. Our parameter space is then {k2, k1, k0: 0 ≤ ki ≤ 1 (i = 0, 1, 2), k0 + k1 + k2 = 1, 4k2k0 <
(Thompson 1976), and we have incorporated this constraint into our estimations. Our parameter space is then {k2, k1, k0: 0 ≤ ki ≤ 1 (i = 0, 1, 2), k0 + k1 + k2 = 1, 4k2k0 < 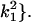
To find the maximum-likelihood estimator for the coancestry coefficient, θXY, for a pair of individuals, we first use the simplex method (Press et al. 2002) to find maximum-likelihood estimators for k2, k1, and k0 and then estimate 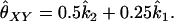
Other estimators:
In this article, we compare our maximum-likelihood estimator to various other popular relationship estimators. These include the maximum-likelihood estimator described by Thompson (1975) and Milligan (2003), as well as a number of other estimators that we briefly describe below.
The Queller–Goodnight (QG) estimator, first introduced in Queller and Goodnight (1989), is one of the earlier relationship estimators. We chose to use the form of the Queller–Goodnight estimator presented in Equation 11 of Lynch and Ritland (1999), where we averaged the single-locus estimates across loci. The similarity index (SIM) (Li et al. 1993) is another popular relationship estimator. Here, we use the version given in Equation 8 of Lynch and Ritland (1999), averaged across loci. The Lynch–Ritland (LR) estimator is known to perform well and is given in Equations 5–7 of Lynch and Ritland (1999), with a weighted average taken across loci. The final moment estimator we use is Wang's estimator, which we denote as W and compute according to Equations 9 and 10 in Wang (2002).
Both the QG and LR estimators are asymmetric with respect to the two individuals, that is, 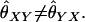 For these two estimators, we use the average of the estimates taken from the different orderings of the two individuals.
For these two estimators, we use the average of the estimates taken from the different orderings of the two individuals.
Assessing uncertainty in the estimators:
An estimation of relatedness is incomplete without an indication of the uncertainty associated with that estimation. Suppose m markers were genotyped in the two individuals being compared. A different estimate of relatedness might occur if a different set of m markers had been chosen. If we think of our set of markers as being randomly chosen from some distribution of possible markers, we want a confidence interval such that a fixed proportion (e.g., 95%) of all random sets of m markers would produce intervals that contain the true parameter value. To do this, we created bootstrap confidence intervals for the estimates, where bootstrapping was done over loci. More specifically, each bootstrap sample consisted of the two individuals' genotypes at m loci, where the loci in the bootstrap sample are chosen at random (with replacement) from the originally genotyped m loci. Each bootstrap sample yielded an estimate for 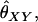 and the final 95% confidence interval for the original pair of individuals was found by taking the middle 95% of the estimates from the bootstrap samples.
and the final 95% confidence interval for the original pair of individuals was found by taking the middle 95% of the estimates from the bootstrap samples.
Simulations:
We ran a series of simulations in which we compared the performance of the various point estimators under a variety of circumstances. Each simulation began with the generation of allele frequencies for the overall population, and allele frequencies in the subpopulation were stochastically generated from these using the Dirichlet distribution as described, for example, in Weir (2003). In our analyses, we used the allele frequencies from the ancestral population in estimating the relatedness between individuals in subpopulations.
In our first set of simulations, we considered an ancestral population with 10 markers, each of which had 10 alleles with frequencies determined by a triangle distribution. For each value of θ (θ = 0.0, 0.03, 0.10), we generated 4000 sets of subpopulation allele frequencies, and, for each of these subpopulations, we estimated the relatedness of 1000 pairs of individuals of each of the following types: parent–offspring (PO), full sibling (FS), half sibling (HS), first cousins (FC), second cousins (SC), and unrelated (UN). The genotypes for relative pairs were generated using the subpopulation allele frequencies and the appropriate IBD probabilities (k0, k1, k2) for the relationship type. In these simulations, we estimated the bias and root mean-square error (RMSE) for each subpopulation and then presented the average of these values across subpopulations.
For the second set of simulations, we were interested in the behavior of the various estimators within a subpopulation and whether the maximum-likelihood estimator was sensitive to misspecification of θ. All simulations were performed with 10 markers, each with 10 alleles. For each value of θ (θ = 0.0, 0.03, 0.10), we generated a set of subpopulation allele frequencies and simulated 1000 pairs of individuals of each relationship type using these frequencies. We estimated the MLE using various assumed values of θ (θ = 0.0, 0.01, 0.02, 0.03, 0.05, 0.10, 0.15) and also obtained relatedness estimates from the moment estimators. To get a sense of whether our results would vary substantially depending on either the allele frequencies in the ancestral population or the particular realization of subpopulation allele frequencies, we carried out the analysis on five replicate subpopulations for each of the following types of allele frequencies in the ancestral population: equally frequent alleles, triangle allele frequencies, and random allele frequencies generated from a Dirichlet distribution with all parameters set to unity. Once we had determined that our results did not vary substantially among these simulations, we ran larger data sets of 5000 relative pairs of each type from subpopulations (θ = 0.0, 0.03, 0.10) of a population in which the overall allele frequencies followed a triangle distribution.
The third set of simulations was designed to investigate the effect of varying the number and type of loci. We considered both diallelic and microsatellite (10 alleles) loci and varied the number of loci from 5 to 100. For both marker types, the ancestral population had triangle allele frequencies. In the case in which loci were diallelic, the QG estimator is undefined for heterozygous individuals, so we did not consider its performance in these simulations. In addition, in the situation with diallelic loci, if all loci have equal assumed allele frequencies, the SIM and Wang estimators are identical, so we presented only one of these in our results. For each combination of number of loci, number of alleles, and θ, we simulated 10,000 relative pairs of each type (full sibling and unrelated).
