

Abstract
The G matrix measures the components of phenotypic variation that are genetically heritable. The structure of G, that is, its principal components and their associated variances, determines, in part, the direction and speed of multivariate trait evolution. In this article we present a framework and results that give the structure of G under the assumption of neutrality. We suggest that a neutral expectation of the structure of G is important because it gives a null expectation for the structure of G from which the unique consequences of selection can be determined. We demonstrate how the processes of mutation, recombination, and drift shape the structure of G. Furthermore, we demonstrate how shared common ancestry between segregating alleles shapes the structure of G. Our results show that shared common ancestry, which manifests itself in the form of a gene genealogy, causes the structure of G to be nonuniform in that the variances associated with the principal components of G decline at an approximately exponential rate. Furthermore we show that the extent of the nonuniformity in the structure of G is enhanced with declines in mutation rates, recombination rates, and numbers of loci and is dependent on the pattern and modality of mutation.
PHENOTYPIC evolution is channeled through patterns of heritable variation. This is exemplified by the breeder's equation, 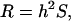 which for a single quantitative trait states that the evolutionary response (R) to selection (S) of the trait is proportional to its heritability, h2 (see, e.g., Falconer and MacKay 1996; Lynch and Walsh 1998). Likewise, the simultaneous evolutionary responses of several potentially correlated quantitative characters are predicted by the multivariate extension of the breeder's equation,
which for a single quantitative trait states that the evolutionary response (R) to selection (S) of the trait is proportional to its heritability, h2 (see, e.g., Falconer and MacKay 1996; Lynch and Walsh 1998). Likewise, the simultaneous evolutionary responses of several potentially correlated quantitative characters are predicted by the multivariate extension of the breeder's equation,  where
where  is a vector describing the responses of each character to selection, β—the selection gradient vector—describes the pattern of directional selection applied to the traits, and G is the additive-genetic covariance matrix describing the heritable patterns of variation and covariation of the traits (Lande 1979).
is a vector describing the responses of each character to selection, β—the selection gradient vector—describes the pattern of directional selection applied to the traits, and G is the additive-genetic covariance matrix describing the heritable patterns of variation and covariation of the traits (Lande 1979).
Quantitative geneticists and evolutionary biologists alike are particularly interested in the principal components of the G matrix, since these indicate independent paths along which evolution can occur, and the variance associated with each principal component, since these modulate the speed of evolution along the direction of each principal component. For instance, the “evolutionary line of least resistance” for adaptive radiation (Schluter 1996) is defined by the principal component of G with the largest variance, i.e., with the greatest amount of heritable variation. At the other extreme, principal components of G with the smallest variances represent evolutionary avenues with the greatest degrees of genetic resistance (Kirkpatrick and Lofsvold 1992; Hine and Blows 2006).
Since the G matrix itself can evolve, it is natural to consider the influences of evolutionary forces (such as selection, mutation, random genetic drift, and recombination) on the patterns of variation associated with the principal components of G. For example, principal components with the smallest eigenvalues might reflect the strongest patterns of multivariate selection (e.g., Blows et al. 2004; Hine et al. 2004). Indeed, this is expected on general theoretical grounds (Lande 1980; Lande and Arnold 1983; Phillips and Arnold 1989) and has been verified by computer simulations (Jones et al. 2003).
A second force that causes the G matrix to evolve is random genetic drift. Phillips et al. (2001) showed that severe inbreeding led to an average reduction in total variation of the G matrix that was proportional to the theoretical expectation 1 − F, where F is the inbreeding coefficient. Furthermore, the principal components of the G matrices differed (in an apparently random way) among replicated inbred lines, although, on average, the G matrices across replicated inbred lines did not differ from those of the outbred control population. Subsequent work by Whitlock et al. (2002) on the same set of inbred lines showed that the G matrix in some lines continued to change over 20 generations of random mating, but there was no evidence that the G matrices were returning to the original state in the outbred control line. On the basis of work by Avery and Hill (1977) and Zeng and Cockerham (1991), Whitlock et al. (2002) hypothesized that the subsequent changes in G matrices in the generations following inbreeding were caused, in part, by the decay of linkage disequilibria that were generated during the inbreeding episode. Besides the force of recombination, theoretical work by Lande (1980) shows that mutational pressure can also cause the G matrix to evolve.
At equilibrium the proximate causes of the structure of a G matrix are linkage disequilibria and the pleiotropic and nonpleiotropic effects of segregating mutations. The ultimate forces that contribute to the structure of the G matrix are recombination, mutation, genetic drift, and selection (e.g., Phillips and McGuigan 2006). To date, no theoretical work presents a neutral expectation for the structure of the G matrix when the only forces shaping its evolution are mutation, recombination, and drift, although Lynch and Hill (1986) presented a neutral model of the evolution of genetic variance for a single character. In the case of multiple characters Lynch (1994) and Phillips and McGuigan (2006) postulate that G will evolve to be proportional to M, the mutational variance–covariance matrix.
The focus of this article is the question of how mutation, recombination, and random genetic drift determine the eigenstructure of the G matrix at equilibrium, where the eigenstructure of the G matrix is measured according to its eigenvectors and eigenvalues. The eigenvectors of G are its principal components and the eigenvalues of G are the variances associated with each eigenvector. Naïvely, for the case of free recombination among loci, one might expect that the eigenvalues of G would be proportional to those of M, with drift-induced linkage disequilibrium and stochastic sampling from M causing nondirectional variation around this expectation. Accordingly, if the eigenvalues of M were all similar in size, then on average so would the eigenvalues of G. We explore the validity of this intuitively reasonable expectation.
One of the main insights of this article is that this intuitively reasonable expectation is not valid. A primary reason is that segregating alleles in a population are related to one another due to shared common ancestry or, to put it another way, alleles are related genealogically through shared common descent via reproduction. Patterns of shared common ancestry can be described for a sample from a population using Kingman's (1982) coalescent and multilocus extensions such as Griffiths and Marjoram's (1997) ancestral-recombination graph. In this article we use the coalescent and the ancestral-recombination graph as a framework to investigate neutral evolution of G.
The neutral expectations developed here aid the interpretation and analysis of recent empirical work that showed significant additive genetic variance for 20 wing-shape characters in Drosophila melanogaster and an approximately exponential decline in the ordered eigenvalues of the G matrix for these characters (Mezey and Houle 2005). The exponential decline in ordered eigenvalues may be interpreted as signatures of either variable selection across characters and/or variable mutational heritabilities across characters. Our analyses show that, under the neutral model, the ordered eigenvalues of G will evolve toward a regular pattern. Surprisingly, even if the ordered eigenvalues of M are perfectly uniform and selection is absent, the expected eigenvalues of G in a sample from a population are predicted to decline at an approximately exponential rate.
METHODS
The model:
We consider the evolution of a multivariate quantitative trait affected by multiple loci in a diploid population of size N with Wright–Fisher random mating. (For nonideal populations, N = Ne, the variance-effective population size.) Evolution is neutral as we assume the trait experiences no selection. Alleles affecting the trait act additively within and across loci (i.e., no dominance, epistasis, or genotype–environment interactions). For example, if 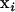 and
and 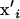 are multivariate effects of maternally and paternally inherited alleles at locus i (e.g., the effects of each allele on m traits), then the genotypic value g of a typical individual is
are multivariate effects of maternally and paternally inherited alleles at locus i (e.g., the effects of each allele on m traits), then the genotypic value g of a typical individual is 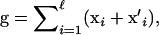 where
where 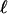 is the number of loci. The phenotype is
is the number of loci. The phenotype is 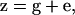 where e is an independent, mean-zero random variable. The G matrix is the m × m covariance matrix of additive values, g. We hereafter ignore e because, in the absence of selection, it plays no role in the evolution of G. It is important to note, however, that e can affect estimates of G, which may affect predictions of the eigenstructure of any particular G, especially for small samples from a population. We do not address this issue in this article and leave it for further study.
where e is an independent, mean-zero random variable. The G matrix is the m × m covariance matrix of additive values, g. We hereafter ignore e because, in the absence of selection, it plays no role in the evolution of G. It is important to note, however, that e can affect estimates of G, which may affect predictions of the eigenstructure of any particular G, especially for small samples from a population. We do not address this issue in this article and leave it for further study.
