

Abstract
Statistical tests for detecting mosaic structure or recombination among nucleotide sequences usually rely on identifying a pattern or a signal that would be unlikely to appear under clonal reproduction. Dozens of such tests have been described, but many are hampered by long running times, confounding of selection and recombination, and/or inability to isolate the mosaic-producing event. We introduce a test that is exact, nonparametric, rapidly computable, free of the infinite-sites assumption, able to distinguish between recombination and variation in mutation/fixation rates, and able to identify the breakpoints and sequences involved in the mosaic-producing event. Our test considers three sequences at a time: two parent sequences that may have recombined, with one or two breakpoints, to form the third sequence (the child sequence). Excess similarity of the child sequence to a candidate recombinant of the parents is a sign of recombination; we take the maximum value of this excess similarity as our test statistic Δm,n,b. We present a method for rapidly calculating the distribution of Δm,n,b and demonstrate that it has comparable power to and a much improved running time over previous methods, especially in detecting recombination in large data sets.
MOSAIC structure exists in a nucleotide sequence if different segments of the sequence descend from different ancestors. A nucleotide sequence can be a mosaic of other sequences as a result of recombination or gene conversion; mosaic structure in bacterial DNA can also result from transduction, transformation, or conjugation, which are collectively referred to as horizontal gene transfer. The detection of mosaic structure has received much attention over the past two decades as a result of both a proliferation of sequence data and leaps in computing power, which together have allowed for the inference of multiple ancestral contributions to a nucleotide sequence. The biological questions at the source of this recent attention range from interest in the evolution of pathogens (Awadalla 2003; Moya et al. 2004; Wilson et al. 2005) and the characterization of linkage disequilibrium in large genomes (Pritchard and Przeworski 2001; Ardlie et al. 2002; Gabriel et al. 2002) to theoretical questions about clonality and the definitions of clonal and nearly clonal organisms (Maynard Smith et al. 1993; Halkett et al. 2005). For reviews on the methods and results in this field, see Posada et al. (2002) and Stumpf and McVean (2003).
Maynard Smith (1999) recognized that the continuum between completely clonal and freely recombining organisms naturally gives rise to two distinct problems: determining whether recombination occurs and measuring its frequency. In this investigation, we focus on the former. Detecting recombination usually involves searching groups of sequences for candidate recombinants or recombination signals and testing whether these represent statistically significant departures from expectation under a null hypothesis of no recombination. Dozens of statistical tests have been developed (Stephens 1985; Sawyer 1989; Balding et al. 1992; Karlin and Brendel 1992; Maynard Smith 1992; Takahata 1994; Sneath 1995; Goss and Lewontin 1996; Jakobsen and Easteal 1996; Grassly and Holmes 1997; Maynard Smith and Smith 1998; Sneath 1998; Awadalla et al. 1999; Crandall and Templeton 1999; Holmes et al. 1999; Maynard Smith 1999; Wall 1999; Gibbs et al. 2000; Martin and Rybicki 2000; Worobey 2001; Bruen et al. 2006) and evaluated (Wall 2000; Brown et al. 2001; Posada and Crandall 2001; Wiuf et al. 2001; Posada 2002) in this endeavor, none of which has yet emerged as the single standard test to be used for identifying recombination. In addition to testing for the existence of recombination, certain methods are also able to locate recombination breakpoints and, sometimes, the parent sequences involved in the recombination event, although the latter can be quite difficult. Methods that do not focus on parent sequences and breakpoints usually rely on detecting a recombination signal—for example, a phylogenetic incongruence or an excess of homoplasies—but may have trouble isolating the actual recombination event, which entails identifying particular parent sequences that recombined at particular breakpoints to form a recombinant offspring sequence.
Some methods (Takahata 1994; Robertson et al. 1995; Crandall and Templeton 1999; Holmes et al. 1999; Gibbs et al. 2000; Martin and Rybicki 2000; Martin et al. 2005) perform tests on three sequences at a time, which allows them to posit candidate parent sequences and candidate breakpoints. The proposed arrangement is then tested with a likelihood analysis, by visual detection of similarity in different sequence regions, or against a null distribution that would be expected under clonal evolution. The most common among these triplet tests—the Chimaera method (Posada and Crandall 2001; Posada 2002), which is based on a χ2-statistic (Maynard Smith 1992), and the Martin–Rybicki (MR) binomial distribution test (Martin and Rybicki 2000)—identify unusually high levels of sequence similarity inside a predefined window or on either side of a candidate breakpoint. We also take this approach by introducing a simple and intuitive statistic describing how identity varies along a sequence within a sequence triple. Our test statistic Δm,n,b is discrete and nonparametric. Describing its distribution, in principle, would require a computing time that grows exponentially with the number of informative sites (a subset of the polymorphisms) in the given sequence triple; to avoid this costly brute-force computation, we introduce a method for computing probabilities and P-values in polynomial time. Our method is memory intensive but very fast: computation of exact P-values takes seconds on a personal computer when there are <250 informative sites in the proposed sequence triple.
Our triplet test represents an advance over Chimaera and the MR method in that we eliminate the need for a sliding window, use a nonparametric statistic, and introduce a computation scheme that is exact and orders of magnitude faster. In evaluating our method's power to detect recombination in sequence triplets, we find that we always have higher power than the MR method and comparable power to Chimaera. In repeated applications of our triplet test to data sets with more than three sequences, we show that our method is among the most powerful of 16 previously tested methods.
STATISTICAL TESTS
We begin with three homologous sequences of the same length. The relationship among these three sequences is similar in practice to the relationship formulated by Crandall and Templeton (1999, pp. 166–167) among networks of sequences. From our three sequences, we designate one as the child sequence and investigate whether it could be a recombinant of the other two sequences, which we call parent sequences. We first present the simple case of a single-breakpoint recombinant but later focus on the more interesting and realistic case of a double-breakpoint recombinant. Considering our sequence triple, we ask whether one can reject the null hypothesis that the evolutionary history among the three sequences was completely clonal.
We call our parent sequences p and q and our child sequence c. For sequence length L, we can represent our three sequences as vectors of nucleotides: p = (p1, p2 , … , pL), q = (q1, q2 , … , qL), and c = (c1, c2 , … , cL). A single-breakpoint recombinant between the parent sequences at position l can be denoted
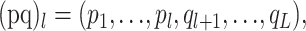 |
with 0 ≤ l ≤ L.
Writing |p − q| as the number of nucleotide differences between sequences p and q, we say that the most likely recombination breakpoint l minimizes |(pq)l − c|, the number of differences between the observed child sequence and a possible recombinant of the parent sequences. If this candidate recombinant is much closer (than either parent) to the child sequence, then we may have reason to believe that the evolutionary history of sequence c is better explained by a recombination or a gene conversion than by strictly clonal reproduction. If the candidate recombinant (pq)l is only slightly closer than the parents to the child sequence, then the candidate recombinant's additional sequence similarity may simply be an accident of how mutations accumulated on either side of the breakpoint l. Assessing whether the locations of the mutations (relative to the breakpoint) are significantly nonrandom is the foundation for the maximum χ2-test (Maynard Smith 1992), the Chimaera method (Posada and Crandall 2001; Posada 2002), the exact test based on the binomial distribution suggested by Martin and Rybicki (2000), and the heuristic test suggested by Crandall and Templeton (1999); it is also the focus of our analysis.
We introduce a nonparametric statistic slightly different from the ones above, but one that is more direct at detecting potential mosaics. Let
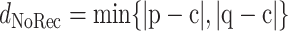 |
(1) |
be the minimum distance from the child to either of the parents, and let
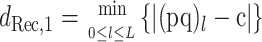 |
(2) |
be the minimum distance from the child to a candidate recombinant of the parents (including the boundary case recombinants, which are just the parents themselves); the subscript “1” indicates that there is just one breakpoint in the recombinant. Then, we define
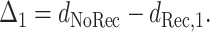 |
(3) |
The quantity Δ1 describes the difference, between clonal evolution and nonclonal evolution, in the number of mutations needed to describe the evolutionary history between the child and the closer parent; by nonclonal evolution we mean, here, an evolutionary history that allows for a single recombination event with a single breakpoint. Clearly Δ1 ≥ 0, and even if there had truly been no recombination or gene conversion among the sequences, a particular sequence triple could give the appearance of recombination with a high value of Δ1 if, by chance, the pattern of mutations was such that the left side of the child sequence appeared to be more closely related to parent p and the right side appeared to be closer to parent q. The distribution of this recombination signal Δ1 under the null hypothesis of clonal reproduction can be easily computed (see next section).