We also performed a set of simulations to investigate the performance of bootstrap confidence intervals for the fMLE. In these simulations, we looked at one subpopulation of an overall population in which each locus had 10 alleles with frequencies determined by a triangle distribution and considered cases in which 5, 10, 15, 20, 30, and 40 loci were used. Within the subpopulation, we generated 1000 relative pairs of each type, and for each pair we used 1000 bootstrap samples of the markers to determine a 95% confidence interval. We also ran a series of simulations to examine the behavior of the confidence intervals for varying assumed values of θ. In this series of simulations, for each combination of θ (θ = 0.00, 0.03, 0.10) and number of markers (10 or 40), we simulated 1000 relative pairs from a single simulated subpopulation and then formed a bootstrap confidence interval for each relative pair under several assumed values of θ. When the data were simulated under θ = 0.00, we analyzed the data with each of the following assumed values of θ: 0.00, 0.01, 0.03, and 0.05. When the true value of θ was 0.03, the data were analyzed under θ = 0.00, 0.01, 0.03, 0.05, and 0.08. When θ = 0.10, we analyzed the data assuming θ = 0.05, 0.08, 0.10, 0.12, and 0.15.
Centre d'Ètude du Polymorphisme Humain data analysis:
To compare the behavior of the estimators in a real setting, we examined relative pairs from Version 10 of the Centre d'Ètude du Polymorphisme Humain (CEPH) database. To this end, we chose a set of 49 widely spaced genetic markers from throughout the genome that were genotyped in the eight CEPH reference families. These families included six families from Utah (CEPH families 1331, 1332, 1347, 1362, 1413, and 1416) as well as an Old Order Amish family (CEPH family 884) and a Venezuelan family (CEPH family 102). We chose our markers to have moderately high gene diversities (expected heterozygosities) in the range of 0.7–0.8. For comparison, and to illustrate the point that a choice of a different set of markers will lead to slightly different results, we repeated all analyses with a second set of 49 loci.
Within each family and for each set of markers, we looked at all possible relative pairs for which both individuals were genotyped at all 49 markers. We chose this approach to give us the largest possible sample size upon which to evaluate the behavior of the estimators, but we are aware that the lack of independence between different pairs of relatives may have an effect on the interpretation of our results. In particular, this dependence will not bias the relatedness estimates for any given family, but we would expect the mean estimates from each family to vary more than if they were based on independent relative pairs.
The allele frequencies we used were those listed in the CEPH data sets, with one exception: Any allele whose allele frequency was listed as zero in the CEPH data set but appeared in that data set was reassigned an allele frequency of 1 × 10−4.
RESULTS
Simulation results:
Figure 2 shows the average bias and RMSE over subpopulations descended from a population that had 10 alleles at each of 10 loci considered. We first compared the performance of the rMLE and moment estimators to examine their robustness to this type of model misspecification. The moment estimators all performed similarly and all showed increasing bias with increasing values of θ. The rMLE also increases its bias with increasing θ, but not as severely as the moment estimators. When θ is as large as 0.10, the moment estimators no longer show less bias than the rMLE.
Figure 2.—

Average behavior in subpopulations of a population in which allele frequencies follow a triangle distribution. Here we show the average bias and RMSE based on 1000 relative pairs of each type drawn from each of 4000 simulated subpopulations. The symbols are as follows: •, rMLE; ▴, fMLE; ▵, Queller–Goodnight; +, Lynch–Ritland; x, similarity index; ⋄, Wang.
We next examined how the bias and RMSE would be affected by using a model that takes population structure into account. In all cases, the fMLE showed reduced bias compared to models that do not take population structure into account. Even when we use the true value of θ in the fMLE, though, the bias still increased with θ. Naturally, though, this bias will be seen to decrease when the number of loci increases.
A plot corresponding to Figure 2 was also produced for the case in which the overall population had all alleles equally frequent, but was similar to the triangle allele-frequency case and so is not shown here. The main difference between the existing Figure 2 and the version with equally frequent alleles is the relative performance of the moment estimators.
Figure 3 shows a more in-depth view of the results from a single subpopulation. As before, we considered 10 loci, each with 10 alleles that had triangle allele frequencies in the overall population. We performed these simulations under three values of θ and compared the four moment estimators as well as maximum-likelihood estimators under various assumed values of θ. The top row of Figure 3 shows the behavior of the estimators when there is no population structure. The plot showing the relatedness estimates for unrelated individuals clearly demonstrates a fundamental difference between the moment and maximum-likelihood estimators: The moment estimators can give relatedness estimates that are less than zero whereas the maximum-likelihood estimates are constrained to give results that lie within the space of possible values for θ. Note that, for distantly related or unrelated individuals, this constraint causes much of the bias seen in the maximum-likelihood estimators: Since it is impossible for the estimator to substantially underestimate the degree of relatedness, but it is possible to overestimate this value, the estimator will, on average, overestimate the degree of relatedness. The unbiasedness of the moment estimators is a result of the undesirable property of allowing estimates that are less than zero. A comparison of the box plots of the actual estimates shows the superior performance of the maximum-likelihood estimator.
Figure 3.—

Box plots showing the distribution of the estimators on a single subpopulation. We simulated 5000 relative pairs of each type for each of three values of θ. The top row of plots shows the results when the relative pair comes from a subpopulation with θ = 0.0, whereas the middle and bottom rows of plots show results when the simulations were run with θ = 0.03 and θ = 0.10, respectively. We estimated the relatedness of the individuals using the four moment estimators as well as the maximum-likelihood estimators assuming various values of θ. The symbols for the maximum-likelihood estimators are: M0, θ = 0.00; M1, θ = 0.01; M2, θ = 0.03; M3, θ = 0.05; M4, θ = 0.10; M5, θ = 0.15. The MLE that assumes the correct value of θ for the subpopulation is shaded. The box plots shown contain boxes that extend from the first to the third quartiles of the relatedness estimates, with a line through the box indicating the median. Whiskers extend from the boxes to the most extreme data point that is within 1.5 times the interquartile range from the box.
The second and third rows of plots in Figure 3 show the effects of increasing the degree of population structure. The results confirm what was seen in Figure 2: Ignoring population structure causes inflation in the relationship estimates, and this effect is reduced when we account for the population structure in our likelihood. In addition, we see that the MLE calculated from the likelihood with population structure is quite robust to misspecification of θ. In particular, analyzing the data with a specified value of θ that is a little too high may actually improve the performance (by helping to counter some of the natural bias in the MLE).
