

Abstract
Human keratinocytes grown in co-culture with fibroblast feeder cells have an extended in vitro lifespan and delayed accumulation of the tumor suppressor protein p16INK4a when compared to the same cells grown on tissue culture plastic alone. Previous studies have indicated that human keratinocytes can be immortalized by telomerase activity alone when grown in co-culture with feeder cells, suggesting that loss of the p16INK4a/Rb pathway is not required for immortalization. Using two independent human keratinocyte cell strains, we found that exogenous telomerase expression and co-culture with feeder cells results in efficient extension of lifespan without an apparent crisis. However, when these cells were transferred from the co-culture environment to plastic alone they experienced only a brief period of slowed growth before continuing to proliferate indefinitely. Examination of immortal cell lines demonstrated p16INK4a promoter methylation had occurred in both the absence and presence of feeder cells. Reintroduction of p16INK4a into immortal cell lines resulted in rapid growth arrest. Our results suggest that p16INK4a/Rb-induced telomere-independent senescence, although delayed in the presence of feeders, still provides a proliferation barrier to human keratinocytes in this culture system and that extended culture of telomerase-transduced keratinocytes on feeders can lead to the methylation of p16INK4a.
Keywords: hTERT , CDKN2A, senescence, epigenetic, Rb, telomeres
Introduction
Previous reports have suggested that exogenous expression of the catalytic component of telomerase (TERT) is capable of immortalizing many different human cell types (fibroblasts, retinal pigmented epithelial cells, vascular endothelial cells and mesothelial cells) without the need for additional genetic alterations (Bodnar et al., 1998; Yang et al., 1999; Dickson et al., 2000). Several human epithelial cell types (keratinocytes, mammary epithelial cells, bladder urothelial cells and prostatic epithelial cells), however, have been shown to experience a telomere-independent growth arrest that is enforced by the p16/pRb pathway (Foster and Galloway, 1996; Brenner et al., 1998; Puthenveettil et al., 1999). For these cells, it has been proposed that the p16/pRb pathway must be inactivated in the context of immortalization by TERT (Kiyono et al., 1998; Jarrard et al., 1999; Dickson et al., 2000; Sandhu et al., 2000; Stampfer and Yaswen, 2003). The p16INK4a/Rb pathway has been shown to be modulated by differences in culture conditions. There is evidence that suggests some epithelial cell types can be immortalized by TERT expression alone when cultured in the presence of feeder cells (Ramirez et al., 2001; Herbert et al., 2002; Harada et al., 2003). Furthermore, it has been suggested that the p16/Rb pathway is still intact in these immortalized epithelial cells and remains responsive to both UV irradiation and transfer to the plastic culture condition. These observations are relevant when considering telomerase-based tissue therapies and imply that human epithelial cell immortalization in vitro does not require the permanent inactivation of the p16/Rb tumor-suppressor pathway.
It has been suggested that controlled telomerase activation should be considered for the treatment of human disease and age-related loss of function disorders. Tissue culture studies have provided evidence that ectopic expression of telomerase can enhance both cellular growth and proliferation and is correlated with the inhibition of several tumor suppressor pathways. Long-term culture of TERT-transduced human mammary epithelial cells has been reported to be associated with increased expression of the proto-oncogene c-myc and resistance to growth inhibition by TGF-β (Wang et al., 2000; Stampfer et al., 2001). Examination of human fibroblasts immortalized by telomerase alone has found a significant repression of both p16 and p53 expression in these cells as they continue to be passaged in vitro (Noble et al., 2004; Taylor et al., 2004). In fact, one recent study has demonstrated that telomerase immortalized human fibroblasts can acquire a transformed phenotype associated with a loss of p16 and p53 function and upregulation of c-myc expression (Zongaro et al., 2005). These combined results strongly suggest that telomerase immortalization of human cells leads to the dysregulation of both tumor suppressor and oncogenic growth pathways which when combined with the ability to maintain telomere length could lead to the development of tumorigenic cells at increased frequency.
When grown in co-culture with post-mitotic fibroblast feeder cells, human keratinocytes, exhibit a delay in passage dependent p16INK4a(p16) expression and have an extended lifespan in culture (Ramirez et al., 2001; Rheinwald et al., 2002; Baek et al., 2003; Fu et al., 2003; Kang et al., 2003; Darbro et al., 2005). We have found that co-culture of keratinocytes with feeder cells delays the accumulation of p16 protein but does not prevent the eventual increase of p16 expression in late passage keratinocytes cultured with feeder cells (Darbro et al., 2005). This observation suggests that p16 expression may still be an important growth regulatory mechanism in keratinocytes despite co-culture with feeders. Previous reports that have shown immortalization of co-cultured human keratinocytes with TERT alone imply that telomere-dependent mechanisms are the sole barrier preventing continued proliferation in this culture system. However, the presence of inflection points in the growth curves of TERT immortalized keratinocytes co-cultured with feeders (Herbert et al., 2002; Rheinwald et al., 2002; Ramirez et al., 2003), combined with the observations that telomeres experience only limited shortening in co-cultured human keratinocytes (Kang et al., 2004), suggest that telomere-independent mechanisms may still be involved in limiting the replicative capacity of these cells. Thus, examination of p16 expression in TERT-transduced human keratinocytes co-cultured with feeder cells should provide vital information concerning not only the potential safety of telomerase-based cell therapies but also the mechanism of epithelial cell senescence in the co-culture environment.
In this study, we demonstrate that telomerase activity alone in human keratinocytes co-cultured with feeder cells results in efficient extension of lifespan of these cells and that switching these cells from the co-culture environment to plastic leads to a brief induction of p16 expression followed by continued proliferation and maintenance of an immortal phenotype. Examination of telomerase-immortalized cell lines by 5-aza-2′deoxycytidine treatment and bisulfite sequencing demonstrated p16 promoter methylation in immortal cells, regardless of whether they were co-cultured with feeders or on plastic alone. Reintroduction of p16 into TERT-immortalized cell lines resulted in a rapid growth arrest and a senescent phenotype. Our results suggest that immortalization of human keratinocytes with TERT is associated with a high incidence of p16 inactivation, regardless of culture conditions, and emphasizes the need to examine p16 status in molecular detail in all human epithelial cells immortalized by telomerase activity over long time periods.
Results
TERT immortalization of co-cultured human keratinocytes
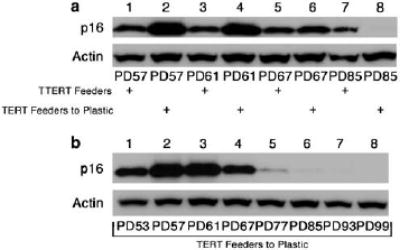
Previous reports have suggested that human keratinocytes can be immortalized by telomerase activity alone when grown in co-culture with fibroblast feeder cells (Ramirez et al., 2001; Harada et al., 2003). Furthermore, these studies suggest that p16 expression in TERT-immortalized keratinocytes is not inactivated and remains inducible by either transfer to plastic culture conditions or exposure to UV irradiation. Recent evidence has shown that p16 expression is permanently inactivated in TERT-immortalized fibroblasts when they are passaged over long periods of time (Noble et al., 2004; Taylor et al., 2004), thus, we sought to determine if a similar result could be seen in TERT immortalized human keratinocytes co-cultured with feeder cells. We observed that Strain C human keratinocytes transduced with TERT and co-cultured with feeder cells do acquire a greatly extended lifespan phenotype without an apparent crisis (Figure 1a). Similar to previous reports (Kiyono et al., 1998), we also observed that TERT-transduced HFKs cultured on plastic alone did not have a significantly extended lifespan and growth arrested at approximately population doublings (PD) 26. As expected, HFKs transduced with pLXSH did not have an extended lifespan and did not become immortal on either plastic or in co-culture with feeders and displayed cellular lifespans similar to our previous observations (Darbro et al., 2005). At PD 22, Strain C TERT-transduced HFKs were transferred from the co-culture environment to tissue culture plastic alone. In contrast to previous reports, transferred HFKs did not undergo rapid growth arrest. In fact, TERT HFKs transferred to the plastic culture condition mirrored the proliferation rate of TERT HFKs remaining in the co-culture environment over PDs 24–48, and after a period of slowed growth spanning PDs 48–54, resumed a rapid proliferation rate and continued to proliferate indefinitely. Expression levels of p16 were examined in each of the transduced cell lines and were found to be consistent with both growth rate and arrest (Figure 2a). In both pLXSH- and TERT-transduced HFKs cultured on plastic, p16 levels were increased before and at the point of growth arrest, suggesting that these cells succumbed to p16 enforced telomere-independent senescence. Protein levels of p16 were increased but to a lesser extent in pLXSH cells co-cultured with feeder cells. TERT-transduced HFKs co-cultured with feeder cells maintained a relatively low level of p16 expression throughout the duration of the experiment. Soon after the transfer of TERT-transduced HFKs from the co-culture environment to plastic, p16 protein levels began to rise and reached a maximal level during the period of slow growth experienced by these cells between PDs 48-54 (Figure 2b). As transferred TERT HFKs began to resume a rapid growth rate p16 protein levels began to decline. No further p16 expression was seen in the transferred TERT HFKs for the duration of this experiment (data not shown). Thus, p16 expression was lost in TERT HFKs transferred from the co-culture environment to plastic alone.