The difference in (3) is affected only by informative sites of the sequence triple (p, q, c). For our purposes, we define informative sites as those where the child's nucleotide matches exactly one of the parents' nucleotides. Uninformative sites are sites where (i) all three sequences agree, (ii) all three sequences differ, or (iii) the parents have matching (i.e., identical) nucleotides that differ from the child's. Our definition of informative sites is identical to that used in the Chimaera method and to the sister groups defined by Takahata (1994).
Suppose that there are m informative sites where p and c match and n informative sites where q and c match. The quantity Δ1 in (3) is then more precisely defined as Δm,n,1. Under the null hypothesis of clonal evolution among sequences p, q, and c, Δm,n,1 is a random variable that describes the maximum number of mutation events one could “explain away” by recombining p with q at a single breakpoint.
A two-breakpoint recombinant of sequences p and q can be described by
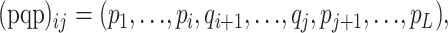 |
where i ≤ j. Letting
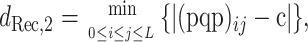 |
(4) |
we define
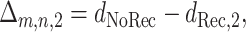 |
(5) |
where m and n are again the numbers of the two types of informative sites.
Δm,n,1 and Δm,n,2 are random variables that describe single-breakpoint and double-breakpoint recombination signals, respectively, under the null hypothesis of no recombination. They are discrete random variables with range 0 ≤ Δm,n,b ≤ min {m, n}, where b is the number of breakpoints. Observed Δ-quantities can be quickly calculated [in 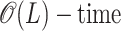 , for any b] from sequence data, and the null hypothesis of clonal evolution can be rejected if they are too high. In the next two sections, we review what is already known about the distribution of Δm,n,1 and present a method for calculating the distribution of Δm,n,2.
, for any b] from sequence data, and the null hypothesis of clonal evolution can be rejected if they are too high. In the next two sections, we review what is already known about the distribution of Δm,n,1 and present a method for calculating the distribution of Δm,n,2.
Single-breakpoint recombinant:
Consider a sequence triple (p, q, c) with m informative sites where p and c match and n informative sites where q and c match. Moving left to right across the informative sites on the child sequence, we can assign each informative site a letter based on probable ancestry (determined by the parent to which it is identical) and obtain a sequence such as PPPQPPPQQQQ, where a P denotes an informative site at which the child sequence and parent p share a nucleotide, and Q denotes an informative site at which the child sequence and parent q share a nucleotide. Under the null hypothesis of clonal reproduction, the placement of P's and Q's in the sequence should be completely random; i.e., each of the (m + n)!/(m!n!) possibilities has equal probability. In the example sequence above, it appears that the P's cluster toward the left side of the sequence and the Q's to the right side; therefore, this sequence may be a true (statistically significant) recombinant.
This sequence of P's and Q's is most easily visualized as a random walk on a set of axes where P is a step up and Q is a step down. This is not a traditional random walk since the number of up steps is known to be m, the number of down steps is known to be n, and the only randomness is the order in which they appear. After s steps, the height Xs of the random walk is distributed quasi-hypergeometrically [the quantity (Xs + s)/2 is distributed hypergeometrically]. The probability of being at height h after s steps, when |h| ≤ s and 0 ≤ s ≤ m + n, is
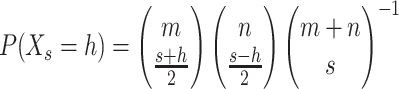 |
if h + s is even; P(Xs = h) = 0 if h + s is odd. This type of finite stochastic process can be called a hypergeometric random walk (HGRW). HGRWs have been previously analyzed in the probability literature in the form of ballot problems (Feller 1957), wherein one candidate in an election receives m votes, the second candidate receives n votes, and the order in which the votes are counted is of interest. We denote a hypergeometric random walk with m up steps and n down steps by the random variable Hm,n. Given data, we refer to an observed walk diagrammed from the informative sites of a sequence triple; examples of observed walks diagrammed from real data are in Figure 1.
Figure 1.—

Observed walks diagrammed from the informative sites of sequence triples. (A) The walk is diagrammed from Neisseria data (from the fourth row of Table 1). (B) The walk is diagrammed from influenza data (from the first row of Table 2). The circles indicate the beginning and end of the maximum descent in each walk, and in both cases the beginning of the maximum descent is also the maximum height of the walk. The dotted line in each diagram denotes the expected location of the hypergeometric random walk. The shaded areas in each diagram show the range of 100 simulated HGRWs.
Given our sequence triple with m + n informative sites and allowing only one breakpoint in a putative recombinant, the observed value Δm,n,1 is related to the maximum height of the walk diagrammed from the informative sites of sequences p, q, and c, by the relation
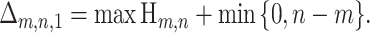 |
Using results from ballot theory (Barton and Mallows 1965) and gambling problems (Whitworth 1901, prop. 39, pp. 116–117), it can be shown that
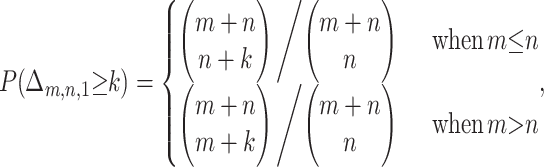 |
(6) |
or equivalently that
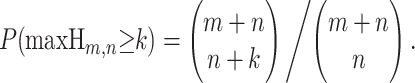 |
(7) |
From the observed maximum height of the diagrammed walk of the informative sites of a sequence triple, the null hypothesis of clonal reproduction can be rejected at the level P as calculated in (6) or (7). This is implicitly a one-tailed test with rejection of the null hypothesis of clonal evolution when the observed Δm,n,1 (or the maximum height of the observed walk) is large relative to m and n. An HGRW with a statistically improbable maximum height will have its up steps clustered toward the beginning (left side) of the walk and its down steps clustered toward the end (right side) of the walk. This is precisely a mosaic pattern in a nucleotide sequence: a child sequence having ancestry in p in the left-hand side of its sequence and ancestry in q in the right-hand side of its sequence.
Double-breakpoint recombinant:
Identifying mosaics with two breakpoints is the more relevant and interesting problem since in long sequence regions, converted tracts of DNA or horizontally transferred segments will usually have both breakpoints present. Identification of two breakpoints also allows for the removal of the horizontally acquired segment; the remaining segment(s) can then be tested again for clonal evolution, and multibreakpoint mosaics could be inferred by repeating such a process. Note that the two-breakpoint case subsumes the one-breakpoint case since a one-breakpoint recombinant can be viewed as having two breakpoints where one breakpoint is on the end of the sequence.
Again, considering only the informative sites of the sequence triple (p, q, c) and viewing their ordering in the context of a hypergeometric random walk, the quantity Δm,n,2 can be calculated by identifying the maximum descent (md) of the walk constructed from the arrangement of informative sites. Letting Xs be the height of Hm,n at step s, the maximum descent is defined as
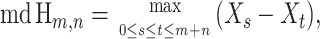 |
and it can be shown that
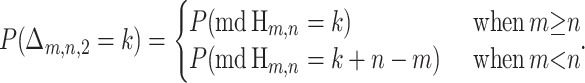 |
Statistical theory underlying a general class of statistics based on partial sum processes (Siegmund 1988; Karlin et al. 1990), change-point problems (Siegmund 1986), and maximal segmental sums (Karlin and Dembo 1992) provides asymptotic approximations that could be applied to calculate the probability that md Hm,n is large relative to m and n. Notably, Lemmas 3 and 4 in Siegmund (1988) and Theorems 2 and 3 in Hogan and Siegmund (1986) contain the appropriate constructions to approximate probabilities of maximum descents in HGRWs. In the theory on ballot problems, the maximum descent of an HGRW represents the maximum lead change (in one direction only) when counting ballots in a two-candidate election; as far as we are aware, this distribution has not been calculated with the combinatorial methods and reflection techniques usually applied in ballot problems. Below, we provide a method for calculating this distribution exactly.