Under neutrality and additivity, three main processes influence G-matrix evolution: spontaneous mutation, recombination, and random genetic drift via stochastic reproduction. In this article we focus on the recombination–mutation–drift equilibrium characteristics of G. For spontaneous mutation we examined two infinite-allele mutation models: the continuum-of-alleles model (Crow and Kimura 1964; Kimura 1965) and the house-of-cards model (Kingman 1977, 1978). The continuum-of-alleles model assumes that each mutation generates a new multivariate effect that is added to the current effect of an allele at a locus, where a new effect is sampled from a mutational distribution. In our simulations we used a zero-mean multivariate normal distribution with covariance matrix A for the distribution of mutational effects. The diagonal elements of A are the variances in mutational effects and the off diagonals are the covariances of mutational effects on different traits. The house-of-cards model (Kingman 1978) assumes that each new mutation completely replaces the current allelic effect at a mutated locus.
Coalescent approaches:
Because the alleles contributing to g are neutral and every mutation is novel, the alleles at individual loci should follow the distribution expected at equilibrium for the infinite-alleles neutral model (see, e.g., Ewens 2004). Rather than sample from this distribution directly, we used a different but equivalent approach for n < N, where n is the sample size—the coalescent. The coalescent is useful in that it retains information about the underlying genealogical history of alleles. Thus it can be used to model how variation in allelic values is determined by patterns of shared common ancestry.
Coalescent with independent loci:
Figure 1 illustrates the approach that we used to generate the genotypic values g of individuals in a sample of size n based on the neutral coalescent (Kingman 1982). Loci were treated independently. For each locus we generated a tree with 2n tips and placed mutations on the tree using the standard neutral coalescent approach. (We employed the algorithm in the Appendix of Hudson 1991.) When 1 unit of coalescent time is scaled to be 2N generations, coalescent events occur at a rate 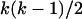 per coalescent time unit using the exponential approximation of a geometric process, where k is the number of lineages present at a particular time back in history. Once a gene tree is drawn, branch segments in the tree have times associated with them as a result of the coalescent process. Mutations were randomly placed along branch segments such that the number of mutations along a branch segment of the coalescent tree was Poisson distributed with mean 2N
per coalescent time unit using the exponential approximation of a geometric process, where k is the number of lineages present at a particular time back in history. Once a gene tree is drawn, branch segments in the tree have times associated with them as a result of the coalescent process. Mutations were randomly placed along branch segments such that the number of mutations along a branch segment of the coalescent tree was Poisson distributed with mean 2N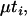 where
where 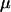 is the haploid per locus mutation rate and ti is the length (in coalescent time units) of branch segment i. With each mutation, we then associated a vector of effects drawn from a multivariate normal distribution with mean 0 and covariance A. A population was generated by randomly assigning two alleles from each locus (without replacement) to each of the n individuals. Note that this coalescent approach to generating individual genotypic values imposes the strict rule that loci are completely independent; i.e., there are no transient linkage disequilibria.
is the haploid per locus mutation rate and ti is the length (in coalescent time units) of branch segment i. With each mutation, we then associated a vector of effects drawn from a multivariate normal distribution with mean 0 and covariance A. A population was generated by randomly assigning two alleles from each locus (without replacement) to each of the n individuals. Note that this coalescent approach to generating individual genotypic values imposes the strict rule that loci are completely independent; i.e., there are no transient linkage disequilibria.
Figure 1.—

Demonstration of how individual genotypic values were generated using the neutral coalescent in which the evolutionary histories of loci are completely independent. It is assumed that there are two individuals in a sample and each individual is composed of two loci. The gene tree on the left is that for locus A and the gene tree on the right is that for locus B. For each individual two alleles from each locus are chosen without replacement. Genotypic values under the continuum-of-alleles (COA) and house-of-cards (HOC) mutation models are given. See main text for further details.
The overall process is illustrated in Figure 1 for a sample of n = 2 individuals, each with two loci. Mutations in the coalescent tree are marked with hatch marks and the associated vector of effects is represented by 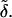 For locus A, alleles a1 and a2 are unmutated relative to the ancestral allele and, accordingly, have the ancestral value
For locus A, alleles a1 and a2 are unmutated relative to the ancestral allele and, accordingly, have the ancestral value 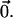 Allele a3 is differentiated from the ancestral allele by one mutation and has the value
Allele a3 is differentiated from the ancestral allele by one mutation and has the value  =
= 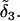 The value of allele a4 depends on the mutation model. For the continuum-of-alleles model, the value of a4 is
The value of allele a4 depends on the mutation model. For the continuum-of-alleles model, the value of a4 is  but for the house-of-cards model, the value of a4 is
but for the house-of-cards model, the value of a4 is 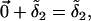 since the later mutational effect
since the later mutational effect 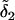 completely replaces the earlier effect
completely replaces the earlier effect 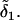 The allelic effects at locus B are determined similarly.
The allelic effects at locus B are determined similarly.
Next, the alleles are randomly assigned two at a time, without replacement, to an individual. In Figure 1, alleles a1 and a4 at locus A and alleles b3 and b4 at locus B were assigned to the first individual. On the basis of the alleles sampled and the mutations in the gene tree, the genotypic value of the first individual is g = 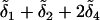 for the continuum-of-alleles model and g =
for the continuum-of-alleles model and g = 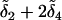 assuming the house-of-cards.
assuming the house-of-cards.
Ancestral recombination graph:
As stated at the outset, linkage disequilibria are also a proximate cause of additive-genetic covariances. Strictly applying independence among loci may therefore eliminate a factor promoting genetic covariances even when there is free recombination among loci. To allow for physical linkage among loci and for the possibility of linkage disequilibria we used the ancestral recombination graph coalescent approach (Griffiths 1981, 1991; Griffiths and Marjoram 1996, 1997; see Nordborg 2001, pp. 195–200, for a useful introduction).
Figure 2 illustrates how a sample of two individuals each consisting of two loci is generated using the ancestral recombination graph. Instead of having two independent loci, the loci are linked. In Figure 2, the rectangles at the top each represent a haplotype in the current generation with the open section corresponding to locus A and the solid section corresponding to locus B. In an ancestral recombination graph, two processes occur when forming a gene tree, namely recombination and coalescent events. Scaling time to be 2N generations, coalescent events occur as before at rate 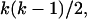 where k is the number of lineages at a given time. Recombination events occur at the rate 4Nrk/2, where r is the per haplotype recombination rate per generation. The total rate of events is
where k is the number of lineages at a given time. Recombination events occur at the rate 4Nrk/2, where r is the per haplotype recombination rate per generation. The total rate of events is 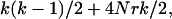 and, given an event, the probability it is a coalescent is
and, given an event, the probability it is a coalescent is 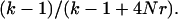 Although the rate of recombination is typically much faster than the rate of coalescence, a common ancestor is guaranteed (Griffiths and Marjoram 1997).