Figure 1.
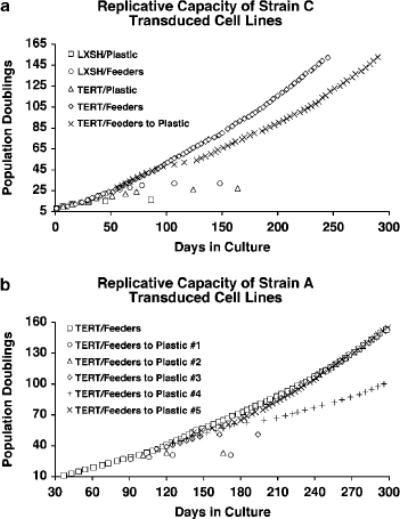
Immortalization of TERT-transduced Strain C and A HFKs co-cultured with feeder cells. (a) Replicative capacities of Strain C HFKs transduced with either LXSH or TERT and cultured on plastic alone (Plastic), in the presence of feeder cells (Feeders), or transferred from the co-culture environment to plastic alone (Feeders to Plastic). TERT/Feeders to Plastic cells were transferred form the co-culture environment to Plastic at PD 26. TERT/Feeders and TERT/Feeders to Plastic HFKs were found to be immortal. The following transduced cell lines senesced at the specified PDs: LXSH/Plastic (~16 PDs), LXSH/Feeders (~32 PDs), TERT/Plastic (~26 PDs). (b) Replicative capacities of Strain A HFKs transduced with TERT and transferred from the co-culture environment to Plastic alone (Feeders to Plastic) at various PDs. TERT/Feeders to Plastic cells were transferred from the co-culture environment to Plastic at the following PDs: PD 25 (#1), PD 29 (#2), PD 35 (#3), PD 41 (#4), PD 51 (#5). TERT/Feeders, TERT/Feeders to Plastic #4, and TERT/Feeders to Plastic #5 HFKs were found to be immortal. The following TERT/Feeders to Plastic cell lines senesced at the specified PDs: ~31 PDs (#1), ~33 PDs (#2), ~49 PDs (#3).
Figure 2.

Expression of p16 in tranduced cell lines. (a) Immunoblots of p16 protein levels in Strain C HFKs transduced with either LXSH or TERT and cultured on plastic alone (Plastic: lanes 1, 2, and 10–12), in the presence of feeder cells (Feeders: lanes 3–6 and 13–15), or transferred from the co-culture environment to plastic alone (Feeders/Plastic: lanes 7–9). (b) Immunoblot of p16 protein levels in Strain C HFKs transduced with TERT and transferred from the co-culture environment to plastic alone (TERT Feeders to Plastic) at PD 26. Protein levels of actin are included as a loading control. Approximate PDs are represented below each lane.
To determine whether the loss of p16 expression seen in Strain C TERT-transduced HFKs transferred from the co-culture environment to plastic was reproducible and dependent on the number of PDs cells had experienced, we repeated TERT retroviral infections in another HFK cell strain derived from a different donor (Strain A). Strain A TERT transduced HFKs were transferred from the feeder culture system to plastic at various points in their lifespan (Figure 1b). Consistent with our previous result, Strain A HFKs co-cultured with feeder cells were immortalized by transduction with TERT without an apparent crisis. TERT transduced HFKs were transferred from the co-culture environment to plastic at PDs 25, 29, 35, 41 and 51 to examine their viability in the absence of feeder cells. TERT-transduced HFKs transferred at PDs 25, 29 and 35 all underwent growth arrest within seven passages after transfer. In constrast, TERT transduced HFKs transferred at PD 41 experienced a slowed growth phase between PDs 61–69 before resuming a regular proliferation rate. TERT-transduced HFKs transferred at PD 51 experienced a slight growth inhibition between PDs 57 and 65 before acquiring a rapid proliferation rate. Protein levels of p16 were elevated above the levels seen in parallel cultures of TERT HFKs left on feeders during the period of slowed growth exhibited by Strain A TERT HFKs transferred from the co-culture environment to plastic at PD 51 (Figure 3a). Loss of p16 expression in these cells correlated with the resumption of a rapid proliferation rate (Figure 3b). Thus, in the human keratinocyte cell strains we examined, the loss of p16 expression appears to be a common event upon transfer of TERT expressing keratinocytes from the co-culture environment to plastic, whereas we did not observe escape from crisis when TERT transduced keratinocytes were cultured only on plastic. Furthermore, maintenance of TERT-induced HFK immortality upon transfer from the co-culture environment to plastic appears to be dependent on the length of time the cells are co-cultured with feeders.
Figure 3.
Expression of p16 in TERT-transduced Strain A HFKs transferred from co-culture with Feeders to Plastic alone. (a) Immunoblot of p16 protein levels in both TERT-transduced Strain A HFKs retained on Feeders (TERT Feeders: odd lanes) and TERT/Feeders to Plastic #5 cells transferred to Plastic from the co-culture environment at PD 51 (TERT Feeders to Plastic: even lanes). (b) Immunoblot of p16 protein levels in TERT/Feeders to Plastic #5 cells. Protein levels of actin are included as a loading control. Approximate PDs are represented below each lane.
Characterization of immortal human keratinocytes cell lines
Analysis of telomerase activity in all HFK cell lines was performed to determine whether TERT-transduced HFKs that continued to divide upon transfer from the co-culture environment to plastic retained telomerase activity. As expected, all HFK cell lines that had been transduced with TERT exhibited high telomerase activity (Figure 4). Cytogenetic analysis was also performed to determine if any gross chromosomal alterations had occurred that might have contributed to maintenance of the immortal phenotype in TERT HFKs transferred from the co-culture environment to plastic. In Strain C TERT HFKs we found no obvious genetic abnormalities associated with transfer from the co-culture system to plastic (Table 1). In fact, the only cytogenetically apparent abnormalities in Strain C TERT HFKs were found in the cells that remained in co-culture with feeder cells. This result suggests that there were no gross genetic abnormalities (such as chromosomal amplification, duplication, deletion or translocations) acquired by Strain C TERT-transduced HFKs transferred to plastic that allowed them to proliferate. In Strain A TERT HFKs transferred from the co-culture environment to plastic we observed duplication of chromosomes 7 and 20. In contrast, Strain A TERT HFKs continuously co-cultured with feeder cells exhibited no apparent cytogenetic abnormalities. Thus, the maintenance of immortality in these TERT transduced HFKs transferred from the co-culture environment to plastic is likely not the result of gross chromosome abnormalities, although other unapparent epigenetic and genetic alterations are possible.
Figure 4.
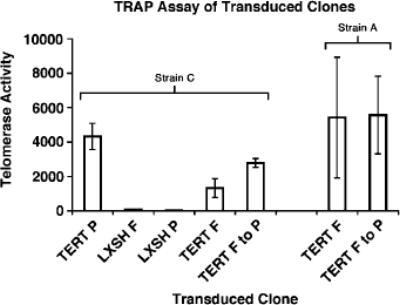
Telomerase activity in transduced Strain C and A HFKs. Real-time quantitative TRAP assays were performed on cell extracts from Strain C and A HFKs transduced with either LXSH or TERT and cultured on plastic alone (P), in the presence of feeder cells (F), or transferred from the co-culture environment to plastic alone (F to P). Strain A TERT/Feeders to Plastic #5 cells, transferred form the co-culture environment to plastic at PD 51, are represented above. Approximate PDs at which telomerase activity was measured were Strain C: PD 12 (TERT P), PD 12 (LXSH F), PD 12 (LXSH P), PD >100 (TERT F), PD >100 (TERT F to P); Strain A: PD >100 (TERT F), PD >100 (TERT F–P). Telomerase activity is measured in standard cell equivalents relative to our control cell line. Error bars represent s.d. of the mean.