We use the shorthand xm,n,k = P(md Hm,n = k), and for j, k ≥ 0, we define
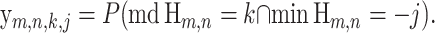 |
Then,
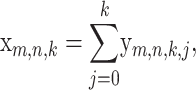 |
(8) |
and the y-probabilities can be obtained by solving the recursions
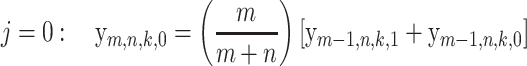 |
(9) |
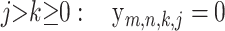 |
(10) |
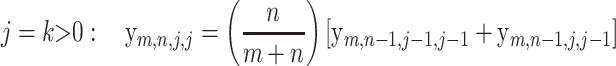 |
(11) |
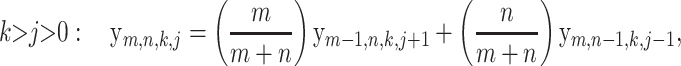 |
(12) |
with boundary conditions
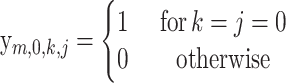 |
(13) |
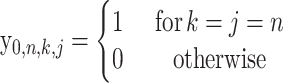 |
(14) |
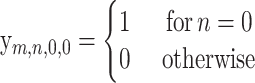 |
(15) |
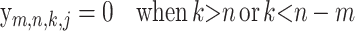 |
(16) |
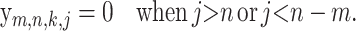 |
(17) |
All of the above recursions can be proved with a simple but careful first-step analysis of the random walk Hm,n. Below, the random variables Hm−1,n and Hm,n−1 refer to the subwalk of Hm,n that starts after the first step of Hm,n.
As an example, recursion (11) can be proved by noting that the event {md Hm,n = j ∩ min Hm,n = −j} implies that the first step of Hm,n must be down (X1 = −1) and that md Hm,n−1 must be either j or j − 1. Thus,
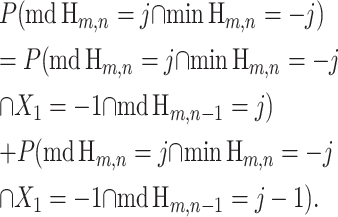 |
(18) |
In both summands of the right-hand side of (18), the last three events imply the first. We can rewrite the right-hand side of (18) as
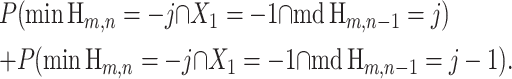 |
(19) |
The events
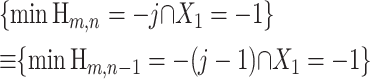 |
(20) |
are identical; one occurs if and only if the other occurs. Using this identity, we substitute into (19) and obtain
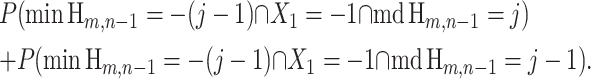 |
(21) |
By independence of the first step X1 = −1 from the subwalk Hm,n−1, this becomes
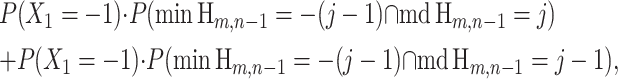 |
(22) |
which is
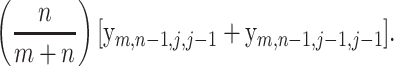 |
The other recursions can be proven similarly, and the boundary cases (13)–(17) are easily verifiable.
The computation time for any ym,n,k,j is bounded above by mn3, which is the maximum table size required in memory to solve recursions (9)–(12); k + 1 y-values must be computed to calculate xm,n,k via Equation 8. On a single 3-GHz processor with access to 2 GB RAM, the worst-case x-calculations for 250 informative sites take <3 sec; most x-probabilities can be calculated in <1 min for up to 400 informative sites. All calculations presented in this article (except where noted) were done on a 3.2-GHz Linux laptop with 1 GB of RAM and 750 MB of virtual memory. C++ source code for calculating the x- and y-variables is available from the authors.
For a given sequence triple in which we observe a Δm,n,2 = k, with a P-value of 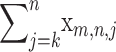 we can reject the null hypothesis of completely clonal reproduction in favor of an evolutionary history that includes a two-breakpoint recombination event.
we can reject the null hypothesis of completely clonal reproduction in favor of an evolutionary history that includes a two-breakpoint recombination event.
APPLICATIONS
The following are two simple examples that use the distributions Δm,n,1 and Δm,n,2 to test for mosaic structure among three sequences.
Neisseria:
We considered a classic example from the genus Neisseria and applied our tests to its argF gene, which is widely believed to have mosaic structure as a result of horizontal gene transfer among different species (Zhou and Spratt 1992; Grassly and Holmes 1997; Husmeier and McGuire 2003). Zhou and Spratt (1992) found regions of clustered polymorphism in a comparison between the argF genes of a Neisseria meningitidis isolate and a N. gonorrhoeae isolate and deduced that this region of clustered polymorphisms had likely ancestry in the species N. cinerea (since N. meningitidis and N. cinerea were nearly identical in this region). The authors noted that there were two regions in N. meningitidis that could have arisen by horizontal gene transfer, one of which might have been the result of variation in mutation rates or fixation rates (usually called “rate variation”). Further studies (Grassly and Holmes 1997; Husmeier and McGuire 2003) suggested that additional regions in the argF gene may have arisen by recombination.
We used three of the Neisseria sequences, one of each species, from the studies mentioned above (GenBank accession nos. X64860, X64866, and X64869; 787 nt in length) and tested whether there is any parent–parent–child relationship among them that lends support to one sequence being a mosaic of the other two. Table 1 shows that of the six possible arrangements, one has a highly significant (P = 10−12) single-breakpoint recombination signal, while the other five have none. This occurs because the first 202 nucleotides of N. meningitidis cluster significantly with N. cinerea (3.5% divergent, while N. meningitidis and N. gonorrhoeae are 13% divergent in this region) and the final 585 nucleotides of N. meningitidis cluster significantly with N. gonorrhoeae (2.9% divergent, while N. meningitidis and N. cinerea are 15% divergent in this region). This indicates that the first 202 nucleotides of N. meningitidis have probable ancestry in N. cinerea while the final 585 nucleotides of N. meningitidis have probable ancestry in N. gonorrhoeae, a view that is supported by the last two columns of Table 1, which allow for two breakpoints in the child sequence's composition but support a mosaic structure almost identical to the one-breakpoint case.
TABLE 1.
Mosaic structure in Neisseria argF gene
| p | q | c | Null | Observed maximum | P-value | Observed maximum descent | P-value |
|---|---|---|---|---|---|---|---|
| N. men. | N. cin. | N. gon. | H84,6 | 78 | 1 | 2 | 0.30 |
| N. cin. | N. men. | N. gon. | H6,84 | 0 | 1 | 78 | 1 |
| N. gon. | N. cin. | N. men. | H84,32 | 52 | 1 | 19 | 8.93 × 10−11 |
| N. cin. | N. gon. | N. men. | H32,84 | 19 | 1.42 × 10−12 | 71 | 2.50 × 10−11 |
| N. gon. | N. men. | N. cin. | H6,32 | 0 | 1 | 26 | 1 |
| N. men. | N. gon. | N. cin. | H32,6 | 26 | 1 | 2 | 0.60 |
The first three columns show a candidate parent–parent–child configuration that is tested for recombination; the fourth column shows the null distribution for the ordering of informative sites in the given sequence triple. The 1-breakpoint recombinant in the fourth row can be achieved with three different breakpoints, at positions 201, 202, and 203 (a breakpoint at position 201 indicates a breakpoint after the 201st nucleotide). The 2-breakpoint recombinant in the fourth row can be achieved with 66 different pairs of breakpoints: the first is always one of 202–204 while the second is one of 742–759/784–787. The 2-breakpoint recombinant in the third row can be achieved with 21 different pairs of breakpoints: the first is always one of 0–6 while the second is one of 202–204. N. men., N. meningitidis; N. cin., N. cinerea; N. gon., N. gonorrhoeae.