In Figures 4 and 5, we examined the effect of number and type of loci for estimating the relatedness of full siblings and unrelated pairs of individuals drawn from one subpopulation. In each case, we show the mean parameter estimate and RMSE from 10,000 relative pairs. For full siblings, we see that, although accounting for population structure reduces the bias associated with the estimators, it provides little reduction in the RMSE unless θ is large. For unrelated pairs, however, the MLE that takes population structure into account shows a notable reduction in RMSE compared to the other estimators for all values of θ examined. Figures 4 and 5 also illustrate the point that increasing the number of loci does not necessarily result in increased accuracy when the allele frequencies are misspecified.
Figure 4.—

Full siblings. Here, we have generated 10,000 full-sibling pairs from a single subpopulation and examined the effect of the number of loci and number of possible alleles on relationship estimation. The symbols for the various estimators are as given in Figure 2.
Figure 5.—

Unrelateds. Here, we have generated 10,000 pairs of unrelated individuals from a single subpopulation and examined the effect of the number of loci and number of possible alleles on relationship estimation. The symbols for the various estimators are as given in Figure 2.
Looking at the behavior of each estimator for unrelated pairs of individuals can give insight into how the various estimators respond to this type of model misspecification. The fMLE approaches zero as the number of loci are increased. The models that do not account for population structure naturally measure relatedness relative to some ancestral population. The parameter θ is the probability that any two alleles drawn from the subpopulation are IBD with respect to that ancestral population. Hence, we might expect an estimator that does not take population structure into account to have a nonzero expected value of θ for “unrelated” individuals. However, under the assumption that any two alleles have a nonzero probability of being IBD, all nine of Jacquard's IBD configurations are possible, so the estimators under consideration (which assume no inbreeding) also have to contend with this type of model misspecification.
For unrelated individuals, as the number of loci increases, the rMLE appears to approach the value of θ used to simulate the data, but we have not looked into its behavior closely. The moment estimators display a variety of behaviors. In the appendix, we derive the expected behavior of the moment estimators as a function of θ in the general diallelic case and in the case in which there are n equally frequent alleles at a locus. In the diallelic case, Wang's estimator has an expected value of θ/2 whereas the Lynch–Ritland estimator has θ/(1 + θ) as its expected value. In the case with n equally frequent alleles, the Lynch–Ritland and Queller–Goodnight estimators are identical and have an expected value of θ/(1 + θ), regardless of the value of n. The similarity index and Wang's estimator have expected values that depend on n and approach θ/(1 + θ) and θ(1 + 3θ − θ2)/(1 + 3θ + 2θ2), respectively, as 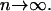
Bootstrap simulation results:
Figures 6 and 7 show Monte Carlo estimates for the coverage probabilities of bootstrap 95% confidence intervals for the rMLE and fMLE, respectively. When population structure is not taken into account (Figure 6), coverage decreases with increased sample size. When the data are analyzed under the correct model, however, the coverage for all relationships examined and all values of θ was at least 88.5% whenever at least 10 loci were used (results not shown). In Figure 7, we see the effects of misspecifying the value of θ assumed in the analysis. For small sample sizes, this method of constructing confidence intervals is robust to misspecification of θ. When the number of markers is large, however, the fact that these confidence intervals do not take uncertainty in θ into account results in reduced coverage, especially for more distantly related individuals.
Figure 6.—

Coverage probabilities based on the reduced-model MLE when relative pairs were generated from a subpopulation with θ = 0.0 (○), θ = 0.03 (▵), and θ = 0.10 (+).
Figure 7.—

Effects of parameter misspecification on confidence interval coverage. Each plot shows the empirical coverage probabilities for bootstrap confidence intervals based on a fixed number of markers (10 or 40) and a set degree of population structure (θ = 0.00, 0.03, 0.10). Within each plot, for each type of relative pair is the coverage of 95% confidence intervals based on 1000 pairs of individuals, where the analysis was performed under various assumed values of θ. When the true value of θ was 0.00, we analyzed each pair of individuals under the assumed values of (left to right) θ = 0.00, 0.01, 0.03, and 0.05. When the true value of θ was 0.03, we analyzed the data under assumed values of θ = 0.00, 0.01, 0.03, 0.05, and 0.08. Finally, when θ was 0.10, we performed analyses under assumed values of θ = 0.05, 0.08, 0.10, 0.12, and 0.15. In all cases, the results when the true value of θ was assumed are represented by a solid circle. All other values of θ are indicated by open circles.
CEPH results:
Figures 8 and 9 show the mean estimates and root mean-square error for the relative pairs of each type (parent–offspring, full siblings, grandparent–grandchild, unrelated) within the CEPH data set for the initial set of 49 markers with gene diversities between 0.7 and 08. The Utah families, where we would expect θ to be small, show a pattern similar to that seen in our simulated data sets with little or no population structure: All estimators show fairly little bias, with the maximum-likelihood estimators generally exhibiting lower RMSE than the other estimators. Figure 10 shows the mean estimates using the second set of 49 markers.
Figure 8.—

Mean estimates for the CEPH data set, based on the first set of 49 loci. The families are denoted as follows: U1, …, U6 refer to Utah families 1331, 1332, 1347, 1362, 1413, and 1416; A refers to the Old Order Amish family 884; V refers to the Venezuelan family 102. For each family, we have plotted the mean estimates in two columns: The left column has the MLE estimates calculated with θ = 0.0, 0.01, 0.02, 0.03, 0.05. All MLEs are indicated by solid circles, with darker shaded circles indicating lower values of θ. The right column has the other estimators with symbols as follows: ▵, Queller–Goodnight; +, Lynch–Ritland; x, similarity index; ⋄, Wang. Above the horizontal axis are values indicating the number of relative pairs evaluated in each family.
Figure 9.—

Root mean-square error for the CEPH data set, based on the first set of 40 loci. The symbols for this plot are the same as those in Figure 8.
Figure 10.—

Mean estimates for the CEPH reference families, as generated by a second set of markers. The symbols in this plot are the same as those in Figure 8.
Although there is some variation between the various families and between marker sets, the MLE does not give mean results that are substantially different from the non-maximum-likelihood estimators for the Utah families. In other words, 49 unlinked markers seem to be enough to make the MLE (with θ = 0) essentially unbiased.