Although the rate of recombination is typically much faster than the rate of coalescence, a common ancestor is guaranteed (Griffiths and Marjoram 1997).
Figure 2.—

Demonstration of how individual genotypic values were generated using the ancestral recombination graph. Loci are linked such that individuals inherit haplotypes as opposed to independent alleles. Haplotypes are presented as rectangles and each haplotype consists of two loci: locus A (open rectangles) and locus B (solid rectangles). Points at which a lineage bifurcates into two lineages (back in time) correspond to recombination events. Where lineages join are coalescent events. In this example, each individual consists of a random sample of two haplotypes (without replacement). Genotypic values under the continuum-of-alleles (COA) and house-of-cards (HOC) mutation models are given. See main text for further details.
Proceeding backward in time from the present, a recombination event is represented by the splitting of a single haplotype lineage into two lineages. For a two-locus model, one locus is retained in one ancestral lineage, whereas the other locus is retained in the second ancestral lineage. In Figure 2, shading corresponds to a locus that is recombined out in the descendant lineage following a recombination event. Often recombination events occur in an ancestral lineage that consists of one locus that is present in the current generation and one that is subsequently recombined out and not present in the current generation. With respect to the coalescence of alleles in the sample, these recombination events are relevant only with respect to incrementing time backward in the gene tree, but do not add additional information. Consequently, the splitting of these types of lineages is not recorded in the gene tree, although the time of the event is.
Once an ancestral recombination graph is drawn, there are times associated with each branch segment (in units of 2N generations) and numbers of mutations are modeled to occur along each branch segment according to a Poisson distribution with mean 2N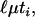 where ti is the time associated with branch segment i. The per locus mutation rate is assumed to be equal across loci, such that for
where ti is the time associated with branch segment i. The per locus mutation rate is assumed to be equal across loci, such that for 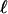 loci and given a mutational event, the probability the mutation occurs at a particular locus is
loci and given a mutational event, the probability the mutation occurs at a particular locus is 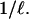 In Figure 2, a hatch mark represents a mutation, the box next to it represents the locus at which the mutation occurred, and
In Figure 2, a hatch mark represents a mutation, the box next to it represents the locus at which the mutation occurred, and 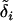 is the vector of effects of mutation i. To get an allele's value, simply trace its history back in the gene tree, being careful to follow the correct ancestral lineage at a recombination event.
is the vector of effects of mutation i. To get an allele's value, simply trace its history back in the gene tree, being careful to follow the correct ancestral lineage at a recombination event.
Because loci can be physically linked, individual genotypes are formed by randomly sampling haplotypes as opposed to alleles at independent loci. With a sample consisting of two individuals there are four haplotypes, each individual consisting of two haplotypes. In the example (Figure 2), individual 1 consists of haplotypes 1 and 4. Following the history of the allele a1 back in time, only the mutation with effect  occurs before reaching the common ancestor of the population so the additive effect of a1 is
occurs before reaching the common ancestor of the population so the additive effect of a1 is 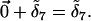 Note that the mutation with effect
Note that the mutation with effect 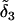 occurred in an ancestral allele not present in the current generation. As with the independent-loci coalescent approach described above, an allele's additive value is the sum of the effects of mutations in a lineage for the continuum-of-alleles mutation model whereas, for the house-of-cards model, the additive effect is equal to the most recent mutation for the allele.
occurred in an ancestral allele not present in the current generation. As with the independent-loci coalescent approach described above, an allele's additive value is the sum of the effects of mutations in a lineage for the continuum-of-alleles mutation model whereas, for the house-of-cards model, the additive effect is equal to the most recent mutation for the allele.
Given the relatively large sample sizes examined in this study combined with a large number of loci and a moderate to fast recombination rate, there were too many possible ancestral haplotypes for computers with 1 GB of memory to simulate the ancestral recombination graph algorithm; hence, there are significant computational limitations when considering recombination while utilizing the coalescent method.
Parameters:
Parameters underlying the results presented in this article are cast in terms of 
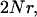 the number of loci, and M (see below for a definition of M). The expression
the number of loci, and M (see below for a definition of M). The expression 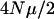 gives the expected number of new mutations that enter a population each generation at each locus. In this article,
gives the expected number of new mutations that enter a population each generation at each locus. In this article, 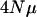 ranges from 0.1 to 10.0. The expression
ranges from 0.1 to 10.0. The expression 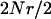 gives the expected number of recombination events per generation during the meiotic phase of the life cycle for an entire population per adjacent pair of loci and it ranges from 0.0 to 1000.0. Numbers of loci vary from 1 to 10 in the results, and it is assumed that each locus has the same value of
gives the expected number of recombination events per generation during the meiotic phase of the life cycle for an entire population per adjacent pair of loci and it ranges from 0.0 to 1000.0. Numbers of loci vary from 1 to 10 in the results, and it is assumed that each locus has the same value of 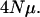
In this article, mutations at a locus act either pleiotropically or nonpleiotropically. When mutations act pleiotropically, a single mutation potentially affects all characters and the covariance matrix of their effects is given by A. For simplicity, we concentrate on diagonal covariance matrices such that 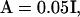 where I is the identity matrix. When mutations at a locus act nonpleiotropically, the mutations affect only a single character and the variance of their effects is 0.05.
where I is the identity matrix. When mutations at a locus act nonpleiotropically, the mutations affect only a single character and the variance of their effects is 0.05.
Throughout this article it is assumed that mutation rates and mutational effects are uniform across characters. Thus, the expected eigenvalues of the mutational covariance matrix, M, are uniform, where  and U is the zygotic mutation rate. We make these simplifying assumptions so that any nonuniformity in the eigenvalues of G arises via factors other than nonuniformity in the eigenvalues of the mutational covariance matrix. In reality, characters of organisms will likely have nonuniform mutational variance and this may be an important factor in determining the ordered eigenvalues of G. But, here we are particularly interested in demonstrating that genealogical history per se is important in shaping the ordered eigenvalues of G. Furthermore, we do not explore cases in which there is a nonorthogonal transformation between the underlying mutational process and the measured characters of an organism. If the transformation is not orthogonal then this will generally affect the eigenvalues of G.
and U is the zygotic mutation rate. We make these simplifying assumptions so that any nonuniformity in the eigenvalues of G arises via factors other than nonuniformity in the eigenvalues of the mutational covariance matrix. In reality, characters of organisms will likely have nonuniform mutational variance and this may be an important factor in determining the ordered eigenvalues of G. But, here we are particularly interested in demonstrating that genealogical history per se is important in shaping the ordered eigenvalues of G. Furthermore, we do not explore cases in which there is a nonorthogonal transformation between the underlying mutational process and the measured characters of an organism. If the transformation is not orthogonal then this will generally affect the eigenvalues of G.