Influenza A:
Gibbs et al. (2001) found evidence for recombination in the hemagglutinin gene of the 1918 “Spanish” influenza strain, but their results were later refuted by Worobey et al. (2002) and Strimmer et al. (2003). We reanalyzed the five sequences presented by Gibbs that were the candidate recombiners and recombinants: two swine sequences (A/swine/Iowa/15/30 and A/swine/Wisconsin/1/61) and three human sequences (A/South Carolina/1/18, A/Kiev/59/79, and A/Alma Ata/1417/84), where the last two numbers in the sequence names indicate the year the sequence was isolated. In Table 2 we show the results obtained using our Δ-method on the significant relationships presented in Figure 1 of Gibbs et al. (2001).
TABLE 2.
Mosaic structure in influenza A hemagglutinin gene
| Dunn–Šidák |
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| p | q | c | Null | Observed maximum | P-value | Observed maximum descent | P-value | Maximum | md |
| 1979h | 1961s | 1918h | H148,159 | 7 | 0.45 | 46a | 4.00 × 10−4 | 0.03 | |
| 1961s | 1979h | 1918h | H159,148 | 39b | 7.85 × 10−4 | 30c | 5.22 × 10−3 | 0.05 | NS |
| 1979h | 1961s | 1930s | H94,218 | 0 | 1 | 124 | 1 | ||
| 1961s | 1979h | 1930s | H218,94 | 124 | 1 | 10d | 9.06 × 10−3 | NS | |
| 1979h | 1961s | 1984h | H92,220 | 0 | 1 | 128 | 1 | ||
| 1961s | 1979h | 1984h | H220,92 | 128 | 1 | 12e | 8.88 × 10−4 | 0.06 | |
The first three columns refer to the five influenza sequences mentioned in the Influenza A section. Here, the sequences are referred to by year and whether the sequence is human (h) or swine (s). The last two columns show the Dunn–Šidák corrected P-values given that without any knowledge about which sequences are recombinant, 60 comparisons would have to be made to test all parent–parent–child combinations. The breakpoint descriptions listed in footnotes a–e refer to a gapped alignment of length 1778 nt; 80 positions are gapped.
There are 90 pairs of breakpoints that result in a maximum descent of 46 units in the diagrammed walk from these three sequences. The first breakpoint is in position 242–247, while the second one is in 953–955/971–982.
The maximum height of 39 for this triple can be attained by 15 different breakpoints, at positions 952–954/970–981.
There are 30 pairs of breakpoints that result in a maximum descent of 30 for this sequence triple. The first breakpoint is in position 953–955/971–982; the second breakpoint is either in position 1653 or in position 1654.
There are 45 pairs of breakpoints that result in a maximum descent of 10 for this sequence triple. The first breakpoint is in position 953–955/971–982; the second breakpoint is in position 1049–1051.
There are 9 pairs of breakpoints that result in a maximum descent of 12 for this sequence triple. The first breakpoint is in position 953–955; the second breakpoint is in position 1049–1051.
With any type of analysis, detecting recombination in ancient influenza sequences is a challenge because of the high mutation rates in RNA viruses. A recombination that occurred 90 years ago would have its recombination signal obscured by mutations that accumulated after the recombination event. The relationship specified by the first two rows in Table 2, for example, requires a minimum of 104 years of evolution after the posited recombination event (61 years between the South Carolina and the Kiev strains and 43 years between the South Carolina and Wisconsin strains). Our five influenza sequences are on average 10% divergent (range: 2.4–18.3%), which means that detecting recombination events should be easy if the events were recent but difficult if they were ancient. On the timescale of influenza evolution, the hypothesized recombination events in Table 2 would be quite ancient.
Nevertheless, our method does detect weak recombination signals in the 1918 and 1984 human influenza strains. It is important to note that we are performing post hoc tests on previously analyzed sequences for which Gibbs et al. (2001) obtained statistically significant recombination signals. Given these same five sequences without any a priori knowledge about their relationships, we might compute P-values for all 60 possible parent–parent–child relationships among these sequences. The last two columns of Table 2 show which of these comparisons would still be significant after a Dunn–Šidák correction for 60 comparisons. The Dunn–Šidák correction is, of course, extremely conservative, especially since the Δ-values from our comparisons are positively correlated. A more accurate correction for multiple comparisons would take into account that we have multiple significant results. Using an exact binomial test, the probability under H0 that ≥3 of 60 comparisons would be significant at the 10−3 level is P = 3.3 × 10−5. To be slightly more conservative, we could say that the two P-values in rows 1 and 2 of Table 2 that are <10−3 are in fact manifestations of the same arrangement of strains (Kiev, Wisconsin, and South Carolina); then, the probability that ≥2 of 60 comparisons would be significant at the 10−3 level is P = 1.7 × 10−3.
Although it has been long believed that intragenic (homologous) recombination does not occur in influenza (Kilbourne 1978), the occurrence of nonhomologous recombination (Khatchikian et al. 1989; Orlich et al. 1994; Suarez et al. 2004) together with the data presented by Gibbs suggests that homologous recombination in influenza may be possible. However, as pointed out by Worobey et al. (2002), the observed substitution pattern in the influenza hemagglutinin can also be explained by within-sequence rate variation that varies across the different branches of the phylogeny (lineage-specific rate variation). Using pairwise comparisons among human sequences of the influenza A hemagglutinin, Worobey et al. described the HA1 region (nucleotide sites 151–920) as evolving more quickly than the HA2 region (sites 1–150 and 921–1695) in humans. If the opposite can be shown to be true for swine hemagglutinin sequences—that the HA2 evolves more quickly than the HA1—then the detected mosaicism in the 1918 human influenza hemagglutinin would be best explained by lineage-specific rate variation. This type of rate variation has also been called heterotachy (Lopez et al. 2002), and it was first introduced in the context of a changing set of concomitantly variable codons by Fitch and Markowitz (1970). It has been suggested that, for influenza A viruses, heterotachous or lineage-specific rate variation is a more likely evolutionary history than an intragenic recombination event (E. C. Holmes, personal communication).
SIMULATIONS
In addition to our Δ-method's theoretical appeal of being exact and nonparametric we show that it has the practical advantages of speed, power, and a low false-positive rate.
Power and false positives:
We compared the power and false-positive rates of our Δ-method to the 14 methods evaluated in Posada and Crandall (2001). Figure 2 duplicates the conditions of Figure 1 in Posada and Crandall (2001); in addition, two of the methods described by Carvajal-Rodríguez et al. (2006) are included in the top two rows of comparisons in Figure 2. Power and false-positive rates are tested for different values of the population-genetic parameter θ = 4NeμL, where Ne is the effective population size, μ is the per site per generation mutation rate, and L is the sequence length. Power is tested across different values of the recombination parameter ρ = 4NerL, where r is the per site per generation recombination rate. False-positive rates are tested for different levels α of rate variation (α is the shape parameter of a fixed-mean Γ-distribution of evolutionary rates as in Yang 1996) since, as noted in the Neisseria and influenza examples, statistical tests for recombination can confound recombination and variation in mutation/fixation rates.
Figure 2.—

Power and false-positive comparisons to the 14 methods tested in Posada and Crandall (2001). The top four graphs include two additional LPT methods described in Carvajal-Rodríguez et al. (2006). The graphs in the left column plot power under different recombination rates, while the right-hand column shows false-positive rates when there is variation in mutation rates but recombination is not present; α = ∞ means that there is no rate variation, while lower values of α indicate higher rate variation. The red line shows the power and false-positive rate of Δm,n,2 in detecting recombination. The gray lines show the power and false-positive rates of 14 (or 16) other methods. α = ∞ in the left column; ρ = 0 in the right column.
The left column of Figure 2 shows the power of 14 (or 16) other methods as well as the power of our Δ-method, which was determined as follows. Each data point corresponds to 100 simulated sequence sets with 10 sequences in each set (details in Posada and Crandall 2001). In a set of 10 sequences, there are 720 unique parent–parent–child arrangements; the quantity Δm,n,2 was calculated for each of these 720 triplets and the P-value associated with that quantity was computed with recursions (9)–(12). The minimum of these 720 P-values was corrected with a Dunn–Šidák correction and then reported as the P-value for rejecting clonal evolution in that 10-sequence set. This procedure was implemented in C++ as a command-line Linux program called 3SEQ; source code is available from the authors. The number of sets in which clonal evolution could be rejected at the 0.05 level was reported as the power of our Δ-method. The false-positive rates in the right-hand column of Figure 2 were computed in the same way.