In the Amish family, relative pairs show inflated relatedness estimates, especially among the grandparent–grandchild and unrelated pairs. The Old Order Amish form a small genetic isolate, so any pair of individuals from this population may be expected to share multiple ancestors in recent generations. Thus, a relative pair from this group will often be more related than the nominal degree of relatedness indicated in the CEPH pedigrees. The genealogy of this particular Amish family is known for more generations than are included in the CEPH database and none of the grandparents in the CEPH pedigree share common ancestors within the three preceding generations (Egeland 1972; Broman and Weber 1999). Nevertheless, with both our marker sets, the mean maximum-likelihood estimate for relatedness for each type of relative pair was higher than the nominal level, even when we allowed θ to be as high as 0.05.
A previous study (Broman and Weber 1999) has shown evidence of excess relatedness (as evidenced by exceptionally long spans of homozygosity within individuals) within the Venezuelan family (CEPH family 102). Because of this and the fact that Venezuelan allele frequencies might well be quite different from the Utah allele frequencies that should have dominated the CEPH allele frequency estimates, we expected the Venezuelan families to show higher than nominal degrees of relatedness, especially with the non-maximum-likelihood estimators. Contrary to our expectations, though, all relatedness estimators performed well for this family using both sets of markers.
A close look at the RMSE values in the various families indicates that the Amish and Venezuelan families differ from the Utah families in an important way. As an overall principle, increasing the value of θ used in the MLE calculations decreases the estimated coancestry coefficient between a pair of individuals. For pairs of unrelated individuals, then, it is clear that increasing the value of θ will always result in a decrease in RMSE. When highly polymorphic markers are used, the same is also true for parent–offspring pairs for the following reason: When two individuals share at least one allele at a marker, the single-locus MLE for the coancestry coefficient at that marker will always be 0.25 or 0.5 unless the pair shares an allele with a population allele frequency >0.25. With highly polymorphic markers, we only rarely see alleles with such high frequencies. Hence, since the maximum-likelihood estimator for several markers should not be less than the minimum of the single-locus MLEs, we see that 0.25 is a lower bound for the MLE for the coancestry coefficient between a parent–offspring pair (note that this is not true for SNP markers as seen in Figure 4 of Weir et al. 2006). An increase in θ thus results in a reduction of the RMSE for such pairs. For other relationships, increasing θ past a certain point will drive the MLE below the correct value and cause an increase in the RMSE. For the Utah families, we see that increasing θ for full siblings and grandparent–grandchild pairs always increases the RMSE, as might be expected if the true value of θ is small. The Amish family shows the opposite pattern: Increasing θ decreases the RMSE. This is consistent with two scenarios: The value of θ is truly very high or the individuals are truly more closely related than the nominal level. For the Venezuelan siblings, the minimum RMSE (among the θ-values examined) is achieved when θ = 0.02.
DISCUSSION
Our purpose here has been to develop methodology for estimating relatedness within a subpopulation of a population in which allele frequencies are known or estimated. In effect, this method measures IBD probabilities with respect to recent generations while filtering out additional allele sharing that comes from the more distant generations during which allele frequencies in the subpopulation drifted to their current values.
The bottom left-hand plot in Figure 5 illustrates this point. Here, θ = 0.10 and the rMLE and other non-maximum-likelihood estimators estimate the relatedness of supposed unrelated individuals in this population at 0.10. From the perspective of the researcher interested in relatedness going back many generations, this is the correct answer: θ represents the degree of relatedness in the individuals relative to approximately the generation when the subpopulation split from the overall population. For a researcher interested in questions regarding relatedness relative to less distant generations (specifically, the degree of relatedness that would be estimated if the researcher had access to allele frequencies from the subpopulation), the correct value for these unrelated individuals is θXY = 0, the value given by the fMLE.
We have looked at relatedness estimation from the perspective of researchers who want to base their estimators on the subpopulation allele frequencies but have access only to population allele frequencies. We have shown that this misspecification of allele frequencies causes positive bias in relatedness estimators that are currently in use. When the subpopulation is quite differentiated from the overall population, as is frequently seen in animal populations, the amount of the degree of bias can be large (for unrelated individuals, the expected amount of bias is approximately equal to the degree of differentiation between the subpopulation and the overall population).
We have proposed a maximum-likelihood estimator that takes population structure into account, but requires the degree of differentiation between the subpopulation and the overall population (θ or FST) to be specified. Our simulations show that this estimator exhibits reduced bias compared to the estimators that ignore the possibility that allele frequencies come from an overall population rather than from the pertinent subpopulation. In addition, we have demonstrated that this estimator is fairly robust to small misspecifications of θ. Note that the value of θ used with this estimator will need to be estimated from a data set that does not contain individuals whose relatedness is in question because θ cannot be estimated from sets of individuals with unspecified relationships. Indeed, commonly used estimators for θ (e.g., Weir and Cockerham 1984) require that it be estimated from data sets consisting of unrelated individuals from various subpopulations.
When we examined the effect of the number of loci on this full-model MLE, we saw that the functional relationship between mean estimate (or RMSE) and number of markers is shaped like a negative exponential. Hence, when few markers are being used, small increases in the number of markers produce large decreases in bias and RMSE. When the number of markers is larger, though, it takes the addition of many more markers to give a substantial improvement in the estimator. Our simulation results indicate that, for highly polymorphic markers such as microsatellites, moderate increases in the number of loci beyond, say, 40 or 60 has little effect. For diallelic loci (e.g., SNPs) substantial improvements in performance are obtained at >100 loci. We did not pursue this beyond 100 loci because the methods presented here are for unlinked loci (where, by unlinked, we mean segregating independently within a single meiosis), and a genome will not contain many more than 50 such loci. We have reason to believe that maximum-likelihood estimation may be fairly robust to this assumption provided that the loci cover a wide region of the genome: Hepler (2005) performed maximum-likelihood estimation of Jacquard's nine Delta parameters (Δ1, …, Δ9), using a large set of tightly linked loci spread over an entire human chromosome, and obtained quite accurate results.