RESULTS
All cases we examined were consistent with the prediction that, at equilibrium, the average G matrix is equal to 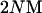 under neutrality (Phillips and McGuigan 2006). However, our results revealed two new, consistent features of neutral evolution of G matrices at recombination–mutation–drift balance. First, the eigenvalues of equilibrium G matrices are expected to be highly nonuniform—declining in an approximately exponential manner—even when the mutation covariance matrix M has perfectly uniform eigenvalues (i.e., when M has a single repeated eigenvalue). Second, we found that genealogical ancestry of alleles in a finite population is particularly important in determining the intensity of decay of the ranked eigenvalues of G matrices under neutrality. In the absence of selection, two forces, namely recombination and random genetic drift, generate genealogies of alleles, and the genotypic values of alleles in a particular genealogy are determined by the pattern of mutation. In what follows we show in turn how mutation, drift, and recombination shape the ordered eigenvalues of G matrices.
under neutrality (Phillips and McGuigan 2006). However, our results revealed two new, consistent features of neutral evolution of G matrices at recombination–mutation–drift balance. First, the eigenvalues of equilibrium G matrices are expected to be highly nonuniform—declining in an approximately exponential manner—even when the mutation covariance matrix M has perfectly uniform eigenvalues (i.e., when M has a single repeated eigenvalue). Second, we found that genealogical ancestry of alleles in a finite population is particularly important in determining the intensity of decay of the ranked eigenvalues of G matrices under neutrality. In the absence of selection, two forces, namely recombination and random genetic drift, generate genealogies of alleles, and the genotypic values of alleles in a particular genealogy are determined by the pattern of mutation. In what follows we show in turn how mutation, drift, and recombination shape the ordered eigenvalues of G matrices.
Genealogical effect on G:
Figure 3 demonstrates how genealogical ancestry can generate nonuniformity in the eigenvalues of G assuming allelic variation at a single locus. In Figure 3a, the top left corner shows a genealogy of a sample of six alleles. On the genealogy are 10 mutations that give rise to five allelic types out of the six sampled alleles. The bivariate effects of these 10 mutations are plotted. The principal components of mutational variation are of similar magnitude, suggesting that variation is evenly distributed across the two dimensions. Figure 3b shows the values of the alleles at the tips of the tree, plotted as solid dots, using the continuum-of-alleles model. What is immediately apparent in this example is that allelic variation, instead of being evenly spread across two dimensions (as in Figure 3a), is primarily concentrated in one dimension. The reason for this is that allelic values are dependent due to their shared common ancestry whereas the mutational effects are independent.
Figure 3.—

The effect of genealogy at a single locus on the eigenstructure of allelic variation. (a and b) In the top left corner is an illustration of a genealogy of six alleles with 10 mutations. (a) The bivariate effects of 10 random mutations are plotted assuming normality, where the average effect of a mutation per character is zero and effects are uncorrelated. The shaded arrows represent the principal components of the variation in mutational effects with the length of each arrow proportional to the variance explained by each direction. The arrows are similar in length, suggesting that mutational variation is evenly distributed across the two dimensions. (b) The effects of the six alleles at the tips of the genealogy are plotted as solid dots. There are only five dots because two alleles are identical in state. Lines of descent, shaded to aid comparison with the genealogy, connect allelic values. As in a, the shaded arrows indicate the principal components of allelic variation. Comparing a and b suggests that shared common ancestry leads to a reduction in the dimensionality of allelic variation relative to mutational variation. Allelic variation is concentrated along one principal component axis, whereas mutational variation is distributed relatively evenly over two principal component axes.
Figure 4 extends the principle illustrated in Figure 3 by replicating the genealogical history of a single locus many times to determine if, on average, there is a greater rate of decline in the eigenvalues associated with breeding values vs. mutational variation. Furthermore, a larger sample is taken from each population and there are more trait dimensions. The boxes in Figure 4 give the average ordered eigenvalues of mutational variation for a single locus (as was done in Figure 3a, except replicated 500 times). Note that variation is nonuniform across the principal components even though mutation is sampled from a distribution with uniform variance across its principal components. The nonuniformity is the result of stochasticity, in that there are a finite number of mutations segregating at a locus and by chance the effects of these mutations are nonuniformly distributed across characters. The triangles in Figure 4 suggest that the eigenvalues associated with the breeding values of individuals decline at a greater rate than the eigenvalues associated with mutational variation. Two processes within the genealogy appear to bring about the enhanced decline in eigenvalues. One is that genealogy builds up statistical dependence among allelic values due to the pattern of shared common ancestry (branching). The second is that along a single branch multiple mutations may occur, which reduces the number of independently acting mutations. Figure 4 suggests that the point illustrated in Figure 3, namely that genealogy potentially enhances the nonuniformity in the eigenvalues associated with variation in breeding values, is general because, on average, there is a faster decline in eigenvalues associated with variation in breeding values than in mutational effects. On occasion, genealogical ancestry may have no effect on eigenvalues, but, on average, Figure 4 suggests that it enhances the nonuniformity in their values.
Figure 4.—

The effects of random mutation and genealogy on the eigenvalues of G. Shown are results for a single locus that pleiotropically affects five characters with 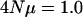 and a sample size of 300. To generate this figure 500 replicate coalescent trees were generated and random mutation was placed on the tree according to a Poisson process. Mutations were drawn from a multivariate normal distribution with zero mean and uncorrelated effects. To determine the eigenvalues of mutational effects, for each tree a covariance matrix was estimated on the basis of the effects of mutations on the tree (as in Figure 3a). Allelic effects were calculated assuming the continuum-of-alleles model. Individual breeding values were calculated by sampling without replacement two alleles from a coalescent tree. Eigenvalues are on a log scale and error bars represent 95% confidence intervals of the mean eigenvalue. The internal hatch mark within an error bar is the mean eigenvalue.
and a sample size of 300. To generate this figure 500 replicate coalescent trees were generated and random mutation was placed on the tree according to a Poisson process. Mutations were drawn from a multivariate normal distribution with zero mean and uncorrelated effects. To determine the eigenvalues of mutational effects, for each tree a covariance matrix was estimated on the basis of the effects of mutations on the tree (as in Figure 3a). Allelic effects were calculated assuming the continuum-of-alleles model. Individual breeding values were calculated by sampling without replacement two alleles from a coalescent tree. Eigenvalues are on a log scale and error bars represent 95% confidence intervals of the mean eigenvalue. The internal hatch mark within an error bar is the mean eigenvalue.
Linkage:
Linkage causes the genealogies of alleles at different loci to be correlated. Our results show that tighter linkage between two loci tends to enhance the nonuniformity in the expected eigenvalues of G (Figure 5). When the loci are completely linked (2Nr = 0) there is greater nonuniformity in eigenvalues than with a higher rate of recombination (2Nr = 1000).
Figure 5.—

The effects of recombination on the eigenvalues of G. A sample of 300 individuals was drawn from a population in which 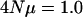 per locus and four loci were modeled using the ancestral recombination graph. 2Nr is given as the rate of recombination per adjacent pair of loci, and thus the total genomic recombination rate is 6Nr. Mutations pleiotropically affected five characters. A total of 500 replicate recombination graphs were simulated. Eigenvalues are on a log scale and error bars represent 95% confidence intervals of the mean eigenvalue. The internal hatch mark within an error bar is the mean eigenvalue.
per locus and four loci were modeled using the ancestral recombination graph. 2Nr is given as the rate of recombination per adjacent pair of loci, and thus the total genomic recombination rate is 6Nr. Mutations pleiotropically affected five characters. A total of 500 replicate recombination graphs were simulated. Eigenvalues are on a log scale and error bars represent 95% confidence intervals of the mean eigenvalue. The internal hatch mark within an error bar is the mean eigenvalue.
Other factors influencing eigenstructure:
Besides finite mutation, genealogy, and linkage, our results show that several other factors contribute to the pattern of eigenvalues of G.
Number of loci:
Alleles at each locus vary in how they contribute to patterns of additive genetic variance. As more loci are added, the relative contribution of each to the variability in G is reduced. Accordingly, there is more nonuniformity in the eigenvalues of G when there are few vs. many loci (Figure 6).
