

Abstract
Interaction between genes, or epistasis, is found to be common and it is a key concept for understanding adaptation and evolution of natural populations, response to selection in breeding programs, and determination of complex disease. Currently, two independent classes of models are used to study epistasis. Statistical models focus on maintaining desired statistical properties for detection and estimation of genetic effects and for the decomposition of genetic variance using average effects of allele substitutions in populations as parameters. Functional models focus on the evolutionary consequences of the attributes of the genotype–phenotype map using natural effects of allele substitutions as parameters. Here we provide a new, general and unified model framework: the natural and orthogonal interactions (NOIA) model. NOIA implements tools for transforming genetic effects measured in one population to the ones of other populations (e.g., between two experimental designs for QTL) and parameters of statistical and functional epistasis into each other (thus enabling us to obtain functional estimates of QTL), as demonstrated numerically. We develop graphical interpretations of functional and statistical models as regressions of the genotypic values on the gene content, which illustrates the difference between the models—the constraint on the slope of the functional regression—and when the models are equivalent. Furthermore, we use our theoretical foundations to conceptually clarify functional and statistical epistasis, discuss the advantages of NOIA over previous theory, and stress the importance of linking functional and statistical models.
TRADITIONALLY, most of the theory to study the evolution and genetic architecture of quantitative traits has been built on the assumption of additivity across the loci that contribute to the expression of a trait (Bürger 2000). Interest in how interacting genes contribute to multifactorial trait expression is increasing in both quantitative and evolutionary genetics as it has been shown that gene effects commonly interact and that the effect of those interactions on the evolution and artificial selection of traits is far from negligible (Carlborg and Haley 2004; Hansen 2006). We use the term epistasis to refer to nonadditivity in the contributions of several genes to a trait, meaning that the effects of the alleles of one gene depend on the genetic background (Phillips 1998; Wagner et al. 1998; Wade et al. 2001). This allows additive effects of genes (or allele substitutions) to evolve and the effect of particular loci to range from being of crucial importance to completely vanishing changing backgrounds (Carlborg and Haley 2004; Carlborg et al. 2006). Epistasis is therefore critical in the understanding of the evolution of natural populations, the response to selection in animal and plant breeding programs, and the genetic factors underlying multifactorial disease (Templeton 2000; Moore and Williams 2005). Thus, theoretical models of evolution including epistasis may become more useful especially in the light of the new molecular and statistical tools available for the study of allelic effects.
The term statistical epistasis refers to the use of statistical tools to analyze gene interactions. Fisher (1918) provided the basis of the study of gene effects of a trait using parameters that represent the average effects of allele substitutions over the population and lead to a decomposition of the genetic variance. Cockerham (1954) and Kempthorne (1954) complemented this work with a subdivision of the epistatic variance into separate components. Fisher (1958) perceived epistasis as a nuisance effect whose evolutionary consequences would thus be equivalent to those of environmental variation. Albeit such an approach could be suitable to study phenotypic change in very large random-mating populations, it might not be reasonable otherwise. In fact, the theory of speciation by hybrid incompatibilities (Dobzhansky 1936; Muller 1942) and the shifting balance theory (Wright 1931, 1977) are two major theories that exemplify the crucial role of epistasis as a driving force in evolution. The evolutionary consequences of epistasis in the context of these theories, and in general in speciation and in adaptation in subdivided populations, have been studied by inspecting the components of the genetic variance (Goodnight 1988, 1995, 2000; Wade and Goodnight 1998; Barton and Turelli 2004; Turelli and Barton 2006).
Cheverud and Routman (1995) and Cheverud (2000) analyze and discuss the efficiency of statistical epistasis for studying the evolution of complex traits. They underline the difference between genotypic and genetic values and suggest to study epistasis by focusing on genotypic values, as they represent natural effects of allele substitutions regardless of the allele frequencies in the population under study. Their view is in accordance with the first definition of the term epistasis by Bateson (1909), and they refer to this as physiological epistasis because the aim is to capture the interactions of the genes at the level of the organism rather than at a population level (see Phillips 1998 for a comprehensive, historical dissection of this duality). Hansen and Wagner (2001b) further inspected the relationship between physiological and statistical epistasis. They prefer to use the term functional epistasis—instead of physiological epistasis—as it reflects the functional properties of the gene interactions in determining the expression of a trait. Their multilinear model incorporates this in the form of a simplified genotype–phenotype map based on genetic values that capture the main role of gene interactions in evolution. The loss of generality of the multilinear model is rewarded by analytical tractability. A key concept in Hansen and Wagner's (2001b) development is their change-of-reference tool, which allows the description of epistatic interactions as allele substitutions made on any reference genotype. In particular, this allows inspection of evolutionary properties of a population by means of describing the (multilinear) epistasis parameters using the mean of the population as a reference point (Hansen and Wagner 2001a; Hermisson et al. 2003; Carter et al. 2005; Hansen et al. 2006). Barton and Turelli (2004) have developed a model to analyze the consequences of epistasis in the presence of genetic drift. Their theoretical framework complements that of Hansen and Wagner (2001b) and implements a new notation with the purpose of providing more transparent results than the previous approaches. Functional—or physiological—epistasis has also been referred to as biological epistasis, and has even been split into genetical epistasis and biological epistasis, when discussing how to integrate systems biology and quantitative trait loci (QTL) analysis (Moore 2005; Moore and Williams 2005).
In the context of QTL analysis, Yang (2004) and Zeng and collaborators (Kao and Zeng 2002; Zeng et al. 2005) have reviewed and analyzed several statistical models used for obtaining estimates of epistasis. For two major reasons, they stress the use of orthogonal–statistical models. First, the measurement of genetic effects of reduced models is consistent in orthogonal models. This enables a straightforward comparison of nested models for performing model selection. Second, each genetic effect in an orthogonal model can be independently estimated and plays a role in the computation of its component of variance alone. Zeng et al. (2005) have developed the G2A model, a multilocus two-allele model that is orthogonal in populations under strict Hardy–Weinberg and linkage equilibrium, regardless of the frequencies of the alleles at each locus. Wang and Zeng (2006) have extended this model to a multiallele framework with linkage disequilibrium, particularly focusing on the decomposition of the genetic variance. Yang (2004) has built an explicit two-locus two-allele model that is generally orthogonal regarding the frequencies of the genotypes in the populations and has implemented it with a tool for measuring the bias in the estimates of genetic effects caused by linkage disequilibrium.
Here we establish a formal link between the models of statistical and functional epistasis through a unified, formal framework—the natural and orthogonal interactions (NOIA) model. We provide a mathematical description of genetic systems that leads to a conceptual interpretation of the relationship between statistical and functional epistasis and a set of explicit expressions to translate between statistical and functional estimates and between genetic effects in different populations. The resulting model incorporates general statistical and functional formulations of genotypic values on genetic effects that improve both the existing statistical and the functional models of gene interactions. We also provide a graphical interpretation of the functional formulation of NOIA, similar to that of the statistical models—as linear weighted regressions of the genotypic values on the gene content (Fisher 1918). The slope of the functional regression is constrained to the one of an unweighted regression, which provides a characterization of when the functional and the statistical formulations are equivalent.
THE NOIA MODEL
Modeling genetic effects as allele substitutions on one specific genotype:
Consider a trait controlled by a number of diallelic loci, n. We begin by using a particular genotype as a reference point to build a genotype–phenotype map for this trait. First we focus in one locus, locus A. Zeng et al. (2005) discuss several maps for one locus and two alleles, and we basically follow their notation and nomenclature here, G = S · E, where G is the vector of genotypic values (G11, G12, G22, the phenotypes of the three genotypes for alleles A1 and A2). S is called the genetic-effect design matrix and its scalars, the natural scales, are the coefficients of the genetic effects present at each genotype. E is the vector of genetic effects, actually accounting for the reference point, R, of the model (i.e., the point from which the genetic effects are deviations) and the genetic effects—the additive effect, a, and the dominance effect, d. Changes in the genetic-effect design matrix, S, lead to alternative descriptions of the genetic system with different reference points when describing the genotypic values of the individuals in terms of the genetic effects.
We begin by using a single genotype, say G11, as a reference point from which to measure the genetic effects, resulting in the following formulation of the model, G
=
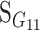 ·
E:
·
E:
 |
(1) |
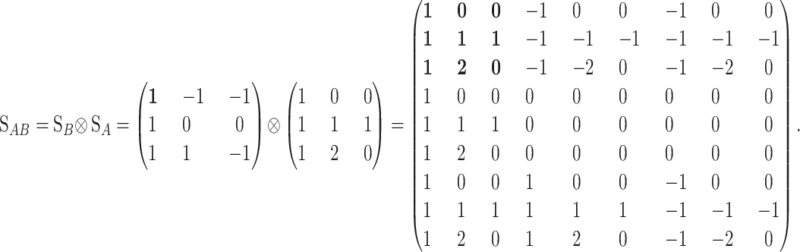
The first column of  illustrates that the phenotypes are measured as deviations from the reference point, here R = G11. The second column illustrates that one additive effect is added to R for each A2 allele and the third column that the dominance effect is added to the heterozygote. The genetic effects are thus effects of allelic substitutions on the reference genotype A1A1. The extension of this NOIA functional formulation to several loci is obtained as the Kronecker product of the S matrices of the single loci (appendix a). For two loci, A and B, with genetic-effect design matrices SA and SB, respectively, this reads
illustrates that the phenotypes are measured as deviations from the reference point, here R = G11. The second column illustrates that one additive effect is added to R for each A2 allele and the third column that the dominance effect is added to the heterozygote. The genetic effects are thus effects of allelic substitutions on the reference genotype A1A1. The extension of this NOIA functional formulation to several loci is obtained as the Kronecker product of the S matrices of the single loci (appendix a). For two loci, A and B, with genetic-effect design matrices SA and SB, respectively, this reads
 |
(2) |
where EAB is the two-locus vector of genetic effects. Let us call 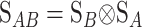 . By using the properties of the Kronecker product we get
. By using the properties of the Kronecker product we get 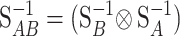 and, hence, the genetic effects can be obtained by solving the system
and, hence, the genetic effects can be obtained by solving the system
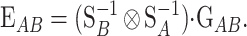 |
(3) |
If 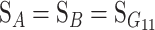 , the formulations (2) and (3) describe the effects of allelic substitutions on the reference genotype A1A1B1B1 (or simply “1111”), as both loci have G11 as their respective reference points. This is convenient for constructing the model, but insufficient for a functional genotype–phenotype map, which should be able to describe the genetic effects as effects of substitutions from any reference point. Therefore, we implement the model with a change-of-reference tool.
