

Abstract
The recent development of Affymetrix chips designed from assembled EST sequences has spawned considerable interest in identifying single-feature polymorphisms (SFPs) from transcriptome data. SFPs are valuable genetic markers that potentially offer a physical link to the structural genes themselves. However, most current SFP prediction methodologies were developed for sequenced species although SFPs are particularly valuable for species with complex and unsequenced genomes. To establish the sensitivity and specificity of prediction, we explored four methods for identifying SFPs from experiments involving two tissues in two commercial barleys and their doubled-haploid progeny. The methods were compared in terms of numbers of SFPs predicted and their ability to identify known sequence polymorphisms in the features, to confirm existing SNP genotypes and to match existing maps and individual haplotypes. We identified >4000 separate SFPs that accurately predicted the SNP genotype of >98% of the doubled-haploid (DH) lines. They were highly enriched for features containing sequence polymorphisms but all methods uniformly identified a majority of SFPs (∼64%) in features for which there was no sequence polymorphism while 5% mapped to different locations, indicating that “SFPs” mainly represent polymorphism in cis-acting regulators. All methods are efficient and robust at predicting markers for gene mapping.
SEVERAL recent studies have explored the possibility of using transcript abundance data from cRNA hybridizations to Affymetrix microarrays (Affymetrix, Santa Clara, CA) to reveal genetic polymorphisms that can be used as markers to genotype individuals in mapping populations (Borevitz et al. 2003; Cui et al. 2005; Ronald et al. 2005; Rostoks et al. 2005a,b; West et al. 2006).
Each gene on an Affymetrix gene chip is typically represented by 11 different 25-bp oligos covering features of the transcribed region of that gene. Each of these features is present as a “so-called” perfect match (PM) and mismatch (MM) oligonucleotide. The PM exactly matches the sequence of a particular standard genotype, often one parent of a cross, while the MM differs from this in a single substitution in the central, 13th base. If two individuals differ in the amount of mRNA produced by the particular tissue under study, this should result in a relatively uniform difference in their hybridizations across the 11 features. Alternatively, if the two individuals produce the same amount of mRNA but contain a genetic polymorphism within their DNA that coincides with one particular feature (or features if they overlap), this may also give rise to differential hybridizations but now confined to that feature alone. Such differences have been termed single-feature polymorphisms (SFPs) (Borevitz et al. 2003). The third and probably most frequent possibility is that the individuals differ both in gene expression and in one or more feature polymorphisms. Thus, in principle, it is possible to explore both general expression effects and specific SFP polymorphisms using the same data set. The former could be the result of genetic polymorphism in that gene or in a trans- or cis-regulator that affects transcription, while the second is most likely, though not exclusively, due to polymorphism in the gene itself that affects hybridization success. Some of these expression differences and SFPs may be distributed bimodally in a population and hence can be “Mendelized” as genetic expression markers (GEMs) or SFP markers, respectively (West et al. 2006).
The ability to recognize SFPs reliably for a large number of genes opens up the possibility of carrying out expression QTL (eQTL) studies (“genetical genomics”) (Jansen and Nap 2001; Brem et al. 2002; Schadt et al. 2003; Morley et al. 2004; Mehrabian et al. 2005) while simultaneously genotyping “immortal,” biparental populations such as recombinant inbred or doubled-haploid lines (RILs or DHLs). Much of the previous work on genetical genomics has been based on sequenced and well-characterized model species such as yeast, mouse, and Arabidopsis (Brem et al. 2002; Borevitz et al. 2003; Bing and Hoeschele 2005; Bystrykh et al. 2005). In these cases the features on the chip are frequently based on the gene sequence of one of the parents while the physical and genetical chromosomal locations of these genes are precisely known. The location of eQTL can then be compared with the gene location to distinguish cis- and trans-regulated genes. Genetical-genomics approaches have also been applied to other species for which marker genotypes of the population (RILs, DHLs, etc.) are known, thus allowing eQTL studies to be performed (Jansen and Nap 2001; Alberts et al. 2005; DeCook et al. 2006). However, in such cases, the physical/genetical linkage relationships between the genes and the probe features on the array will not normally be known. The ability to map the features as SFPs and the eQTL using the same data set would be particularly valuable because it could provide this physical link between marker (SFP) and gene. Such studies could be complemented with additional, preexisting and mapped markers, providing potential causal links between markers and SFPs as well as anchoring SFPs to particular chromosomes. The comparisons between SFPs, eQTL, and QTL for conventional phenotypic traits provide a powerful route to identify QTL candidate genes and study gene networks.
Various methods have been proposed for identifying sequence polymorphisms. Winzeler et al. (1998) demonstrated that hybridization of labeled genomic DNA to oligonucleotide microarrays could identify sequence polymorphisms. The technique has been successfully employed to identify and genotype genetic markers across the whole genome of budding yeast (Brem et al. 2002; Steinmetz et al. 2002). Borevitz et al. (2003) succeeded in employing the approach to identify the polymorphism embedded in single probe sequences in more complex species such as Arabidopsis thaliana and termed them “single feature polymorphism.” The idea was extended to hybridize cRNA to expression microarrays for detecting SFP markers in barley (Cui et al. 2005; Rostoks et al. 2005a,b).
Ronald et al. (2005) were probably the first to use expression data assayed for a segregating population of budding yeast from Affymetrix microarrays to identify SFPs between two parental strains and to genotype the segregants at the SFPs. The approach has relied heavily on information about the match between probe and transcript sequences (for details, see materials and methods). Its use may thus be limited to the cases where transcript sequences are largely known by making use of a reference strain or variety in the analysis, such as in yeast and Arabidopsis. Second, some of the inferred SFPs may be due more to differential expression between two parental genotypes, i.e., GEMs (West et al. 2006), than to genuine sequence polymorphisms.
In this article we are concerned with the relative efficiencies of four different SFP prediction methods and explore the nature of the polymorphisms they are detecting. We develop two new statistical methods for identifying SFPs by modeling expression data from replicated Affymetrix microarrays on two commercial barley varieties Steptoe (St) and Morex (Mx) and for genotyping and mapping SFPs in a doubled-haploid population from a cross of these two parental lines. The new methods are compared with the approaches proposed by Ronald et al. (2005) for predicting SFPs by use of cRNA microarray data and that by Winzeler et al. (1998), which is appropriate for screening for sequence polymorphisms by use of genomic DNA microarray data. The expression analysis was performed on Affymetrix Barley1 chips using RNA taken from two tissues, seedling leaves and embryo-derived tissue from the germinating grain. We explore these four approaches to identify and map SFPs and test the reliability of our genotype predictions (i) using existing sequence information of the features in the parents of the cross, (ii) using SNP genotype information for the predicted polymorphic genes among the doubled-haploid (DH) lines, and (iii) by constructing DH graphical genotypes produced from mapping SFPs to compare with those from known SNPs. Our major aim is to define and validate a robust procedure for fast and reliable SFP genotyping in mapping populations that is appropriate to model organisms as well as agriculturally important but less tractable species. In this context, validation involves identifying the sources of the polymorphisms that are being recognized by each method.