Table 1.
Chromosomal analyses of TERT-transduced cell lines
| Transduced Cell Line | Karyotype |
|---|---|
| Strain C HFKs | |
| TERT Feeders | 46, XY, iso (8)(q10)a |
| TERT Feeders to Plastic | 46, XY |
| Strain A HFKs | |
| TERT Feeders | 46, XY |
| TERT Feeders to Plastic #5 | 48, XY, +7, +20 |
| (transferred at PD 51) |
These cells contain one copy of the short arm of chromosome 8 and three copies of the long arm of chromosome 8.
Inactivation of p16 by promoter methylation in keratinocytes immortalized by TERT and co-cultured with feeder cells
Growing evidence has suggested that hypermethylation of CpG islands is a common mechanism employed by cancer to permanently silence expression of tumor suppressor genes (Herman and Baylin, 2003; Momparler, 2003; Paz et al., 2003; Das and Singal, 2004; Curtis and Goggins, 2005). Furthermore, in vitro immortalized human fibroblast cell lines have been found to methylate a wide array of genes some of which are involved in cell cycle regulation (Liu et al., 2005). The promoter region of the p16 gene contains a 5′ CpG island that has been found to exhibit increased levels of methylation in various cancers ( Liggett and Sidransky, 1998; Rocco and Sidransky, 2001; Sharpless, 2005). In addition, methylation has been found to be responsible for p16 inactivation in several human keratinocyte cell lines immortalized by telomerase alone in the absence of feeder cells (Farwell et al., 2000). To examine the status of p16 promoter methylation in immortal TERT-transduced HFKs, we examined the inducibility of p16 by the DNA methyltransferase inhibitor 5-aza-2′deoxycytidine as has been shown previously in oral squamous cell carcinomas that had downregulated p16 by promoter methylation (Timmermann et al., 1998). Following treatment with 5-aza-2′deoxycytidine, re-expression of p16 at both the mRNA and protein levels was observed in TERT-transduced HFKs that had lost p16 expression upon being transferred from the co-culture environment to plastic (Figure 5a and b). This result is highly suggestive of p16 promoter methylation in these cells. Inducibility of p16 expression to 5-aza- 2′ deoxycytidine was also examined in TERT-transduced HFKs that remained in the co-culture environment. We also observed an increase in p16 expression at both the mRNA and protein levels when these cells were treated with 5-aza-2′ deoxycytidine (Figure 5 c and d). This result suggests p16 promoter methylation may be occurring in TERT-transduced keratinocytes co-cultured with feeder cells and is not simply an effect of transfer to the plastic culture condition.
Figure 5.
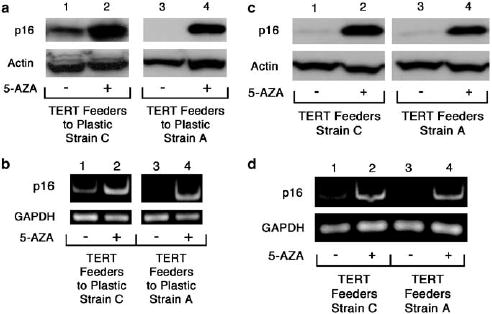
Induction of p16 expression upon treatment with 5-aza-2′ deoxycytidine. (a, b) Induction in immortalized TERT-transduced HFKs transferred from the co-culture environment to plastic alone. (a) Immunoblot of p16 protein levels in immortalized Strain C (lanes 1 and 2) and A (3 and 4) TERT Feeders to Plastic cells treated with or without 5-aza-2′ deoxycytidine (5-AZA). Protein levels of actin are included as a loading control. (b) Semi-quantitative RT–PCR of p16 mRNA levels in immortalized Strain C (lanes 1 and 2) and A (3 and 4) TERT Feeders to Plastic cells treated with or without 5-aza-2′ deoxycytidine (5-AZA). GAPDH, a housekeeping gene, is included as an internal control. In both (a) and (b) above, Strain A TERT/Feeders to Plastic #5 cells are represented. Protein and RNA were collected at PD 56 and PD >100 for Strain C and A TERT Feeder to Plastic cells, respectively. (c, d) Induction in immortalized TERT transduced HFKs maintained in co-culture with feeder cells. (c) Immunoblot of p16 protein levels in immortalized Strain C (lanes 1 and 2) and A (3 and 4) TERT Feeders cells treated with or without 5-aza-20deoxycytidine. Protein levels of actin are included as a loading control. (d) Semi-quantitative RT–PCR of p16 mRNA levels in immortalized Strain C (lanes 1 and 2) and A (3 and 4) TERT Feeders cells treated with or without 5-aza-2′ deoxycytidine. GAPDH, a housekeeping gene, is included as an internal control. Protein and RNA were collected at PD >100 for Strain C and A TERT Feeder cells.
To confirm that p16 promoter methylation had occurred in both TERT-transduced HFKs co-cultured with feeders and those transferred to plastic we performed bisulfite sequencing on the p16 promoter region spanning from –351 to –73. This region of the p16 promoter has been shown previously to be important for p16 downregulation by DNA methylation (Wong et al., 1997; Cody et al., 1999; Farwell et al., 2000). Bisulfite sequencing relies on the ability of bisulfite to induce modifications to genomic DNA, specifically, the conversion of unmethylated cytosines to uracils. Following treatment of genomic DNA with bisulfite, the specific region of the p16 promoter was amplified and PCR products were cloned into pGEM vectors and transformed into competent bacterial cells. Subsequent sequencing revealed which specific cytosines had been methylated in the HFK cell lines tested. As controls, bisulfite sequencing was performed on Strain C HFKs transduced with pLXSH and grown in both culture conditions, Strain C HFKs transduced with TERT and cultured on plastic alone, and Strain A primary HFKs grown in both culture conditions. Furthermore, bisulfite sequencing was performed on both early and late passage TERT-transduced HFKs co-cultured with feeder cells to determine if promoter methylation occurred as a result of time in culture. We observed little, if any, p16 promoter methylation in Strain C HFKs transduced with pLXSN (in either culture condition), Strain C TERT HFKs cultured on plastic alone, or Strain A primary HFKs (in either culture condition) (Figures 6 and 7). Little p16 promoter methylation was detected in early passage Strain A or C TERT HFKs co-cultured with feeder cells. However, a remarkable increase in p16 promoter methylation was observed in late passage Strain A and C TERT HFKs co-cultured with feeders. The degree of methylation was also increased in both Strain A and C TERT HFKs that had lost p16 expression upon transfer from the co-culture environment to plastic. Thus, the loss of p16 expression in TERT-transduced HFKs transferred from co-culture with feeders to plastic is most likely attributed to promoter methylation. Furthermore, p16 promoter methylation also occurs in TERT-transduced HFKs co-cultured with feeders suggesting that, during prolonged culture, TERT-transduced HFKs co-cultured with feeders are susceptible to progressive inactivation of p16 expression. It is interesting to note that methylation of the examined region was incomplete and even absent in some clones. This is in contrast to nearly complete methylation of this region observed in squamous cell carcinoma cell lines (Cody et al., 1999) and suggest that some cells in the population remain unmethylated at the p16 promoter.
Figure 6.
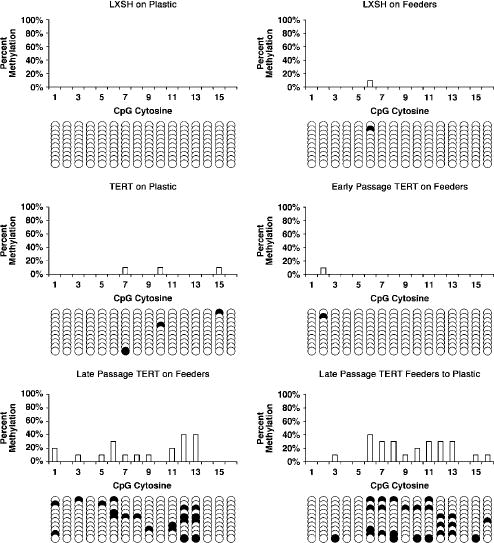
Bisulfite sequencing analysis of the p16 promoter in LXSH and TERT transduced Strain C HFKs. Numbers on the x axis represent CpG sites in the p16 promoter region spanning from –351 to –73. Methylation status of CpG sites in each individual epigenotype is indicated as either unmethylated (open circle) or methylated (closed circle). DNA from LXSH on Plastic, LXSH on Feeders, and TERT on Plastic Strain A HFKs was collected from cells at PDs ~12–16. Early passage TERT on Feeders cells were at PD 14, and both late passage TERT on Feeders and TERT Feeders to Plastic cells were at PD >100.