, the formulations (2) and (3) describe the effects of allelic substitutions on the reference genotype A1A1B1B1 (or simply “1111”), as both loci have G11 as their respective reference points. This is convenient for constructing the model, but insufficient for a functional genotype–phenotype map, which should be able to describe the genetic effects as effects of substitutions from any reference point. Therefore, we implement the model with a change-of-reference tool.
Modeling genetic effects as allele substitutions on any genotype:
Here we provide a simple way to compute the genetic-effect design matrix for using any individual of the population as a reference point of the genetic system. This enables us to describe all genotypic values in the genetic system as sets of allele substitutions on any particular (reference) individual in the population and also to use the mean of the population under study as the reference point.
The general expression for the one-locus functional genetic-effect design matrix, SF, is
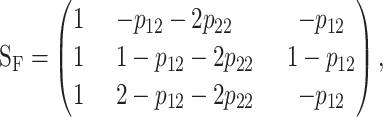 |
(4) |
where p11, p12, and p22 are the genotypic frequencies. This expression is derived in appendix b and its (generally fictitious) reference point is R = p11G11 + p12G12 + p22G22. Expression (4) makes complete sense when R is the phenotype of any single genotype, e.g., G11 by setting p11 = 1, p12 = 0, p22 = 0. The extension to the general multilocus case is obtained by using the Kronecker product of the genetic-effect matrices of the single loci, as in (2).
The inverse of matrix (4) is
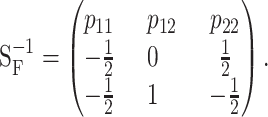 |
(5) |
This expression is very useful to inspect some particularities of the one-locus and multilocus NOIA functional formulations. By equating E in (1), the general expression of the genetic effects of the one-locus system is  . From this expression and (5) it becomes clear that the reference point is in fact R = p11G11 + p12G12 + p22G22, and that the genetic effects are always defined in the same way, regardless of the reference point used, as
. From this expression and (5) it becomes clear that the reference point is in fact R = p11G11 + p12G12 + p22G22, and that the genetic effects are always defined in the same way, regardless of the reference point used, as  (G22 − G11),
(G22 − G11), 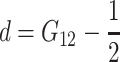 (G11 + G22). This is the same definition of genetic effects as in, for instance, Cockerham's F2 model (Zeng
et al. 2005). The general two-locus functional formulation of the NOIA model can be obtained by inserting two single-locus genetic-effect design matrices (4) in expression (2). In this expression (not shown), the frequencies at each locus affect the single-locus effects at the other locus. This is in accordance with the definition of epistasis—the effects of the allele substitutions at one gene depend on the genetic background. The (pairwise) epistatic effects in the two-locus case, on the other hand, are independent of the frequencies. This logic, only the highest-order effects being independent of the frequencies, extends to higher-order terms of epistasis when more loci are involved.
(G11 + G22). This is the same definition of genetic effects as in, for instance, Cockerham's F2 model (Zeng
et al. 2005). The general two-locus functional formulation of the NOIA model can be obtained by inserting two single-locus genetic-effect design matrices (4) in expression (2). In this expression (not shown), the frequencies at each locus affect the single-locus effects at the other locus. This is in accordance with the definition of epistasis—the effects of the allele substitutions at one gene depend on the genetic background. The (pairwise) epistatic effects in the two-locus case, on the other hand, are independent of the frequencies. This logic, only the highest-order effects being independent of the frequencies, extends to higher-order terms of epistasis when more loci are involved.
Translating genetic effects from one to another reference genotype:
Expressions (4) and (5) enable us to change the reference point from which to describe the genetic effects. Given a description of the genetic system from reference point R1, 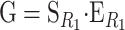 , and a description of the same genetic system from a different reference point R2,
, and a description of the same genetic system from a different reference point R2,  , it is straightforward to get to the expression
, it is straightforward to get to the expression
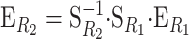 |
(6) |
by just inserting G from the first description into the second one and equating  . This expression is useful to change the reference of the genetic effects, i.e., to translate the genetic effects associated with a reference point to the genetic effects associated with a different reference point.
. This expression is useful to change the reference of the genetic effects, i.e., to translate the genetic effects associated with a reference point to the genetic effects associated with a different reference point.
When are the genetic effects of allele substitutions orthogonal?
The NOIA functional formulation is orthogonal for several populations, by just using the mean of these populations as a reference point of the model. These populations fulfill
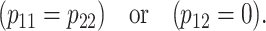 |
(7) |
This expression is derived in appendix c and its graphical interpretation is in the next section. For the populations fulfilling (7), the NOIA functional formulation is an orthogonal statistical formulation that can therefore be used to properly estimate genetic effects in QTL studies as justified by Yang (2004) and Zeng and collaborators (Kao and Zeng 2002; Zeng et al. 2005).
A general orthogonal–statistical model:
The explicit and general orthogonal [regardless of whether or not condition (7) holds] expression of the statistical one-locus genetic-effect design matrix, SS, is
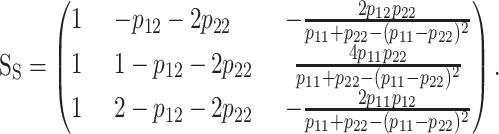 |
(8) |
The scalars of the SS matrix fulfill the conditions to be orthogonal scales sensu Cockerham (1954) (appendix c). The first two columns of the functional (4) and statistical (8) genetic-effect design matrices are the scalars of the reference point and the scales related to additive effects and are identical in the two formulations. The differences between the two one-locus formulations are in the third column, the scales for dominance. The expressions for these dominance orthogonal scales can be obtained by computing the values of the dominance deviations in the graphical interpretation (appendix c). In the same way as in the functional formulation, the general one-locus statistical formulation of the NOIA model (8) can be easily extended to a general multilocus case by taking the Kronecker product of single-locus genetic-effect design matrices (2). This resembles the way it has been done for particular cases of statistical formulations (Zeng et al. 2005). The statistical formulation (8) reduces to the functional one (4) whenever the conditions for orthogonality of the functional formulation (7) hold. The only exception is when the frequency of one of the genotypes is one, where the denominators in the third column of the statistical genetic-effect design matrix (8) are zero. This intuitively makes sense as no meaningful statistical formulation can be expected in a population in which only one genotype is present.
The inverse of matrix (8) is
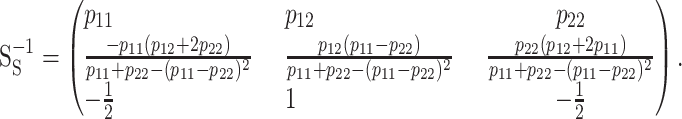 |
(9) |
Unlike in the general functional formulation (5), the additive effects, reflected in the second row of this inverse matrix, change depending on the allele frequencies in the population. This is a consequence of the parameters of the model no longer being natural effects of allele substitutions, but instead average effects of allele substitutions over the population. To clarify the difference between the meaning of the parameters in the statistical and the functional model formulations, we use ES = (μ, α, δ)T for the genetic effects vector in the one-locus statistical formulation instead of EF = (R, a, d)T that was used in the functional formulation (1). In ES we use μ for denoting the mean of the population as in other statistical epistasis models (e.g., Zeng et al. 2005). However, we prefer to denote the statistical genetic effects as Greek letters for making a clear distinction between statistical and functional genetic effects. The vector EF follows the notation of the unweighted regression model by Cheverud and Routman (1995) regarding the functional genetic effects, although we prefer to use R instead of C for the reference point (Cheverud 2000).
Taking into account this notation of the vectors of genetic effects, and interpreting the genetic-effect design matrices as statistical matrices instead of functional matrices, expression (6) holds for the statistical formulation, and therefore it enables us to translate statistical genetic effects of one population into how they would look in a different population. The statistical formulation of the NOIA model (8) can be used for estimating multilocus genetic effects in the exact same way as previous models (appendix c).
Obtaining functional estimates of genetic effects from statistical estimates:
Let us denote by SF and EF the genetic-effect design matrix and the vector of genetic effects in the functional formulation and by SS and ES the corresponding ones in the statistical formulation. In the one-locus case, the vectors of genetic effects are EF = (R, a, d)T and ES = (μ, α, δ)T. By implementing this notation in (1) we have G = SF · EF and G = SS · ES. Hence, the expressions for the transformations of genetic effects between the two formulations of the NOIA model are
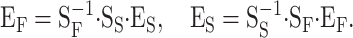 |
(10) |
These expressions resemble the translations of genetic effects between different reference points (6), but they have a different meaning. In fact, the transformations in (10) do not change the reference point of the system at all. Expressions for simultaneous translations of genetic effects, regarding both the reference point and the model formulation, can be easily obtained by combining expressions (6) and (10).