To give a measure of confidence in our estimates, we proposed forming bootstrap confidence intervals for our estimates, where the bootstrapping is performed over the loci. When population structure exists and is not taken into account, we showed that the performance of these confidence intervals (as measured by their coverage probabilities) decreased dramatically with increasing numbers of loci. When population structure was taken into account by using the full-model MLE with the correct value of θ, the confidence intervals performed well whenever the number of loci was > ∼10. With larger sample sizes, though, the performance of the confidence intervals depended on the specification of θ; a reduction in coverage occurred when analyses were performed with incorrect values of θ. Not unsurprisingly, the greater the number of markers, the closer the assumed value of θ needed to be to the true value for the confidence intervals to maintain adequate coverage.
We concluded our study by looking at the performance of various estimators based on 49 unlinked (or loosely linked) microsatellite loci genotyped on the eight CEPH reference families. Six of the families were from Utah and, for these, we would expect to have θ close to 0. Hence, this amounted to a comparison of previous methods on a real data set. With 49 loci, all estimators were essentially unbiased and the MLE was shown to outperform the others in terms of RMSE (by virtue of performing better on parent–offspring and unrelated pairs and performing no worse on full siblings and grandparent–grandchild pairs).
Acknowledgments
This work was supported in part by National Institutes of Health grants GM45344 and GM75091.
APPENDIX
Here we derive the expected values of the Wang, Lynch–Ritland, similarity index, and Queller–Goodnight estimators for the general diallelic case and the case in which there are n equally frequent alleles at a locus. This is done for the situation in which the relative pair is drawn from a subpopulation of the population from which the allele frequencies are taken and the estimators are not modified to take this into account. The relative pairs in these calculations are not inbred except through the background relatedness, θ, so their relatedness within the subpopulation can be summarized by the values of k0, k1, and k2.
Wang's estimator:
Wang's estimator (Wang 2002) is based on the proportion of loci for which the relative pair is in each of four IBS categories. Category 1 includes any case in which the two individuals share two alleles IBS, category 2 includes cases in which three of the four alleles are IBS, category 3 consists of genotype pairs of the type AiAj, AiAk, where each of Ai, Aj, and Ak represents a distinct allele, and category 4 contains any genotype pair for which the two individuals share no alleles identical in state. Pi denotes the proportion of loci for which the relative pair falls into category i. Wang adopts the following notational convention: 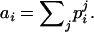
In the diallelic case, Wang's Equation 8 implies the following:
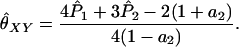 |
(A1) |
Joint genotype probabilities for the diallelic case are given in Table A1. The expected values of 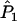 and
and 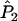 are as follows:
are as follows:
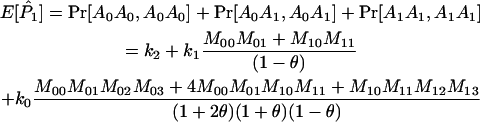 |
(A2) |
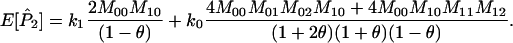 |
(A3) |
TABLE A1.
Joint genotype probabilities for diallelic loci for individuals that are not inbred except for background inbreeding captured by θ
| Genotype | Probability | Genotype category | LR |
|---|---|---|---|
| A0A0, A0A0 | 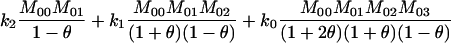 |
1 | 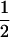 |
| A0A0, A0A1 | 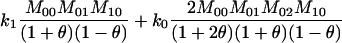 |
2 | 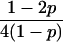 |
| A0A0, A1A1 | 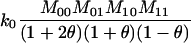 |
4 | 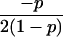 |
| A0A1, A0A0 | 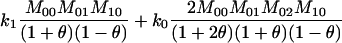 |
2 | 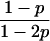 |
| A0A1, A0A1 | 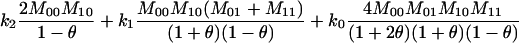 |
1 | 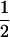 |
| A0A1, A1A1 | 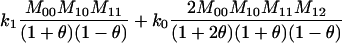 |
2 | 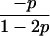 |
| A1A1, A0A0 | 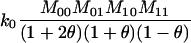 |
4 | 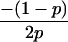 |
| A1A1, A0A1 | 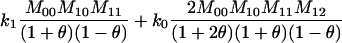 |
2 | 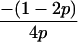 |
| A1A1, A1A1 | 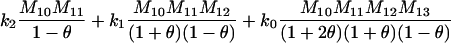 |
1 | 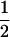 |
The single-locus Lynch–Ritland (LR) estimate of θXY for each joint genotype is also given.
At a single locus, the expected value of 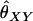 is
is
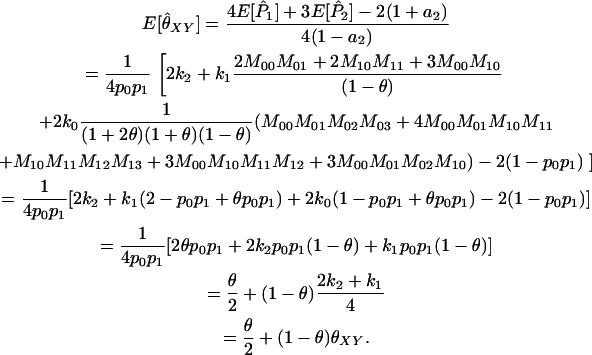 |
(A4) |
For multiple loci, Wang replaces each of 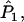
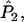 and
and 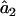 in Equation A1 with a weighted average of its value across all loci. The expected value of
in Equation A1 with a weighted average of its value across all loci. The expected value of 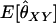 remains the same as that in the single-locus case.
remains the same as that in the single-locus case.
Note that, when θ = 0 (as it is in Wang's model), the estimator is unbiased for θXY. When θ > 0, however, we might expect that an estimator that does not take population structure into account might have the property that 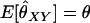 for unrelated individuals. Wang's estimator instead gives
for unrelated individuals. Wang's estimator instead gives 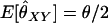 for diallelic loci in this case.
for diallelic loci in this case.
For the case in which the locus has n equally frequent alleles, Table A2 lists the possible IBS modes and their probabilities. Note that, with all alleles equally frequent, Mij = M0j for all i and j. Since each IBS mode corresponds to several genotypes, we have also listed the number of genotypes included in each IBS mode. For example, if there are n alleles, there are n genotypes of the form (AiAi, AiAi).
TABLE A2.