Figure 6.—

The effects of varying the number of loci on the eigenvalues of G. A sample of 300 individuals was drawn from a population in which 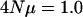 per locus. For the 10- and 100-loci cases, independent gene trees for each locus were simulated using Hudson's (1991) algorithm. Mean eigenvalues and their confidence intervals are based on 500 replicates. Eigenvalues are on a log scale and error bars represent 95% confidence intervals of the mean eigenvalue. The internal hatch mark within an error bar is the mean eigenvalue.
per locus. For the 10- and 100-loci cases, independent gene trees for each locus were simulated using Hudson's (1991) algorithm. Mean eigenvalues and their confidence intervals are based on 500 replicates. Eigenvalues are on a log scale and error bars represent 95% confidence intervals of the mean eigenvalue. The internal hatch mark within an error bar is the mean eigenvalue.
Mutation rate and effective population size:
The rate of decline in eigenvalues is greater with smaller values of 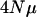 (Figure 7). In fact, there is a limit to the expected number of principal components that will have nonzero variance associated with it (Table 1). In Table 1, the M matrix has 25 dimensions with equal variances across dimensions and no covariance, yet beyond 12 principal components for a sample size of 75 and 15 principal components for a sample size of 300, principal components are expected to have zero variance associated with them. As the mutation rate or effective population size increases, the number of principal components with nonzero variance will increase (data not shown). Figure 7 illustrates another important point, namely that the decline in eigenvalues is not log linear for high-dimensional cases.
(Figure 7). In fact, there is a limit to the expected number of principal components that will have nonzero variance associated with it (Table 1). In Table 1, the M matrix has 25 dimensions with equal variances across dimensions and no covariance, yet beyond 12 principal components for a sample size of 75 and 15 principal components for a sample size of 300, principal components are expected to have zero variance associated with them. As the mutation rate or effective population size increases, the number of principal components with nonzero variance will increase (data not shown). Figure 7 illustrates another important point, namely that the decline in eigenvalues is not log linear for high-dimensional cases.
Figure 7.—

The consequence of varying 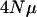 (per locus) with a fixed number of loci (10). To aid direct comparison between the different
(per locus) with a fixed number of loci (10). To aid direct comparison between the different 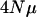 values with respect to how rank eigenvalues decline, the points for
values with respect to how rank eigenvalues decline, the points for 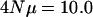 were all shifted down an equal amount such that the average leading eigenvalues for
were all shifted down an equal amount such that the average leading eigenvalues for 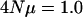 and
and 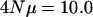 coincide. The figure demonstrates that different per locus mutation rates (or effective population sizes) lead to different rates of decrease in the eigenvalues, with smaller
coincide. The figure demonstrates that different per locus mutation rates (or effective population sizes) lead to different rates of decrease in the eigenvalues, with smaller 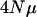 values showing a faster decline. The independent locus coalescent approach was used and n = 300. Eigenvalues are on a log scale and error bars represent 95% confidence intervals of the mean eigenvalue. The internal hatch mark within an error bar is the mean eigenvalue.
values showing a faster decline. The independent locus coalescent approach was used and n = 300. Eigenvalues are on a log scale and error bars represent 95% confidence intervals of the mean eigenvalue. The internal hatch mark within an error bar is the mean eigenvalue.
TABLE 1.
Limits to multivariate trait variation
| Sample size
|
||
|---|---|---|
| Eigenvalue rank | 75 | 300 |
| 1 | 6.41e-1 ± 3.20e-2 | 6.22e-1 ± 2.80e-2 |
| 2 | 3.09e-1 ± 1.86e-2 | 3.08e-1 ± 1.74e-2 |
| 3 | 1.56e-1 ± 1.25e-2 | 1.56e-1 ± 1.17e-2 |
| 4 | 7.37e-2 ± 7.32e-3 | 7.67e-2 ± 7.22e-3 |
| 5 | 3.63e-2 ± 4.69e-3 | 3.87e-2 ± 4.62e-3 |
| 6 | 1.72e-2 ± 2.71e-3 | 1.92e-2 ± 2.78e-3 |
| 7 | 7.85e-3 ± 1.60e-3 | 9.48e-3 ± 1.69e-3 |
| 8 | 3.31e-3 ± 8.07e-4 | 4.38e-3 ± 9.05e-4 |
| 9 | 1.27e-3 ± 4.42e-4 | 1.91e-3 ± 5.04e-4 |
| 10 | 5.23e-4 ± 2.44e-4 | 8.12e-4 ± 2.95e-4 |
| 11 | 2.02e-4 ± 1.34e-4 | 3.33e-4 ± 1.74e-4 |
| 12 | 9.08e-6 ± 1.78e-5 | 1.27e-4 ± 9.90e-5 |
| 13 | 0.0 | 6.02e-5 ± 5.00e-5 |
| 14 | 0.0 | 1.49e-5 ± 1.60e-5 |
| 15 | 0.0 | 1.19e-6 ± 2.34e-6 |
| 16 | 0.0 | 0.0 |
When the effective population size or genomic mutation rate for a set of characters is low enough, some principal components are expected to have zero variance associated with them. For instance, the eigenvalues are given ranked in order from largest to smallest when 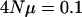 per locus and the number of loci is 10. The M matrix had dimensionality of 25 with the same variance for each principal component.
per locus and the number of loci is 10. The M matrix had dimensionality of 25 with the same variance for each principal component.
Mutational matrices with variable eigenvalues or nonzero correlations:
Until now, we have discussed only cases in which the expected mutational variance–covariance matrix has perfectly uniform eigenvalues with completely uncorrelated mutational effects on different traits. This is clearly a hypothetical extreme. In reality, we expect that the eigenvalues of M itself will vary and that mutational effects on different traits will be correlated (e.g., see Ajie et al. 2005; Estes et al. 2005). Results suggest that nonuniformity in the eigenvalues of M increases nonuniformity in the eigenvalues of G (data not shown).
We also found that introducing nonuniformity in the eigenvalues of M had an important influence on the eigenvectors of G. Simulations for two traits show that, when M has uniform eigenvalues, the leading eigenvector of G is uniformly distributed between 0° and 360°. In contrast, when the eigenvalues of M differ, the leading eigenvector of G follows a unimodal, symmetric distribution approximately centered at the leading eigenvector of M (results not shown). Thus, uneven eigenvalues of M affect the entire expected eigenstructure (i.e., eigenvalues and eigenvectors) of G under neutrality.
As for nonzero mutational correlations, their impacts on the eigenstructure of G are channeled through their effects on the eigenstructure of M. For example, consider a pair of traits each with mutation variance Vm and mutational correlation ρ (as in Jones et al. 2003). That is, 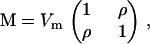 which has eigenvalues
which has eigenvalues 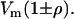 The expected eigenvalues of G given this M are exactly the same as those produced by the mutational covariance matrix
The expected eigenvalues of G given this M are exactly the same as those produced by the mutational covariance matrix 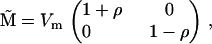 which has the same eigenvalues as M but with uncorrelated mutational effects (results not shown). This suggests that the eigenvalues of M—and not the specific patterns of mutational variances and covariances—determine the expected eigenvalue distribution of G at equilibrium. Note, however, that the distributions of expected eigenvectors of G would differ for the two mutational matrices since the eigenvectors of M and
which has the same eigenvalues as M but with uncorrelated mutational effects (results not shown). This suggests that the eigenvalues of M—and not the specific patterns of mutational variances and covariances—determine the expected eigenvalue distribution of G at equilibrium. Note, however, that the distributions of expected eigenvectors of G would differ for the two mutational matrices since the eigenvectors of M and 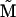 differ.
differ.