PREVIOUS MODELS AS PARTICULAR CASES OF NOIA
The F2 and the F∞ models:
One of the most commonly used populations for QTL analysis is the F2 population, ideally with genotype frequencies 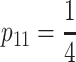 ,
,  ,
, 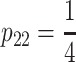 . The genetic-effects design matrix of the F2 model can be obtained by inserting the genotype frequencies of an ideal F2 population in the NOIA statistical formulation (8), and its reference point μ is, thus, the mean of an F2 population. For the multilocus case, the description of the system is obtained by first computing the correct genetic-effect design matrices for the individual loci and then computing the Kronecker product of the single-locus genetic-effect design matrices, as shown in (2). The F∞ model, which is orthogonal for—and thus adapts to the mean of—a population with frequencies
. The genetic-effects design matrix of the F2 model can be obtained by inserting the genotype frequencies of an ideal F2 population in the NOIA statistical formulation (8), and its reference point μ is, thus, the mean of an F2 population. For the multilocus case, the description of the system is obtained by first computing the correct genetic-effect design matrices for the individual loci and then computing the Kronecker product of the single-locus genetic-effect design matrices, as shown in (2). The F∞ model, which is orthogonal for—and thus adapts to the mean of—a population with frequencies  , p12 = 0,
, p12 = 0, 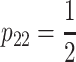 , is also a particular case of the general NOIA statistical formulation that can be explicitly obtained in the same way as explained for the F2 model above. One unsurprising remark about the F∞ population is that it fails in offering estimates of dominance effects, due to the absence of heterozygotes.
, is also a particular case of the general NOIA statistical formulation that can be explicitly obtained in the same way as explained for the F2 model above. One unsurprising remark about the F∞ population is that it fails in offering estimates of dominance effects, due to the absence of heterozygotes.
The G2A model:
Zeng et al. (2005) provided the genetic-effect design matrix of the G2A model for the one-locus case as
 |
(11) |
where p is the gene frequency of allele “1.” This model is a statistical formulation of genetic effects that is orthogonal for populations under Hardy–Weinberg proportions. From (8), we can obtain a genetic-effect design matrix for a population under Hardy–Weinberg as a particular case of the NOIA statistical formulation. This reads
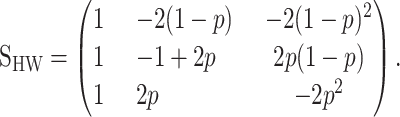 |
(12) |
Matrices (11) and (12) differ only in the sign of the values of their second columns. This sign difference occurs because Zeng et al. (2005) assume in all their models—following the notation by Falconer and MacKay (1996)—that allele “2” always leads to a lower genotypic value than allele 1. Therefore their estimates reflect the absolute value of the additive effects, by reporting the positive decrement of the genotypic value of an allele 1 to allele 2 substitution. In principle, this fits to the context of a QTL mapping experiment, in which allele 1 comes from the high line and allele 2 comes from the low line. It is, however, not a generally consistent formulation. Transgressive alleles are known to exist (Tanksley 1993) and in an extension of the model to several loci with epistasis, the effect of the alleles could switch signs at two different genetic backgrounds—a phenomenon known as sign epistasis (Weinreich et al. 2005). In such situations, the estimates of genetic effects from the G2A model (Equation 11) remain positive when the allele substitutions decrease the genotypic value, and they become negative when they increase it. On the contrary, the Hardy–Weinberg statistical formulation we obtain as a particular case of NOIA (Equation 12) is consistent with the direction of the allele substitution, as NOIA always leads to adding values that are positive when they increase the genotypic values from allele 1 and negative when they decrease them.
The unweighted regression model:
Cheverud and Routman's (1995) unweighted regression model (see also Cheverud 2000; Zeng
et al. 2005) is a particular case of NOIA in which 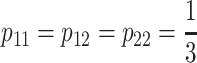 for each locus. Since—as well as for the F2 and the F∞ models—these frequencies fulfill criterion (7), the unweighted regression model can be considered as a particular case of both the functional (Equation 4) and the statistical (Equation 8) formulations of NOIA. The reference point of this model is the unweighted mean of the genotypic values of all genotypes, R = (1/3)G11 + (1/3)G12 + (1/3)G22 and the definition of genetic effects is the same as in the F2 model, as explained in relation to expression (5).
for each locus. Since—as well as for the F2 and the F∞ models—these frequencies fulfill criterion (7), the unweighted regression model can be considered as a particular case of both the functional (Equation 4) and the statistical (Equation 8) formulations of NOIA. The reference point of this model is the unweighted mean of the genotypic values of all genotypes, R = (1/3)G11 + (1/3)G12 + (1/3)G22 and the definition of genetic effects is the same as in the F2 model, as explained in relation to expression (5).
GRAPHICAL INTERPRETATION OF NOIA
Ideograph representing one-locus functional and statistical formulations:
The main foundations of the NOIA model are presented in the ideograph in Figure 1. The functional (Equation 4) and the statistical (Equation 8) formulations of genetic systems are represented by solid and shaded lines, respectively. The arrows pointing to the right or up represent the way in which the model is developed in the previous section of this article. Starting from a single genotype, G11, as a reference point, the model can be extended to a general functional formulation (solid line). Whenever criterion (7) holds—to the left of the vertical dashed line—the functional formulation is also an orthogonal–statistical formulation (shaded line). The F2 model is one example of such a model. An orthogonal–statistical formulation exists also when criterion (7) does not hold—to the right of the dashed line. The HW3 population (see below) is an example of this other case. The genetic effects can be easily translated between models by using the change-of-reference and transformation tools (6) and (10). The arrows pointing down or to the left represent how we translate genetic effects in a numerical example below.
Figure 1.—

Ideograph showing the main foundations of the NOIA model for the one-locus case. The starting point is a description of the genetic effects as allele substitutions on the reference genotype G11 (solid circle). This description is extended to a general functional formulation (thick solid line) by means of the change-of-reference tool represented by the horizontal and the nearly horizontal arrows. The reference points for which criterion (7) holds are represented to the left of the vertical dashed line. For populations with those reference points as mean phenotype, the functional formulation is orthogonal, and it coincides with the statistical formulation (thick shaded line). Other reference points may be represented to the right of the vertical dashed line. For populations with those reference points as mean phenotype, the two formulations do not coincide, and the transformation tool, represented by the vertical arrow, can be used to transform the functional formulation into the statistical formulation and vice versa. The F2 and the HW3 populations, represented as labeled vertical bars on the functional and statistical formulations, are examples of these two situations (see text for details).
The statistical formulation and the decomposition of the genetic variance:
We provide a graphical interpretation of the parameters in the one-locus formulations of the NOIA model based on the classical linear least-squares regression of the genotypic values of a single locus on the gene content (Fisher 1918; see Figures 4.6 and 4.7 in Lynch and Walsh 1998). This is the graphical interpretation of the NOIA statistical formulation, in which the slope of the regression determines the additive values of the allele substitutions, and the dominance deviations—the dominance effects of allele substitutions—are the vertical distances from the regression line to the real values (Figures 2 and 3A). Together with the graphical interpretation of the average effects and the dominance deviations (αi and δij, which are directly related to the decomposition of the genetic variance), we provide the expressions that give those values as functions of the parameters of the NOIA functional formulation—Greek letters. The additive variance is the variance of the average effects, αi, and the dominance variance is the variance of the dominance deviations, δij (Cockerham 1954; Falconer and MacKay 1996; Lynch and Walsh 1998). The extension of this to several loci is straightforward. The additive-by-additive variance, for instance, is the variance of the additive-by-additive average effects, ααij, and these would be obtained in a multilocus genetic system as the products of the additive-by-additive genetic effects and the corresponding orthogonal scales in the multilocus genetic-effect design matrix.
Figure 2.—

Graphical interpretation of the parameters of the NOIA model for an F2 population in the one-locus case. The values of the parameters come from a regression of the genotypic values (G11, G12, G22) on the gene content. These genotypic values are represented as solid circles, and their size is determined by their frequency in the population. We show a case of strong overdominance because it allows us to better visualize the parameters of interest. The functional regression (thick solid line) is constrained to have the same slope as the (dashed) line through G11 and G22. The statistical regression (thick shaded line) is a weighted linear regression on the gene content, and under condition (7) it has the same slope as the line through G11 and G22. In this case, therefore, the functional and statistical regressions coincide. The elements of the S = (sij) matrix are the natural and the orthogonal scales, in the functional and the statistical model formulations, respectively. Latin letters are the functional genetic effects, and Greek letters are the statistical genetic effects. The reference point, R = 1.25, is represented by a triangle. It is the intercept of the regressions and it occurs at the average gene content, which in this case is one. It is the starting point from which to measure the additive effects (αI = ai). The deviations of the regression, the dominance deviations (δij = dij) would be zero if there were no dominance. We show their relationship to the parameters of the model.
Figure 3.—

Graphical interpretation of the parameters of the NOIA model for an HW3 population, with frequencies p11 = 0.09, p12 = 0.42, p22 = 0.49, in the one-locus case. (A) Statistical formulation. All the symbols have the same meaning as in Figure 2. The regression on the gene content (thick shaded line) does not have the same slope as the (dashed) line through G11 and G22, and therefore it does not coincide with the functional regression of the same population (shown below). This happens when the frequencies of the population do not fulfill condition (7), as shown in the ideograph (above). The reference point, R = 1.33, occurs at gene content 1.4. (B) Functional formulation. All the symbols have the same meaning as in Figures 2 and 3A. The regression on the gene content (thick solid line) is forced to be parallel to the (dashed) line through G11 and G22 and would have a different slope otherwise (see statistical regression of the same population above). However, the reference point is the same as in the statistical regression.
The criterion for overlapping functional and statistical formulations:
The slope of the regression in Figure 2 equals the slope of the line defined by the genotypic values of G11 and G22. This is a result of the regression being made on an ideal F2 population, where both homozygotes have the same weight in the regression, p11 = p22 (=  ), thus making criterion (7) hold. Consequently, the regression is parallel to the line through G11 and G22 only when the functional formulation of the NOIA model is orthogonal. In Figure 3A, the regression is made on what we label as an HW3 population, in which p1 = 0.3 and the Hardy–Weinberg proportions hold, thus leading to p11 = 0.09, p12 = 0.42, p22 = 0.49. In this case criterion (7) does not hold, and the slope of the regression differs from the line defined by G11 and G22, leading to a change in the additive values. In this particular case they even change signs.
