

Abstract
Patients with Smith-Magenis syndrome have a high incidence of congenital heart disease that requires open-heart surgery. These patients may have gene deletions that affect cholesterol homeostasis, although no previous association has been made with premature atherosclerosis. Herein, we report a case of such a patient, who experienced a stroke after cardiac surgery because of what we believe to be premature intracerebral atherosclerosis.
Key words: Abnormalities, multiple/genetics; arteriosclerosis/complications; cardiac surgery; cerebrovascular accident; cholesterol/blood; chromosome deletion; chromosomes, human, pair 17; heart valve prosthesis; perfusion
Smith-Magenis syndrome (SMS), a genetic disorder resulting from a deletion of chromosome 17 band p11.2, is characterized by distinctive facial features, developmental delay, and cognitive and behavioral abnormalities.1 Congenital heart disorders have been described in 37% of SMS patients; most defects have been atrial septal defects, ventricular septal defects, mitral valve prolapse, tricuspid and mitral valve stenosis or regurgitation, supravalvular pulmonary stenosis, and subvalvular aortic stenosis.1,2 These defects often necessitate open-heart surgery early in life, and may require further surgery later in life. We report the case of a young woman with SMS who underwent repeat cardiac surgery and suffered a postoperative stoke.
Case Report
In October 2005, a 32-year-old woman presented at our institution so that we could investigate her recently developed pulmonary regurgitation, which was marked by episodes of dizziness and decreased exercise tolerance. At the age of 4 years, our patient, who was known to have SMS, had undergone uneventful open-heart repair of an atrial septal defect and a ventricular septal defect, excision of the pulmonary valve, and enlargement of the pulmonary outflow tract for a dysplastic pulmonary valve. She had done well until her recent onset of symptoms.
Physical examination on this admission revealed a woman 148 cm in height, 45 kg in weight, with facial features typical of SMS—a broad, square-shaped face, brachycephaly, a broad nasal bridge, and a prominent jaw. Loud systolic and diastolic murmurs were auscultated over the precordium. A recent echocardiogram had suggested right ventricular dilatation with severe pulmonary regurgitation. Cardiac magnetic resonance angiography (MRA) confirmed severe pulmonary regurgitation (regurgitant fraction, 0.40), a right ventricular ejection fraction of 0.47, and a right ventricle that was double the size of the left ventricle. The right ventricular end-diastolic volume was 145 cc, compared with a left ventricular end-diastolic volume of 76 cc. Residual atrial and ventricular shunting were ruled out by transesophageal echocardiography.
The patient underwent repeat heart surgery, with normothermic perfusion (36 °C) on the beating heart. To avoid injury to her enlarged right ventricle, we used femoro–femoral bypass. Blood flow was maintained during bypass at 2.4 L/m2, with a prime hematocrit of 25%. Her left heart was never vented or otherwise entered. Enlargement of the right ventricular outflow tract with a bovine pericardial patch, together with placement of a 25-mm bovine pericardial valve (Model 3000 TFX, Edwards Lifesciences; Irvine, Calif) in the pulmonic position, went smoothly. Intraoperatively, her mean arterial pressures were maintained at 40 to 50 mmHg during perfusion, and her metabolic profile and urine output were normal. The total time on cardiopulmonary bypass was 157 minutes.
Postoperatively, she was noted to be slow to wake, and she developed left-sided hemiparesis. Her electrolyte levels, arterial blood gases, and vital signs were unremarkable. The next day, a computed tomographic scan of the head revealed no hemorrhage, an electroencephalogram revealed no seizure activity, and a carotid Doppler study revealed normal carotid bifurcations. However, magnetic resonance imaging of the brain (Fig. 1) revealed bilateral acute cortical infarcts, and MRA (Fig. 2) revealed severe intracranial atherosclerosis. Our patient had a cholesterol level of 171 mg/dL (normal range, 110–199 mg/dL); triglyceride level of 142 mg/dL (normal range, 40–150 mg/dL); high-density lipoprotein (HDL) level of 31 mg/dL (normal range, 40–75 mg/dL); low-density lipoprotein (LDL) level of 112 mg/dL (normal range, 70–129); and a cholesterol-to-HDL ratio of 5.5 (normal range, 2.0–5.0). Gradually, the patient's neurologic function improved. On postoperative day 3, she was fully awake and following commands, so she was extubated. A rigorous physical-therapy regimen was instituted, and she was started on aspirin. On postoperative day 18, due to improvement in her left-sided weakness, she was discharged to a rehabilitation facility, and she is now able to walk.