MATERIALS AND METHODS
Mapping population:
We used mRNA from seedling leaves and embryo-derived tissue from germinating grains for expression profiling from 35 recombinant lines of a St × Mx doubled-haploid population (Kleinhofs et al. 1993). These lines (the “minimapper” set) were selected from a larger population of 150 DH lines on the basis of informative recombination events, allowing markers to be positioned evenly across all chromosomes. Of the 35 DH lines, 5 were removed for technical reasons explained in the discussion. The remaining 30 DH lines plus three replicates of each parent are referred to as the “trial set” of lines.
Plant material, RNA isolation, and GeneChip hybridizations:
Plant material was generated essentially as described previously (Druka et al. 2006) but with some modifications specific to these studies. To obtain seedling leaf tissue, 10 sterilized seeds per line were sown in each of three replicate 13-cm2 pots. One pot of every member of the trial set was randomized in each of three randomized blocks and each block was placed in a separate Snijders growth cabinet set at 17° with 16-hr light/12° 8-hr dark periods at a light intensity of 400 μE m−1 sec−1. After 12 days, leaves of 7–8 seedlings from each pot were collected, bulked, and flash frozen in liquid nitrogen; tissues from all three replicate pots of each line were bulked for RNA isolation. To obtain embryo-derived tissue from the germinating grain, 30–50 sterilized seeds per line of the trial set were germinated on a petri plate between three layers of wet 3-mm filter paper in the dark, for 16 hr at 17° and 8 hr at 12°, for 96 hr total. Embryo-derived tissue [mesocotyl, coleoptile, and seminal roots: 1.3-radicle and coleoptile emergence stage (GRO:0007236)] from three grains was dissected as a single tissue piece and flash frozen in liquid nitrogen. Germination and collection were repeated for all lines with complete randomization of the petri plates on each of three occasions. For each line, tissues from all three occasions were bulked before RNA isolation.
RNA was isolated, processed, and hybridized to the Barley1 GeneChip (complete description and references at http://www.affymetrix.com/products/arrays/specific/barley.affx Affymetrix product no. 900515 GeneChip Barley Genome Array), using previously described Trizol procedures (Caldo et al. 2004). The labeling, hybridization, and GeneChip data acquisition were conducted at the GeneChip facility at Iowa State University (http://www.biotech.iastate.edu/facilities/genechip/Genechip.htm). Forty-one Affymetrix Barley1 GeneChips were allocated to the trial set for each tissue. For simplicity these two tissues are referred to as “leaf” and “embryo” in the text.
Altogether there were 22,840 different probe sets on every chip. Each probe set was represented by 11 features (each of 25-bp oligos) present both as a perfect match (PM) and as a mismatch (MM), giving a total of 501,622 probe features.
Data access:
All detailed data and protocols from these experiments have been deposited in BarleyBase/PLEXdb (http://barleybase.org; http://plexdb.org/), a MIAME-compliant expression database for plant GeneChips (Shen et al. 2005). Data files have also been deposited in ArrayExpress (http://www.ebi.ac.uk/arrayexpress) as accession nos. E-TABM-111 (leaf) and E-TABM-112 (embryo).
Sources of check data:
Tests of the accuracy and reliability of the SFP predictions were made using two sources of data derived from a recently developed Barley SNP database (Rostoks et al. 2005a,b). Partial DNA sequence was available for a sample of 518 genes on the Affymetrix chip for both parents, St and Mx. These provided SNP information upon which the set of 129 DH lines, including our subset of 35, were genotyped. This sequence information also allowed us to identify individual oligonucleotide probes on the Affymetrix chip that contained sequences that differed between St and Mx. There were 167 features of 5698 (518 genes × 11 features) that contained a SNP and these came from 123 genes. Of these genes, 95 had just 1 feature containing a SNP but some had ≥2 (e.g., 1 had 9 and 2 had 5).
ANALYTICAL METHODS
Method 1:
Consider one probe pair of a probe set that represents any gene on an Affymetrix microarray. The perfect-match and mismatch sequences of this probe pair are  and
and 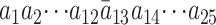 , where
, where 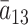 differs from
differs from 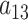 . The corresponding transcript sequence is denoted by
. The corresponding transcript sequence is denoted by 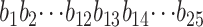 , which is known to have a certain degree of similarity with the probe sequences but is usually unknown exactly. The binding affinity between the transcript and probe nucleotides is parameterized as
, which is known to have a certain degree of similarity with the probe sequences but is usually unknown exactly. The binding affinity between the transcript and probe nucleotides is parameterized as 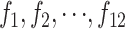 ,
,  , where
, where
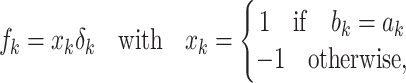 |
(1) |
where 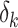 represents the molecular binding affinity between the transcript and probe at nucleotide k. The hybridization intensity values of the jth probe pair of the ith gene, PMij and MMij for perfect match and mismatch, respectively, can be written as
represents the molecular binding affinity between the transcript and probe at nucleotide k. The hybridization intensity values of the jth probe pair of the ith gene, PMij and MMij for perfect match and mismatch, respectively, can be written as 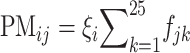 and
and 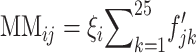 with
with 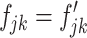 when
when 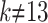 , where
, where 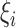 is the abundance parameter of the transcript. Thus, the expected difference between the perfect-match and mismatch intensities has the form
is the abundance parameter of the transcript. Thus, the expected difference between the perfect-match and mismatch intensities has the form
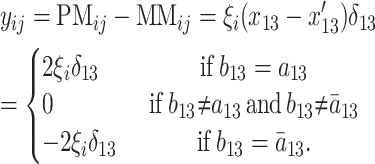 |
(2) |
Equation 2 explains the multiplicative model proposed by Li and Wong (2001) in which 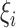 is defined as the model-based expression index and
is defined as the model-based expression index and 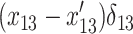 as the probe intensity index. One of the important features revealed by this model is that the difference between the PM and MM hybridization intensity values is largely determined by the match between transcript and probe sequences at the nucleotide where the two (perfect and mismatch) probe sequences differ. This is useful for the present analysis in at least two respects. First, it holds regardless of whether the transcript sequence perfectly matches either of the probe sequences. Second, variation in the PM–MM difference is largely explained by the extent to which the transcript sequence matches either of the two probe sequences at the nucleotide site where the two probe sequences differ. Thus, this information can be used in the following analysis.
as the probe intensity index. One of the important features revealed by this model is that the difference between the PM and MM hybridization intensity values is largely determined by the match between transcript and probe sequences at the nucleotide where the two (perfect and mismatch) probe sequences differ. This is useful for the present analysis in at least two respects. First, it holds regardless of whether the transcript sequence perfectly matches either of the probe sequences. Second, variation in the PM–MM difference is largely explained by the extent to which the transcript sequence matches either of the two probe sequences at the nucleotide site where the two probe sequences differ. Thus, this information can be used in the following analysis.