), thus making criterion (7) hold. Consequently, the regression is parallel to the line through G11 and G22 only when the functional formulation of the NOIA model is orthogonal. In Figure 3A, the regression is made on what we label as an HW3 population, in which p1 = 0.3 and the Hardy–Weinberg proportions hold, thus leading to p11 = 0.09, p12 = 0.42, p22 = 0.49. In this case criterion (7) does not hold, and the slope of the regression differs from the line defined by G11 and G22, leading to a change in the additive values. In this particular case they even change signs.
A graphical interpretation of the functional formulation:
The NOIA functional formulation can also be interpreted as a regression on the gene content, albeit this is not a typical linear regression anymore. Here, the slope of the regression remains constant regardless of the allele frequencies. In particular, it always remains at the same value as in the cases in which it is orthogonal—i.e., the slope of the line defined by G11 and G22 (Figures 2 and 3B). This is actually the same slope as for an unweighted regression on the gene content. This constraint of the functional regression becomes apparent when comparing Figure 3A with 3B. Figure 3A represents the statistical formulation, showing a normal least-squares linear regression in an HW3 population, as defined above, which is not parallel to the line through G11 and G22. Figure 3B represents the functional regression (for the same population) that fits the data under the constraint of retaining the same slope as the regression in Figure 2, i.e., by being parallel to the line defined by G11 and G22. This constraint enables us to perform the regression in populations in which one only genotype is present; i.e., it allows us to use one single genotype as a reference point of the functional formulation. This is not possible for the statistical formulation, as already commented above in relation to expression (8). The equivalent parameters to αi and δij are, in the functional formulation, the additive effects of natural allele substitutions in individuals, ai, and the deviations from those, dij.
NUMERICAL EXAMPLE
Here we apply the NOIA model to estimates from a QTL analysis on simulated data by Zeng et al. (2005). The data consist of a single trait controlled by three biallelic loci with defined underlying additive and dominance effects and pairwise gene interactions. Two populations are simulated: an F2 population and a population (HW347 or H as in subscripts) where the three loci follow Hardy–Weinberg proportions, with the frequencies of the 2 alleles being 0.3, 0.4, and 0.7 (see Tables 3 and 5 in Zeng et al. 2005). Zeng et al. (2005) report different estimates of genetic effects for the same trait in the F2 and HW347 simulated populations, explained by the fact that they report statistical estimates representing the average effects of allele substitutions in the two populations. Those estimates are indeed properties of the populations as well as of the underlying genetic system.
The statistical formulation of NOIA in QTL analysis:
The statistical formulation of NOIA can be used in QTL analysis in the same way as other statistical models of epistasis (appendix c). Here we show how to use NOIA to translate statistical estimates, as they come from the analysis of experimental data, into what would come from other experimental designs and into estimates of functional epistasis. Let us first consider the estimates of genetic effects Zeng
et al. (2005) obtained for a HW347 simulated population using the G2A model as a starting point. Following the logic of expressions (6) and (10), from G
=
SG2A · EG2A and G
=
SHW · EHW, we obtain 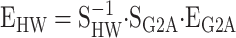 to translate the G2A estimates into what they would have been if the statistical formulation of NOIA had been used instead. The simulated populations in Zeng
et al. (2005) consisted of 100,000 individuals, meaning that the random departures from the Hardy–Weinberg proportions are certainly negligible and the G2A model is, thus, virtually orthogonal in the population under study. The only differences between the G2A estimates and the NOIA estimates are, therefore, the signs of the additive effects (as illustrated when obtaining the G2A model as a particular case of NOIA). This can be seen in the first row of Table 1—the genetic effects obtained from Zeng
et al. (2005) are all positive.
to translate the G2A estimates into what they would have been if the statistical formulation of NOIA had been used instead. The simulated populations in Zeng
et al. (2005) consisted of 100,000 individuals, meaning that the random departures from the Hardy–Weinberg proportions are certainly negligible and the G2A model is, thus, virtually orthogonal in the population under study. The only differences between the G2A estimates and the NOIA estimates are, therefore, the signs of the additive effects (as illustrated when obtaining the G2A model as a particular case of NOIA). This can be seen in the first row of Table 1—the genetic effects obtained from Zeng
et al. (2005) are all positive.
TABLE 1.
E vectors of the NOIA functional and statistical formulations of a genetic system from different reference points
| Vector of genetic effects, Ea |
|||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Caseb | R | μ | aA | αA | dA | δA | aB | αB | dB | δB | aC | αC | dC | δC | aaAB | ααAB | adAB | αδAB | daAB | δαAB | ddAB | δδAB | aaAC | ααAC | adAC | αδAC | daAC | δαAC | ddAC | δδAC | aaBC | ααBC | adBC | αδBC | daBC | δαBC | ddBC | δδBC |
| HW347S | −0.16 | −0.42 | 0.70 | −0.67 | 0.84 | −2.10 | 1.50 | 0.48 | −0.60 | −0.80 | 1.00 | 0.84 | −0.60 | −1.40 | 1.00 | 1.12 | −0.80 | −1.40 | 1.00 |
| HW347F | −0.16 | −0.70 | 0.70 | −0.84 | 0.84 | −1.50 | 1.50 | 1.00 | −1.00 | −1.00 | 1.00 | 1.00 | −1.00 | −1.00 | 1.00 | 1.00 | −1.00 | −1.00 | 1.00 |
| F2 | 0.00 | −1.00 | 1.00 | −1.00 | 1.00 | −1.00 | 1.00 | 1.00 | −1.00 | −1.00 | 1.00 | 1.00 | −1.00 | −1.00 | 1.00 | 1.00 | −1.00 | −1.00 | 1.00 |
| G111111 | 2.25 | −2.00 | 2.00 | −2.00 | 2.00 | −2.00 | 2.00 | 1.00 | −1.00 | −1.00 | 1.00 | 1.00 | −1.00 | −1.00 | 1.00 | 1.00 | −1.00 | −1.00 | 1.00 |
Statistical genetic effects (Greek letters) apply to HW347S, functional genetic effects (Latin letters) apply to HW347F and G111111, and both apply to F2.
HW347S and HW347F are the statistical and functional estimates of the parameters for the simulated population of Zeng et al. (2005). F2 are the corresponding joint statistical and functional estimates for an F2 population we obtained from the previous ones using the NOIA model, and they are identical to those obtained by Zeng et al. (2005) when analyzing a simulated F2 population, except for some sign differences (as justified in the text). G111111 are the functional estimates described using the phenotypic value of the genotype “111111” as a reference point. See text for details.
Transformation from statistical into functional estimates:
In the NOIA model, the statistical genetic effects of HW347 can be transformed into functional genetic effects (“HW347F,” second row in Table 1), as depicted in the ideograph by the arrow labeled “transformation” in Figure 1. This is done using (10), which for this particular case becomes  , where H represents HW347, F represents functional, and S represents statistical in the subscripts. The genetic-effect design matrices needed for the operation are the Kronecker products of the matrices for the individual loci as in (2) and as in (B10) in appendix b. A, B, and C are the three biallelic loci affecting the trait and the frequencies of the A2, B2, and C2 alleles are qA = 0.3, qB = 0.4, and qC = 0.7. Thus, we have
, where H represents HW347, F represents functional, and S represents statistical in the subscripts. The genetic-effect design matrices needed for the operation are the Kronecker products of the matrices for the individual loci as in (2) and as in (B10) in appendix b. A, B, and C are the three biallelic loci affecting the trait and the frequencies of the A2, B2, and C2 alleles are qA = 0.3, qB = 0.4, and qC = 0.7. Thus, we have 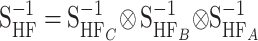 (utilizing that the Kronecker product is interchangeable with the inverse operation) and
(utilizing that the Kronecker product is interchangeable with the inverse operation) and 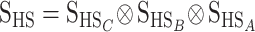 , where the matrices for the individual loci are derived using expressions (4) and (5) for the functional formulation and (8) and (9) for the statistical formulation. The functional genetic effects in the resulting vector EHF are the effects of allele substitutions performed on a fictitious genotype whose genotypic value would be the mean of the HW347 population. Thus, the reference point of the functional description has not changed after the transformation from the statistical description. We change the reference to the genotypic value of a real individual below in this section.
, where the matrices for the individual loci are derived using expressions (4) and (5) for the functional formulation and (8) and (9) for the statistical formulation. The functional genetic effects in the resulting vector EHF are the effects of allele substitutions performed on a fictitious genotype whose genotypic value would be the mean of the HW347 population. Thus, the reference point of the functional description has not changed after the transformation from the statistical description. We change the reference to the genotypic value of a real individual below in this section.
Translating genetic effects into an ideal F2 population:
In the HW347 population, we do not consider the functional genetic effects as being meaningful per se. Here we use them as an intermediate step to compute—as depicted in one of the change-of-reference arrows in the ideograph (Figure 1)—the genetic effects as they would appear in an ideal F2 population (third row in Table 1). These calculations are done using (6), here taking the form 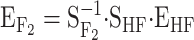 . The genetic-effect design matrices are computed as in (2) and (3), or more explicitly as in (B3) in appendix b, by
. The genetic-effect design matrices are computed as in (2) and (3), or more explicitly as in (B3) in appendix b, by 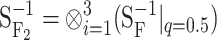 and
and  , where the matrices of the individual loci are again computed from (4) and (5). The vector
, where the matrices of the individual loci are again computed from (4) and (5). The vector  (third row in Table 1) gives the average effects of substitutions in an F2 population. These are, therefore, the values that would be obtained in a QTL experiment by means of the F2 model in an ideal F2 population. And, in fact, these values are the same values Zeng
et al. (2005) estimate from an F2 simulated population, built on the same genetic system (except for the sign differences in the genetic effects involving one additive effect, as explained above).