Figure 7.
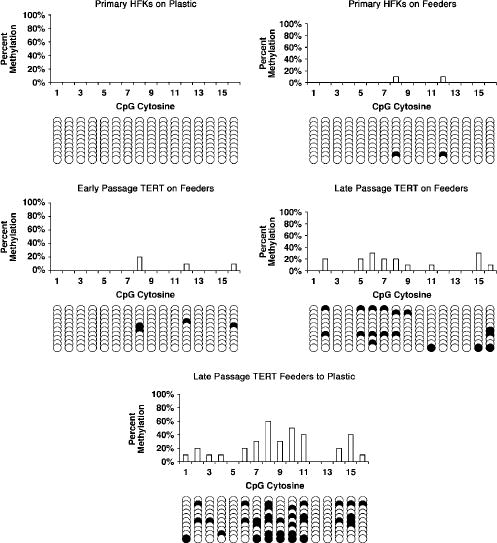
Bisulfite sequencing analysis of the p16 promoter in primary and TERT-transduced Strain A HFKs. Numbers on thexaxis represent CpG sites in the p16 promoter region spanning from –351 to –73. Methylation status of CpG sites in each individual epigenotype is indicated as either unmethylated (open circle) or methylated (closed circle). DNA from primary Strain A HFKs cultured under both conditions was collected from cells at PD ~10, early passage TERT on Feeders cells at PD 20, and both late passage TERT on Feeders and TERT Feeders to Plastic cells at PD >100. Strain A TERT/Feeders to Plastic #5 cells are represented above.
Reintroduction of p16 expression into TERT-immortalized keratinocytes on plastic causes growth arrest
To determine whether the loss of p16 expression was a significant event that allowed TERT expressing cells to proliferative indefinitely, as opposed to a mutation in some other component of the Rb pathway, we infected TERT-transduced HFKs transferred to plastic with a retrovirus containing p16. Following retroviral infection, cell cultures were exposed to a brief period of selection with puromycin. This period of selection provided for the vast majority of nontransduced HFKs to be selected against but allowed for a small population of uninfected cells to remain viable. Following the brief selection period, we fixed infected cultures and subjected them to immunocytochemical staining for both p16 and BrdU. We observed that in both Strain A and C TERT HFKs that remained immortal following transfer to plastic, p16 expression caused both growth arrest and morphological changes consistent with senescence (Figure 8). We detected a lack of BrdU incorporation in cells expressing p16 consistent with p16-induced growth arrest. Cells that did not express p16 exhibited positive BrdU staining, suggesting they had entered the S phase of the cell cycle during the 12 h BrdU treatment period before fixation. Morphologically, HFKs expressing p16 were also much larger than those cells in which p16 expression was absent. This observation is consistent with the morphology of HFKs that have undergone p16-induced, telomere-independent senescence following serial passage in the plastic culture condition. Control cultures infected with the pBABE vector alone also exhibited BrdU staining with a lack of any noticeable p16 expression. Thus, reintroduction of p16 expression into immortal TERT-transduced HFKs transferred from the co-culture environment to plastic is sufficient to cause both growth arrest and acquisition of a senescent morphology.
Figure 8.
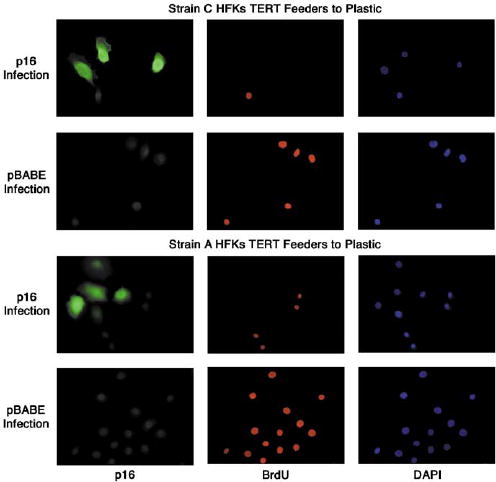
Reintroduction of p16 expression causes growth arrest and a senescent morphology in immortalized Strain C and A TERT-transduced HFKs transferred from co-culture with Feeders to Plastic. Late passage (PD>100) Strain C and A TERT Feeders to Plastic cells were infected with retrovirus carrying either pBABE-p16 or pBABE alone. Cells were incubated with BrdU before fixation and stained with antibodies to both p16 and BrdU. DAPI staining was performed for visualization of all HFK nuclei in the microscopic field. Strain A TERT/Feeders to Plastic #5 cells are represented above.
Discussion
It has been proposed previously that human keratino-cytes can be immortalized by exogenous expression of TERT alone if they are co-cultured with fibroblast feeder cells. Furthermore, it has been suggested that these immortalized keratinocytes retain inducibility and function of the p16 tumor suppressor gene. In this study, we have shown that human keratinocytes have a significantly extended lifespan with TERT expression alone when co-cultured with feeder cells, however, loss of p16 expression by promoter methylation occurs in these cell lines overtime. These results suggest that the p16/Rb pathway is inactivated in telomerase immortalized human keratinocytes regardless of culture conditions and provides more evidence that immortalization of human cells with telomerase may eventually lead to loss of tumor suppressor gene expression.
Several studies have suggested that p16 inactivation is required for human epithelial cell immortalization in vitro (Reznikoff et al., 1996; Brenner et al., 1998; Kiyono et al., 1998; Jarrard et al., 1999; Dickson et al., 2000; Rheinwald et al., 2002; Tsutsui et al., 2002). Most of these studies, however, have involved human epithelial cells cultured in the absence of feeder cells. In the absence of feeder cells, cultures of human epithelial cells accumulate p16 protein in a passage-dependent manner. This increase in p16 expression eventually leads to growth arrest by telomere-independent mechanisms. There is a growing body of evidence that suggests induction of epithelial cell migration is associated with upregulation of p16 expression (Jung et al., 2001; Natarajan et al., 2003; Svensson et al., 2003; Nilsson et al., 2004; Darbro et al., 2005). Previously, we have shown that human keratinocytes cultured in the absence of feeder cells acquire a phenotype consistent with a migratory response and that this migratory stimulus may be responsible for inducing p16-mediated, telomere-independent senescence in this culture condition (Darbro et al., 2005). Co-culture with post-mitotic fibroblast feeder cells has been shown to increase human epithelial cell lifespan in culture but appears to only delay or reduce the upregulation of p16 that precedes growth arrest. In the co-culture environment, we have shown that human keratinocytes downregulate the expression of genes involved in migration, suggesting that p16 expression is regulated by different mechanisms in this culture system. Whereas it has been suggested that p16 expression can be induced by telomere dysfunction (Jacobs and de Lange, 2004), it has also been shown that human keratinocytes experienced very limited shortening of telomeres in the co-culture environment (Kang et al., 2004). These observations suggest that a telomere-independent mechanism, separate from a migratory stimulus, may be responsible for upregulation of p16 expression in human keratinocytes co-cultured with feeder cells. If telomere-independent mechanisms are responsible for p16 induction in human keratinocytes co-cultured with feeder cells, one would expect to observe immortalization of these cells by exogenous expression of TERT alone if either p16 expression is inactivated or induction of p16 expression is not responsible for the growth arrest seen in human keratinocytes co-cultured with feeder cells. Interestingly, the results of this study would seem to suggest that neither condition is entirely true.