Figure 2 shows that for a high enough mutation rate, our method is among the most powerful available for detecting recombination. For the sequence sets where θ = 10, the mean pairwise distance within each set of 10 sequences ranges from 1 to 30 nt. Using Δm,n,2 to test for recombination requires a minimum of nine informative sites to reject clonality at the 0.05 level; when correcting with a Dunn–Šidák correction for 720 comparisons, a minimum of 20 informative sites is needed. For this reason, our method has low power for data sets with little polymorphism. For the tested parameter combinations, our false-positive rate is at most 2% and among the lowest of all methods tested. It is important to note that some of the more powerful methods in the left-hand column had high false-positive rates in the right column. The plots in supplemental Figure S1 (http://www.genetics.org/supplemental/) show the ratios of power to false-positive rate for the 16 methods from Figure 2.
Supplemental Figure S2 at http://www.genetics.org/supplemental/ shows an additional false-positive analysis in data sets generated with autocorrelated mutation rates (from Figure 5c of Bruen et al. 2006); our false-positive rate was never >3.2% for these data sets. Supplemental Figure S3 at http://www.genetics.org/supplemental/ shows a power analysis under conditions with population growth, using the simulated data from Figure 4 of Bruen et al. (2006). Δm,n,2 is quite powerful under a scenario of population growth (as long as sequence diversity is high enough), and it retains very high power even when the recombination parameter ρ is small.
Figure 4.—
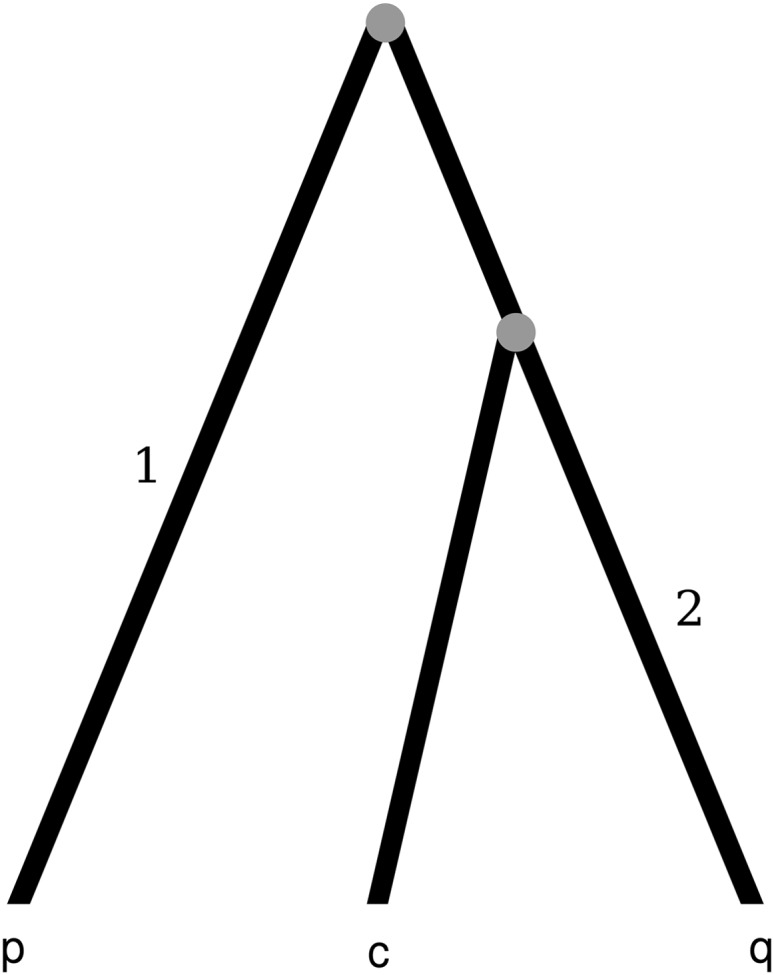
Phylogenetic tree that shows a possible clonal evolutionary history for the sequences p, q, and c. Mutations occurring in branch 1 will result in an informative site of type Q, while mutations occurring in branch 2 will result in an informative site of type P. The distributions describing the probability that the mutations in branch 1 or 2 cluster on either side of a breakpoint or between some pair of breakpoints are those of Δm,n,1 and Δm,n,2.
Since our statistical test is designed for sequence triplets we perform an additional power analysis that focuses exclusively on detecting recombination in sets of three sequences. We compare Δm,n,2 to three other common statistical tests designed to identify recombination in sequence triplets (a total of eight methods were tested of which the three most powerful are shown in Figure 3; details of and results for all eight methods are in the supplemental materials at http://www.genetics.org/supplemental/). For each data point in Figure 3, the program TREEVOLVE (Grassly et al. 1999) was used to generate 100 replicates of three sequences with the given population-genetic parameters, using the F84 model of nucleotide substitution (Felsenstein and Churchill 1996) with πA = 0.4, πC = 0.2, πG = 0.1, πT = 0.3, and a transition/transversion ratio of two. The black line in Figure 3 denotes the power and false-positive rate of a single-breakpoint version of Chimaera with exact P-value computations (Posada and Crandall 2001; Spencer 2003), the gray line corresponds to the most recent version of Chimaera (Chim-2006), and the blue line corresponds to the Martin–Rybicki method with window size 30 nt and step size 1 nt.
Figure 3.—

Power and false-positive comparisons with MR and Chimaera on sequence triplets. The red line shows power and false-positive rates for Δm,n,2. The black line shows the power and false-positive rates for Chim-Sp, a single-breakpoint no-window Chimaera implementation (described on p. 14 of the supplemental materials of Posada and Crandall 2001) whose P-values were calculated using the method of Spencer (2003). The gray line shows the power and false-positive rates of Chim-2006, a new Chimaera implementation with a sliding-window and sliding-breakpoint scheme; P-values were computed by permuting alignment columns 1000 times. The blue line shows the power and false-positive rates for MR-30,1 (Martin–Rybicki method with window size 30 nt and step size 1 nt). The third column shows ratio of power to false-positive rate at α = ∞. False-positive rates at α = ∞ were calculated with 1000 simulated triplets; all other data points were calculated with 100 simulated triplets. α = ∞ in the left column; ρ = 0 in the middle column.
For statistical identification of mosaic structure in sequence triplets, our Δ-method is as powerful as the most powerful methods available. All four methods in Figure 3 have similar power and false-positive rates, with the distinguishing feature that Chimaera is the least conservative method, MR is the most conservative, and Δm,n,2 is somewhere in between. For θ ≥ 50, Δm,n,2 has the best combination of power and false-positive rate.
Speed:
Table 3 shows the computation times of our method compared to MR and Chimaera. Our method has a clear advantage, especially in large data sets, since P-values are simply read from memory once a table of ym,n,k,j-values is built. For example, analysis of the influenza data (Boni 2007) requires reading ∼29 million P-values from memory, which is not a time-consuming task for a 3.2-GHz processor. Likewise, computing exact P-values using the method described by Spencer (2003) is quite fast; this is slightly slower than our Δ-method since a new table needs to be built for each P-value computation. On the other hand, performing 14.5 million sliding-window χ2-computations on each of 1000 randomized data sets (Chim-2006) or computing 9.6 million P-values from a binomial distribution for each of 287 possible windows (MR-30,1) can be quite computationally expensive.
TABLE 3.
Computation times (last four columns) for computing recombination statistics and P-values in large data sets
| Data set | Segregating sites | No. sequences | P-value | Δm,n,2 (min) | MR-30,1 | Chim-Sp | Chim-2006 |
|---|---|---|---|---|---|---|---|
| Dengue E | 618 | 69 | 3.3 × 10−5 | 2 | 86 min | 24 min | ∼100 hr |
| Human mtDNA | 1079 | 262 | 4.6 × 10−3 | 6 | ∼180 hr | ∼48 hr | ∼550 days |
| Influenza HA | 316 | 308 | 1 | 4 | ∼43 hr | 9 hr | ∼105 days |
All times and estimates are for a single 3.2-GHz processor. Dengue data are serotype 2 from Holmes et al. (1999); human mitochondrial DNA sequences are a subset of distinct strains from Kivisild et al. (2006); influenza seqeunces are New Zealand H3N2 isolates from 2000–2005 analyzed in Boni (2007). The P-value reported in this table is the minimum P-value (testing with Δm,n,2) from all comparisons in a data set, corrected with a Dunn–Šidák correction.