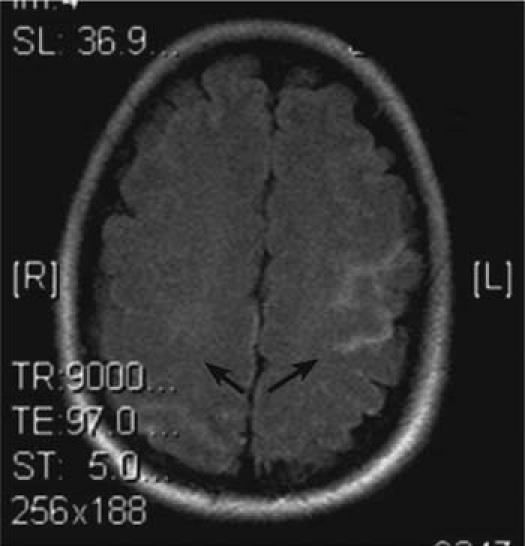
Fig. 1 Magnetic resonance image of the brain shows bilateral acute cortical infarcts in a watershed distribution (arrows).
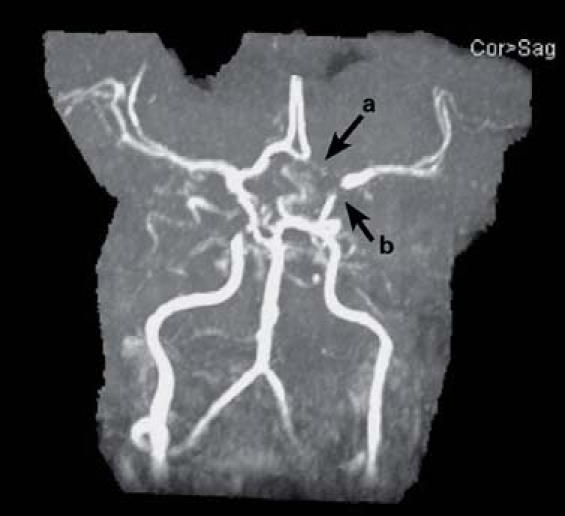
Fig. 2 Magnetic resonance angiogram of the brain shows a hypoplastic A1 segment of the left anterior cerebral artery (a) and nearly complete narrowing of the distal left internal carotid artery at the bifurcation (b).
Discussion
The incidence of SMS is unknown but may be as high as 1 in 25,000 births.3 Most SMS patients have a deletion of the SREBF1 (sterol regulatory element binding protein 1) gene for a transmembrane transcription factor that regulates the LDL receptor involved in cholesterol homeostasis.4 However, this deletion has not previously been associated with premature atherosclerosis. Smith and colleagues4 studied fasting lipid profiles in children with SMS and found that 57% of SMS patients had lipid values greater than the 95th percentile for age and sex. They concluded that hypercholesterolemia is common in SMS and may serve as a useful early clinical biochemical marker of the syndrome. Our patient's lipid levels were not particularly high, and she did not have any other risk factors associated with atherosclerosis, such as diabetes, hypertension, or a habit of smoking. We believe that there may be something unique about lipid metabolism in these patients that results in accelerated cerebrovascular atherosclerosis, despite apparently normal triglyceride levels. Our patient suffered an ischemic infarct secondary to the severe atherosclerotic disease that was found in her intracranial circulation. She was likely hypoperfused during the operation despite our maintaining a mean arterial pressure of 40 to 50 mmHg during perfusion.
We suggest that all patients with SMS who require open-heart surgery in adolescence or adulthood be evaluated for possible premature cerebrovascular disease before the surgery. If evidence of significant atherosclerotic disease is found, higher pressures should be maintained to perfuse vital organs. Further, because SMS patients can have a normal life expectancy (the oldest known person with SMS was 80 years old), screening for atherosclerotic disease and subsequent intervention may be crucial in achieving full life expectancy for these patients.
Footnotes
Address for reprints: Alyas P. Chaudhry, MD, Brown University Division of Cardiothoracic Surgery, Rhode Island Hospital, 593 Eddy St., Providence, RI 02903. E-mail: calyas@hotmail.com
References
- 1.Greenberg F, Lewis RA, Potocki L, Glaze D, Parke J, Killian J, et al. Multi-disciplinary clinical study of Smith-Magenis syndrome (deletion 17p11.2). Am J Med Genet 1996;62:247–54. [DOI] [PubMed]
- 2.Smith AC, McGavran L, Robinson J, Waldstein G, Macfarlane J, Zonona J, et al. Interstitial deletion of (17)(p11.2p11.2) in nine patients. Am J Med Genet 1986;24:393–414. [DOI] [PubMed]
- 3.Greenberg F, Guzzetta V, Montes de Oca-Luna R, Magenis RE, Smith AC, Richter SF, et al. Molecular analysis of the Smith-Magenis syndrome: a possible contiguous-gene syndrome associated with del(17)(p11.2). Am J Hum Genet 1991; 49:1207–18. [PMC free article] [PubMed]
- 4.Smith AC, Gropman AL, Bailey-Wilson JE, Goker-Alpan O, Elsea SH, Blancato J, et al. Hypercholesterolemia in children with Smith-Magenis syndrome: del (17) (p11.2p11.2). Genet Med 2002;4:118–25. [DOI] [PubMed]