We found that human foreskin keratinocytes immortalized by exogenous TERT expression and co-cultured with feeder cells exhibited p16 promoter methylation. Previous studies have indicated that p16 loss is a necessary event for immortalization of keratinocytes when TERT expressing cells are cultured on plastic alone (Dickson et al., 2000). Downregulation of p16 in TERT immortalized keratinocytes was found to occur by a variety of methods including homozygous and heterozygous deletions and promoter methylation, all of which have also been observed in squamous cell carcinoma cell lines. In the current study, we were unable to obtain immortal keratinocytes when they were cultured on plastic alone. On the other hand, TERT expressing cells cultured with feeders eventually gave rise to immortal cells that exhibited p16 promoter methylation, perhaps indicating that extension of lifespan on feeders can more easily lead to epigenetic alterations that result in downregulation of tumor suppressor genes such as p16. At first glance, our results would suggest that p16 inactivation is required to immortalize human keratinocytes regardless of culture conditions. It should be noted, however, that bisulfite sequencing indicated that not all p16 alleles exhibited methylation, which is in contrast to an oral squamous cell carcinoma (SCC-15) that exhibited nearly 100% methylation in this region of the p16 promoter (Cody et al., 1999). The presence of several unmethylated p16 promoter sequences in late passage TERT-transduced HFKs co-cultured with feeders and the observation that p16 expression is induced when these cells are transferred from the co-culture environment to plastic suggest that a subpopulation of cells with functional p16 may exist in the population. Whereas it is possible that other epigenetic mechanisms of gene regulation may be responsible for p16 silencing in the absence of any detectable DNA methylation, such as DNA methylation-independent histone modification (Thiagalingam et al., 2003; Lewis et al., 2004; Zhao et al., 2005), the consistent induction of p16 expression following transfer from the co-culture environment to plastic is highly suggestive of a mixed population of both p16 positive and negative TERT-transduced HFKs. It is reasonable to conclude that upon transfer to the plastic culture condition, those keratinocytes that had retained functional p16 immediately upregulated p16 expression and senesced. Following the senescence of p16 positive cells, those keratinocytes that had inactivated p16 expression in the co-culture environment continued to proliferate and remained immortal. We can be reasonably confident that methylation of the p16 promoter occurred as a result of continued co-culture of TERT-transduced HFKs, and was not the result of a pre-existing epigenetic mutation, since TERT HFKs cultured only on plastic alone induced p16 expression and failed to become immortal. We can also conclude that the loss of p16 expression is the primary abrogation in the Rb pathway since reintroduction of p16 caused a rapid growth arrest.
The observation that a p16 negative population of cells is frequently selected for during continuous co-culture of TERT-transduced HFKs suggests that p16 expression does limit proliferation of human keratinocytes in this culture environment. If p16 expression were not exerting a growth inhibitory effect, cells would acquire no selective growth advantage by inactivating p16. Thus, the inactivation of p16 expression in a subpopulation of TERT immortalized keratinocytes co-cultured with feeder cells suggests that a telomere-independent induction of p16 expression in late passage co-cultured keratinocytes does contribute to their growth arrest in this culture system. However, the retention of p16 expression in a subpopulation of TERT immortalized keratinocytes co-cultured with feeder cells may suggest that p16 inactivation is not absolutely required to immortalize human keratinocytes in this culture system. That being said, we cannot exclude the possibility that p16 negative HFKs may, in some way, be contributing to the survival of their p16 competent counterparts. Experiments in which primary keratinocytes are co-cultured with both feeder cells and p16 negative, TERT-immortalized keratinocytes could provide useful information as to whether a p16 negative population of keratinocytes enhances the replicative capacity of p16 positive ones.
Methylation of the p16 promoter in TERT-transduced keratinocytes cultured with feeders is of extreme significance when considering telomerase based cell therapies. The loss of p16 expression has been observed in a myriad of human malignancies and is a negative prognostic marker in many of them (Groeger et al., 1999; Tsihlias et al., 1999; Esteller et al., 2001; Korkolopoulou et al., 2001; Chakravarti et al., 2003; Weinberger et al., 2004; Partridge et al., 2005). Thus, p16 inactivation in TERT engineered cells is an event that needs to be addressed before any therapeutic interventions. It is conceivable that shorter periods in culture might be sufficient to reduce the incidence of tumor suppressor gene inactivation in TERT-transduced human cells as our results suggest that early passage TERT-transduced HFKs co-cultured with feeders did not possess a significant number of p16 negative clones. Furthermore, transfer of early passage TERT-transduced HFKs from the co-culture environment to plastic did not result in subsequent immortalization in the new culture environment. Culture of TERT-transduced human cells in more physiologic oxygen conditions may also reduce the necessity for p16 downregulation and reduce the incidence of epigenetic mutations in tumor suppressor genes. This strategy may be particularly relevant to p16 mutations since several studies have found an association between increased levels of reactive oxygen species and p16 methylation during tumor progression (Govindarajan et al., 2002; Romanenko et al., 2002; Jarmalaite et al., 2003). Other studies have shown that oxygen accelerates accumulation of mutations and may even promote telomere attrition, thus providing further impetus to perform immortalization experiments in low oxidative stress conditions. Another factor that might influence keratinocyte immortalization is calcium concentration. In our studies, we used a calcium concentration in our E-media that was similar to that used by Ramirez et al. but different from that used by Dickson et al. (2000) (i.e. 1.28 vs 0.4 mM). Cells grown in K-SFM on plastic without feeders are generally grown in a low concentration of calcium (i.e. 0.09 mM). Different calcium concentrations could significantly affect the differentiation state of the cells and their ability to be immortalized by telomerase. Along these lines, recent studies have suggested that certain subsets of keratinocytes (stem cells versus transit amplifying cells) might be more prone to immortalization with TERT. In addition, it has been suggested that donor age might affect ability to be immortalized by telomerase activation alone. In our studies using neonatal keratinocytes, it is unknown whether cells that eventually became immortal were originally derived from stem cells or not. Isolation and transduction of pure stem cell populations might provide another means to generate immortal cells that could be used safely for therapeutic intervention.
In summary, we have shown that exogenous expression of TERT in human keratinocytes co-cultured with feeder cells is sufficient to induce immortalization, however, these cells have an increased frequency of p16 promoter methylation and subsequent inactivation. These results suggest that inactivation of the p16/Rb tumor suppressor pathway occurs in TERT-immortalized human keratinocytes regardless of culture conditions and emphasizes the need for further experimentation to determine the precise reason for and mechanism of p16 induction in human epithelial cells.
Materials and methods
Cell culture
Human foreskin keratinocytes (HFKs) were isolated as previously described (Blanton et al., 1991). Foreskin tissue was obtained using a protocol approved by the University of Iowa Institutional Review Board in accordance with HIPAA guidelines. HFKs were grown under two different culture conditions as previously described (Darbro et al., 2005). In the ‘plastic alone’ condition, HFKs were grown on tissue culture plastic in keratinocyte-serum free media (KSFM; Invitrogen, Carlsbad, CA, USA: 10724-011) supplemented with 0.2 ng/ml human recombinant epidermal growth factor (EGF), 30 μg/ml bovine pituitary extract (BPE), and 1% Penicillin–Streptomycin (Invitrogen: 15140–122). In the ‘feeder cell’ condition, HFKs were grown on tissue culture plastic in co-culture with post-mitotic, gamma irradiated J2 3T3 fibroblasts in E-media (Darbro et al., 2005). Final calcium concentrations in the plastic alone and with feeders were 0.09 and 1.28 mM, respectively. In the co-culture condition, irradiated fibroblasts were removed by incubation with a solution of 0.05% trypsin (Invitrogen: 15400–054), 50 mM HEPES buffer solution (Invitrogen: 15630-080) in Hanks balanced salt solution (Invitrogen: 14170–112). This protocol effectively removed the fibroblast feeder cells while leaving the keratinocytes attached (Darbro et al., 2005). HFKs were removed by further incubation in 0.05% trypsin/EDTA solution (Invitrogen: 25300–054). Trypsinized HFKs were pelleted by centrifugation and replated onto plastic alone or into co-culture with a new population of irradiated J2 3T3 fibroblasts. All cells were passaged when 70–80% confluent. All cells were maintained at 37°C with 5% CO2 at atmospheric (~20%) oxygen levels. PDs were calculated using the following equation:PDn=PD(n-1)log10[split ratio]/log10[2](Reznikoff et al., 1987).
