

Abstract
Friedreich's ataxia is the most common hereditary neurodegenerative disorder, and more than half of all patients show echocardiographic evidence of cardiomyopathy. Although angina has been reported in these patients, the role of coronary artery disease has previously been dismissed and is therefore underestimated. Premature obstructive coronary disease has rarely been angiographically demonstrated in patients with Friedreich's ataxia. We present an unusual case of a 35-year-old woman with Friedreich's ataxia who presented with intermittent chest pressure associated with dyspnea and diaphoresis. Cardiac catheterization revealed a chronically occluded left circumflex coronary artery and a high-grade stenosis of the left anterior descending coronary artery. A Cypher® stent, placed within the left anterior descending artery, left no residual stenosis. This case illustrates the importance of fully investigating anginal symptoms in patients with Friedreich's ataxia, because coronary artery disease is likely underdiagnosed in this population. Early diagnosis may permit aggressive management and may delay the progression to end-stage cardiomyopathy.
Key words: Cardiomyopathy, ischemic; coronary artery disease, premature/etiology; disease progression; European continental ancestry group/genetics; Friedreich ataxia/complications/genetics/physiopathology; stent, drug-eluting; trinucleotide repeats
Friedreich's ataxia is the most common hereditary neurodegenerative disorder, and more than half of all patients show echocardiographic evidence of cardiac involvement.1 Although angina has been reported in these patients, the role of coronary artery disease has previously been dismissed and is therefore underestimated. Rarely has premature obstructive coronary artery disease been angiographically demonstrated in patients with Friedreich's ataxia. We present an unusual case of a 35-year-old woman with Friedreich's ataxia who presented with intermittent chest pressure associated with dyspnea and diaphoresis.
Case Report
The patient was a 35-year-old white woman with a history of Friedreich's ataxia and associated quadriplegia, hypertension, diabetes mellitus, and dyslipidemia (this last controlled with medical therapy). She was wheelchair-bound and a nonsmoker, with no family history of premature coronary artery disease. In December 2005, she presented to her primary care physician with a 3-day history of intermittent chest discomfort, characterized as a nonradiating sensation of pressure in association with dyspnea and diaphoresis, which had lasted for 5 minutes. She was afebrile, with a heart rate of 65 beats/min and blood pressure of 115/70 mmHg. The cardiac examination revealed normal 1st and 2nd heart sounds, a positive 4th heart sound, and no appreciable murmurs. The 12-lead electrocardiogram showed an ectopic atrial rhythm with low-voltage QRS, a rightward axis, a slightly prolonged QT interval, and nonspecific ST-T wave changes. Evaluation of the patient's lipids revealed total cholesterol of 153 mg/dL, triglycerides of 85 mg/dL, high-density lipoprotein cholesterol of 58 mg/dL, and low-density lipoprotein cholesterol of 78 mg/dL. The hemoglobin A1C (glycated hemoglobin) result was 8%. The rest of her laboratory results were normal.
Echocardiography, performed at an outlying hospital, revealed concentric left ventricular hypertrophy with inferoposterior hypokinesis. Left ventricular function was mildly depressed, with an overall estimated ejection fraction of 0.50. The patient then underwent an outpatient dipyridamole-sestamibi myocardial perfusion study, which revealed reversible inferolateral and anterolateral stress defects.
She was subsequently referred to our facility for elective cardiac catheterization. Angiography revealed 2 serial 90% lesions in the proximal mid portion of the left anterior descending coronary artery (LAD), chronic total occlusion of the proximal circumflex coronary artery, and luminal irregularities in the dominant right coronary artery (Fig. 1). The circumflex artery was filled by extensive, grade-III, right-to-left collateral vessels. We successfully opened the lesion in the proximal mid LAD by means of a 3.0-mm × 23-mm Cypher® stent (Cordis Corporation, a Johnson & Johnson company; Miami Lakes, Fla), leaving no residual stenosis (Fig. 2). The patient's medical management was optimized with aspirin, clopidogrel, metoprolol, lisinopril, and high-dose atorvastatin. She was discharged after a brief, uneventful hospital stay.