We consider two genotypes, Mx (Morex) and St (Steptoe). A general form for the difference between PM and MM hybridization intensities at the jth probe for gene i can be written as
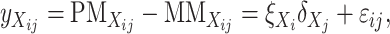 |
(3) |
with X = Mx or St and ɛij being a normally distributed residual variable. In the design of the expression experiment described above, there are three (replicate) expression profiles for each of the two parental genotypes. The parameters in Equation 3 can be estimated from 3 × 2 ×11 hybridization intensity values for each of the two genotypes by implementing the restrained iterative least-squares algorithm as first proposed by Li and Wong (2001). On the basis of the estimates 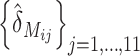 and
and 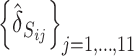 , we calculate
, we calculate 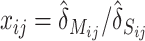 (j =1, 2, · · · , 11) and sort them into an ascending order
(j =1, 2, · · · , 11) and sort them into an ascending order 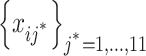 with j* being the permutated value of j. The jth probe is chosen as a candidate of a SFP if
with j* being the permutated value of j. The jth probe is chosen as a candidate of a SFP if  , where
, where 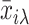 and
and 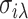 are the mean and standard deviation of all those
are the mean and standard deviation of all those  that exclude j* and do not satisfy the inequality for a prior given constant λ. It can readily be seen that inference of the SFP candidates has effectively avoided the influence of differential expression level between the two genotypes.
that exclude j* and do not satisfy the inequality for a prior given constant λ. It can readily be seen that inference of the SFP candidates has effectively avoided the influence of differential expression level between the two genotypes.
To integrate expression data from the doubled haploid lines into further confirmation and prediction of genotypes of the candidate SFPs diagnosed from the above, we consider the following three different forms of transformation,
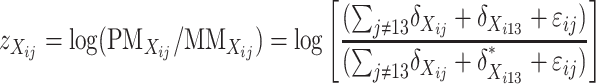 |
(4.1) |
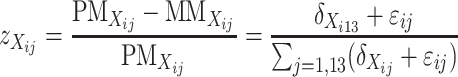 |
(4.2) |
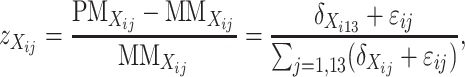 |
(4.3) |
which are common in at least two aspects. First, they are independent of expression level of the gene in question, and, second, those individuals having the same genotype at the transcript sequence are expected to have the same value of 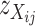 . Thus,
. Thus, 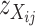 can be used as a discrimination function to predict the genotypes of the DH lines at the candidate SFPs. For any given candidate SFP probe,
can be used as a discrimination function to predict the genotypes of the DH lines at the candidate SFPs. For any given candidate SFP probe, 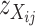 can be calculated for each of the parental genotypes and each of n DH individuals on the basis of each of Equations 4.1–4.3, yielding a series
can be calculated for each of the parental genotypes and each of n DH individuals on the basis of each of Equations 4.1–4.3, yielding a series 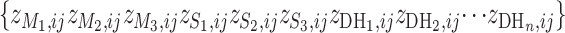 . The SFP probe is inferred if the three observations of
. The SFP probe is inferred if the three observations of 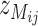 and three observations of
and three observations of 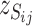 form two clusters when a two-mean clustering analysis is carried out with the sample. At the same time,
form two clusters when a two-mean clustering analysis is carried out with the sample. At the same time, 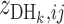 is inferred to have a Mx genotype if
is inferred to have a Mx genotype if 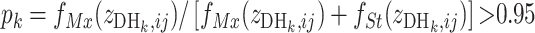 or a St genotype if
or a St genotype if 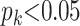 ; otherwise its genotype is uncertain, where
; otherwise its genotype is uncertain, where 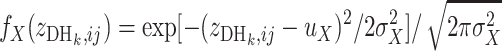 and
and 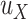 and
and 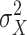 are, respectively, the mean and variance of the three values of
are, respectively, the mean and variance of the three values of  (X = Mx or St). With the predicted genotypes of the doubled-haploid individuals, we calculate a t-statistic for the difference between two genotype samples of
(X = Mx or St). With the predicted genotypes of the doubled-haploid individuals, we calculate a t-statistic for the difference between two genotype samples of 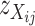 -values. The P-value of the t-test is used to assess the statistical efficiency of the genotype prediction.
-values. The P-value of the t-test is used to assess the statistical efficiency of the genotype prediction.
Method 2:
Ronald et al. (2005) developed an approach for predicting SFP and genotypes at the SFP in a yeast segregating population on the basis of the proposition that the binding affinity of a transcript sequence to its complementary probe sequence can be adequately predicted from the positional-dependent-nearest-neighbor (PDNN) model (Zhang et al. 2003) as
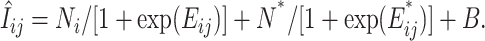 |
(5) |
For those species such as yeast considered by Ronald
et al. (2005), perfect-match probe sequences are known to exactly match their corresponding transcript sequences in one of the parental strains from which the segregating population was created. In the standard strain, 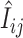 may be recognized as the expected value of perfect-match hybridization intensity of the jth probe for the ith gene. Under the PDNN model, Ni is defined as the expression index for the gene i and has a form of
may be recognized as the expected value of perfect-match hybridization intensity of the jth probe for the ith gene. Under the PDNN model, Ni is defined as the expression index for the gene i and has a form of
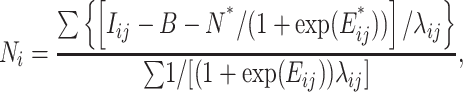 |
(6) |
where 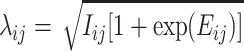 and
and 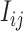 is the observed perfect-match value.
is the observed perfect-match value. 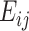 and
and 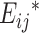 are energy parameters depicting, respectively, specific and nonspecific RNA–DNA binding and depend on nucleotide sequence of the target probe. Each of the energy parameters involves 40 unknown parameters (see Zhang
et al. 2003 for details). Together with
are energy parameters depicting, respectively, specific and nonspecific RNA–DNA binding and depend on nucleotide sequence of the target probe. Each of the energy parameters involves 40 unknown parameters (see Zhang
et al. 2003 for details). Together with 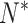 , the nonspecific binding parameter, and B, the constant background parameter, Equations 5 and 6 involve a total of 82 unknown parameters to be estimated from n × 11 perfect-match intensity values and probe sequences for each of the arrays in question by minimizing the so-called fitness function
, the nonspecific binding parameter, and B, the constant background parameter, Equations 5 and 6 involve a total of 82 unknown parameters to be estimated from n × 11 perfect-match intensity values and probe sequences for each of the arrays in question by minimizing the so-called fitness function
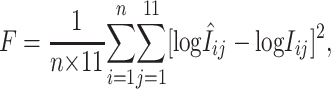 |
(7) |
where n is the number of genes interrogated. Ronald
et al. (2005) compared 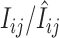 of a yeast strain against that of the reference yeast strain. Significance of the comparison was taken as evidence to support inference of SFP associated with the probe.
of a yeast strain against that of the reference yeast strain. Significance of the comparison was taken as evidence to support inference of SFP associated with the probe.