RNA and protein isolation
Both RNA and protein were collected at selected PDs when HFKs reached 70-80% confluence. RNA and protein were collected from HFKs grown with feeders after the removal of the irradiated fibroblasts. Total RNA was collected using TriReagent (Molecular Research Center, Cincinnati, OH, USA) and purified according to the manufacturer’s instructions. Total protein was collected using WE16 lysis buffer, as previously described (Foster and Galloway, 1996).
Immunoblot analysis
Immunoblot analyses of cellular protein levels were performed as previously described (Foster et al., 1994; Darbro et al., 2005). The following antibodies were used in this study: p16 (Pharmingen, San Diego, CA, USA: G175-405) and Actin (Santa Cruz Biotechnology Inc.: I-19). Blots were probed with the appropriate HRP-conjugated secondary antibody: goat anti-mouse IgG (Jackson ImmunoResearch Laboratories, West Grove, PA, USA) or donkey anti-goat IgG (Santa Cruz Biotechnology Inc.). Detection was performed using the Western Lightning chemiluminescence kit (Perkin Elmer Life Sciences, Boston, MA, USA).
Semi-quantitative RT–PCR analysis
cDNA synthesis reactions were carried out using the RETRO-script kit (Ambion, Austin, TX, USA). RNA samples (2 μg) were used as the template for single-stranded cDNA synthesis reactions primed with the oligo dT primers included in the RETROscript kit. PCR analysis was carried out using the SuperTaq kit (Ambion) according to the manufacturer’s instructions. RT–PCR reaction products were separated by agarose gel electrophoresis, stained with ethidium bromide, and visualized by UV light. The following primers and conditions were used for RT–PCR amplification of the indicated genes: p16, sense primer 5′-CAACGCACCGAA TAGTTACGG-3′, antisense primer 5′-TGCCCATCATCAT GACCTG-3′, annealing temperature of 62°C, 28 cycles (Fu et al., 2003); GAPDH, sense primer 5′-TGAAGGTCG GAGTCAACGGATTTGGT-3′, antisense primer 5′-CATG TGGGCCATGACCTCCACCAC-3′, annealing temperature of 60°C, 30 cycles (cDNA was diluted 1:100 primer to PCR amplification).
Retroviral constructs and infections
The TERT-neo, TERT-hygro (both gifts from R Weinberg, Whitehead Institute, Cambridge, MA, USA), and pLXSH (gift from D Galloway, Fred Hutchinson Cancer Research Center, Seattle, WA, USA) vectors have all been described previously (Kiyono et al., 1998; Dickson et al., 2000; Farwell et al., 2000). The p16 retroviral construct was made by excising p16 cDNA out of a p16-pBluescript construct (gift from F Gourronc, University of Iowa) with EcoRI and cloning it directly into pBABE-puro. DNA sequencing performed by the University of Iowa DNA Sequencing Facility confirmed proper orientation and sequence of the p16 gene. Generation of virus with Phoenix amphotropic packaging lines has been previously described (Der et al., 1986; Whitehead et al., 1995; Zohn et al., 1998).
For TERT-neo, TERT-hygro, and pLXSH infections, exponentially growing HFKs were infected overnight in 4 μg/ml polybrene. Infected cells were washed in regular media the following morning. Cells were passed the following day, and selective media containing either 50 μg/ml neomycin (G418) (Invitrogen: 11811–031) or 10 μg/ml hygromycin (Invitrogen: 10687–010) was added on the next day. All infections were performed in the plastic culture condition and selection was carried out over 7 days. Following selection, clonal populations were pooled and split into either the feeder co-culture environment or left on tissue culture plastic in the absence of feeder cells. We performed two separate series of infections on two independent HFK cell strains (Strain A and Strain C). In the first infection, Strain C primary HFKs cultured on plastic alone were infected with TERT-hygro or pLXSH at approximately four PDs. Following the selection on plastic, clones were pooled and divided between the two culture conditions. When Strain C TERT-transduced HFKs on feeders reached PD 22 they were split normally and half the cells were replated into the co-culture environment and the other half were plated onto tissue culture plastic alone. In the second infection, Strain A primary HFKs cultured on plastic alone were infected with TERT-neo at approximately four PDs. Following selection on plastic, clones were pooled and divided between the two culture conditions. When Strain A TERT-transduced HFKs on feeders reached PDs 25, 29, 35, 41 and 51 they were split normally and half the cells were replated into the co-culture environment and the other half were plated onto tissue culture plastic alone.
For p16 and pBABE infections, late passage (PD>100) exponentially growing TERT-transduced HFKs, that had been switched from the feeder co-culture environment to plastic, were infected overnight in 4 μg/ml polybrene. Infected cells were washed in regular media the following morning. Cells were passed the following day onto glass coverslips, and selective media containing 1 μg/ml puromycin (Sigma: P8833) was added on the next day. Control experiments were conducted by infecting parallel cultures of HFKs with the pBABE vector alone. All infections were performed in the plastic culture condition and selection was carried out over 4 days. Before the end of the selection period, cultures were incubated with 100 μM BrdU (Sigma: B5002) for 12 h. Following incubation with BrdU, cells were fixed in 3.7% formaldehyde for future immunocytochemical staining.
Real-time quantitative TRAP analysis and cytogenetics
Real-time quantitative TRAP analysis (RQ-TRAP) was performed as previously described by Wege et al. (2003).Briefly, extracts from ~1000 cells were mixed with 0.1 μg of telomerase primer TS (5′-AATCCGTCGAGCAGATGG-3′), 0.05 μg of anchored return primer ACX (5′-GCGCG G(CTTACC)3CTAACC-3′), and Syber green PCR master mix (Applied Biosystems: 4309155). Reaction samples were incubated at 25°C for 20 min before amplification to allow telomerase to elongate the TS primer by adding TTAGGG repeat sequences. Real-time PCR amplification was carried out in a 7700 Sequence Detection System real-time thermalcycler (Applied Biosystems) with the following reaction conditions: Cycles at 95°C for 10 min, 95°C for 15 s, and 60°C for 1 min. Threshold cycle values were determined from amplification plots and compared with standard curves generated from serial dilutions (1000, 100, 10, 1 cell) of a telomerase-positive cell line (HPV16 E6/E7 HFKs) . Telomerase activity is reported as standard cell equivalents as compared to the telomerase positive control cell line. All RQ-TRAP reactions were carried out in triplicate and error bars created using standard deviation of the mean.
Chromosomal analyses were performed as previously described at the University of Iowa Cytogenetics Facility (Klingelhutz et al., 2005) on late passage (PD>100) Strain A and C TERT transduced HFKs continuously cultured with feeders or transferred to plastic. Briefly, cells growing on coverslips were arrested in metaphase by adding ethidium bromide followed by colcemid. Cell were incubated at room temperature with hypotonic solution (3:1 mixture of 0.8% sodium citrate and 0.075 M potassium chloride) and then fixed three times with a 3:1 methanol/acetic acid solution. Ten to twenty G-banded metaphases were analysed for each cell type.
5-aza-2′deoxycytidine treatment and bisulfite sequencing
TERT-transduced HFKs continuously co-cultured with feeders or transferred to plastic were treated with 1.5 μM 5-aza-2′ deoxycytidine (Sigma: A3656) at subconfluency for approximately 7 days before cells were collected for both protein and RNA. Untreated, parallel cultures were also collected for both protein and RNA at the same passage.
For bisulfite sequencing, DNA was collected at selected time points from either primary or transduced HFKs using the DNeasy Kit (Qiagen, Valencia, CA, USA: 69504) according to the manufacturer’s instructions. Bisulfite sequencing was performed according to the method of Cody et al. (1999). Briefly, 5 μg of genomic DNA was digested with EcoRI overnight at 37°C. Digested DNA was diluted and denatured followed by bisulfite treatment. Bisulfite treated DNA was subsequently desalted, desulphonated, and precipitated. PCR amplification of the p16 promoter region from –351 to –73 was performed with the sense primer 5′-GTGGGGAGGAGTT TAGTTTTTTTTTTTTG-3′ and antisense primer 5′-TCTAA TAACCAACCAACCCCTCCTCTTTC-3′ (Cody et al., 1999). Reaction conditions were 40 cycles at 95°C for 30 s, 60°C for 30 s and 72°C for 30 s. Isolated PCR products were cloned into the pGEM-T cloning vector (Promega: A3600) according to the manufacturer’s instructions. Ligation products were transformed into DH5α max efficiency competent cells (Invitrogen: 18258–012) and selection of bacterial clones was performed by blue/white colony screening. At least 15 bacterial clones were selected, grown and subjected to plasmid minipreps (Qiagen: 27106) to ensure the recovery of at least 10 epigenotypes per sample. Plasmid DNA was submitted to the University of Iowa DNA Sequencing Facility for sequence analysis. Analysis of non-CpG cytosines indicated the efficiency of bisulfite conversion at ~99%.