Note that nontriplet methods can be much faster than triplet methods. For example, analyzing the data in Table 3 with Φw (Bruen et al. 2006) takes seconds, but the recombinant sequences cannot be isolated.
DISCUSSION
Comparison:
Many statistical methods have already been developed for detecting recombination from sequence data. The usual recombination signals that these methods attempt to identify are (i) varying patterns of sequence identity, (ii) phylogenetic incongruencies, (iii) excess homoplasies, (iv) clustered polymorphism, and (v) low linkage disequilibrium; our method is of the first type. Here, we summarize the main similarities/differences between and advantages/disadvantages of our method and previous ones.
Most importantly, our method considers three sequences at a time using the appropriate mechanistic framework in which to view mosaic structure: the existence of one sequence that is a mosaic of a second and a third. Maynard Smith (1992) also acknowledged this as the appropriate framework, although the test he developed is designed for two sequences. Maynard Smith's maximum χ2-method was later reformulated as a proper three-sequence problem and is now called maximum-match χ2 or Chimaera (Posada and Crandall 2001; Posada 2002). Takahata (1994) recognized that one needed to look at a minimum of three sequences by focusing on sites that support a particular sister-group status where exactly two of three nucleotides agree. The BOOTSCAN search method (Salminem et al. 1995) examines candidate recombinants to see how different regions cluster with either of two parental sequences; bootstrap support, rather than a significance test, provides a measure of reliability of the proposed clustering. Recently, Martin et al. (2005) modified the BOOTSCAN method to search only sequence triples and to find recombinants statistically using the binomial test in Martin and Rybicki (2000). Finally, Holmes et al. (1999) describe a phylogenetic method called LARD that considers three sequences at a time and tests the hypothesis of completely clonal evolution vs. the hypothesis of clonal evolution for segments on either side of a breakpoint; their problem is formulated similarly to ours, the main difference being that their method focuses on phylogeny. It should be noted that some methods (Robertson et al. 1995; Gibbs et al. 2000) require four sequences: three involved in a recombination event and a fourth used as an outgroup.
The mechanistic three-sequence approach contrasts with approaches that attempt to identify indirect signals from sequence data, such as an excess of homoplasies (Hudson and Kaplan 1985; Jakobsen and Easteal 1996; Maynard Smith and Smith 1998; Maynard Smith 1999; Bruen et al. 2006) or a clustering of polymorphisms (Stephens 1985; Maynard Smith 1992; Martin and Rybicki 2000) that would be indicative of a recent recombination or gene conversion. While these methods can be quite effective, one must keep in mind that polymorphism clustering can be caused by selection or mutational hotspots and that an excess of homoplasies can be quite difficult to detect in rapidly mutating organisms such as RNA viruses.
Our method has several technical advantages. First, we do not use Monte Carlo methods to generate P-values, which makes our P-value computations very fast. Moreover, once a table is built in memory to calculate a particular xm,n,k, successive P-values can simply be extracted from the table; this means that repeated application of our Δ-tests is limited only by how quickly the computer's memory can be accessed. Monte Carlo methods have the additional disadvantage that the precision of computed P-values is limited by the number of permutations that can be done; this could be problematic in large data sets where precise P-values may be needed to survive multiple-comparisons corrections. Second, we avoid the widely used sliding-window approaches (Salminem et al. 1995; Siepel et al. 1995; Grassly and Holmes 1997; Lole et al. 1999; Martin and Rybicki 2000; Strimmer et al. 2003; Martin et al. 2005) that require the user to define a window size at the scale at which recombination is believed to have occurred. By considering all possible breakpoints in expression (4), we find the optimal “window size” that should be used for inferring recombination in a particular sequence triplet. This allows for the detection of recombinant segments at any scale.
By removing uninformative sites, our Δ-method should not confound variation in mutation/fixation rates with recombination; indeed, the middle column of Figure 3 and supplemental Figure S2 at http://www.genetics.org/supplemental/ show that even under high rate variation our false-positive rate is at most 5% (and usually <3%). However, lineage-specific or heterotachous rate variation can, in the absence of recombination, produce the pattern that is meant to be rejected by our Δ-distributions. Consider the tree in Figure 4. Branch 1 connects the root to sequence p while branch 2 connects the q–c common ancestor to sequence q. Differential environmental pressures on branches 1 and 2 can create the impression of mosaic structure. Suppose that the organism, during its evolution along branch 1, experiences an environment where the right-hand side of the sequence evolves rapidly and accumulates many substitutions while the left-hand side is either conserved or mutates neutrally. Suppose further that the organism, during evolution along branch 2, experiences an environment where the left-hand side of the sequence evolves rapidly and accumulates substitutions while the right-hand side is conserved or mutates neutrally. Under this scenario of clonal evolution, where environmental pressure increases substitution rates in the right part of the sequence on branch 1 and in the left part of the sequence on branch 2, the resulting sequence triple (p, q, c) will give the appearance that a recombination event occurred. In this case, the right part of sequence c will be very similar to sequence q while the left part will be very similar to sequence p. This type of sequence identity in different sequence regions is exactly what our Δ-statistics are designed to reveal.
While this combination of events may seem unlikely, the influenza sequences described here may have undergone just such evolutionary pressures. A key component in this scenario where mosaic structure is generated without recombination is that the organism experiences different selective environments on different branches of its phylogeny.
General conclusions:
We have introduced exact, nonparametric statistical tests for identifying nucleotide sequence mosaic structure with one or two breakpoints. Our test statistic is a function of a given sequence triple where one sequence is hypothesized to be a recombinant of the other two. Given a sequence triple, we calculate the difference in proximity (to the child sequence) between the closer parent sequence and the closest candidate recombinant sequence. This difference is denoted Δm,n,b—where m and n describe the numbers of informative sites at which the child sequence clusters with one or the other parent, and b denotes the number of breakpoints allowed in a candidate recombinant—and it is studied as a random variable under the null hypothesis of clonal evolution. The distribution of Δm,n,1 has been described in the probability literature on ballot problems, while the distribution of Δm,n,2 has been approximated but not described exactly. With brute-force methods, exact probabilities of the distribution of Δm,n,2 would require exponentially growing computation times that would become unmanageable once m + n > 35. To remedy this problem, we derive a set of recursive equations to calculate the probability mass function of Δm,n,2 in 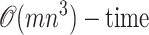 . These calculations can be performed in seconds on a single-processor personal computer (3 GHz, 2 GB RAM) as long as m + n < 250. When 250 < m + n < 400, most computations are equally quick although some may require additional memory or the use of virtual memory.
. These calculations can be performed in seconds on a single-processor personal computer (3 GHz, 2 GB RAM) as long as m + n < 250. When 250 < m + n < 400, most computations are equally quick although some may require additional memory or the use of virtual memory.
Our method relies on deducing parent–child sequence identity for different parents in different sequence regions. If a recombination occurred between sequences p and q to create the sequence c, then one segment of sequence c should be more similar to parent p while the remaining segment(s) of sequence c should be more similar to parent q. If this pattern is statistically significant—i.e., if it appears in the far right-hand tail of the distribution of Δm,n,b—we deduce that a recombination occurred.
Our Δ-method is among the most powerful available for detecting recombination in sequence data, even in highly recombinant data sets (generating data sets as in Figure 2 with ρ = 128, our method had 100% power for θ ≥ 50) or in data sets generated under conditions of population growth (see supplemental Figure S3 at http://www.genetics.org/supplemental/). For many of the simulated data sets in this article, Δm,n,2 appears to have the best combination of power and low false-positive rate. With comparable power to the best available methods, the most immediate practical advantage of using Δm,n,2 over other methods is its speed in large data sets. As can be seen in Table 3, computing P-values from Δm,n,2 can be many orders of magnitude faster than other triplet methods, depending on the number of sequences and the amount of polymorphism in the data set. For N sequences, triplet methods will make on the order of N3 comparisons, which for N > 1000 can be quite a large number for a personal computer. For example, 1000 influenza sequences with a similar level of polymorphism as in Table 3 would take 137 min to analyze with Δm,n,2, while 2000 sequences would take 18 hr. Fortunately, our method (along with most triplet methods) is completely parallelizable, which means that as sequence databases grow we can take advantage of parallel computing to search for recombinants in very large data sets. Note that if we have a particular query sequence that we would like to test for recombination, the number of comparisons is of order N2.