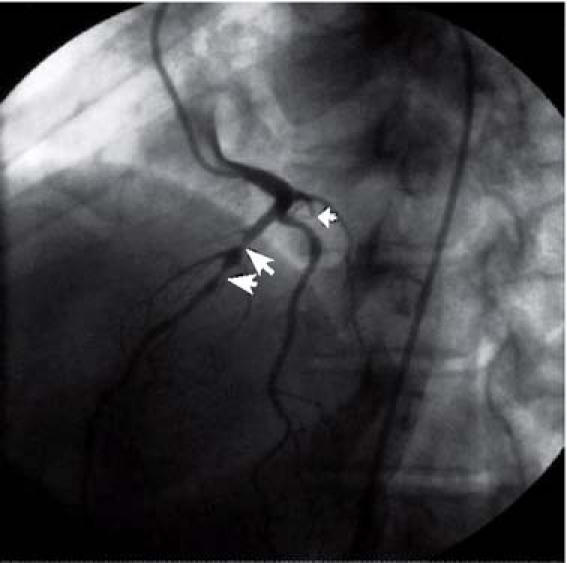
Fig. 1 Angiography revealed 2 serial 90% lesions in the proximal mid portion of the left anterior descending coronary artery (large arrows), chronic total occlusion of the proximal circumflex coronary artery (small arrow), and luminal irregularities in the dominant right coronary artery.
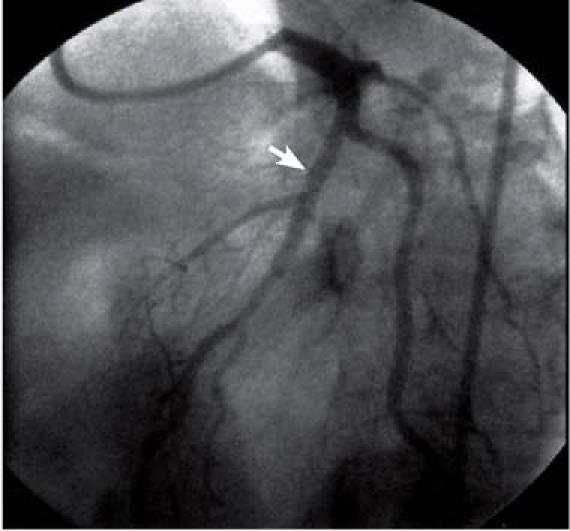
Fig. 2 The sequential lesions in the proximal-to-mid portion of the left anterior descending coronary artery were treated successfully by means of a single 3.0-mm × 23-mm Cypher® stent (arrow), which left no residual stenosis.
Two months later, the patient presented to the emergency department, again with chest discomfort. On this occasion, laboratory evaluation revealed an elevated troponin T level of 0.16 ng/mL (normal, <0.02 ng/mL). Her electrocardiogram was unchanged from her post-treatment results upon discharge. A repeat echocardiogram revealed global hypokinesis and a left ventricular ejection fraction of approximately 0.35. There was marked asymmetric septal hypertrophy (2 cm). Repeat coronary angiography revealed a widely patent LAD stent and no substantial change in the coronary anatomy (Fig. 3). Due to the rapid deterioration of left ventricular function, the patient was referred to a tertiary center for a 2nd opinion. There, she was thought to have a “burnt-out cardiomyopathy” with a very poor prognosis. Medical therapy alone was recommended, because it was believed, after a review of the current literature, that further aggressive measures, such as placement of an automatic implantable cardioverter-defibrillator (AICD), were not warranted due to poor life expectancy.
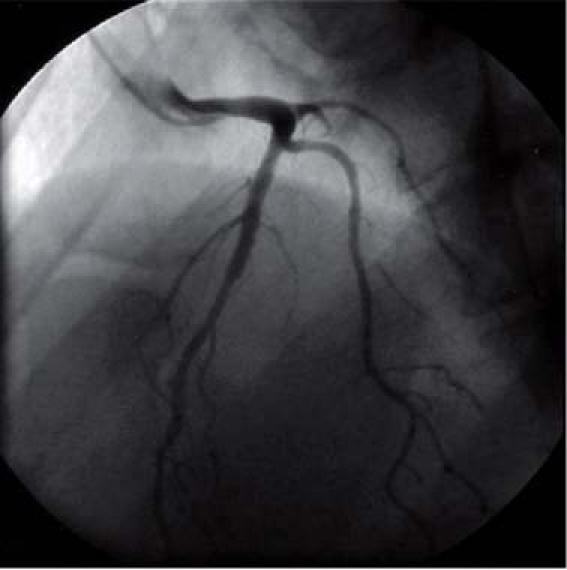
Fig. 3 Repeat coronary angiography, 3 months after the initial procedure, revealed patency of the stent in the left anterior descending coronary artery and no interim changes in the other coronary arteries.
Discussion
Friedreich's ataxia is a neurodegenerative disease with autosomal recessive inheritance. It is the most common hereditary ataxia, and it is characterized by progressive gait and limb ataxia, loss of lower-extremity tendon reflexes, loss of proprioception, and dysarthria.2 Cardiac involvement is highly prevalent in these patients. Onset of Friedreich's ataxia usually occurs during puberty, and survival beyond the 3rd decade is unusual. Trials with antioxidants have demonstrated reductions in left ventricular mass, but no net survival benefit.3 Most patients succumb to respiratory failure before their 4th decade of life.