Joint genotype probabilities for general loci when all loci have n equally frequent alleles
| IBS mode | Count | Probability | Genotype category | LR | SXY | SIM |
|---|---|---|---|---|---|---|
| AiAi, AiAi | n | 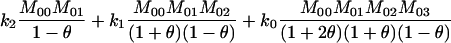 |
1 | 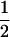 |
1 | 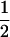 |
| AiAi, AiAj | n(n − 1) | 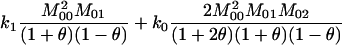 |
2 | 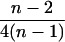 |
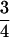 |
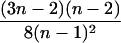 |
| AiAi, AjAj | n(n − 1) | 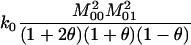 |
4 | 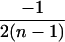 |
0 | 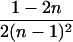 |
| AiAi, AjAk | 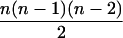 |
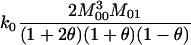 |
4 | 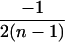 |
0 | 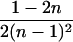 |
| AiAj, AiAi | n(n − 1) | 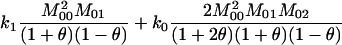 |
2 | 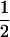 |
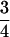 |
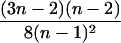 |
| AiAj, AiAj | 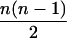 |
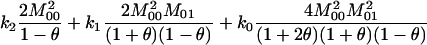 |
1 | 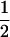 |
1 | 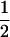 |
| AiAj, AiAk | n(n − 1)(n − 2) | 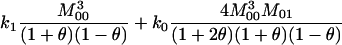 |
3 | 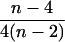 |
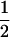 |
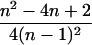 |
| AiAj, AkAk | 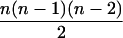 |
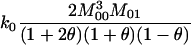 |
4 | 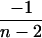 |
0 | 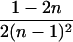 |
| AiAj, AkAl | 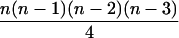 |
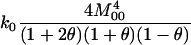 |
4 | 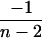 |
0 | 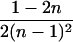 |
For each IBS mode, the number of different genotypes possible in that mode is indicated in the column labeled “count.” The genotypic category to which each IBS mode belongs is listed, as are the mode's value of SXY and single-locus Lynch–Ritland (LR) and similarity index (SIM) estimates for the coancestry coefficient based on that mode.
The expected values of P1, P2, and P3 are
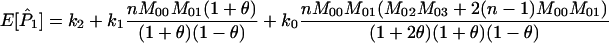 |
(A5) |
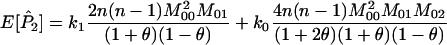 |
(A6) |
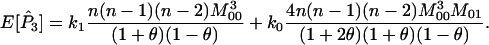 |
(A7) |
Wang's equations for the multiallelic case are written in terms of some functions of the allele frequencies. These functions, and their values in the equally frequent allele case, are as follows:
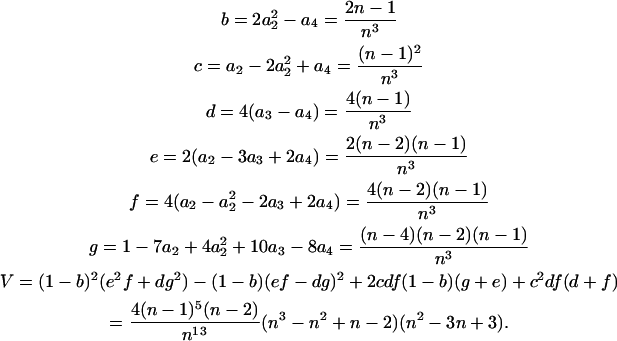 |
Wang writes equations for 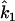 and
and 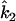 (which he denotes
(which he denotes 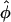 and
and 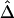 ) in terms of the above functions. Using Wang's Equations 9 and 10, the expected values of the estimates of k1 and k2 are
) in terms of the above functions. Using Wang's Equations 9 and 10, the expected values of the estimates of k1 and k2 are
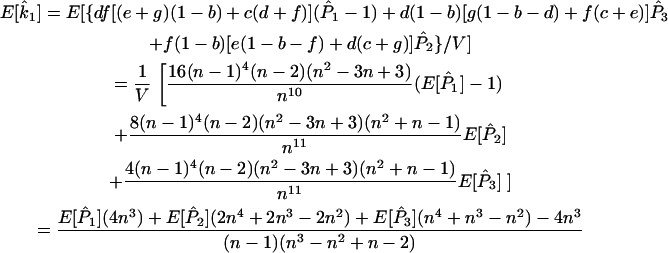 |
(A8) |
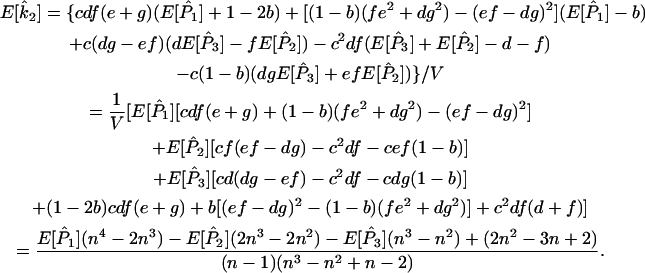 |
(A9) |
This gives
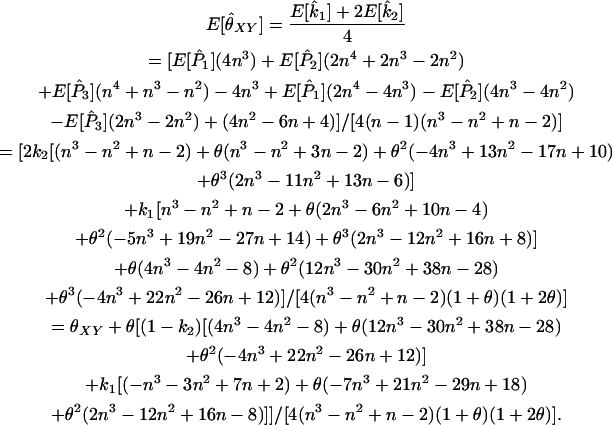 |
(A10) |
Wang's approach for the multilocus case with multiple alleles is the same as that for the diallelic case. Each of 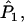
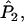
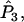 a1, a2, a3, a4, and
a1, a2, a3, a4, and 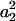 is replaced by a weighted average taken over the loci. Since we are considering the case in which allele frequencies at all loci are the same, the average value of ai (i = 1, 2, 3, 4) is the same as the single-locus value. Hence, the only difference between the single-locus and multilocus estimates is that
is replaced by a weighted average taken over the loci. Since we are considering the case in which allele frequencies at all loci are the same, the average value of ai (i = 1, 2, 3, 4) is the same as the single-locus value. Hence, the only difference between the single-locus and multilocus estimates is that 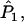
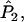 and
and 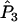 in Equations A8 and A9 are replaced by weighted averages. However, since Equations A8 and A9 are linear in these terms, the multilocus estimator has the same expected value as the single-locus estimator.
in Equations A8 and A9 are replaced by weighted averages. However, since Equations A8 and A9 are linear in these terms, the multilocus estimator has the same expected value as the single-locus estimator.