Our choice of applications here represents only a small sample of the clonal or nearly clonal sequences we could analyze with our Δ-statistics. They would also be quite useful in finding recombinants in human immunodeficiency virus databases and in larger dengue virus data sets and in analyzing the recently suggested recombinants in measles (Schierup et al. 2005). Human mitochondrial DNA is generally believed to evolve clonally, although the data set in Table 3 has quite strong mosaic signals; a reanalysis of other mtDNA data sets (Piganeau and Eyre-Walker 2004; Piganeau et al. 2004) would help determine whether recombination occurred during the evolution of the mitochondrion. For the influenza virus, our test could be used on whole (concatenated) influenza genomes, as in Holmes et al. (2005), to detect possible reassortment; hundreds of sequenced whole influenza genomes have already been analyzed (Nelson et al. 2006) and thousands more have been deposited in GenBank. As sequence databases expand in the genomic era, the Δ-method presented here could become one of the most efficient methods for detecting recombination and finding recombinants in large data sets.
Acknowledgments
We thank E. C. Holmes for many discussions especially on the rate variation scenario for influenza; we thank T. C. Bruen for providing data sets for power analysis and false-positive analysis; and we thank N. A. Rosenberg, J. M. Macpherson, and J. Van Cleve for helpful comments and suggestions. An anonymous editor pointed us to the known result in Equation 7. This work was funded in part by National Institutes of Health grants GM28016 (M.F.B., M.W.F.) and HG000205 (M.F.B.). D.P. is funded by grant BFU2004-02700 of the Spanish Ministry of Education and Science and by the Ramón y Cajal program of the Spanish government.
References
- Ardlie, K. G., L. Kruglyak and M. Seielstad, 2002. Patterns of linkage disequilbrium in the human genome. Nat. Rev. Genet. 3 299–309. [DOI] [PubMed] [Google Scholar]
- Awadalla, P., 2003. The evolutionary genomics of pathogen recombination. Nat. Rev. Genet. 4 50–60. [DOI] [PubMed] [Google Scholar]
- Awadalla, P., A. Eyre-Walker and J. Maynard Smith, 1999. Linkage disequilibrium and recombination in hominid mitochondrial DNA. Science 286 2524–2525. [DOI] [PubMed] [Google Scholar]
- Balding, D. J., R. A. Nichols and D. M. Hunt, 1992. Detecting gene conversion: primate visual pigment genes. Proc. R. Soc. Lond. Ser. B 249 275–280. [DOI] [PubMed] [Google Scholar]
- Barton, D. E., and C. L. Mallows, 1965. Some aspects of the random sequence. Ann. Math. Stat. 36 236–260. [Google Scholar]
- Boni, M. F., 2007. Vaccination and antigenic drift in influenza. Vaccine (in press). [DOI] [PMC free article] [PubMed]
- Brown, C., E. C. Garner, A. K. Dunker and P. Joyce, 2001. The power to detect recombination using the coalescent. Mol. Biol. Evol. 18 1421–1424. [DOI] [PubMed] [Google Scholar]
- Bruen, T. C., H. Philippe and D. Bryant, 2006. A simple and robust statistical test detecting the presence of recombination. Genetics 172 2665–2681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvajal-Rodríguez, A., K. A. Crandall and D. Posada, 2006. Recombination estimation under complex evolutionary models with the coalescent composite-likelihood method. Mol. Biol. Evol. 23 817–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crandall, K. A., and A. R. Templeton, 1999. Statistical methods for detecting recombination, pp. 153–176 in The Evolution of HIV, edited by K. A. Crandall. Johns Hopkins University Press, Baltimore.
- Feller, W., 1957. An Introduction to Probability Theory and Its Applications, Vol. I. John Wiley & Sons, New York.
- Felsenstein, J., and G. A. Churchill, 1996. A hidden Markov model approach to variation among sites in rate of evolution. Mol. Biol. Evol. 13 93–104. [DOI] [PubMed] [Google Scholar]
- Fitch, W. M., and E. Markowitz, 1970. An improved method for determining codon variability in a gene and its application to the rate of fixation of mutations in evolution. Biochem. Genet. 4 579–593. [DOI] [PubMed] [Google Scholar]
- Gabriel, S. B., S. F. Schaffner, H. Nguyen, J. M. Moore, J. Roy et al., 2002. The structure of haplotype blocks in the human genome. Science 296 2225–2229. [DOI] [PubMed] [Google Scholar]
- Gibbs, M. J., J. S. Armstrong and A. J. Gibbs, 2000. Sister-scanning: a Monte Carlo procedure for assessing signals in recombinant sequences. Bioinformatics 16 573–582. [DOI] [PubMed] [Google Scholar]
- Gibbs, M. J., J. S. Armstrong and A. J. Gibbs, 2001. Recombination in the hemagglutinin gene of the 1918 “Spanish flu.” Science 293 1842–1845. [DOI] [PubMed] [Google Scholar]
- Goss, P. J. E., and R. C. Lewontin, 1996. Detecting heterogeneity of substitution along DNA and protein sequences. Genetics 143 589–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grassly, N. C., and E. C. Holmes, 1997. A likelihood method for the detection of selection and recombination using nucleotide sequences. Mol. Biol. Evol. 14 239–247. [DOI] [PubMed] [Google Scholar]
- Grassly, N. C., P. H. Harvey and E. C. Holmes, 1999. Population dynamics of HIV-1 inferred from gene sequences. Genetics 151 427–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halkett, F., J.-C. Simon and F. Balloux, 2005. Tackling the population genetics of clonal and partially clonal organisms. Trends Ecol. Evol. 20 194–201. [DOI] [PubMed] [Google Scholar]
- Hogan, M. L., and D. Siegmund, 1986. Large deviations for the maxima of some random fields. Adv. Appl. Math. 7 2–22. [Google Scholar]
- Holmes, E. C., M. Worobey and A. Rambaut, 1999. Phylogenetic evidence for recombination in dengue virus. Mol. Biol. Evol. 16 405–409. [DOI] [PubMed] [Google Scholar]
- Holmes, E. C., E. Ghedin, N. Miller, J. Taylor, Y. Bao et al., 2005. Whole-genome analysis of human influenza A virus reveals multiple persistent lineages and reassortment among recent H3N2 viruses. PLoS Biol. 3 e300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson, R. R., and N. L. Kaplan, 1985. Statistical properties of the number of recombination events in the history of a sample of DNA sequences. Genetics 111 147–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Husmeier, D., and G. McGuire, 2003. Detecting recombination in 4-taxa DNA sequence alignments with Bayesian hidden Markov models and Markov chain Monte Carlo. Mol. Biol. Evol. 20 315–337. [DOI] [PubMed] [Google Scholar]
- Jakobsen, I. B., and S. Easteal, 1996. A program for calculating and displaying compatibility matrices as an aid in determining reticulate evolution in molecular sequences. Comput. Appl. Biosci 12 291–295. [DOI] [PubMed] [Google Scholar]
- Karlin, S., and V. Brendel, 1992. Chance and statistical significance in protein and DNA sequence analysis. Science 257 39–49. [DOI] [PubMed] [Google Scholar]
- Karlin, S., and A. Dembo, 1992. Limit distributions of maximal segmental score among Markov-dependent partial sums. Adv. Appl. Probab. 24 113–140. [Google Scholar]
- Karlin, S., A. Dembo and T. Kawabata, 1990. Statistical composition of high-scoring segments from molecular sequences. Ann. Stat. 18 571–581. [Google Scholar]
- Khatchikian, D., M. Orlich and R. Rott, 1989. Increased viral pathogenicity after insertion of a 28S ribosomal RNA sequence into the hemagglutinin gene of an influenza virus. Nature 340 156–157. [DOI] [PubMed] [Google Scholar]
- Kilbourne, E. D., 1978. Molecular epidemiology—influenza as archetype. Harvey Lect. 73 225–258. [PubMed] [Google Scholar]