(third row in Table 1) gives the average effects of substitutions in an F2 population. These are, therefore, the values that would be obtained in a QTL experiment by means of the F2 model in an ideal F2 population. And, in fact, these values are the same values Zeng
et al. (2005) estimate from an F2 simulated population, built on the same genetic system (except for the sign differences in the genetic effects involving one additive effect, as explained above).
Genetic effects as allele substitutions on a particular genotype:
Here we use the NOIA model to obtain estimates at the reference point of the phenotypic value of a real genotype, G111111. In this way, the functional parameters get a direct genetic interpretation as natural effects of allele substitutions made on one particular individual. We shorten G111111 to read R1 in the subscripts, and hence expression (6) takes the form 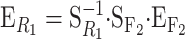 , where
, where 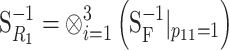 and
and  . The functional estimates of genetic effects as the natural effects of allele substitutions on the reference genotype G111111 (i.e., the resulting
. The functional estimates of genetic effects as the natural effects of allele substitutions on the reference genotype G111111 (i.e., the resulting  vector) are shown in the last row of Table 1. These can of course be easily transformed into natural effects of allele substitutions from any other reference genotype by means of another change of reference operation.
vector) are shown in the last row of Table 1. These can of course be easily transformed into natural effects of allele substitutions from any other reference genotype by means of another change of reference operation.
General remarks:
All cases in Table 1, except form HW347S in the first row, are either purely functional or both functional and statistical descriptions of a genetic system in which the highest level of epistasis present is pairwise epistasis. This is why, as pointed out above in relation to expression (5), all the genetic effects of the interactions remain constant throughout these cases. The additive and dominance effects, on the other hand, do not necessarily remain constant between cases in Table 1. The values of the genetic effects of a functional and a statistical description of the same population are different because of the different meaning of the parameters in the functional and the statistical formulations of the NOIA model. The values of the genetic effects of (functional or statistical) descriptions of the system from different reference points are different because the single-locus genetic effects depend on the genetic background—i.e., because of epistasis.
DISCUSSION
Conceptualizing and unifying functional and statistical epistasis:
In our opinion, the use of the concepts of statistical epistasis and functional epistasis has sometimes been misleading. Models of statistical epistasis were developed for performing orthogonal decompositions of variance in populations and for QTL detection and estimation. On the other hand, models of functional epistasis—also called physiological epistasis, biological epistasis, and genetical epistasis—were proposed to better analyze the role of gene interactions in evolution and to understand the genetic architecture underlying multifactorial disease, but its relationship to statistical epistasis has not been entirely explored. The NOIA model provides the necessary theory to unite these concepts and allows us to gain new insights into how epistasis should be modeled and inspected. In the light of the two formulations of our model, the terms functional epistasis and statistical epistasis can be viewed as two shadows cast from one object. We have characterized the situations under which these two shadows completely coincide and developed tools to transform them into each other when they do not. Statistical formulations of genetic systems are built on orthogonal parameters that represent average effects of allele substitutions over populations, whereas functional formulations of genetic systems are genotype–phenotype maps built on parameters that represent natural effects of allele substitutions on real or fictitious genotypes. Both of them are core models of genetic effects (gene effects and gene interactions) that describe the genetic architecture underlying a trait in two different ways and that are, therefore, suitable for different purposes.
Since NOIA overcomes the duality of functional and statistical models of epistasis, it enables us to obtain estimates of both functional and statistical genetic effects from data. The NOIA statistical formulation achieves orthogonality regardless of the genotype frequencies in the population and is therefore convenient for QTL detection and estimation and for an orthogonal decomposition of the genetic variance. The NOIA model is implemented with a tool to transform those orthogonal estimates into functional estimates. When expressed from the mean of the population under study, these functional estimates represent effects of allele substitutions performed on a fictitious genotype. Using the change-of-reference tool of the NOIA model, the reference point of the functional formulation can be changed to any real genotype, and therefore the NOIA model handles natural effects of allele substitutions on those genotypes, which is the genuine point of functional models. All these possibilities are represented in Table 1 as the result of a numerical example that illustrates the practical use of the theory provided within this article. The transformations in Table 1 can be explained using the classical concepts of cell means and factor effects (Searle 1971; Coffman et al. 2005). Indeed, expressions (6) and (10) are based on the fact that the genetic values (cell means) remain constant and they can therefore be used for linking and translating between genetic (factor) effects that entail different interpretations (statistical, functional, and both from different reference points).
The NOIA statistical formulation:
The statistical formulation of the NOIA model is an explicit, orthogonal description of multilocus two-allele models. Previous statistical epistasis models can thus be obtained as particular cases of NOIA. Orthogonality is a key property for statistical epistasis models to be appropriate for QTL analysis methods based on model selection. The F∞ model, for instance, lacks this property in commonly used experimental populations (Kao and Zeng 2002; Yang 2004; Zeng
et al. 2005). The classical F2 model, on the other hand, is orthogonal in ideal F2 populations in which the frequencies are 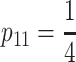 ,
, 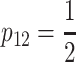 ,
, 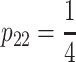 . However, in QTL studies there are always deviations from these genotype frequencies due to sampling errors, leading to a number of problems related to QTL detection and estimation as thoroughly pointed out by Yang (2004) and by Zeng and collaborators (Kao and Zeng 2002; Zeng
et al. 2005). These problems involve a bias in the estimates of genetic effects that will dramatically increase whenever segregation distortion affects at least one of the loci of the genetic system. The generality of the NOIA statistical formulation allows us to describe gene interactions of multilocus genetic systems in populations regardless of the gene frequencies of the alleles at the loci affecting the trait under study, thus avoiding the bias caused by sampling errors and segregation distortion. Furthermore, by changing the reference of the orthogonal–statistical estimates to a common reference point in NOIA, it is possible to compare the estimates of genetic effects coming from different QTL experiments affected by specific sampling errors or carried out using different experimental designs. This is an original feature of NOIA and we have proved its validity and accuracy by successfully transforming genetic effects between two simulated populations with different genotype frequencies but the same underlying genetics (Table 1).
. However, in QTL studies there are always deviations from these genotype frequencies due to sampling errors, leading to a number of problems related to QTL detection and estimation as thoroughly pointed out by Yang (2004) and by Zeng and collaborators (Kao and Zeng 2002; Zeng
et al. 2005). These problems involve a bias in the estimates of genetic effects that will dramatically increase whenever segregation distortion affects at least one of the loci of the genetic system. The generality of the NOIA statistical formulation allows us to describe gene interactions of multilocus genetic systems in populations regardless of the gene frequencies of the alleles at the loci affecting the trait under study, thus avoiding the bias caused by sampling errors and segregation distortion. Furthermore, by changing the reference of the orthogonal–statistical estimates to a common reference point in NOIA, it is possible to compare the estimates of genetic effects coming from different QTL experiments affected by specific sampling errors or carried out using different experimental designs. This is an original feature of NOIA and we have proved its validity and accuracy by successfully transforming genetic effects between two simulated populations with different genotype frequencies but the same underlying genetics (Table 1).
Yang's (2004) genetic effects model can, like NOIA, deal with departures from the Hardy–Weinberg proportions, but his model is explicitly developed only for the two-locus case, whereas NOIA is not constrained regarding the number of loci. The epistasis model of Wang and Zeng (2006) is particularly focused on the decomposition of the genetic variance. Their model is more general than the current NOIA statistical formulation regarding the number of alleles and the computation of genetic covariances due to linkage disequilibrium. However, this model is valid only for populations under strict Hardy–Weinberg proportions and not developed using the convenient algebraic notation that simplifies the computation of the model for the particular population under study. This notation (together with the generality regarding genotype frequencies) allows us, in particular, to implement in NOIA a tool to translate, and therefore to compare, statistical estimates of genetic effects, as explained above. Finally, Wang and Zeng's (2006) model, the F2 model, and the G2A model do not provide a link between statistical epistasis and functional epistasis, which is the main motivation for the NOIA model.
The NOIA functional formulation:
The algebraic structure of the NOIA functional formulation resembles the statistical formulation but instead of being based on average effects of allele substitutions in populations, it uses natural (nonaverage) effects of allele substitutions as parameters. The graphical interpretation of these parameters is also akin to the classical linear regression of the genotypic values on the gene content that defines the average effects (see Figures 2 and 3B). The connection we provide between the functional and the statistical formulations enables us to feed the first one with estimates of genetic effects obtained by means of QTL mapping studies on biallelic systems, as explained in the text and illustrated by means of the numerical example. Several studies have analyzed general key properties of gene interactions using functional epistasis models. Hansen and collaborators, for instance, have found directionality of gene interactions to determine the way in which short- and long-term genetic architecture evolves in the face of selection (Carter et al. 2005; Hansen 2006; Hansen et al. 2006). The NOIA model enables us now to study directionality in particular traits of particular populations, by using just data on orthogonal gene interactions from QTL studies and transforming them into functional estimates in which directionality can be inspected.
Cheverud and Routman (1995; Cheverud 2000) made a challenging attempt in the direction of linking statistical and functional (physiological) epistasis. Their unweighted regression model can be understood as a simultaneously functional and statistical description of genetic effects for a specific reference point and can be obtained as a particular case of NOIA. However, their model is not implemented with a change-of-reference tool, which causes two major practical problems. First, as a statistical model of epistasis, it is only orthogonal (and therefore appropriate for QTL detection and estimation) in populations in which every single genotype is present in the same quantity. Second, as a functional model, it cannot deal with natural effects of allele substitutions in real genotypes. In addition, several errors in the use and interpretation of the unweighted regression model have been pointed out (Zeng et al. 2005). Hansen and Wagner's (2001b) and Barton and Turelli's (2004) functional epistasis models do incorporate change-of-reference tools. The first one is formulated for multiple alleles and for constrained gene effects and interactions and the second one, like the current NOIA formulation, is a general formulation for two alleles. We find the algebraic notation of the NOIA functional formulation to be an advantage over these functional epistasis models. It is in fact by means of a parallel notation in the functional and the statistical formulations of the NOIA model that we developed both a graphical interpretation of functional epistasis and a transformation tool that enables us to feed the NOIA formulation with estimates of genetic effects from real data.