Mutational history:
We examined two models of mutation: the continuum-of-alleles model (Crow and Kimura 1964; Kimura 1965) and the house-of-cards model (Kingman 1977, 1978). The two models represent opposing extremes of how to model the effects of mutation. With respect to a genealogy of alleles, under the house-of-cards model the genealogy is important only in shaping allele frequencies, whereas in the continuum-of-alleles model the genealogy shapes both allele frequencies as well as the genotypic values of alleles. Thus, by comparing results from house-of-cards and continuum-of-alleles models, it is possible to disentangle, in part, the effects of variable allele frequency from the historical sequence of mutations on the eigenstructure of G.
Both the house-of-cards and the continuum-of-alleles models yield, on average, nonuniform eigenvalues at equilibrium (Figure 8). Therefore, it appears that variable allele frequencies contribute to the nonuniformity of eigenvalues of G and the mutational history of alleles further increases this nonuniformity.
Figure 8.—

The effect of different mutational models: house-of-cards vs. continuum-of-alleles. Three hundred individuals were sampled and 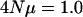 for each locus. The independent locus coalescent approach was used. Estimates are based on 500 replicates. Eigenvalues are on a log scale and error bars represent 95% confidence intervals of the mean eigenvalue. The internal hatch mark within an error bar is the mean eigenvalue.
for each locus. The independent locus coalescent approach was used. Estimates are based on 500 replicates. Eigenvalues are on a log scale and error bars represent 95% confidence intervals of the mean eigenvalue. The internal hatch mark within an error bar is the mean eigenvalue.
Pleiotropic vs. nonpleiotropic mutation:
All of the results discussed thus far have assumed universal pleiotropy. For example, if five characters are modeled then each random mutation could potentially affect all five characters. Under universal pleiotropy variation at each character is determined by the same set of gene trees. At the other extreme, when mutations at a locus are not pleiotropic, each character has a unique set of gene trees underlying its variation, provided that there is some recombination between loci. Figure 9 shows that pleiotropic mutation enhances nonuniformity in eigenvalues of G (particularly for lower mutation rates), presumably because the allelic ancestries that underlie variation of different characters overlap. If mutations act nonpleiotropically, then, to the extent that loci freely recombine, the allelic ancestries underlying each character's variation will be independent.
Figure 9.—

The effects of pleiotropy on the nonuniformity in G. Three characters and loci were modeled. In the pleiotropy cases, mutations at the three loci affect all characters, whereas in the no pleiotropy case a single locus affects only one of the characters. The top set of points corresponds to a per character 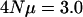 and the bottom set of points corresponds to a per character
and the bottom set of points corresponds to a per character  Mutation rate was modeled on a per character basis so that a direct comparison between the pleiotropy and the no pleiotropy case can be made. The ancestral-recombination graph was simulated in which the genomic recombination rate was
Mutation rate was modeled on a per character basis so that a direct comparison between the pleiotropy and the no pleiotropy case can be made. The ancestral-recombination graph was simulated in which the genomic recombination rate was 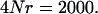 Eigenvalues are on a log scale and error bars represent 95% confidence intervals of the mean eigenvalue. The internal hatch mark within an error bar is the mean eigenvalue.
Eigenvalues are on a log scale and error bars represent 95% confidence intervals of the mean eigenvalue. The internal hatch mark within an error bar is the mean eigenvalue.
Whole-population properties of G:
The results have focused on the properties of G based on a sample of size n from a population of size N, where n < N. An important question is the extent to which the nonuniformity in G is the result of taking a sample from a population. In the appendix, we describe results for the eigenstructure of G for an entire population based on individual-based simulations. The results suggest that the eigenvalues of G decline at an approximately exponential rate and that the decline is slightly less than the decline based on taking a subsample from the population to estimate the properties of G.
DISCUSSION
Our results are consistent with the equilibrium expectation of G,  predicted by Phillips and McGuigan (2006) under neutral evolution. However, our results demonstrate that the expected eigenvalues of G at equilibrium and the eigenvalues of E(G) are highly discordant. For instance, when M has uniform eigenvalues associated with it, then E(G) has uniform eigenvalues, but the expected eigenvalues of G are nonuniform. The practical consequence of this is that although E(G) may imply that variation in breeding values is evenly distributed across characters, in fact due to stochasticity, variation in breeding values is never evenly distributed. Some characters by chance will have less genetic variation than others and furthermore, due to stochasticity and the pleiotropic nature of mutation and/or linkage, breeding values will typically covary among characters even if the expectation is independence. Our results suggest that the nonuniformity in the eigenvalues of G is not trivial because the nonuniformity is approximately exponential. This approximate exponential decline in eigenvalues may lead to potentially strong constraints on the freedom of a multivariate phenotype to respond to selection.
predicted by Phillips and McGuigan (2006) under neutral evolution. However, our results demonstrate that the expected eigenvalues of G at equilibrium and the eigenvalues of E(G) are highly discordant. For instance, when M has uniform eigenvalues associated with it, then E(G) has uniform eigenvalues, but the expected eigenvalues of G are nonuniform. The practical consequence of this is that although E(G) may imply that variation in breeding values is evenly distributed across characters, in fact due to stochasticity, variation in breeding values is never evenly distributed. Some characters by chance will have less genetic variation than others and furthermore, due to stochasticity and the pleiotropic nature of mutation and/or linkage, breeding values will typically covary among characters even if the expectation is independence. Our results suggest that the nonuniformity in the eigenvalues of G is not trivial because the nonuniformity is approximately exponential. This approximate exponential decline in eigenvalues may lead to potentially strong constraints on the freedom of a multivariate phenotype to respond to selection.
A key factor shaping the nonuniformity in the eigenvalues of G is genealogy. Genealogy causes the effects of alleles at a locus to be dependent. The principle that genealogy causes dependencies is not new and has been a key basis of analyses in phylogenetics (e.g., Piazza and Cavalli-Sforza 1974 and Felsenstein 1985). That genealogy is important in shaping the eigenstructure of G has been until now unrecognized. Shared common descent enhances the nonuniformity in the eigenvalues of G due to dependencies generated by branching patterns within the genealogy. In addition, multiple mutations may occur within a single branch of a genealogy, and this too can enhance the nonuniformity in the eigenvalues of G because there are fewer independently acting mutations. The genealogical ancestry of alleles is intrinsic to biology. The only way to partially overcome the covariance in allelic values that is induced by genealogy is for the value of an allele to be decoupled from its ancestry. The house-of-cards mutation model is one where the value of an allele is decoupled from its ancestry and our results show that this causes the decline in eigenvalues to be less than if the value of an allele is coupled to its ancestry (such as in the continuum-of-alleles model). Tighter linkage enhances nonuniformity in the eigenvalues of G because alleles at different loci do not segregate independently, thus decreasing the number of independent gene genealogies contributing to the structure of G.
In general, then, the nonuniformity in the eigenvalues of G is exaggerated with more dependency among allelic effects. We found that these dependencies can be affected by a variety of other factors as well. For instance, when there are fewer loci or the per locus mutation rate is lower the observed nonuniformity in the eigenvalues of G is increased. The degree of pleiotropy of new mutations affects the dependency of allelic effects. Under universal pleiotropy all characters are influenced by the same set of gene trees and this overlap increases the observed nonuniformity in the eigenvalues of G. In contrast, in the absence of pleiotropy, each character has a unique set of gene trees (as long as there is some recombination), and this independence decreases correlations between genotypic values and reduces nonuniformity in the expected eigenvalues of G.