Method 3:
This was first proposed by Winzeler et al. (1998) to identify SFP from genomic DNA microarrays. Constancy in abundance of molecules interrogated for all genes is probably the most distinct feature of genomic DNA microarray data when compared to RNA microarray data. However, the two methods share a common principle in screening SFP, i.e., identification of the probes whose signal intensities contrast with the uniformity between the two genotypes for the remaining probes in the same set. The method would be appropriate to survey SFPs at least for those genes whose expression is not so different that the effect of the SFP-associated probe will be hidden by variation in gene expression between the two genotypes. Thus, an obvious risk of using this method to predict SFPs is that genes differentially expressed between two genotypes are likely to be predicted as SFP-associated genes even though there is no genetic polymorphism in the coding sequence of the genotypes. A detailed description of the method can be found elsewhere (Brem et al. 2002).
Method 4:
The background adjusted, normalized PM values for each probe from all 30 DH lines and the three replicates of each parent (36 in total) are separated into two clusters by k-mean clustering. Probes with nonoverlapping clusters are identified as follows. The means and standard deviations of each cluster are determined and the probability of every member of each cluster belonging to the other cluster is estimated using the normal deviate zi = (xi −mj)/sj, where xi is the score of an individual from cluster i and mj and sj are the mean and standard deviation of cluster j. We used a zi ≥ 2.576 (P ≤ 0.01) to indicate 99% probability that probe i does not belong to the other cluster, otherwise it is treated as a missing datum. This is repeated for all members of both clusters and the total number of missing data is calculated. We accepted only those probes that had no more than 1 missing individual of 36 and for which the parents were consistently different in all three replicates.
SFP mapping:
SFP genetic linkage maps were constructed using JoinMap version 3.0 (Van Ooijen and Voorrips 2001). The SFP markers were assigned to linkage groups using anchor markers with minimal LOD = 3.0.
The programs developed to carry out the SFP predictions presented in this article are written in FORTRAN-90 and we are willing to provide executable versions with instructions on request. We hope to provide more user-friendly Windows-based applications in the longer term.
RESULTS
We explored two new (methods 1 and 4) and two existing methods (Winzeler et al. 1998; Ronald et al. 2005) for identifying SFPs and genotyping lines on the basis of the SFPs predicted from Affymetrix gene expression profiled on two tissues (leaf and embryo) of two commercial varieties of barley (Morex × Steptoe) and their doubled-haploid progeny. Method 1 was developed to separate hybridization affinity between probe and transcript sequences (i.e., probe effect) from transcript abundance (gene expression). Candidacy of an SFP-associated probe was determined by three criteria: (i) its estimated probe effect deviated significantly from the distribution of estimates of the probe effect for the remaining probes of the same probe set, (ii) the estimated probe effect differed significantly between the two parental lines, and (iii) the difference was stably inherited and segregated in the offspring of the parents. Method 4, on the other hand, makes no attempt to separate SFPs from GEMs and simply identifies features that can be Mendelized across the DH lines. It was used as a final control against which to assess the SFP predictions of the other three. In all cases the association of genotypes to DH lines is achieved using k-means clustering with consistent separation of the two parents across replicates. The methods are described in more detail in materials and methods.
A single probe set on the array generally, but not always, represents 11 features of a single gene. So for simplicity in the text, we refer to probe sets as genes and the individual probes as features. The initial predictions of possible SFP containing features and genes are given in supplemental Table S1 at http://www.genetics.org/supplemental/. The numbers predicted by methods 1–3 to contain SFPs vary with the method of prediction, with method 1 identifying the most genes but fewer features per gene than the other two. Moreover, method 1 has the highest proportion of genes represented by just a single feature while the other two methods yield many genes with multiple features, some identifying as many as 11. The very high numbers of features in genes identified by methods 2 (Ronald et al. 2005) and 3 (Winzeler et al. 1998) suggest that they may be detecting GEMs. More genes are identified with SFPs from embryo than leaf tissue but this is partly a reflection that more genes were expressed in this tissue (17,218 for embryo and 16,004 for leaf). Overall, ∼25% of possible genes on the chip are identified as containing features with SFPs. The percentage of SFP-bearing genes in which just one feature was identified is high, 55–76%.
SFP genotype predictions compared to information from 518 gene sequences:
We first consider the SFP predictions for the genotypes of the 30 DH lines and compare them with the known SNP genotypes that are used as the “gold standard” of genotype assignment. For each method and tissue, the genes containing the predicted SFPs were compared with the 518 genes for which SNP data were available and all matching genes were identified. The numbers of matching genes and features for the three methods and tissues are given in Table 1 for the 30 DH lines. The genes sequenced for SNP identification represent only 2.3% of the genes on the chip (518/22,801); therefore we would expected this same percentage of matching genes if the prediction methods were identifying SFP-containing genes at random. Table 1 shows that, in fact, they identify between 29 and 53% of the SNP-bearing genes.
TABLE 1.
Number of SFPs detected by the three methods that match genes for which SNPs have been identified (518 in total)
| Leaf |
Embryo |
|||||
|---|---|---|---|---|---|---|
| Methods | 1 | 2 | 3 | 1 | 2 | 3 |
| Features | 253 | 381 | 227 | 438 | 672 | 455 |
| Genes | 185 | 204 | 149 | 277 | 259 | 236 |
| Genes as % of maximum (518) | 36 | 39 | 29 | 53 | 50 | 45 |
A more useful test of the accuracy of the SFP predictions is the extent of their agreement with the known SNP genotypes of the DH lines using those features/genes shown in Table 1. The predicted DH genotypes (St or Mx) for each of these identified SFPs were compared with the corresponding SNP genotypes for the same genes using two criteria: (A) the percentage of the possible 30 DH genotypes predicted (for some features the genotype cannot be unambiguously determined in certain lines) and (B) the percentage of these predictions that agree with the SNP genotype. These values were computed for all SFPs predicted by each method on both tissues.
When we looked at the accuracy of these initial SNP genotype predictions across methods and tissues we found the following classes: (i) 40% of SFPs matched all the DH SNP genotypes exactly, (ii) 21% failed to match between 3.3 and 10% (i.e., 1–3 wrong of 30), (iii) 11% failed between 10 and 19.9%, while (iv) 28% incorrectly predict ≥ 20% of the DH genotypes. This supports the view that the class iv and possibly class iii SFPs are due to variation in trans-acting factors and are probably gene expression markers. Thus, 61% of SFPs (groups i and ii) are probably due to polymorphism in the gene itself or a closely linked regulator, while 28–39% are due to different, loosely linked genes, of indeterminate origin. We removed the class iv genes from the next stage of the analysis, on the grounds that they were probably not erroneous genotype predictions but instead were genuine polymorphisms, albeit in different genes such as trans-acting regulators. However, we consider them again later.
Using prediction criteria A and B above, we explored how the success rates varied as more stringent significance thresholds were used to assess the SFP predictions. Each method provided a P-value based on the discriminant function for every SFP prediction and the predictions were sorted against these P-values. We then asked the question, “If we choose all predictions with a P ≤ PT (where PT is a given threshold), what are the overall success rates of the predictions?” Plots of these success rates (three methods and two tissues) for varying PT-values are given in Figure 1. They show that all three methods rapidly approach very high success rates for both criteria, although method 1 is more conservative in terms of numbers of SFPs predicted. At a threshold of P ≤ 10−18, all methods predict ∼98% of the genotypes with ∼99% accuracy, which is more than comparable to many conventional marker-based methods. At a lower threshold of P ≤ 10−15, the prediction rate drops to ∼95% but accuracy is still high at ∼98%. We decided to use the more stringent threshold for all further analyses.