Future extensions of NOIA:
As discussed above, the theoretical framework of the NOIA model presents considerable advantages over the previous formulations of epistasis, in particular in analysis of real QTL experiments. Consequently, we are in the process of implementing NOIA in the context of QTL interval mapping with Haley–Knott regressions (Haley and Knott 1992). We also aim to extend NOIA to multiple alleles and linkage disequilibrium, this last implementation motivated by the fact that even for unlinked loci, there is nonrandom association of alleles due to sampling in the experimental populations used in QTL mapping, resulting in biased estimates.
Closing perspective:
The formal framework we propose in this article—together with the implementations we currently pursue—comprises theoretical developments and conceptual elucidations on the mathematical description of the genetic effects underlying a trait. Such a fundamental framework is reflected in graphical interpretations analogous to the classical regressions on the gene content provided by Fisher (1918) and will aid in the study of epistasis at different levels, including the role of epistasis in evolution, the response to selection in animal and plant breeding programs, and the analysis of multifactorial disease. Marker-assisted selection is a promising strategy for improving selection response for traits that are difficult to measure in individuals used for breeding or that manifest themselves late in life. The efficiency of marker-assisted selection relies on the precision with which estimates of genetic effects of individual or combinations of loci obtained in one genetic background can predict their effect in another. The generality of the NOIA model as well as its transformation and change-of-reference tools can allow the breeders to estimate the genetic effects in one experimental design and use these estimates to predict the effect of the same locus or loci in a particular genotype of a breeding individual or an average effect in any breeding population. This cannot be done with the currently available models. Another example where the NOIA model will fundamentally change the way science could proceed is in the mapping of loci underlying multifactorial disease. For example, we are on the verge of performing massive association studies on a grand scale. In these studies, deviations from ideal population conditions include sampling errors, segregation distortion, linkage disequilibrium, and (when the association studies are based on haplotypes) multiple alleles. The aim of these studies is to statistically detect loci affecting disease, but to functionally predict the effects of allele substitutions on an individual genotype basis to be able to suggest appropriate treatments or develop treatment regimes. The currently available models are far from suitable for this purpose, whereas the NOIA model is designed to do just this.
Acknowledgments
The authors thank Lars Rönnegård and Thomas Hansen for fruitful discussion. Örjan Carlborg acknowledges funding from the Knut and Alice Wallenberg Foundation.
APPENDIX A: THE NOIA MODEL FOR THE GENERAL MULTILOCUS CASE
To clarify the details of the general multilocus case, we first deal separately with the different components of the model, the G and E vectors and the S matrix, and then combine them using an example. Although we use a functional formulation in the example, the guidelines for constructing G, E, and S are valid for both the functional and the statistical formulations.
The vector of genotypic values, G:
The way in which the scalars of the vector G are sorted can be obtained by means of the Kronecker product of the vectors of the single-locus genotypic vectors, in which we then substitute the products of single-locus genotypic values by the correspondent multilocus genotypic values, for instance,  (
( by
by  , or simply G1112). It is worth stressing that the Kronecker product of subsequent loci added to the genetic system must be computed to the left of the previous ones, as shown in the example below, which makes the vector expand downward as new loci are considered.
, or simply G1112). It is worth stressing that the Kronecker product of subsequent loci added to the genetic system must be computed to the left of the previous ones, as shown in the example below, which makes the vector expand downward as new loci are considered.
The vector of genetic effects, E:
This is obtained in a similar way as the G vector, by the Kronecker product of single-locus genetic effects vectors. In this case, we first replace the reference point by a one in the single-locus vectors and next compute the Kronecker product of the subsequent loci to the left of the previous ones. Then, to obtain E from the resulting vector, we just replace the products of the genetic effects by the corresponding interactions, for instance, dB (aA by adAB or by just ad in the two-locus case), and the first scalar of the vector, which shall be one, by the reference point R. Greek letters are used instead of the Latin letters in the statistical formulation. As was the case for G, to add new loci makes the vector E expand downward.
The genetic-effect design matrix, S:
Once the single-locus genetic-effects design matrices are expressed at the desired single-locus reference point, the multilocus S matrix for the complete system can be obtained as the Kronecker product (for subsequent loci, to the left of the previous ones) of the single loci, as already explained in the text using expressions (2) and (3) and also in appendix b using (B3). We could also describe the system by multiplying the S matrices of subsequent loci to the right of the previous ones. In this case the vectors of genotypic values and genetic effects would need to be sorted in a different way, in which the new scalars that appear due to considering new loci would have to be inserted before the previous ones, instead of afterward.
Example:
Here we develop an example of a functional formulation using a real genotype as a reference point. Let us consider the simplest multilocus case, consisting of two loci, A and B, as in expression (2). This example deals with a very similar case to this expression, 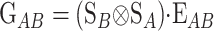 , the only difference being that there we assumed that both genetic-effect design matrices came from expression (1), hence leading to
, the only difference being that there we assumed that both genetic-effect design matrices came from expression (1), hence leading to  or simply G1111, as reference, whereas in this example we use as a reference the phenotypic value G1112 instead. We follow the same order as above, and therefore we begin by building the vector of genotypic values:
or simply G1111, as reference, whereas in this example we use as a reference the phenotypic value G1112 instead. We follow the same order as above, and therefore we begin by building the vector of genotypic values:
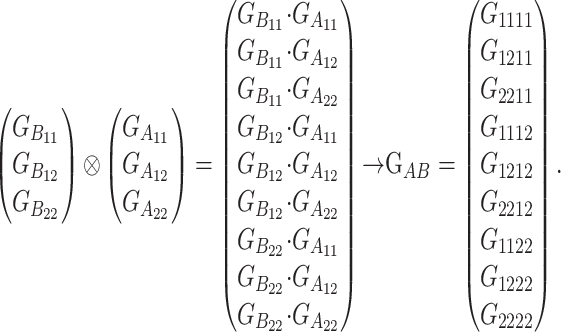 |
(A1) |
Next, the vector of genetic effects is obtained in a similar way:
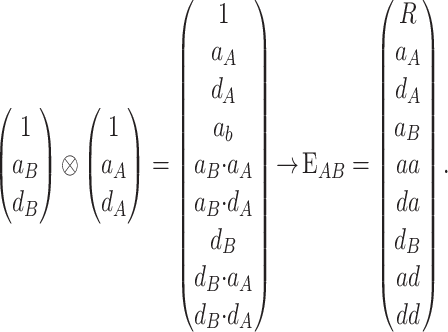 |
(A2) |
Here we use Latin letters, as in the functional formulation, but it works exactly the same for the statistical formulation, in which Greek letters are used instead. (A1) and (A2) are not the common ways in which the genotypic values and the genetic effects are sorted (see Table 1), but we find it convenient to use this configuration in our model for two main reasons. First, this way the vectors just extend downward whenever new loci are added to the genetic system. Second, it allows for a straightforward computation of the genetic-effect design matrix of the system, in which no rearrangement of its rows or columns is needed after computing the Kronecker product, as shown below. The single-locus genetic-effect design matrices for loci A and B are SA =  and SB =
and SB =  and are given in (1) and in (B7) in appendix b, respectively. Therefore, the two-locus genetic-effect design matrix is, by computing just the Kronecker product of these two matrices,
and are given in (1) and in (B7) in appendix b, respectively. Therefore, the two-locus genetic-effect design matrix is, by computing just the Kronecker product of these two matrices,
 |
(A3) |
Here we can observe how the Kronecker product defines the natural scales of the gene interactions in a logical and structured manner. The scalars in boldface type in the SAB matrix come from multiplying the scalar in boldface type in the SB matrix—at the column of the reference point, R—times the SA matrix. The columns of the resulting submatrix have the same meaning as in the SA matrix: they are coefficients of R, aA, and dA for the homozygotes for the 1 allele at locus B. The scalars in italics in the SAB matrix come from multiplying the scalar in italics in the SB matrix—at the column of the additive effect, aB—times the SA matrix. The first column of the resulting submatrix has the same meaning as the scalar in italics in the SB matrix: it is the additive effect aB. The other two columns are the scalars (the natural scales) of the interactions of aB with the genetic effects in the second and third columns of the SA matrix: they are coefficients of aa and da for the same genotypes mentioned above. The interaction effect exists in an individual whenever the two interacting effects have nonzero natural scales in the one-locus matrices. This same logic applies to the other seven submatrices of dimension three in matrix SAB. The places in which the natural scales appear in the SAB matrix determine the way in which we sort the scalars of the G and E vectors.
From (A1), (A2), and (A3) we have GAB = SAB · EAB, which describes every genotypic value in a two-locus two-alleles genetic system as the result of a set of allele substitutions from the reference genotype G1112.
APPENDIX B: THE CHANGE-OF-REFERENCE OPERATION
Here we go into the details of the change-of-reference operation, and we also present some expressions for the transformation of genetic-effect design matrices between the two formulations of the NOIA model. The change-of-reference operation consists of computing the S matrices that lead to describing the genetic system from any reference point. These matrices take the general form (4), as derived in this appendix, and can be used to translate genetic effects both between and inside model formulations by means of (6) and (10). We first describe in detail the change-of-reference operation of the functional formulation and prove that it is transitive. Then we illustrate the logic behind this operation using an example. Finally we obtain algebraic expressions for the change-of-reference operation of the statistical formulation.