- Kivisild, T., P. Shen, D. P. Wall, B. Do, R. Sung et al., 2006. The role of selection in the evolution of human mitochondrial genomes. Genetics 172 373–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lole, K. S., R. C. Bollinger, R. S. Paranjape, D. Gadkari, S. S. Kulkarni et al., 1999. Full-length immunodeficiency virus type 1 genomes from subtype c–infected seroconverters in india, with evidence of intersubtype recombination. J. Virol. 73 152–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez, P., D. Casane and H. Philippe, 2002. Heterotachy, an important process of protein evolution. Mol. Biol. Evol. 19 1–7. [DOI] [PubMed] [Google Scholar]
- Martin, D., and E. Rybicki, 2000. RDP: detection of recombination amongst aligned sequences. Bioinformatics 16 562–563. [DOI] [PubMed] [Google Scholar]
- Martin, D. P., D. Posada, K. A. Crandall and C. Williamson, 2005. A modified bootscan algorithm for automated identification of recombinant sequences and recombination breakpoints. AIDS Res. Hum. Retroviruses 21 98–102. [DOI] [PubMed] [Google Scholar]
- Maynard Smith, J., 1992. Analyzing the mosaic structure of genes. J. Mol. Evol. 34 126–129. [DOI] [PubMed] [Google Scholar]
- Maynard Smith, J., 1999. The detection and measurement of recombination from sequence data. Genetics 153 1021–1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maynard Smith, J., and N. H. Smith, 1998. Detecting recombination from gene trees. Mol. Biol. Evol. 15 590–599. [DOI] [PubMed] [Google Scholar]
- Maynard Smith, J., N. H. Smith, M. O'Rourke and B. G. Spratt, 1993. How clonal are bacteria? Proc. Natl. Acad. Sci. USA 90 4384–4388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moya, A., E. C. Holmes and F. González-Candelas, 2004. The population genetics and evolutionary epidemiology of RNA viruses. Nat. Rev. Microbiol. 2 279–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson, M. I., L. Simonsen, C. Viboud, M. A. Miller, J. Taylor et al., 2006. Stochastic processes are key determinants of the short-term evolution of influenza A virus. PLoS Pathog. 2 e125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orlich, M., H. Gottwald and R. Rott, 1994. Nonhomologous recombination between the hemagglutinin gene and the nucleoprotein gene of an influenza virus. Virology 204 462–465. [DOI] [PubMed] [Google Scholar]
- Piganeau, G., and A. Eyre-Walker, 2004. A reanalysis of the indirect evidence for recombination in human mitochondrial DNA. Heredity 92 282–288. [DOI] [PubMed] [Google Scholar]
- Piganeau, G., M. Gardner and A. Eyre-Walker, 2004. A broad survey of recombination in animal mitochondria. Mol. Biol. Evol. 21 2319–2325. [DOI] [PubMed] [Google Scholar]
- Posada, D., 2002. Evaluation of methods for detecting recombination from DNA sequences: empirical data. Mol. Biol. Evol. 19 708–717. [DOI] [PubMed] [Google Scholar]
- Posada, D., and K. A. Crandall, 2001. Evaluation of methods for detecting recombination from DNA: computer simulations. Proc. Natl. Acad. Sci. USA 98 13757–13762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posada, D., K. A. Crandall and E. C. Holmes, 2002. Recombination in evolutionary genomics. Annu. Rev. Genet. 36 75–97. [DOI] [PubMed] [Google Scholar]
- Pritchard, J. K., and M. Przeworski, 2001. Linkage disequilibrium in humans: models and data. Am. J. Hum. Genet. 69 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson, D. L., B. H. Hahn and P. M. Sharp, 1995. Recombination in AIDS viruses. J. Mol. Evol. 40 249–259. [DOI] [PubMed] [Google Scholar]
- Salminem, M. O., J. K. Carr, D. S. Burke and F. E. McCutchan, 1995. Identification of breakpoints in intergenotypic recombinants of HIV type 1 by bootscanning. AIDS Res. Hum. Retroviruses 11 1423–1425. [DOI] [PubMed] [Google Scholar]
- Sawyer, S., 1989. Statistical tests for detecting gene conversion. Mol. Biol. Evol. 6 526–538. [DOI] [PubMed] [Google Scholar]
- Schierup, M. H., C. H. Mordhorst, C. P. Muller and L. S. Christensen, 2005. Evidence of recombination among early-vaccination era measles virus strains. BMC Evol. Biol. 5 52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegmund, D., 1986. Boundary crossing probabilities and statistical applications. Ann. Stat. 14 361–404. [Google Scholar]
- Siegmund, D., 1988. Approximate tail probabilities for the maxima of some random fields. Ann. Probab. 16 487–501. [Google Scholar]
- Siepel, A. C., A. L. Halpern, C. Macken and B. T. M. Korber, 1995. A computer program designed to screen rapidly for HIV type 1 intersubtype recombinant sequences. AIDS Res. Hum. Retroviruses 11 1413–1416. [DOI] [PubMed] [Google Scholar]
- Sneath, P. H. A., 1995. The distribution of the random division of a molecular sequence. Binary Comput. Microbiol. 7 148–152. [Google Scholar]
- Sneath, P. H. A., 1998. The effect of evenly spaced constant sites on the distribution of the random division of a molecular sequence. Bioinformatics 14 608–616. [DOI] [PubMed] [Google Scholar]
- Spencer, M., 2003. Exact significance levels for the maximum χ2 method of detecting recombination. Bioinformatics 19 1368–1370. [DOI] [PubMed] [Google Scholar]
- Stephens, J. C., 1985. Statistical methods of DNA sequence analysis: detection of intragenic recombination or gene conversion. Mol. Biol. Evol. 2 539–556. [DOI] [PubMed] [Google Scholar]
- Strimmer, K., K. Forslund, B. Holland and V. Moulton, 2003. A novel exploratory method for visual recombination detection. Genome Biol. 4 R33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stumpf, M. P. H., and G. A. T. McVean, 2003. Estimating recombination rates from population-genetic data. Nat. Rev. Genet. 4 959–968. [DOI] [PubMed] [Google Scholar]
- Suarez, D. L., D. A. Senne, J. Banks, I. H. Brown, S. C. Essen et al., 2004. Recombination resulting in virulence shift in avian influenza outbreak, Chile. Emerg. Infect. Dis. 10 693–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahata, N., 1994. Comments on the detection of reciprocal recombination or gene conversion. Immunogenetics 39 146–149. [DOI] [PubMed] [Google Scholar]
- Wall, J. D., 1999. Recombination and the power of statistical tests of neutrality. Genet. Res. 74 65–79. [Google Scholar]
- Wall, J. D., 2000. A comparison of estimators of the population recombination rate. Mol. Biol. Evol. 17 156–163. [DOI] [PubMed] [Google Scholar]
- Whitworth, W. A., 1901. Choice and Chance, Ed. 5. Hafner Publishing, New York.
- Wilson, D. J., D. Falush and G. McVean, 2005. Germs, genomes, and genealogies. Trends Ecol. Evol. 20 39–45. [DOI] [PubMed] [Google Scholar]
- Wiuf, C., T. Christensen and J. Hein, 2001. A simulation study of the reliability of recombination detection methods. Mol. Biol. Evol. 18 1929–1939. [DOI] [PubMed] [Google Scholar]
- Worobey, M., 2001. A novel approach to detecting and measuring recombination: new insights into evolution of viruses, bacteria, and mitochondria. Mol. Biol. Evol. 18 1425–1434. [DOI] [PubMed] [Google Scholar]
- Worobey, M., A. Rambaut, O. G. Pybus and D. L. Robertson, 2002. Questioning the evidence for genetic recombination in the 1918 “Spanish flu” virus. Science 296 211a. [DOI] [PubMed] [Google Scholar]
- Yang, Z., 1996. Among-site variation and its impact on phylogenetic analyses. Trends Ecol. Evol. 11 367–371. [DOI] [PubMed] [Google Scholar]
- Zhou, J., and B. G. Spratt, 1992. Sequence diversity within the argF, fbp and recA genes of natural isolates of Neisseria meningitidis: interspecies recombination within the argF gene. Mol. Microbiol. 6 2135–2146. [DOI] [PubMed] [Google Scholar]