Figure 1.—

Change in (A) percentage of DH genotypes predicted by SFPs and (B) percentage of accuracy of predictions across the three methods and two tissues (leaf and embryo). P is exponent, n in P ≤ 10−n.
We looked at all predicted SFPs (including the putative SFP predictions) at this stringent threshold and checked the accuracy with which they predict the SNP genotypes of the 30 DH lines as we did above. We found that the overall proportion of genes that disagreed with the SNP genotypes by >10% across all three methods and both tissues was entirely consistent at ∼6% (χ2 [5 d.f.] = 7.75, P = 0.17), less than one-third of those predicted earlier before we applied the threshold. Thus, 94% of SFPs cosegregate well with their SNP while 6% do not.
Using the available partial sequences of 518 genes in St and Mx together with the known sequence of all 11 features on the Affymetrix chip for these same genes, it was possible to explore the overlap between the predicted SFPs and identifiable differences in the sequence of these features between St and Mx. The results of this comparison are shown in Table 2.
TABLE 2.
The numbers of predicted SFPs compared to the presence of sequence polymorphism in features from leaf and embryo tissues
| Leaf |
Embryo |
||||||
|---|---|---|---|---|---|---|---|
| Methods | Sequence of all probes | 1 | 2 | 3 | 1 | 2 | 3 |
| SNP present | 167 (4) | 11 (30) | 50 (37) | 16 (27) | 52 (35) | 84 (31) | 46 (36) |
| No SNP | 4513 | 26 | 85 | 45 | 93 | 189 | 83 |
The percentages of SFPs containing SNPs among the informative features are shown in parentheses. 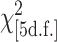 -test of homogeneity across tissues and methods = 398; P = 0.55.
-test of homogeneity across tissues and methods = 398; P = 0.55.
Partial sequences were available for 518 genes for both St and Mx and these sequences fully overlap 4680 features present on the Affy chip: 82% of all possible 5698 (= 518 × 11) features. Of these 4680 “informative” sequences, only 167 (4%) actually contained a sequence difference between St and Mx (Table 2, column 2). We now look at the probes predicted (at P ≤ 10−18) to contain SFPs by each of the three methods in both tissues and that also overlap the set of 4680 informative features (Table 2). We find complete consistency in the proportion of SNP-bearing features identified (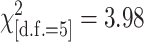 , P = 0.55) and all are highly enriched with features containing SNPs (33% overall compared to 4% by chance alone). So, all methods preferentially identify features in which the matching probe sequence does differ between St and Mx. Very significantly, however, all methods consistently identify a high proportion of SFPs, ∼67%, in features for which there is no sequence polymorphism of any type between St and Mx.
, P = 0.55) and all are highly enriched with features containing SNPs (33% overall compared to 4% by chance alone). So, all methods preferentially identify features in which the matching probe sequence does differ between St and Mx. Very significantly, however, all methods consistently identify a high proportion of SFPs, ∼67%, in features for which there is no sequence polymorphism of any type between St and Mx.
Predictions based on whole data set:
The total numbers of SFPs matching the two thresholds are shown in Table 3 for each method and tissue separately, together with the total number predicted initially. All methods predict a large number of SFPs, but method 2 always predicts the most while method 1 is more conservative. Because we showed above that methods 1–3 were entirely consistent in their predictions based on SNPs and on feature sequence, we combined the predictions across methods and, where the same gene had been predicted by more than one method, we chose the one with smallest P-value. This resulted in identifying between 1853 and 4374 unique SFPs depending on tissue and threshold (Table 3).
TABLE 3.
Total numbers of SFP predictions (gene–feature combinations) and those unique genes matching the two thresholds for each method and tissue
| Threshold |
|||
|---|---|---|---|
| All predictions | ≤ 10−15 | ≤ 10−18 | |
| Leaf tissue | |||
| Method 1 | 7,870 | 1,714 | 698 |
| Method 2 | 8,426 | 4,089 | 2,425 |
| Method 3 | 6,980 | 2,687 | 1,549 |
| Total | 23,276 | 8,490 | 4,672 |
| Total unique genes across all methods | — | 2,791 | 1,853 |
| Embryo tissue | |||
| Method 1 | 12,137 | 4,590 | 3,243 |
| Method 2 | 14,420 | 7,968 | 5,655 |
| Method 3 | 12,791 | 5,688 | 3,883 |
| Total | 39,348 | 18,246 | 12,781 |
| Total unique genes across all methods | — | 4,374 | 3,283 |
The number of genes detected as containing SFPs that are common to the three methods for all predicted probes and under the two thresholds is illustrated in Figure 2. It shows quite clearly that method 2 alone accounts for most of the total SFP-containing genes (80–85%, depending on the tissue or threshold), while methods 1 and 3 lag considerably behind. If we consider combining two methods, then method (M)2 plus M1 or M2 plus M3 raise the total percentage of genes identified to the mid- to high 90s. However, given that method 3 involves considerably more computer space and time, it would appear that it makes practical sense to combine the predictions of methods 1 and 2, which together identify between 91 and 97% of possible informative genes.
Figure 2.—

Venn diagrams illustrating overlap between the three methods on both tissues at the 10−15 and 10−18 thresholds in terms of predicted genes.
Many genes (42%) are common to the two tissues when we pool all methods and identify unique probe sets. However, there are many more unique SFPs identified for the embryo tissue (49% vs. 9% for the leaf tissue) at a threshold of P ≤ 10−18. At this threshold, 91% of all SFPs (3282) were identified in embryo tissue, 14% of all genes on the chip.
The final two approaches to verifying the SFPs involved mapping them and constructing haplotypes (graphical genotypes) for all chromosomes of the 30 DH lines. These were compared with those from the corresponding gold standard obtained from SNPs. We combined the genes and features, together with their SFP genotype predictions, identified by all methods, which had a P ≤ 10−18. Where there were several probes from the same gene or duplicate features in this list, we selected the single feature from each gene across all methods with the smallest P-value. This yielded 1853 and 3283 unique genes from leaf and embryo, respectively (Table 3). We attempted to map these using the small population of 30 DH lines, deliberately using a different subset of genes for leaf (1853) and embryo tissue (1754) of which 1504 and 1523, respectively, mapped successfully; the ∼16% with mapping problems is typical of such a small mapping population. Of these, ∼62% cosegregated largely because of the small population, but >400 individual marker “bins” were mapped for each tissue (supplemental Table S2 at http://www.genetics.org/supplemental/).
We added 21 SNP markers that had previously been mapped in a population of 129 St × Mx DH individuals to these sets of SFP markers to act as anchors for identifying and orientating each chromosome. The total map length of the seven barley chromosomes was estimated as a little over 1100 cM (Kosambi) with comparable lengths for individual chromosomes from both tissues (supplemental Table S2 at http://www.genetics.org/supplemental/). As expected given the small mapping population, map lengths were biased upward relative to the SNP-based map from 129 DH lines. The haplotypes of the individual chromosomes indicated that there were ∼2% double recombinants involving single loci scattered across these 210 “line-by-chromosome” haplotypes. This was as expected given that the threshold for accepting the SFPs initially was chosen for 99% accuracy. Such double recombinants are readily detected and replaced as “missing” genotypes. Comparison of each of the 210 SFP haplotypes with those based on SNPs shows almost complete congruence with both crossover numbers and locations. Figure 3 illustrates this comparison for the seven chromosomes of a randomly chosen line (SM135) (the full set is available on request).