The functional change-of-reference operation:
Recall that we have described a one-locus biallelic genetic system using G11 as a reference (Equation 1). Now, the genetic-effect design matrix that leads to a reference point R2 = p11G11 + p12G12 + p22G22,  , can be obtained from the genetic-effect design matrix for any other reference point R1,
, can be obtained from the genetic-effect design matrix for any other reference point R1, 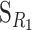 , as
, as
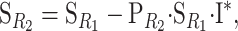 |
(B1) |
where the asterisk means that the first scalar of the identity matrix has been replaced by a zero, and  is the change-of-reference matrix for the reference point R2, a square matrix in which each column is filled with one of the coefficients of the linear combination of genotypes that equals the new reference:
is the change-of-reference matrix for the reference point R2, a square matrix in which each column is filled with one of the coefficients of the linear combination of genotypes that equals the new reference:
 |
(B2) |
It is worth pointing out that we consider only the cases in which p11 + p12 + p22 = 1, so that the scalars of the matrix can be interpreted as frequencies of the genotypes in a population. We show below in this appendix that the change-of-reference operation (B1) consistently leads to the same  matrix, independently of the starting reference point R1 and, immediately afterward, we use an example to illustrate the logic that led us to this operation.
matrix, independently of the starting reference point R1 and, immediately afterward, we use an example to illustrate the logic that led us to this operation.
We obtained expression (4) by performing a change-of-reference operation as shown in (B1), with  =
=  as in (1) and without specifying the values of the frequencies in the change-of-reference matrix
as in (1) and without specifying the values of the frequencies in the change-of-reference matrix 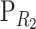 (B2). An extension to the general multilocus change-of-reference operation is straightforward. First the change of reference is performed separately for each locus, and then the S matrix of the complete system is obtained from taking the Kronecker product of the new single-locus reference matrices, in reverse order. For n loci this reads
(B2). An extension to the general multilocus change-of-reference operation is straightforward. First the change of reference is performed separately for each locus, and then the S matrix of the complete system is obtained from taking the Kronecker product of the new single-locus reference matrices, in reverse order. For n loci this reads
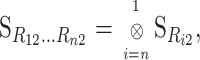 |
(B3) |
where Ri2 is the reference points of locus i, and 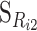 , i = 1, …, n can be obtained as in (B1).
, i = 1, …, n can be obtained as in (B1).
The transitive property:
For the change-of-reference operation to be consistent, the resulting matrix of the particular reference point must remain the same, independently of the starting point from which it is computed. To show this we now prove the transitive property for the operation in Equation B1.
Let R1, R2, and R3 be three reference points, and let 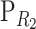 and
and 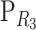 be the change-of-reference matrices of R2 and R3. We shall prove that changing the reference from R1 to R2, and afterward to R3, is the same as changing directly from R1 to R3; that is, given
be the change-of-reference matrices of R2 and R3. We shall prove that changing the reference from R1 to R2, and afterward to R3, is the same as changing directly from R1 to R3; that is, given
 |
(B4) |
and
 |
(B5) |
we want to prove that
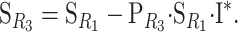 |
(B6) |
By inserting (B4) into (B5), we get
 |
Given that 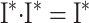 ,
,
 |
Since the scalars in each row in  sum to one, then the scalars in each row in (
sum to one, then the scalars in each row in ( − I) sum to zero. Since all the scalars in
− I) sum to zero. Since all the scalars in  are equal inside columns (say, λi at column i), all the scalars in column i of the matrix (
are equal inside columns (say, λi at column i), all the scalars in column i of the matrix (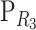 − I) ·
− I) ·  are 0(λi). Hence, the matrix (
are 0(λi). Hence, the matrix (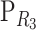 − I) ·
− I) ·  is the zero matrix, and therefore (B6) is proved.
is the zero matrix, and therefore (B6) is proved.
Example:
We now proceed to show how the change-of-reference tool works in the simple case of changing the reference from one homozygote to the heterozygote in a one-locus, two-allele genetic system, which clarifies the motivation for the operation (B1). Using (B2), the change-of-reference matrix is in this case the 3 × 3 matrix with all rows equal to (0, 1, 0). Therefore, (1) and (B1) give
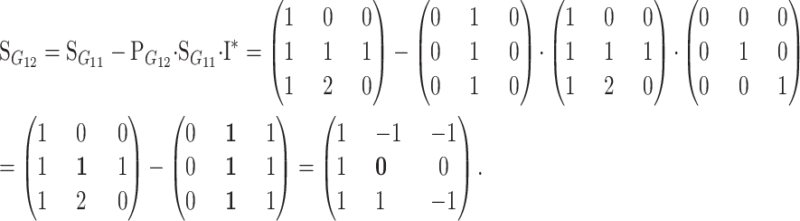 |
(B7) |
The new matrix,  , is the result of subtracting to
, is the result of subtracting to  , a matrix in which scalars are equal inside columns. The scalar of the first column is zero, so that
, a matrix in which scalars are equal inside columns. The scalar of the first column is zero, so that 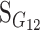 has a first column of ones as well as
has a first column of ones as well as  . The other scalars are the ones at the same column in the row of the new reference point in
. The other scalars are the ones at the same column in the row of the new reference point in  (which is the second row, the one of G12), so that these columns have zeros at the row of the reference point in the resulting matrix,
(which is the second row, the one of G12), so that these columns have zeros at the row of the reference point in the resulting matrix, 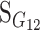 . We show this by using scalars in boldface type and italics in (B4). As expected,
. We show this by using scalars in boldface type and italics in (B4). As expected, 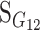 has zeros at the second and third positions of the second row, from which it can be deduced that G12 is the new reference point, and the rest of the scalars at those columns have been modified accordingly.
has zeros at the second and third positions of the second row, from which it can be deduced that G12 is the new reference point, and the rest of the scalars at those columns have been modified accordingly.
The statistical change-of-reference operation:
The genetic-effect design matrix of the multilocus statistical formulation can be obtained for every population as the Kronecker product of one-locus matrices (Equation 8). Nonetheless, and as a final point in this appendix, we derive an explicit algebraic expression to perform the change-of-reference operation in the statistical formulation of the NOIA model. To this end we first provide an explicit algebraic way of performing the transformation tool to obtain a genetic-effect design matrix of the functional formulation of the NOIA model, SF, from the genetic-effect design matrix of the statistical formulation, SS, and vice versa, in a way that resembles the functional change-of-reference tool (B1). In the one-locus case the expressions for performing those transformations are
 |
(B8) |
where the transformation matrices are
 |
(B9) |
From these expressions it follows that TFS = TSF = I whenever condition (7) holds, as expected. The extension to the multilocus case is, as in the functional change of reference, straightforward. First, the transformations have to be computed separately at each of the loci, using (B8) and (B9), and then the genetic-effect design matrix of the complete system is obtained from the Kronecker product of the single-locus matrices. That is, in an n-locus genetic system, the genetic-effect design matrix of the functional formulation, SF, can be obtained from the statistical formulation as
 |
(B10) |
where the subscript i stands for the locus and  , i = 1, …, n can be obtained as in (B8).
, i = 1, …, n can be obtained as in (B8).
We implement the reference points in the notation of the transformation tool as subscripts in (B8), and from that and (B1) we obtain
 |
(B11) |
This is the one-locus statistical change-of-reference operation from the reference point R1 to R2. The I* matrix and the change-of-reference matrix are as in (B1) and (B2). The Kronecker product of the single-locus matrices provides the extension to the multilocus (n-locus) case as in (B3), where for each locus i the matrices 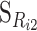 , at reference point Ri2, are obtained from matrices
, at reference point Ri2, are obtained from matrices  , at reference point Ri1, as in (B11).
, at reference point Ri1, as in (B11).
APPENDIX C: ORTHOGONALITY
Here we derive the orthogonal–statistical formulation of NOIA. To do so we first recall the standard regression model of genetic effects and explain when it is orthogonal. Then we show how we obtained criterion (7) for the orthogonality of the functional formulation of NOIA. Finally, we derive the generally orthogonal statistical formulation of NOIA from the classical regression of the genotypic values on the gene content.
The standard regression model of genetic effects:
We consider a one-locus two-allele genetic system like in (1). Let us assume that we have information about n individuals. This information consists of their observed phenotypic value for a trait and their genotype at the locus controlling the trait. We call G* the vector of the observed phenotypes. Ideally, those observations would perfectly fit the genotypic values of their genotypes. This can be expressed in an algebraic way as G* = Z · G,
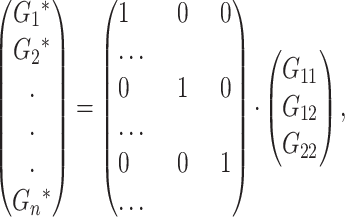 |
(C1) |
where the n rows of the matrix Z reflect the genotype of the corresponding observed phenotypes in G*.
Now let us consider a genetic-effect design matrix S, e.g., matrix (8). Since G = S · E, from (C1) we have G* = Z · S · E. We call X = Z · S and thus the standard regression of genetic effects takes the form
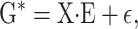 |
(C2) |
where ε is the vector of errors. This is the way in which the NOIA statistical formulation is used for estimating one-locus genetic effects. The extension to several loci is straightforward, by just extending G, S, and E as explained in appendix a and building the matrix Z accordingly to the particular number of loci to be considered. This can be done as a rowwise Kronecker product of the Z matrices of the single loci.