The molecular defect in Friedreich's ataxia is a mutation in the frataxin gene localized to chromosome 9q13.2 Frataxin is a mitochondrial protein found in higher amounts in the brain, heart, and pancreas than in other organs of the body. It is involved in iron homeostasis and is an antioxidant. The mutation results in the expansion of trinucleotide (GAA) repeats in the intron 1.4 The GAA repeat expansion is found chiefly in persons of European, North African, Middle Eastern, and Indian descent.5 Among whites, the disease occurs with a frequency of 1 in 50,000.4 The number of repeats is variable and appears to correlate with the severity of clinical manifestations. At increased repeat expansions, there is depressed transcription and expression of the frataxin protein.6 Its deficiency results in mitochondrial iron accumulation and in reduced antioxidant defenses.7,8 Patients with small repeat expansions do not have cardiomyopathy. Their disease is characterized by early-onset cerebellar ataxia and retained reflexes.6
Clinically, patients most commonly present with progressive neurodegeneration, as stated above. The disease first manifests in the dorsal root ganglia, progresses to the sensory posterior columns, spinocerebellar tracts, and corticospinal tracts, and finally affects the large sensory fibers in the peripheral nerves.2 Cardiac involvement is seen in more than 90% of patients9 and is thought to be independent of the primary neurodegenerative process (that is, the degree of neurodegeneration does not predict cardiac involvement). In fact, 5 of the 6 patients originally described by Friedreich had cardiac involvement. Such involvement can be detected by electrocardiography and echocardiography long before clinical manifestation. Common electrocardiographic findings include left ventricular hypertrophy, abnormal ventricular repolarization, ischemic changes, atrial tachycardia, and atrioventricular block.9 Three disease manifestations may be seen on echocardiographic examination: concentric left ventricular hypertrophy, asymmetric septal hypertrophy, and dilated cardiomyopathy with depressed left ventricular function.1,10 The most common cardiac manifestation is a hypertrophic cardiomyopathy.11 The progression of cardiomyopathy to heart failure is the most common cause of death in these patients. Arrhythmias occur less often than might be expected and are usually supraventricular in origin. Ventricular tachycardia has been reported with the much less common dilated cardiomyopathy but is not known to be associated with the hypertrophic form. The disparity between Friedreich's ataxia and other forms of hypertrophic cardiomyopathy in the occurrence of ventricular tachycardia may relate to the differences in histologic abnormalities.12
Angina has been reported in patients with Friedreich's ataxia, although no correlation with coronary disease has been shown.9,13 Initial necroscopic studies found luminal narrowing in only 9% of coronary arteries examined.14 This led to the dismissal of coronary artery disease as a contributor to cardiac symptoms or to the progression of the cardiomyopathy. Subsequent studies, however, have found evidence of coronary artery occlusions, mainly in small-caliber vessels, that were thought to be caused mainly by fibromuscular dysplasia.15 The functional significance of coronary lesions in Friedreich's ataxia, therefore, has yet to be clearly delineated.
We have presented the case of a young woman with typical anginal symptoms, multiple coronary artery disease risk factors, and Friedreich's ataxia. She was found, on coronary angiography, to have severe epicardial coronary artery disease and subsequently to have rapid, progressive deterioration of her cardiac function, despite restoration of epicardial blood flow. To the best of our knowledge, only 4 cases of acute myocardial infarction in association with Friedreich's ataxia have been described in the world literature,16–19 and only 1 of these cases was associated with angiographically proven epicardial coronary lesions.19 Although the development of premature epicardial coronary disease in our young patient might be attributable to her traditional coronary risk profile, Friedreich's ataxia cannot be dismissed as a possible cause. It is quite possible that the disease exponentially augments the effect of other traditional cardiac risk factors. The question of Friedreich's ataxia as a cause of coronary artery disease was raised in the case reported by Sharma and colleagues,19 but they suspected that dyslipidemia and hyperhomocysteinemia were also strong contributors to the disease in their patient. A causal relationship between coronary artery disease and Friedreich's ataxia would be nearly impossible to demonstrate statistically, due to the low number of cases. However, it is important to identify traditional risk factors in patients with Friedreich's ataxia and to initiate aggressive medical therapy. Although sudden cardiac death may occur, it is an infrequently described cause of death. The role of prophylactic placement of AICDs in this subgroup of patients has not yet been studied, but such placement may warrant investigation.