Figure 3.—

Haplotypes of chromosomes from line SM135 drawn to compare SFP (left) and SNP (right) predictions. Solid bars, St; open bars, Mx.
Finally, we used method 4, which makes no attempt to separate GEMs from true SFPs, as a control against which to compare the other three methods. Methods 1–3 were combined for this comparison because they were consistent in their ability to identify SFPs as judged by their match to SNP genotypes and to known feature polymorphisms, and unique SFPs were identified. The predictions from methods 1–3 (test set) vs. method 4 (control) are shown in Table 4. We see that the relative numbers of polymorphisms detected in leaf and embryo are almost identical in test and control sets and method 4 identifies 93% (3354/3608) of those detected by the other three methods. Likewise, the split between polymorphisms common or unique to each tissue are very similar (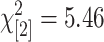 ; P = 0.07, NS). The proportion of polymorphisms that provide good agreement to their corresponding SNP genotypes is not significantly different between test and control (
; P = 0.07, NS). The proportion of polymorphisms that provide good agreement to their corresponding SNP genotypes is not significantly different between test and control (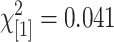 ; P = 0.84) nor are the proportions of identified features that match known polymorphic features between St and Mx (
; P = 0.84) nor are the proportions of identified features that match known polymorphic features between St and Mx (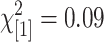 ; P = 0.77). This indicates that the feature polymorphisms predicted are a mixture of true SFPs and GEMs with a large proportion of GEMs. This is a reflection of poor performance of methods 2 and 3 in separating true SFPs from GEMs. However, it should be noted that method 1 conferred an improved resolvability in separating the SFPs from the GEMs because it identified the most genes but far fewer features per gene than the other methods.
; P = 0.77). This indicates that the feature polymorphisms predicted are a mixture of true SFPs and GEMs with a large proportion of GEMs. This is a reflection of poor performance of methods 2 and 3 in separating true SFPs from GEMs. However, it should be noted that method 1 conferred an improved resolvability in separating the SFPs from the GEMs because it identified the most genes but far fewer features per gene than the other methods.
TABLE 4.
Summary of predictions from “test” methods 1–3 designed to identify SFPs against “control” method 4 designed to identify all expression-based single-locus polymorphisms (SFPs and GEMs)
| Initial predictions | Test methods 1–3 (P ≤ 10−18) | Control method 4 |
|---|---|---|
| Leaf tissue SFPs | 1853 | 2131 |
| Embryo tissue SFPs | 3283 | 3598 |
| Combined and unique SFPs | 3608 | 3953 |
| SFPs common to test and control | 3354 | |
| SFPs common to both leaf and embryo | 1527 (42%) | 1776 (45%) |
| SFPs unique to leaf | 325 (9%) | 355 (9%) |
| SFPs unique to embryo | 1755 (49%) | 1822 (46%) |
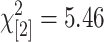 ; P = 0.07, NS ; P = 0.07, NS |
||
| SFPs match to SNP genotypes of 30 DHLs | 208 | 205 |
| Good agreement (>90%) | 198 (95%) | 196 (95%) |
| Poor agreement (≤90%) | 10 (5%) | 9 (5%) |
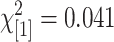 ; P = 0.84, NS ; P = 0.84, NS |
||
| SFPs match to St/Mx informative features | 175 | 188 |
| Feature has sequence polymorphism | 64 (37%) | 66 (35%) |
| Feature has no sequence polymorphism | 111 (63%) | 122 (65%) |
 ; P = 0.77, NS ; P = 0.77, NS |
||
DISCUSSION
We have attempted to test the principle that useful gene-based molecular genetic markers could be easily and reliably obtained from expression data even in nonsequenced species using populations for which the chip features were not specifically designed. The availability of such an approach has wide value in genetical analysis of crop and ecologically important plants and of some farm animals and also in complementing information available in sequenced model organisms. It is particularly useful in supporting genetical genomic analyses.
Method 1, developed in this article, differs methodologically in several respects from its rivals in the earlier and most recent literature on the subject. First, this method has made proper use of information extractable from Affymetrix expression microarrays without relying heavily on the match between transcript and probe sequences as in Ronald et al. (2005). The latter requires prediction of PM hybridization intensity for every probe interrogated on the arrays from their sequence information through the PDNN model (Zhang et al. 2003). Obviously, an accurate prediction of the PM intensities is the basis for an accurate diagnosis of SFPs and in turn reliable genotyping at the SFPs. The less well a transcript sequence matches its probe counterpart, the more seriously the prediction will be biased and hence the greater risk of a false SFP prediction. Furthermore, the PDNN model, which involves as many as 82 unknown parameters, might be recognized as far less robust statistically than the multiplicative model depicted in Equation 3 of this article. Second, method 1 was developed to distinguish variation in the hybridization intensity due to genuine sequence polymorphism from that due to differential gene expression. This is particularly important for an accurate assessment of expression level of a gene by removing probe(s) that contains SFPs and also for effectively avoiding potential autocorrelation between SFP detected within a gene and expression of the gene. Third, instead of using PM information alone (Cui et al. 2005; Ronald et al. 2005; West et al. 2006), diagnosis of SFPs and prediction of genotypes at the SFP markers were based on the differences between PM and MM intensities based on the multiplicative model that has proved adequate in capturing the essential features of Affymetrix microarray data (Li and Wong 2001). The PM–MM model is usually superior to the model with PM alone because of its better control of nonbiological, systematic variation (Harr and Schlotterer 2006). It is clear that method 1 is conservative in its predictability of the number of genes containing SFPs in comparison to method 2 but it is also clear that the method is much more efficient in avoiding identifying GEMs as expected from its design.
Method 1 developed in this study was compared to only two of the approaches available in the current literature. Method 3, originally developed by Winzeler et al. (1998), was the first attempt to screen for SFP by making use of high-density oligonucleotide arrays. It is appropriate for SFP predication only from genomic DNA microarray data. With DNA microarray data, one can expect uniformity in the amount of DNA molecules hybridized onto a microarray chip across all genes. The method was chosen for comparison here because it provides a direct assessment of the added difficulties and bias in modeling expression microarray data. Analytically quite similar to method 1, method 2 (Ronald et al. 2005) was the first designed to predict SFP from RNA microarray data but it did not incorporate sufficiently effective analytical mechanisms to account for possible large variation in abundance of transcripts among different genes. We addressed this problem and made significant improvements to overcome it.
There have been several recent reports on developing statistical methods for SFP prediction from RNA microarray data. Cui et al. (2005) considered a different design of microarray experiment for identification of SFP between different inbred genotypes. Their analysis was designed for the circumstance where each of the inbred genotypes was repeated several times in the microarray experiment. Although their analysis was also based on an estimate of probe affinity effect from a simple additive linear model of log-scaled perfect-match signals, a question remains whether the probe affinity parameter is estimated with a comparable adequacy to that from the multiplicative regression analysis that combines information from both perfect- and mismatch signals as in the present study. Li and Wong (2001) demonstrate that the additive model shows a systematic pattern indicating lack of fit whereas the multiplicative model is able to capture the essential pattern of variation in observed hybridization signals across different probes of a probe set surrogating a gene.