We have seen that Wang's estimator gives unexpected results when the number of alleles is small. We also have observed that, with 10 alleles at a locus, this undesirable behavior appears to have been corrected (see, e.g., Figure 5). We next examine the behavior of Wang's estimator for unrelated individuals as we increase the number of (equally frequent) alleles:
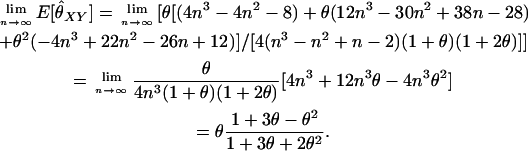 |
(A11) |
Thus, for reasonable values of θ, 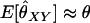 for unrelated individuals when the number of alleles per locus is large.
for unrelated individuals when the number of alleles per locus is large.
Lynch and Ritland's (1999) estimator:
If (a, b) is the genotype of the first individual, (c, d) is the genotype of the second individual, and Sij is an indicator of whether alleles i and j are identical by state, then Lynch and Ritland's single-locus estimator is
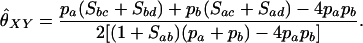 |
(A12) |
In contrast to Wang's estimator, the same expected estimate is derived in both the general diallelic case and the case with n equally frequent alleles, as well as in a general three-allele case (not shown).
For the diallelic case, let p be the frequency of allele A0. The single-locus estimates for θXY for each possible joint genotype are given in Table A1.
If G is the set of all possible joint genotypes, we have
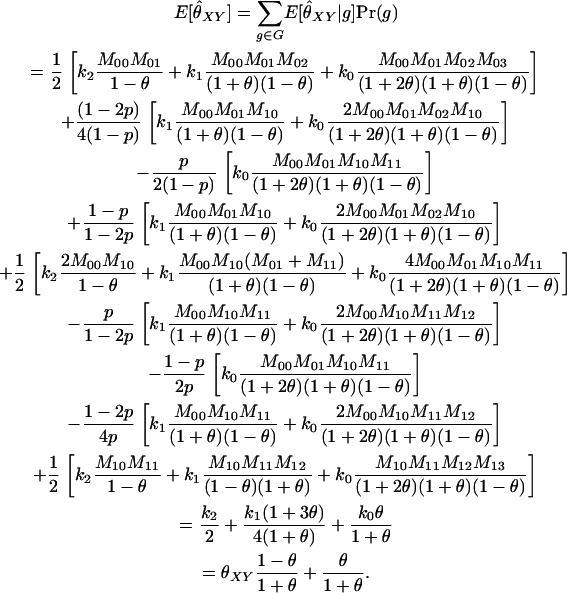 |
(A13) |
The expected value of 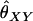 for the case with n equally frequent alleles can be derived using the nine IBS genotype classes, their counts, and their probabilities, which are listed in Table A2. Then
for the case with n equally frequent alleles can be derived using the nine IBS genotype classes, their counts, and their probabilities, which are listed in Table A2. Then
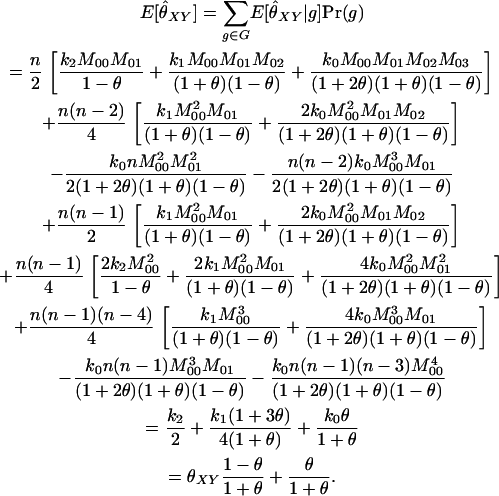 |
(A14) |
For the Lynch–Ritland estimator, the multilocus relatedness estimate is simply a weighted average of the single-locus estimates. Since each of the single-locus estimators has the same expected value, the value for the multilocus case is identical to that for the single-locus case.
The similarity index:
The similarity index is based upon the average proportion of alleles shared IBS in the two individuals, as measured by the quantity SXY = 0.5 · (proportion of X's alleles that are present in Y) + 0.5 · (proportion of Y's alleles that are present in X). The values of SXY for each possible joint genotype are given in Table A2.
Let (a, b) and (c, d) be the genotypes of the two individuals. The equation for the similarity index is then
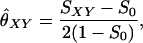 |
(A15) |
where S0 is the expected proportion of alleles shared IBS in unrelated individuals, given by 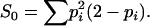
For the diallelic case, the similarity index and Wang's estimators are identical at a single locus and so have the same expected value. Table A2 lists the single-locus estimates for θXY for the case in which there are n equally frequent alleles at a locus. In this case, the expected value of the estimator for a single locus is derived as follows:
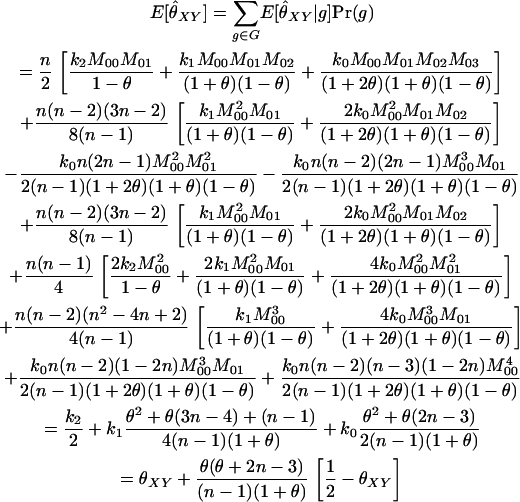 |
(A16) |
The multilocus estimate is the average of the single-locus estimates and so has the same expected value as the single-locus estimator.