Footnotes
Address for reprints: Gregory R. Giugliano, MD, Division of Cardiology, Baystate Medical Center, 759 Chestnut St., Springfield, MA 01199. E-mail: gregory.giugliano@bhs.org
References
- 1.Alboliras ET, Shub C, Gomez MR, Edwards WD, Hagler DJ, Reeder GS, et al. Spectrum of cardiac involvement in Friedreich's ataxia: clinical, electrocardiographic and echocardiographic observations. Am J Cardiol 1986;58:518–24. [DOI] [PubMed]
- 2.Campuzano V, Montermini L, Molto MD, Pianese L, Cossee M, Cavalcanti F, et al. Friedreich's ataxia: autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science 1996;271:1423–7. [DOI] [PubMed]
- 3.Schols L, Meyer CH, Schmid G, Wilhelms I, Przuntek H. Therapeutic strategies in Friedreich's ataxia. J Neural Transm Suppl 2004;68:135–45. [DOI] [PubMed]
- 4.Groh WJ, Zipes DP. Neurological disorders and cardiovascular disease. In: Zipes DP, Libby P, Bonow RO, Braunwald E, editors. Braunwald's heart disease: a textbook of cardiovascular medicine. 7th ed. Philadelphia: Elsevier Saunders; 2005. p. 2153–4.
- 5.Labuda M, Labuda D, Miranda C, Poirier J, Soong BW, Barucha NE, Pandolfo M. Unique origin and specific ethnic distribution of the Friedreich ataxia GAA expansion. Neurology 2000;54:2322–4. [DOI] [PubMed]
- 6.Durr A, Cossee M, Agid Y, Campuzano V, Mignard C, Penet C, et al. Clinical and genetic abnormalities in patients with Friedreich's ataxia. N Engl J Med 1996;335:1169–75. [DOI] [PubMed]
- 7.Schulz JB, Dehmer T, Schols L, Mende H, Hardt C, Vorgerd M, et al. Oxidative stress in patients with Friedreich ataxia. Neurology 2000;55:1719–21. [DOI] [PubMed]
- 8.Babcock M, de Silva D, Oaks R, Davis-Kaplan S, Jiralerspong S, Montermini L, et al. Regulation of mitochondrial iron accumulation by Yfh1p, a putative homolog of frataxin. Science 1997;276:1709–12. [DOI] [PubMed]
- 9.Albano LM, Nishioka SA, Moyses RL, Wagenfuhr J, Bertola D, Sugayama SM, Chong AK. Friedreich's ataxia: cardiac evaluation of 25 patients with clinical diagnosis and literature review [in English, Portuguese]. Arq Bras Cardiol 2002; 78:444–51. [DOI] [PubMed]
- 10.Morvan D, Komajda M, Doan LD, Brice A, Isnard R, Seck A, et al. Cardiomyopathy in Friedreich's ataxia: a Doppler-echocardiographic study. Eur Heart J 1992;13:1393–8. [DOI] [PubMed]
- 11.Child JS, Perloff JK, Bach PM, Wolfe AD, Perlman S, Kark RA. Cardiac involvement in Friedreich's ataxia: a clinical study of 75 patients. J Am Coll Cardiol 1986;7:1370–8. [DOI] [PubMed]
- 12.Zimmermann M, Gabathuler J, Adamec R, Pinget L. Unusual manifestations of heart involvement in Friedreich's ataxia. Am Heart J 1986;111:184–7. [DOI] [PubMed]
- 13.Ferres-Sanchez P, Subirana-Domenech M, Torner-Soler M. Chest pain during exercise as first manifestation of Friedreich's ataxia. Br Heart J 1995;74:464–7. [DOI] [PMC free article] [PubMed]
- 14.Hewer R. The heart in Friedreich's ataxia. Br Heart J 1969; 31:5–14. [DOI] [PMC free article] [PubMed]
- 15.James TN, Cobbs BW, Coghlan HC, McCoy WC, Fisch C. Coronary disease, cardioneuropathy, and conduction system abnormalities in the cardiomyopathy of Friedreich's ataxia. Br Heart J 1987;57:446–57. [DOI] [PMC free article] [PubMed]
- 16.Krongrad E, Joos HA. Friedreich's ataxia in childhood: case report with possible myocardial infarction, cerebrovascular thromboembolization, and persistent elevation of cardiac specific LDH. Chest 1972;61:644–8. [DOI] [PubMed]
- 17.Hewer RL. Study of fatal cases of Friedreich's ataxia. Br Med J 1968;3:649–52. [DOI] [PMC free article] [PubMed]
- 18.Calvo Iglesias FE, Permanyer Miralda G, Blanco Castineiras J, Rodes Cabau J, Anivarro Blanco I, Soler Soler J. Acute myocardial infarct and Friedreich's disease [in Spanish]. Rev Esp Cardiol 1995;48:145–7. [PubMed]
- 19.Sharma AK, Kiyokawa M, Kim ET, Lee DT, Kasuya R. Acute myocardial infarction and Friedreich's ataxia. Hawaii Med J 2002;61:199–201, 212. [PubMed]