West
et al. (2006) attempted to identify SFP independent of a gene expression level by calculating a summary measure, 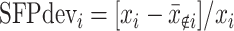 , where xi is the perfect-match value of the ith probe in a given probe set and
, where xi is the perfect-match value of the ith probe in a given probe set and 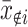 is the mean perfect-match values of all remaining probes excluding the ith probe. An SFP was declared if two parental genotypes had nonoverlapping ranges of SFPdev values separated by an empirically chosen distance. When an SFP was inferred between parental genotypes, genotypes of RILs initiated from the parental lines were to be inferred if SFPdev values of the RILs showed a bimodal distribution. The algorithm was demonstrated by analyzing an experiment profiling gene expression of Arabidopsis in which the two parental strains were respectively replicated 16 times and each of the 148 RILs was repeated twice. It has been demonstrated by Li and Wong (2001) that the perfect hybridization value of a probe from Affymetrix expression microarrays is a complex compound of two major effects: expression level of the gene and level of hybridization success between transcript and probe sequences. The SFPdev measure does not reflect the essential components of the information about the probe-based hybridization signal and ignores the use of mismatch information even though very stringent discrimination criteria were invoked in searching for probes with outlying hybridization signals. Thus, the SFP prediction based on the SFPdev measure may neither take appropriate account of the influence of expression level nor use all the available information from a microarray experiment effectively.
is the mean perfect-match values of all remaining probes excluding the ith probe. An SFP was declared if two parental genotypes had nonoverlapping ranges of SFPdev values separated by an empirically chosen distance. When an SFP was inferred between parental genotypes, genotypes of RILs initiated from the parental lines were to be inferred if SFPdev values of the RILs showed a bimodal distribution. The algorithm was demonstrated by analyzing an experiment profiling gene expression of Arabidopsis in which the two parental strains were respectively replicated 16 times and each of the 148 RILs was repeated twice. It has been demonstrated by Li and Wong (2001) that the perfect hybridization value of a probe from Affymetrix expression microarrays is a complex compound of two major effects: expression level of the gene and level of hybridization success between transcript and probe sequences. The SFPdev measure does not reflect the essential components of the information about the probe-based hybridization signal and ignores the use of mismatch information even though very stringent discrimination criteria were invoked in searching for probes with outlying hybridization signals. Thus, the SFP prediction based on the SFPdev measure may neither take appropriate account of the influence of expression level nor use all the available information from a microarray experiment effectively.
This study considered a much less demanding design of expression microarray experiments than those aforementioned. Without setting replication for offspring individuals (DH lines here) in the barley microarray experiment, we suggested use of Equations 4.1–4.3 as proxy for the probe-effect estimates. When expression profiling is repeated for the offspring as for parental lines, the probe-binding affinity parameters can be directly estimated and used in the next clustering analysis. We anticipate that this will improve performance of the method developed in this study.
We have shown that it is easily possible to use the information from Affymetrix expression arrays to accurately identify >4000 robust polymorphic molecular genetic markers. These SFPs represent ∼18% of the total barley genes on the chip and we show how they can be used to predict the genotypes in an F1-derived, doubled-haploid population. We have produced threshold criteria that guarantee a genotyping accuracy of these SFPs in such a population of at least 98% with 99% of genotypes being predicted. We also show how these rates decline with less stringent thresholds so that users can choose a suitable one for their particular situation. The approach is robust and works with transcripts derived from different tissues, although the number of identified SFPs is partly correlated with the number of genes active in a particular tissue, as would be expected. The ≤2% genotyping errors largely result in double recombinants around a SFP in a single haplotype during genetic mapping and so can be easily identified and replaced as missing data points.
These SFPs have been shown to be highly represented among SNP-containing genes and in chip features for which the parent strains differ in sequence. They result in maps and chromosomal haplotypes that are coincident with those produced with the current gold-standard SNP markers. Using the high stringency threshold of P ≤ 10−18, ∼95% of SFPs cosegregate in the DH population with SNP markers in the same gene while a further ∼5% are the result of polymorphism elsewhere in the genome. The latter could be due to duplicate genes, chance sequence alignments with RNA from elsewhere, or they may be the product of polymorphic trans-acting regulators. Predictably, these latter SFPs show no relation to the presence of parental polymorphism in the sequence represented by the probe feature. When we try to map the 10 SFPs from methods 1–3 that do not match the SNP genotypes we find that all except one easily map elsewhere on the genome. Significantly, 2 of them map to the precise position occupied by the SNP identified in a different mapping population, Oregon/Wolfe (Contig54187/10 and -7811/7), and hence could indicate duplicate genes. Of the 9 poorly fitting SFPs identified by method 4, 7 are in genes that map well to other locations. Five of these 9 were also detected by methods 1–3, including 1 that failed to map to any chromosome and the 2 above found in Oregon/Wolfe.
Of the 95% of SFPs that map to the same location as the SNP, ∼36% match a feature known to have a sequence polymorphism as opposed to the 4% expected by chance alone. These probably represent true SFPs in the structural genes. The remaining ∼64% occur in the absence of sequence polymorphism in any of the identified features and thus are probably GEMs. The 5% of SFPs that do not map to the known position of the gene clearly are part of these, leaving ∼59% that must be polymorphic in regions so close to the genes as to be cosegregating, probably cis-acting expression regulators. This suggests that over all predicted SFPs, ∼36% are true SFPs, ∼59% are cis-acting expression regulators, and ∼5% are trans-acting regulators or duplicate genes. It also appears that the number of trans-acting genes identified reduced as our detection stringency increased. Such an effect was recently reported in two tissues in rats, where generally the cis eQTL detected had much greater LOD scores than the trans-acting eQTL (Yamashita et al. 2005).
The causes of the polymorphism are not important if one simply wants to generate robust genetic markers that are useful both for high-density mapping and to provide additional markers in species such as wheat and some Solanaceae where polymorphism is low. All methods accurately detect true polymorphisms. However, great caution should be exercised in assuming that the polymorphism is independent of overall expression or indeed due to sequence differences in the gene itself given that ∼64% of SFPs do not coincide with polymorphic features in the target genes, irrespective of the method used. This is also a concern for eQTL analysis because features containing SFPs should be removed to avoid autocorrelation.
The ability to genotype a population, while simultaneously measuring gene expression, is extremely valuable, particularly in an agricultural context where mislabeling and other quality assurance issues can easily occur. They can confirm the identity of the individual source material because the SFP genotype can be checked against previously obtained SNP genotypes. Thus they provide a simple “fingerprinting” method that can also be used for intellectual property issues or distinctiveness diagnostics. We were able to use this feature to unambiguously identify and remove data from 5 of the original 35 DH lines that had been wrongly labeled at some stage prior to our receiving the expression data.