What leads to the discrepancy between the expected eigenvalues of G and the eigenvalues of E(G)? Mathematically, the discordance traces to the fact that eigenvalues are nonlinear functions of the elements of G, and hence the expectations of the eigenvalues of G will not generally be equal to the eigenvalues of the expectation of G. This can be seen more directly in the following two-trait case. Suppose the expected G matrix is diagonal with the repeated eigenvalue 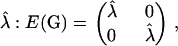 where
where 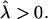 A realized G under neutral evolution can be viewed as a perturbation of E(G), say,
A realized G under neutral evolution can be viewed as a perturbation of E(G), say,  where the
where the 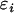 's are random variables with zero means and variances much smaller than
's are random variables with zero means and variances much smaller than 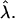 If we imagine that the perturbations are drawn, say, from a zero-mean normal distribution with variance
If we imagine that the perturbations are drawn, say, from a zero-mean normal distribution with variance 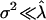 then it is not hard to show that the expected eigenvalues of G are not uniform (i.e., not repeated). Indeed, the expected leading eigenvalue of G exceeds
then it is not hard to show that the expected eigenvalues of G are not uniform (i.e., not repeated). Indeed, the expected leading eigenvalue of G exceeds 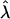 by at least
by at least 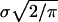 whereas the smaller eigenvalue of G is expected to fall below
whereas the smaller eigenvalue of G is expected to fall below 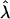 by at least the same amount. This suggests that nonuniformity among the expected eigenvalues of G is driven by the variability of perturbations from E(G). This variability arises from the assorted sources of stochasticity inherent to evolution in finite populations.
by at least the same amount. This suggests that nonuniformity among the expected eigenvalues of G is driven by the variability of perturbations from E(G). This variability arises from the assorted sources of stochasticity inherent to evolution in finite populations.
That the eigenvalues of random covariance matrices (such as a G matrix) are nonuniform is not a new result (e.g., see a review by Edelman and Rao 2005 and Wagner's 1984 random pleiotropy model). Additionally, similar patterns found in this article for the average of ranked eigenvalues are observed if it is assumed that the variances for characters are independently drawn from a chi square for a given degrees of freedom. In particular, a deviation from a purely exponential decline occurs as was observed in Figure 7. Broadly, stochasticity causes the nonuniformity in eigenvalues, but there are biologically unique processes that contribute to this stochasticity. This article suggests that genealogical ancestry is one important process.
Our neutral theoretical predictions for the expected structure of G at recombination–mutation–drift balance form an important set of null expectations for empirical studies of multivariate evolution. In particular, our finding that, under neutrality, the eigenvalues of G tend to be unequal and decay approximately exponentially from largest to smallest is consistent with empirical estimates of G (e.g., Mezey and Houle 2005). However, our results also show that the rate of decay is sensitive in predictable ways to numerous factors including population size, the number and patterns of linkage among loci, and the rate of and modality of mutation. If one includes selection as well (as in Jones et al. 2003), current theoretical considerations suggest that inferring underlying process from an equilibrium estimate of G may be difficult because different underlying processes have similar effects on the eigenvalues associated with G. For instance, it may be difficult to disentangle mutation rates from numbers of loci and the pattern of recombination among loci. Single-locus expectations and statistics may be developed, but this ignores the likely possibility that more than one locus contributes variation to G. Rather, to infer processes that influence the evolution of G may require direct approaches (see Estes et al. 2004) or possibly the study of transient changes in G (Phillips et al. 2001; Whitlock et al. 2002).
Acknowledgments
Comments by Ruth Shaw, Joe Felsenstein, and an anonymous reviewer improved the presentation and content of this article. We thank the anonymous reviewer for suggesting that we examine the case in which the variance for each character is independent and chi-square distributed. We thank P. Phillips and K. McGuigan for allowing us to cite their then unpublished work and the National Science Foundation (NSF) (grants DEB 0337582, EF 0328594, and EF 0531870 to RG) for financial support. The Initiative for Bioinformatics and Evolutionary Studies program at the University of Idaho kindly provided computer resources and they are supported by NSF grant EPS 0080935 and National Institutes of Health grants P20 RR16454 and P20 RR16448 from the Center for Biomedical Research Excellence and Idea Networks of Biomedical Research Excellence programs of the National Center for Research Resources.
APPENDIX
Whole-population predictions for G:
To investigate neutral evolution of G for an entire population we used forward time, individual-based simulations. Our approach is similar to that of Jones et al. (2003) except for the absence of selection and our use of Wright–Fisher mating (i.e., sampling parents with replacement). The individual-based simulations were founded on the same model assumptions as the two coalescent-based methods. Events in the individual-based simulations were ordered mutation → recombination → random mating within generations. At the beginning of a replicate simulation run, each individual was initialized with a genotypic value of zero by assigning allelic values of zero to all loci. Population sizes of 140 and 1366 were simulated. An inspection of simulations suggests that populations reach quasi-recombination–mutation–drift balance by 12,000 generations for populations consisting of 140 and 1366 individuals given our mutation rate and rates of recombination. The basis for this inference was that the average G matrix from simulations was not significantly different from 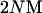 the expected G matrix. For each replicate run, we calculated the G matrix at generation 12,000 using the genotypic values of all individuals in the population. We ran 20 replicates for a particular parameter set when using 50 loci and 200 replicates when using 2 and 5 loci.
the expected G matrix. For each replicate run, we calculated the G matrix at generation 12,000 using the genotypic values of all individuals in the population. We ran 20 replicates for a particular parameter set when using 50 loci and 200 replicates when using 2 and 5 loci.
Some of the parameters used in both the individual-based simulations were similar to those utilized by Jones et al. (2003). For example, the per haploid locus mutation rate utilized was 0.0002 and all traits were uncorrelated with an expected per character variance in the effects of mutations of 0.05.
Results:
Figure A1 illustrates that when the whole population is modeled, the eigenvalues of G decline at an approximately exponential rate and that the rate of decline is greater for smaller populations. Figure A2 suggests that nonindependent segregation of alleles among loci enhances the nonuniformity in the eigenvalues of G when the whole population is modeled. Overall these whole-population simulations combined with the coalescent simulations suggest that the nonuniformity in the eigenvalues of G is a property of both an entire population and subsamples from the population. Furthermore, a comparison of individual-based and coalescent results in Figures A1 and A2 suggests that estimating the eigenstructure of G on the basis of a subsample from a population (as is done with the coalescent) results in slightly enhanced nonuniformity in the eigenvalues of G relative to the true value of the whole population.
Figure A1.—

The eigenvalues of G for an entire population based on individual-based simulations (squares) for (a) small and (b) large population sizes. Mutations pleiotropically affected five characters at five loci. The continuum-of-alleles mutation model was used. In the individual-based simulations, the recombination rate (r) was 0.5 per locus pair. For comparison, results based on the independent locus coalescent are provided for a sample of 10 (a) and 30 (b) individuals from populations of size 140 and 1366, respectively. The per locus mutation rate and number of loci were the same in the coalescent and the individual-based simulations. Results are based on 200 replicates for both the individual-based and 500 replicates for the coalescent simulations. Eigenvalues are on a log scale and error bars represent 95% confidence intervals of the mean eigenvalue. The internal hatch mark within an error bar is the mean eigenvalue. Points using the coalescent approach are offset relative to points using the individual-based approach to aid comparison.