Orthogonal estimation of genetic effects:
For the estimates performed in (C2) to be independent of each other in a statistical sense, i.e., for them to be orthogonal, matrix X has to satisfy XT · X as a diagonal matrix. Given that the three genotype frequencies in the sample of n individuals are p11, p12, and p22, we obtain
 |
(C3) |
where
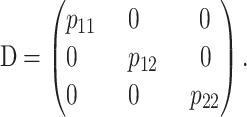 |
(C4) |
Thus, given that S = (sij) with si1 = 1, from (C3) and (C4) we obtain the criteria for orthogonality as derived by Cockerham (1954; Kao and Zeng 2002), which in our notation reads
 |
(C5) |
Orthogonality in NOIA:
The functional genetic-effect design matrix (4) fulfills the first of criteria (C5). From the remaining two conditions, some basic algebra leads to criterion (7), which characterizes the cases when expression (4) is orthogonal. For a generally orthogonal description of a one-locus genetic system, different dominance scales, si3, are needed.
To obtain the orthogonal dominance scales of NOIA we derived expressions for the dominance deviations of the classical regression of genotypic values on the gene content (Figure 1, B and C). We call N the number of “2” alleles (the gene content) and write the expression of this regression as
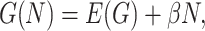 |
(C6) |
where
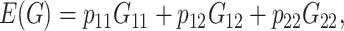 |
and
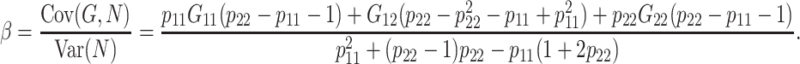 |
Note that the gene contents are N = 0 for G11, N = 1 for G12, and N = 2 for G22. Therefore, from (C6) and some algebra we compute the distances from the genotypic values to the values predicted by the regression as
 |
Finally, by just dividing these values by the dominance genetic effect δ = G12 −  (G11 + G22) (the value the dominance scales are coefficients for in the model) we get the dominance orthogonal scales in (8). Orthogonality of (8) can be tested from (C5) by just applying some basic algebra. Once the orthogonality of the one-locus formulation is proved, the orthogonal scales for the interactions in the multilocus case can be generated by the Kronecker product, as detailed in appendix a. The extension of the model using the Kronecker product guarantees the orthogonality of the multilocus formulations, as for the models presented by Zeng
et al. (2005).
(G11 + G22) (the value the dominance scales are coefficients for in the model) we get the dominance orthogonal scales in (8). Orthogonality of (8) can be tested from (C5) by just applying some basic algebra. Once the orthogonality of the one-locus formulation is proved, the orthogonal scales for the interactions in the multilocus case can be generated by the Kronecker product, as detailed in appendix a. The extension of the model using the Kronecker product guarantees the orthogonality of the multilocus formulations, as for the models presented by Zeng
et al. (2005).
References
- Barton, N. H., and M. Turelli, 2004. Effects of genetic drift on variance components under a general model of epistasis. Evolution 58 2111–2132. [DOI] [PubMed] [Google Scholar]
- Bateson, W., 1909. Mendel's Principles of Heredity. Cambridge University Press, Cambridge.
- Bürger, R., 2000. The Mathematical Theory of Selection, Recombination and Mutation. Wiley, Chichester, UK.
- Carlborg, O., and C. S. Haley, 2004. Epistasis: Too often neglected in complex trait studies? Nat. Rev. Genet. 5 618–625. [DOI] [PubMed] [Google Scholar]
- Carlborg, O., L. Jacobsson, P. Ahgren, P. Siegel and L. Andersson, 2006. Epistasis and the release of genetic variation during long-term selection. Nat. Genet. 38 418–420. [DOI] [PubMed] [Google Scholar]
- Carter, A. J., J. Hermisson and T. F. Hansen, 2005. The role of epistatic gene interactions in the response to selection and the evolution of evolvability. Theor. Popul. Biol. 68 179–196. [DOI] [PubMed] [Google Scholar]
- Cheverud, J. M., 2000. Detecting epistasis among quantitative trait loci, pp. 58–81 in Epistasis and the Evolutionary Process, edited by J. B. Wolf, E. D. Brodie and M. J. Wade. Oxford University Press, Oxford.
- Cheverud, J. M., and E. J. Routman, 1995. Epistasis and its contribution to genetic variance components. Genetics 139 1455–1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cockerham, C. C., 1954. An extension of the concept of partitioning hereditary variance for analysis of covariances among relatives when epistasis is present. Genetics 39 859–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coffman, C. J., R. W. Doerge, K. L. Simonsen, K. M. Nichols, C. K. Duarte et al., 2005. Model selection in binary trait locus mapping. Genetics 170 1281–1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobzhansky, T., 1936. Studies on hybrid sterility. II. Localization of sterility factors in Drosophila pseudoobscura hybrids. Genetics 21 113–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falconer, D. S., and T. F. C. MacKay, 1996. Quantitative Genetics. Prentice-Hall, Harlow, UK.
- Fisher, R. A., 1918. The correlation between relatives on the supposition of Mendelian inheritance. Trans. R. Soc. Edinb. 52 339–433. [Google Scholar]
- Fisher, R. A., 1958. The Genetical Theory of Natural Selection. Dover, New York.
- Goodnight, C. J., 1988. Epistasis and the effect of founder events on the additive genetic variance. Evolution 42 441–454. [DOI] [PubMed] [Google Scholar]
- Goodnight, C. J., 1995. Epistasis and the increase in additive genetic variance: implications for phase 1 of Wright's shifting-balance theory. Evolution 49 502–511. [DOI] [PubMed] [Google Scholar]
- Goodnight, C. J., 2000. Modeling gene interaction in structured populations, pp. 129–145 in Epistasis and the Evolutionary Process, edited by J. B. Wolf, E. D. Brodie and M. J. Wade. Oxford University Press, Oxford.
- Haley, C. S., and S. A. Knott, 1992. A simple regression method for mapping quantitative trait loci in line crosses using flanking markers. Heredity 69 315–324. [DOI] [PubMed] [Google Scholar]
- Hansen, T. F., 2006. The evolution of genetic architecture. Annu. Rev. Ecol. Evol. Syst. 37 123–157. [Google Scholar]
- Hansen, T. F., and G. P. Wagner, 2001. a Epistasis and the mutation load: a measurement-theoretical approach. Genetics 158 477–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen, T. F., and G. P. Wagner, 2001. b Modeling genetic architecture: a multilinear theory of gene interaction. Theor. Popul. Biol. 59 61–86. [DOI] [PubMed] [Google Scholar]
- Hansen, T. F., J. M. Álvarez-Castro, A. J. Carter, J. Hermisson and G. P. Wagner, 2006. Evolution of genetic architecture under directional selection. Evolution 60 1523–1536. [PubMed] [Google Scholar]
- Hermisson, J., T. F. Hansen and G. P. Wagner, 2003. Epistasis in polygenic traits and the evolution of genetic architecture under stabilizing selection. Am. Nat. 161 708–734. [DOI] [PubMed] [Google Scholar]
- Kao, C. H., and Z-B. Zeng, 2002. Modeling epistasis of quantitative trait loci using Cockerham's model. Genetics 160 1243–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kempthorne, O., 1954. The correlation between relatives in a random mating population. Proc. R. Soc. Lond. B Biol. Sci. 143 102–113. [PubMed] [Google Scholar]
- Lynch, M., and B. Walsh, 1998. Genetics and Analysis of Quantitative Traits. Sinauer, Sunderland, MA.
- Moore, J. H., 2005. A global view of epistasis. Nat. Genet. 37 13–14. [DOI] [PubMed] [Google Scholar]
- Moore, J. H., and S. M. Williams, 2005. Traversing the conceptual divide between biological and statistical epistasis: systems biology and a more modern synthesis. BioEssays 27 637–646. [DOI] [PubMed] [Google Scholar]
- Muller, H. J., 1942. Isolating mechanisms, evolution, and temperature. Biol. Symp. 6 71–125. [Google Scholar]
- Phillips, P. C., 1998. The language of gene interaction. Genetics 149 1167–1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Searle, S. R., 1971. Linear Models. Wiley, New York.
- Tanksley, S. D., 1993. Mapping polygenes. Annu. Rev. Genet. 27 205–233. [DOI] [PubMed] [Google Scholar]
- Templeton, A. R., 2000. Epistasis and complex traits, pp. 41–57 in Epistasis and the Evolutionary Process, edited by J. B. Wolf, E. D. Brodie and M. J. Wade. Oxford University Press, Oxford.
- Turelli, M., and N. H. Barton, 2006. Will population bottlenecks and multilocus epistasis increase additive genetic variance? Evolution 60 1763–1776. [PubMed] [Google Scholar]
- Wade, M. J., and C. J. Goodnight, 1998. Genetics and adaptation in metapopulations: when nature does many small experiments. Evolution 52 1537–1553. [DOI] [PubMed] [Google Scholar]
- Wade, M. J., R. G. Winther, A. F. Agrawal and C. J. Goodnight, 2001. Alternative definitions of epistasis: dependence and interaction. Trends Ecol. Evol. 16 498–504. [Google Scholar]
- Wagner, G. P., M. D. Laubichler and H. Bagheri-Chaichian, 1998. Genetic measurement of theory of epistatic effects. Genetica 102–103 569–580. [PubMed] [Google Scholar]
- Wang, T., and Z-B. Zeng, 2006. Models and partition of variance for quantitative trait loci with epistasis and linkage disequilibrium. BMC Genet. 7 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinreich, D. M., R. A. Watson and L. Chao, 2005. Perspective: sign epistasis and genetic constraint on evolutionary trajectories. Evolution 59 1165–1174. [PubMed] [Google Scholar]
- Wright, S., 1931. Evolution in Mendelian populations. Genetics 16 93–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright, S., 1977. Experimental Results and Evolutionary Deductions (Evolution and Genetics of Populations, Vol. III). University of Chicago Press, Chicago.
- Yang, R.-C., 2004. Epistasis of quantitative trait loci under different gene action models. Genetics 167 1493–1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng, Z-B., T. Wang and W. Zou, 2005. Modeling quantitative trait loci and interpretation of models. Genetics 169 1711–1725. [DOI] [PMC free article] [PubMed] [Google Scholar]