Using a species-designed chip, the SFP approach can be used to map and carry out genetical genomics and eQTL analysis on any novel population, even though a previous map is unavailable. The identity of individual chromosomes may be determined through SFP synteny in crosses with chromosomal anchor markers. It could be used, for example, to explore novel populations produced from wide crosses among Arabidopsis accessions, using a generic Arabidopsis chip.
Obviously this same approach can be, and has been (West et al. 2006), applied to RILs, and there should be no major difficulty in extending the basic principles of SFP prediction to heterozygous populations such as F2's, given the evidence that differential expression for most genes is indeed consistent with Mendelian inheritance (Knight 2004). Providing there are expression data from the F1, the candidates for SFP can be screened in the same way as in this article and prediction of individual SFP genotypes in the segregating F2 population could be treated as a mixed-population model. However, the power may be low compared to DHs or RILs because the contrasts between the three subpopulation means are decreased and the expression-based markers may well be dominant.
Acknowledgments
This research was supported by a research grant from the Biotechnology and Biological Sciences Research Council and the National Environmental Research Council of the United Kingdom. Z.W.L. is also supported by the National Natural Science Foundation and the Basic Science Research Program “973” of China.
References
- Alberts, R., P. Terpstra, L. V. Bystrykh, G. de Haan and R. C. Jansen, 2005. A statistical multiprobe model for analyzing cis and trans genes in genetical genomics experiments with short-oligonucleotide arrays. Genetics 171 1437–1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bing, N., and I. Hoeschele, 2005. Genetical genomics analysis of a yeast segregant population for transcription network inference. Genetics 170 533–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borevitz, J. O., D. Liang, D. Plouffe, H. S. Chang, T. Zhu et al., 2003. Large-scale identification of single-feature polymorphisms in complex genomes. Genome Res. 13 513–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brem, R. B., G. Yvert, R. Clinton and L. Kruglyak, 2002. Genetic dissection of transcriptional regulation in budding yeast. Science 296 752–755. [DOI] [PubMed] [Google Scholar]
- Bystrykh, L., E. Weersing, B. Dontje, B. Sutton, M. T. Pletcher et al., 2005. Uncovering regulatory pathways that affect hematopoietic stem cell function using ‘genetical genomics’. Nat. Genet. 37 225–232. [DOI] [PubMed] [Google Scholar]
- Caldo, R. A., D. Nettleton and R. P. Wise, 2004. Interaction-dependent gene expression in Mla-specified response to barley powdery mildew. Plant Cell 16 2514–2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui, X., J. Xu, R. Asghar, P. Condamine, J. T. Svensson et al., 2005. Detecting single-feature polymorphisms using oligonucleotide arrays and robustified projection pursuit. Bioinformatics 21 3852–3858. [DOI] [PubMed] [Google Scholar]
- DeCook, R., S. Lall, D. Nettleton and S. H. Howell, 2006. Genetic regulation of gene expression during shoot development in Arabidopsis. Genetics 172 1155–1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Druka, A., G. Muehlbauer, I. Druka, R. Caldo, U. Baumann et al., 2006. An atlas of gene expression from seed to seed through barley development. Funct. Integr. Genomics 6 202–211. [DOI] [PubMed] [Google Scholar]
- Harr, B., and C. Schlotterer, 2006. Comparison of algorithms for the analysis of Affymetrix microarray data as evaluated by co-expression of genes in known operons. Nucleic Acids Res. 34 e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen, R. A., and J. P. Nap, 2001. Genetical genomics: the added value from segregation. Trends Genet. 17 388–391. [DOI] [PubMed] [Google Scholar]
- Kleinhofs, A., A. Kilian, M. A. Saghai-Maroof, R. M. Biyashev, P. Hayes et al., 1993. A molecular, isozyme and morphological map of the barley genome. Theor. Appl. Genet. 86 705–712. [DOI] [PubMed] [Google Scholar]
- Knight, J. C., 2004. Allele-specific gene expression uncovered. Trends Genet. 20 113–116. [DOI] [PubMed] [Google Scholar]
- Li, C., and W. H. Wong, 2001. Model-based analysis of oligonucleotide arrays: expression index computation and outlier detection. Proc. Natl. Acad. Sci. USA 98 31–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehrabian, M., H. Allayee, J. Stockton, P. Y. Lum, T. A. Drake et al., 2005. Integrating genotypic and expression data in a segregating mouse population to identify 5-lipoxygenase as a susceptibility gene for obesity and bone traits. Nat. Genet. 37 1224–1233. [DOI] [PubMed] [Google Scholar]
- Morley, M., C. M. Molony, T. M. Weber, J. L. Devlin, K. G. Ewens et al., 2004. Genetic analysis of genome-wide variation in human gene expression. Nature 430 744–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronald, J., J. M. Akey, J. Whittle, E. N. Smith, G. Yvert et al., 2005. Simultaneous genotyping gene-expression measurement and detection of allele-specific expression with oligonucleotide arrays. Genome Res. 15 284–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rostoks, N., J. O. Borevitz, P. E. Hedley, J. Russell, S. Mudie et al., 2005. a Single-feature polymorphism discovery in the barley transcriptome. Genome Biol. 6 R54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rostoks, N., S. Mudie, L. Cardle, J. Russell, L. Ramsay et al., 2005. b Genome-wide SNP discovery and linkage analysis in barley based on genes responsive to abiotic stress. Mol. Genet. Genomics 274 515–527. [DOI] [PubMed] [Google Scholar]
- Schadt, E. E., S. A. Monks, T. A. Drake, A. J. Lusis, N. Che et al., 2003. Genetics of gene expression surveyed in maize, mouse and man. Nature 422 297–302. [DOI] [PubMed] [Google Scholar]
- Shen, L., J. Gong, R. A. Caldo, D. Nettleton, D. Cook et al., 2005. BarleyBase—an expression profiling database for plant genomics. Nucleic Acids Res. 33 614–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinmetz, L. M., H. Sinha, D. R. Richards, J. I. Spiegelman, P. J. Oefner et al., 2002. Dissecting the architecture of a quantitative trait locus in yeast. Nature 416 326–330. [DOI] [PubMed] [Google Scholar]
- Van Ooijen, J. W., and R. E. Voorrips, 2001. JoinMap 3.0. Software for the Calculation of Genetic Linkage Maps. Plant Research International, Wageningen, The Netherlands.
- West, M. A. L., H. Leeuwen, A. Kozik, D. K. Kliebenstein, R. W. Doerge et al., 2006. High-density haplotyping with microarray-based expression and single feature polymorphism markers in Arabidopsis. Genome Res. 16 787–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winzeler, E. A., D. R. Richards, A. R. Conway, A. L. Goldstein, S. Kalman et al., 1998. Direct allelic variation scanning of the yeast genome. Science 281 1194–1197. [DOI] [PubMed] [Google Scholar]
- Yamashita, S., K. Wakazono, T. Nomoto, Y. Tsujino and T. Kuramoto, 2005. Expression quantitative trait loci analysis of 13 genes in the rat prostate. Genetics 171 1231–1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, L., M. F. Milles and K. D. Aldape, 2003. A model of molecular interactions on short oligonucleotide microarrays. Nat. Biotech. 21 818–821. [DOI] [PubMed] [Google Scholar]