Immunocytochemistry
Strain A and C TERT-transduced HFKs switched from feeders to plastic and infected with either the p16 or pBABE retroviruses were fixed with 3.7% formaldehyde in PBS for 10 min at room temperature. Cells were permeabilized with 0.5% Triton X-100 in PBS for 7 min. Infected cells were treated with 100 μM BrdU for 12 h before fixation to label cells in the S phase of the cell cycle. After fixation and permeabilization, cultures that had received BrdU were treated for 1 h with 0.2 M HCl to make DNA containing BrdU accessible to the antibody. HCl-treated cultures were then neutralized with 0.1 M borate buffer (pH 8.5) and washed with 1 × PBS before proceeding with immunostaining (Natarajan et al., 2003). Coverslips were blocked in 10% normal goat serum, 1% BSA, and 0.1% Tween-20 in PBS for 45 min. Coverslips were incubated for 1 h with a mixture of both BrdU (Becton Dickinson, Franklin Lakes, NJ, USA: 347580) and p16 (Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA: C-20) primary antibodies. BrdU and p16 primary antibodies were diluted to a ratio of 1:20 and 1:100 in blocking buffer, respectively. A mixture of donkey anti-mouse Texas-red conjugated (Jackson ImmunoResearch Laboratories) and goat anti-rabbit fluorescein conjugated (Chemicon International) secondary antibodies, both diluted 1:100 in blocking buffer, was incubated with the coverslips for 45 min. The coverslips were mounted onto glass slides with VECTASHIELD mounting medium with DAPI (Vector Laboratories, Burlingame, CA, USA) to provide for visualization of HFK nuclei. Images were collected on a Nikon Eclipse E800 fluorescence microscope (Nikon Corporation, Tokyo, Japan). PBS washes were performed between each step of this immunocytochemical staining protocol.
Acknowledgments
We thank Robert Weinberg for providing the TERT-neo and TERT-hygro retroviral constructs, Denise Galloway for providing the pLXSH retroviral construct, and Francoise Gourronc for providing the p16-Bluescript vector, as well as Joseph Zabner for the use of his immunofluorescent microscope, Shiva Patil for assistance with cytogenetic analysis, and members of the Frederick Domann laboratory for help with bisulfite sequencing protocols. We are grateful to the other members of the Klingelhutz laboratory for helpful discussions. This work was supported by a grant to AJK from the National Institute on Aging (NIA), R01 AG18265 and a grant from the National Cancer Institute (NCI), R01 CA73612 to FED. Benjamin Darbro was supported by training grants from the National Heart, Lung, and Blood Institute (NHLBI), T32 HL07638, and the University of Iowa Medical Scientist Training Program (MSTP), T32 GM07337.
References
- Baek JH, Lee G, Kim SN, Kim JM, Kim M, Chung SC, et al. Int J Mol Med. 2003;12:319–325. [PubMed] [Google Scholar]
- Blanton RA, Perez-Reyes N, Merrick DT, McDougall JK. Am J Pathol. 1991;138:673–685. [PMC free article] [PubMed] [Google Scholar]
- Bodnar AG, Ouellette M, Frolkis M, Holt SE, Chiu CP, Morin GB, et al. Science. 1998;279:349–352. doi: 10.1126/science.279.5349.349. [DOI] [PubMed] [Google Scholar]
- Brenner AJ, Stampfer MR, Aldaz CM. Oncogene. 1998;17:199–205. doi: 10.1038/sj.onc.1201919. [DOI] [PubMed] [Google Scholar]
- Chakravarti A, Heydon K, Wu CL, Hammond E, Pollack A, Roach M, et al. J Clin Oncol. 2003;21:3328, 3334. doi: 10.1200/JCO.2003.12.151. [DOI] [PubMed] [Google Scholar]
- Cody DT, 2nd, Huang Y, Darby CJ, Johnson GK, Domann FE. Oral Oncol. 1999;35:516–522. doi: 10.1016/s1368-8375(99)00026-3. [DOI] [PubMed] [Google Scholar]
- Curtis CD, Goggins M. Methods Mol Med. 2005;103:123–136. doi: 10.1385/1-59259-780-7:123. [DOI] [PubMed] [Google Scholar]
- Darbro BW, Schneider GB, Klingelhutz AJ. J Invest Dermatol. 2005;125:499–509. doi: 10.1111/j.0022-202X.2005.23844.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das PM, Singal R. J Clin Oncol. 2004;22:4632–4642. doi: 10.1200/JCO.2004.07.151. [DOI] [PubMed] [Google Scholar]
- Der CJ, Pan BT, Cooper GM. Mol Cell Biol. 1986;6:3291–3294. doi: 10.1128/mcb.6.9.3291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickson MA, Hahn WC, Ino Y, Ronfard V, Wu JY, Weinberg RA, et al. Mol Cell Biol. 2000;20:1436–1447. doi: 10.1128/mcb.20.4.1436-1447.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esteller M, Gonzalez S, Risques RA, Marcuello E, Mangues R, Germa JR, et al. J Clin Oncol. 2001;19:299–304. doi: 10.1200/JCO.2001.19.2.299. [DOI] [PubMed] [Google Scholar]
- Farwell DG, Shera KA, Koop JI, Bonnet GA, Matthews CP, Reuther GW, et al. Am J Pathol. 2000;156:1537–1547. doi: 10.1016/S0002-9440(10)65025-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster SA, Demers GW, Etscheid BG, Galloway DA. J Virol. 1994;68:5698–5705. doi: 10.1128/jvi.68.9.5698-5705.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster SA, Galloway DA. Oncogene. 1996;12:1773–1779. [PubMed] [Google Scholar]
- Fu B, Quintero J, Baker CC. Cancer Res. 2003;63:7815–7824. [PubMed] [Google Scholar]
- Govindarajan B, Klafter R, Miller MS, Mansur C, Mizesko M, Bai X, et al. Mol Med. 2002;8:1–8. [PMC free article] [PubMed] [Google Scholar]
- Groeger AM, Caputi M, Esposito V, De Luca A, Bagella L, Pacilio C, et al. J Thorac Cardiovasc Surg. 1999;118:529–535. doi: 10.1016/s0022-5223(99)70192-3. [DOI] [PubMed] [Google Scholar]
- Harada H, Nakagawa H, Oyama K, Takaoka M, Andl CD, Jacobmeier B, et al. Mol Cancer Res. 2003;1:729–738. [PubMed] [Google Scholar]
- Herbert BS, Wright WE, Shay JW. Oncogene. 2002;21:7897–7900. doi: 10.1038/sj.onc.1205902. [DOI] [PubMed] [Google Scholar]
- Herman JG, Baylin SB. N Engl J Med. 2003;349:2042–2054. doi: 10.1056/NEJMra023075. [DOI] [PubMed] [Google Scholar]
- Jacobs JJ, de Lange T. Curr Biol. 2004;14:2302–2308. doi: 10.1016/j.cub.2004.12.025. [DOI] [PubMed] [Google Scholar]
- Jarmalaite S, Kannio A, Anttila S, Lazutka JR, Husgafvel-Pursiainen K. Int J Cancer. 2003;106:913–918. doi: 10.1002/ijc.11322. [DOI] [PubMed] [Google Scholar]
- Jarrard DF, Sarkar S, Shi Y, Yeager TR, Magrane G, Kinoshita H, et al. Cancer Res. 1999;59:2957–2964. [PubMed] [Google Scholar]