For unrelated individuals, the limit as n grows large is
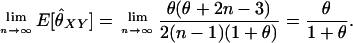 |
(A17) |
The Queller–Goodnight estimator:
The Queller–Goodnight estimator has the following form:
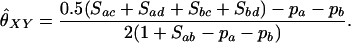 |
(A18) |
This is undefined in the diallelic case when the first individual is heterozygous. For the case in which there are n equally frequent alleles, this estimator is the same as the Lynch–Ritland estimator.
References
- Ayres, K. L., 2000. Relatedness testing in subdivided populations. Forensic Sci. Int. 114: 107–115. [DOI] [PubMed] [Google Scholar]
- Balding, D. J., and R. A. Nichols, 1994. DNA profile match probability calculation: how to allow for population stratification, relatedness, database selection and single bands. Forensic Sci. Int. 64: 125–140. [DOI] [PubMed] [Google Scholar]
- Balding, D. J., and R. A. Nichols, 1997. Significant genetic correlations among Caucasians at forensic DNA loci. Heredity 78: 583–589. [DOI] [PubMed] [Google Scholar]
- Broman, K. W., and J. L. Weber, 1999. Long homozygous chromosomal segments in reference families from the Centre d'Ètude du Polymorphisme Humain. Am. J. Hum. Genet. 65: 1493–1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budowle, B., and K. L. Monson, 1994. Greater differences in forensic DNA profile frequencies estimated from racial groups than from ethnic subgroups. Clin. Chim. Acta 228: 3–18. [DOI] [PubMed] [Google Scholar]
- Egeland, J. A. (Editor), 1972. Descendents of Christian Fisher and Other Amish-Mennonite Pioneer Families. Johns Hopkins Hospital, Baltimore.
- Griffiths, R. C., 1979. A transition density expansion for a multiallele diffusion model. Adv. Appl. Probab. 11: 310–325. [Google Scholar]
- Hepler, A. B., 2005. Improving forensic identification using Bayesian networks and relatedness estimation. Ph.D. Thesis, North Carolina State University, Raleigh, NC.
- Hinds, D., L. Stuve, G. Nilsen, E. Halperin, E. Eskin et al., 2005. Whole-genome patterns of common DNA variation in three human populations. Science 307: 1072–1079. [DOI] [PubMed] [Google Scholar]
- International HapMap Consortium, 2005. A haplotype map of the human genome. Nature 437: 1299–1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacquard, A., 1972. Genetic information given by a relative. Biometrics 28: 1101–1114. [PubMed] [Google Scholar]
- Krane, D. E., R. W. Allen, S. A. Sawyer, D. A. Petrov and D. L. Hartl, 1992. Genetic differences at four DNA typing loci in Finnish, Italian, and mixed Caucasian populations. Proc. Natl. Acad. Sci. USA 89: 10583–10587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kretzmann, M. B., N. Capote, B. Bautschi, J. A. Godoy, J. A. Donázar et al., 2003. Genetically distinct island populations of the Egyptian vulture (Neophron percnopterus). Conserv. Genet. 4: 697–706. [Google Scholar]
- Li, C. C., D. E. Weeks and A. Chakravarti, 1993. Similarity of DNA fingerprints due to chance and relatedness. Hum. Hered. 43: 45–52. [DOI] [PubMed] [Google Scholar]
- Lynch, M., and K. Ritland, 1999. Estimation of pairwise relatedness with molecular markers. Genetics 152: 1753–1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall, H. D., and K. Ritland, 2002. Genetic diversity and differentiation of Kermode bear populations. Mol. Ecol. 11: 685–697. [DOI] [PubMed] [Google Scholar]
- Milligan, B. G., 2003. Maximum-likelihood estimation of relatedness. Genetics 163: 1153–1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Press, W. H., S. A. Teukolsky, W. T. Vetterling and B. P. Flannery, 2002. Numerical Recipes in C++: The Art of Scientific Computing, Ed. 2. Cambridge University Press, Cambridge, UK.
- Queller, D. C., and K. F. Goodnight, 1989. Estimating relatedness using genetic markers. Evolution 43: 258–275. [DOI] [PubMed] [Google Scholar]
- Ritland, K., 1996. Estimators for pairwise relatedness and inbreeding coefficients. Genet. Res. 67: 175–186. [Google Scholar]
- Thompson, E. A., 1975. The estimation of pairwise relationships. Ann. Hum. Genet. 39: 173–188. [DOI] [PubMed] [Google Scholar]
- Thompson, E. A., 1976. A restriction on the space of genetic relationships. Ann. Hum. Genet. 40: 201–204. [DOI] [PubMed] [Google Scholar]
- Wang, J., 2002. An estimator for pairwise relatedness using molecular markers. Genetics 160: 1203–1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weir, B. S., 1994. The effects of inbreeding on forensic calculations. Annu. Rev. Genet. 28: 597–621. [DOI] [PubMed] [Google Scholar]
- Weir, B. S., 2003. Forensics, pp. 830–852 in Handbook of Statistical Genetics, edited by D. Balding, M. Bishop and C. Cannings. John Wiley & Sons, Chichester, UK.
- Weir, B. S., and C. C. Cockerham, 1984. Estimating F-statistics for analysis of population-structure. Evolution 38: 1358–1370. [DOI] [PubMed] [Google Scholar]
- Weir, B. S., L. R. Cardon, A. D. Anderson, D. M. Nielsen and W. G. Hill, 2005. Measures of human population structure show heterogeneity among genomic regions. Genome Res. 15: 1468–1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weir, B. S., A. D. Anderson and A. B. Hepler, 2006. Genetic relatedness analysis: modern data and new challenges. Nat. Rev. Genet. 7: 771–780. [DOI] [PubMed] [Google Scholar]
- Wright, S., 1951. The genetical structure of populations. Ann. Eugen. 15: 323–354. [DOI] [PubMed] [Google Scholar]