Figure A2.—

The eigenvalues of G for an entire population based on individual-based simulations and when the recombination rate varies between loci (squares). The population size in all cases was N = 1366. Mutations pleiotropically affected five characters at two loci. The continuum-of-alleles mutation model was used. For comparison, results based on the ancestral recombination graph are provided (triangles) in which the same number of loci, population size, per locus mutation rate, and recombination rate is modeled, except now results are based on a sample of 30 individuals from the population. Results are based on 200 replicates for the individual-based and 500 replicates for the coalescent simulations. Eigenvalues are on a log scale and error bars represent 95% confidence intervals of the mean eigenvalue. The internal hatch mark within an error bar is the mean eigenvalue. Points using the ancestral recombination graph approach are offset relative to points using the individual-based approach to aid comparison.
References
- Ajie, B. C., S. Estes, M. Lynch and P. C. Phillips, 2005. Behavioral degradation under mutation accumulation in Caenorhabditis elegans. Genetics 170: 655–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avery, P. J., and W. G. Hill, 1977. Variability in genetic parameters among small populations. Genet. Res. 29: 193–213. [DOI] [PubMed] [Google Scholar]
- Blows, M. W., S. F. Chenoweth and E. Hine, 2004. Orientation of the genetic variance-covariance matrix and the fitness surface for multiple male sexually selected traits. Am. Nat. 163: E329–E340. [DOI] [PubMed] [Google Scholar]
- Crow, J. F., and M. Kimura, 1964. The theory of genetic loads, pp. 495–505 in Proceedings of the XI International Congress of Genetics. Pergamon Press, Oxford.
- Edelman, A., and N. R. Rao, 2005. Random matrix theory. Acta Numerica 14: 233–297. [Google Scholar]
- Estes, S., P. C. Phillips, D. R. Denver, W. K. Thomas and M. Lynch, 2004. Mutation accumulation in populations of varying size: the distribution of mutational effects for fitness correlates in Caenorhabditis elegans. Genetics 166: 1269–1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estes, S., B. C. Ajie, M. Lynch and P. C. Phillips, 2005. Spontaneous mutational correlations for life-history, morphological and behavioral characters in Caenorhabditis elegans. Genetics 170: 645–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewens, W. J., 2004. Mathematical Population Genetics. I. Theoretical Introduction. Springer, New York.
- Falconer, D. S., and T. F. C. MacKay, 1996. Introduction to Quantitative Genetics. Longman, Essex, UK.
- Griffiths, R. C., 1981. Neutral two-locus multiple allele models with recombination. Theor. Popul. Biol. 22: 169–186. [Google Scholar]
- Griffiths, R. C., 1991. The two-locus ancestor graph, pp. 100–117 in Selected Proceedings of the Symposium of Applied Probability, edited by I. V. Basawa and R. L. Taylor. Institute of Mathematical Statistics, Hayward, CA.
- Griffiths, R. C., and P. Marjoram, 1996. Ancestral inference from samples of DNA sequences with recombination. J. Comput. Biol. 3: 479–502. [DOI] [PubMed] [Google Scholar]
- Griffiths, R. C., and P. Marjoram, 1997. An ancestral recombination graph, pp. 257–270 in Progress in Population Genetics and Human Evolution, edited by P. Donnelly and S. Tavaré. Springer Verlag, New York.
- Hine, E., and M. W. Blows, 2006. Determining the effective dimensionality of the genetic variance–covariance matrix. Genetics 173: 1135–1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hine, E., S. F. Chenoweth and M. W. Blows, 2004. Multivariate quantitative genetics and the lek paradox: genetic variance in male sexually selected traits of Drosophila serrata under field conditions. Evolution 58: 2754–2762. [DOI] [PubMed] [Google Scholar]
- Hudson, R. R., 1991. Gene genealogies and the coalescent process. Oxf. Surv. Evol. Biol. 7: 1–44. [Google Scholar]
- Jones, A. G., S. J. Arnold and R. Bürger, 2003. Stability of the G-matrix in a population experiencing pleiotropic mutation, stabilizing selection, and genetic drift. Evolution 57: 1747–1760. [DOI] [PubMed] [Google Scholar]
- Kimura, M., 1965. A stochastic model concerning the maintenance of genetic variability in quantitative characters. Proc. Natl. Acad. Sci. USA 54: 731–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kingman, J. F. C., 1977. On the properties of bilinear models for the balance between genetic mutation and selection. Proc. Camb. Philos. Soc. 81: 443–453. [Google Scholar]
- Kingman, J. F. C., 1978. A simple model for the balance between selection and mutation. J. Appl. Probab. 15: 1–12. [Google Scholar]
- Kingman, J. F. C., 1982. The coalescent. Stoch. Proc. Appl. 13: 235–248. [Google Scholar]
- Kirkpatrick, M., and D. Lofsvold, 1992. Measuring selection and constraint in the evolution of growth. Evolution 46: 954–971. [DOI] [PubMed] [Google Scholar]
- Lande, R., 1979. Quantitative genetics analysis of multivariate evolution, applied to brain:body size allometry. Evolution 33: 402–416. [DOI] [PubMed] [Google Scholar]
- Lande, R., 1980. The genetic covariance between characters maintained by pleiotropic mutations. Genetics 94: 203–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lande, R., and S. J. Arnold, 1983. The measurement of selection on correlated characters. Evolution 37: 1210–1226. [DOI] [PubMed] [Google Scholar]
- Lynch, M., and W. G. Hill, 1986. Phenotypic evolution by neutral mutation. Evolution 40: 915–935. [DOI] [PubMed] [Google Scholar]
- Lynch, M., and B. Walsh, 1998. Genetics and Analysis of Quantitative Traits. Sinauer Associates, Sunderland, MA.
- Mezey, J. G., and D. Houle, 2005. The dimensionality of genetic variation for wing shape in Drosophila melanogaster. Evolution 59: 1027–1038. [PubMed] [Google Scholar]
- Nordborg, M., 2001. Coalescent theory, pp. 179–212 in Handbook of Statistical Genetics, edited by D. J. Balding, M. Bishop and C. Cannings. John Wiley & Sons, Chichester, UK.
- Piazza, A., and L. L. Cavalli-Sforza, 1974. Spectral analysis of patterned covariance matrices and evolutionary relationships, pp. 76–105 in Proceedings of the Eighth International Conference on Numerical Taxonomy, edited by G. F. Estabrook. W. H. Freeman, San Francisco.
- Phillips, P. C., and S. J. Arnold, 1989. Visualizing multivariate selection. Evolution 43: 1209–1222. [DOI] [PubMed] [Google Scholar]
- Phillips, P. C., and K. L. McGuigan, 2006. Evolution of genetic variance-covariance structure, pp. 310–325 in Evolutionary Genetics: Concepts and Case Studies, edited by C. W. Fox and J. B. Wolf. Oxford University Press, Oxford.
- Phillips, P. C., M. C. Whitlock and K. Fowler, 2001. Inbreeding changes the shape of the genetic covariance matrix in Drosophila melanogaster. Genetics 158: 1137–1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schluter, D., 1996. Adaptive radiation along genetic lines of least resistance. Evolution 50: 1766–1774. [DOI] [PubMed] [Google Scholar]
- Wagner, G. P., 1984. On the eigenvalue distribution of genetic and phenotypic dispersion matrices: evidence for a nonrandom organization of quantitative character variation. J. Math. Biol. 21: 77–95. [Google Scholar]
- Whitlock, M. C., P. C. Phillips and K. Fowler, 2002. Persistence of changes in the genetic covariance matrix after a bottleneck. Evolution 56: 1968–1975. [DOI] [PubMed] [Google Scholar]
- Zeng, Z.-B., and C. C. Cockerham, 1991. Variance of neutral genetic variances within and between populations for a quantitative character. Genetics 129: 535–553. [DOI] [PMC free article] [PubMed] [Google Scholar]