- Jung A, Schrauder M, Oswald U, Knoll C, Sellberg P, Palmqvist R, et al. Am J Pathol. 2001;159:1613–1617. doi: 10.1016/s0002-9440(10)63007-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang MK, Kameta A, Shin KH, Baluda MA, Kim HR, Park NH. Exp Cell Res. 2003;287:272–281. doi: 10.1016/s0014-4827(03)00061-2. [DOI] [PubMed] [Google Scholar]
- Kang MK, Kameta A, Shin KH, Baluda MA, Park NH. J Cell Physiol. 2004;199:364–370. doi: 10.1002/jcp.10410. [DOI] [PubMed] [Google Scholar]
- Kiyono T, Foster SA, Koop JI, McDougall JK, Galloway DA, Klingelhutz AJ. Nature. 1998;396:84–88. doi: 10.1038/23962. [DOI] [PubMed] [Google Scholar]
- Klingelhutz AJ, Qian Q, Phillips SL, Gourronc FA, Darbro BW, Patil SR. Virology. 2005;340:237–244. doi: 10.1016/j.virol.2005.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korkolopoulou P, Christodoulou P, Lazaris A, Thomas-Tsagli E, Kapralos P, Papanikolaou A, et al. Eur Urol. 2001;39:167–177. doi: 10.1159/000052432. [DOI] [PubMed] [Google Scholar]
- Lewis A, Mitsuya K, Umlauf D, Smith P, Dean W, Walter J, et al. Nat Genet. 2004;36:1291–1295. doi: 10.1038/ng1468. [DOI] [PubMed] [Google Scholar]
- Liggett Jr WH, Sidransky D. J Clin Oncol. 1998;16:1197–1206. doi: 10.1200/JCO.1998.16.3.1197. [DOI] [PubMed] [Google Scholar]
- Liu L, Zhang J, Bates S, Li JJ, Peehl DM, Rhim JS, et al. Int J Oncol. 2005;26:275–285. [PubMed] [Google Scholar]
- Momparler RL. Oncogene. 2003;22:6479–6483. doi: 10.1038/sj.onc.1206774. [DOI] [PubMed] [Google Scholar]
- Natarajan E, Saeb M, Crum CP, Woo SB, McKee PH, Rheinwald JG. Am J Pathol. 2003;163:477–491. doi: 10.1016/s0002-9440(10)63677-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson K, Svensson S, Landberg G. Mod Pathol. 2004;17:1464–1474. doi: 10.1038/modpathol.3800220. [DOI] [PubMed] [Google Scholar]
- Noble JR, Zhong ZH, Neumann AA, Melki JR, Clark SJ, Reddel RR. Oncogene. 2004;23:3116–3121. doi: 10.1038/sj.onc.1207440. [DOI] [PubMed] [Google Scholar]
- Partridge M, Gaballah K, Huang X. Cancer Metastasis Rev. 2005;24:71–85. doi: 10.1007/s10555-005-5048-0. [DOI] [PubMed] [Google Scholar]
- Paz MF, Fraga MF, Avila S, Guo M, Pollan M, Herman JG, et al. Cancer Res. 2003;63:1114–1121. [PubMed] [Google Scholar]
- Puthenveettil JA, Burger MS, Reznikoff CA. Adv Exp Med Biol. 1999;462:83–91. doi: 10.1007/978-1-4615-4737-2_7. [DOI] [PubMed] [Google Scholar]
- Ramirez RD, Herbert BS, Vaughan MB, Zou Y, Gandia K, Morales CP, et al. Oncogene. 2003;22:433–444. doi: 10.1038/sj.onc.1206046. [DOI] [PubMed] [Google Scholar]
- Ramirez RD, Morales CP, Herbert BS, Rohde JM, Passons C, Shay JW, et al. Genes Dev. 2001;15:398–403. doi: 10.1101/gad.859201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reznikoff CA, Loretz LJ, Pesciotta DM, Oberley TD, Ignjatovic MM. J Cell Physiol. 1987;131:285–301. doi: 10.1002/jcp.1041310302. [DOI] [PubMed] [Google Scholar]
- Reznikoff CA, Yeager TR, Belair CD, Savelieva E, Puthen-veettil JA, Stadler WM. Cancer Res. 1996;56:2886–2890. [PubMed] [Google Scholar]
- Rheinwald JG, Hahn WC, Ramsey MR, Wu JY, Guo Z, Tsao H, et al. Mol Cell Biol. 2002;22:5157–5172. doi: 10.1128/MCB.22.14.5157-5172.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocco JW, Sidransky D. Exp Cell Res. 2001;264:42–55. doi: 10.1006/excr.2000.5149. [DOI] [PubMed] [Google Scholar]
- Romanenko A, Morell-Quadreny L, Lopez-Guerrero JA, Pellin A, Nepomnyaschy V, Vozianov A, et al. Diagn Mol Pathol. 2002;11:163–169. doi: 10.1097/00019606-200209000-00007. [DOI] [PubMed] [Google Scholar]
- Sandhu C, Peehl DM, Slingerland J. Cancer Res. 2000;60:2616–2622. [PubMed] [Google Scholar]
- Sharpless NE. Mutat Res. 2005;576:22–38. doi: 10.1016/j.mrfmmm.2004.08.021. [DOI] [PubMed] [Google Scholar]
- Stampfer MR, Garbe J, Levine G, Lichtsteiner S, Vasserot AP, Yaswen P. Proc Natl Acad Sci USA. 2001;98:4498–4503. doi: 10.1073/pnas.071483998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stampfer MR, Yaswen P. Cancer Lett. 2003;194:199–208. doi: 10.1016/s0304-3835(02)00707-3. [DOI] [PubMed] [Google Scholar]
- Svensson S, Nilsson K, Ringberg A, Landberg G. Cancer Res. 2003;63:1737–1742. [PubMed] [Google Scholar]
- Taylor LM, James A, Schuller CE, Brce J, Lock RB, Mackenzie KL. J Biol Chem. 2004;279:43634–43645. doi: 10.1074/jbc.M402388200. [DOI] [PubMed] [Google Scholar]
- Thiagalingam S, Cheng KH, Lee HJ, Mineva N, Thiagalingam A, Ponte JF. Ann NY Acad Sci. 2003;983:84–100. doi: 10.1111/j.1749-6632.2003.tb05964.x. [DOI] [PubMed] [Google Scholar]
- Timmermann S, Hinds PW, Munger K. Oncogene. 1998;17:3445–3453. doi: 10.1038/sj.onc.1202244. [DOI] [PubMed] [Google Scholar]
- Tsihlias J, Kapusta L, Slingerland J. Annu Rev Med. 1999;50:401–423. doi: 10.1146/annurev.med.50.1.401. [DOI] [PubMed] [Google Scholar]
- Tsutsui T, Kumakura S, Yamamoto A, Kanai H, Tamura Y, Kato T, et al. Carcinogenesis. 2002;23:2111–2117. doi: 10.1093/carcin/23.12.2111. [DOI] [PubMed] [Google Scholar]
- Wang J, Hannon GJ, Beach DH. Nature. 2000;405:755–756. doi: 10.1038/35015674. [DOI] [PubMed] [Google Scholar]
- Wege H, Chui MS, Le HT, Tran JM, Zern MA. Nucleic Acids Res. 2003;31:E3–E3. doi: 10.1093/nar/gng003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberger PM, Yu Z, Haffty BG, Kowalski D, Harigopal M, Sasaki C, et al. Clin Cancer Res. 2004;10:5684–5691. doi: 10.1158/1078-0432.CCR-04-0448. [DOI] [PubMed] [Google Scholar]
- Whitehead I, Kirk H, Kay R. Mol Cell Biol. 1995;15:704–710. doi: 10.1128/mcb.15.2.704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong DJ, Barrett MT, Stoger R, Emond MJ, Reid BJ. Cancer Res. 1997;57:2619–2622. [PubMed] [Google Scholar]
- Yang J, Chang E, Cherry AM, Bangs CD, Oei Y, Bodnar A, et al. J Biol Chem. 1999;274:26141–26148. doi: 10.1074/jbc.274.37.26141. [DOI] [PubMed] [Google Scholar]
- Zhao W, Soejima H, Higashimoto K, Nakagawachi T, Urano T, Kudo S, et al. J Biochem (Tokyo) 2005;137:431–440. doi: 10.1093/jb/mvi048. [DOI] [PubMed] [Google Scholar]
- Zohn IE, Symons M, Chrzanowska-Wodnicka M, Westwick JK, Der CJ. Mol Cell Biol. 1998;18:1225–1235. doi: 10.1128/mcb.18.3.1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zongaro S, de Stanchina E, Colombo T, D’Incalci M, Giulotto E, Mondello C. Cancer Res. 2005;65:11411–11418. doi: 10.1158/0008-5472.CAN-05-1140. [DOI] [PubMed] [Google Scholar]