

Abstract
Parkinson’s disease (PD) is a neurodegenerative disorder characterized by the loss of dopaminergic neurons in the substantia nigra pars compacta (SNpc). With the exception of a few rare familial forms of the disease, the precise molecular mechanisms underlying PD are unknown. Inflammation is a common finding in the PD brain, but due to the limitation of post mortem analysis its relationship to disease progression cannot be established. However, studies using the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) model of PD have also identified inflammatory responses in the nigrostriatal pathway that precede neuronal degeneration in the SNpc. To assess the pathological relevance of these inflammatory responses and to identify candidate genes that might contribute to neuronal vulnerability, we used quantitative real-time PCR to measure mRNA levels of 11 cytokine and chemokine encoding genes in the striatum of MPTP-sensitive (C57BL/6J) and MPTP-insensitive (SWR) mice following administration of MPTP. The mRNA levels of all 11 genes changed following MPTP treatment, indicating the presence of inflammatory responses in both strains. Furthermore, of the 11 genes examined only three, Il-6, Mip-1α/Ccl3 and Mip-1β/Ccl4, were differentially regulated between C57BL/6J and SWR mice. In both mouse strains, the level of Mcp-1/Ccl2 mRNA was the first to increase following MPTP administration, and might represent a key initiating component of the inflammatory response. Using Mcp-1/Ccl2 knockout mice backcrossed onto a C57BL/6J background we found that MPTP-stimulated Mip-1α/Ccl3 and Mip-1β/Ccl4 mRNA expression was significantly lower in the knockout mice; suggesting that Mcp-1/Ccl2 contributes to MPTP-enhanced expression of Mip-1α/Ccl3 and Mip-1β/Ccl4. However, stereological analysis of SNpc neuronal loss in Mcp-1/Ccl2 knockout and wild-type mice showed no differences. These findings suggests that it is the ability of dopaminergic SNpc neurons to survive an inflammatory insult, rather than genetically determined differences in the inflammatory response itself, that underlie the molecular basis of MPTP resistance.
Keywords: cytokine/chemokine, tumor necrosis factor α, stromal derived factor 1 macrophage inflammatory protein 1, Tnf-like weak inducer of apoptosis, fibroblast growth factor-inducible 14, interleukin 6, fractalkine, fractalkine receptor, monocyte chemoattractant protein 1
Parkinson’s disease (PD) is a progressive age-related neurodegenerative disorder characterized by the loss of dopaminergic neurons in the substantia nigra pars compacta (SNpc) (Trétiakoff, 1919, Hassler, 1938, Greenfield and Bosanquet, 1953). Although PD was described almost two centuries ago (Parkinson, 1817), the precise mechanisms underlying its pathology remain enigmatic. Studies of PD patients as well as experimental animal models of PD employing the neurotoxins 6-hydroxydopamine (6-OHDA) and 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), have suggested an etiological mechanism that involves inflammatory processes (Kurkowska-Jastrzebska et al., 1999, Barcia et al., 2003, Carvey et al., 2005).
Cytokines and chemokines are small molecules (8–12 kDa) whose function is to recruit immunocompetent cells to the site of inflammation and modulate their activity (Bacon and Harrison, 2000, Murphy et al., 2000, Bajetto et al., 2002, Laing and Secombes, 2004, Adler et al., 2005). Although these molecules were first identified in the immune system they are also present in the central nervous system (CNS), where they are secreted from microglia, neurons and astrocytes (Banisadr et al., 2005c, Cartier et al., 2005, Biber et al., 2006, Minami et al., 2006). Moreover elevated levels of cytokines such as tumor necrosis factor alpha (Tnf-α), interleukin 6 (Il-6), interleukin 1 beta and interferon gamma (Ifn-γ) have been observed in the SNpc of PD patients and rodents treated with 6-OHDA and MPTP (Grunblatt et al., 2000, Mandel et al., 2000, Nagatsu et al., 2000, Nagatsu and Sawada, 2005, Sriram et al., 2006b). Therefore, these molecules may be actively involved in disease progression.
Several studies also sought to correlate polymorphisms in the promoters of several cytokine genes to the risk of developing PD (Nishimura et al., 2000, Schulte et al., 2002, Nishimura et al., 2003, Huerta et al., 2004, Ross et al., 2004, Hakansson et al., 2005). Furthermore, retrospective clinical studies in populations subjected to chronic use of non steroidal anti-inflammatory drugs (NSAIDs) suggest that some of these agents could lower the incidence of PD (Chen et al., 2003, Schiess, 2003, Chen et al., 2005, Hernan et al., 2006). Animal experiments with NSAIDs came to similar conclusions (Ferger et al., 1999, Sairam et al., 2003, Maharaj et al., 2004), and microglia inactivation was also shown to be neuroprotective in the MPTP model of PD (Wu et al., 2002). Despite accumulating evidence of inflammatory responses in PD and its animal models, the question of whether inflammation is a negative or positive contributor to the progression of the disorder, or simply a reaction to neurodegeneration remains unanswered (Marchetti and Abbracchio, 2005).
To evaluate the potential role of cytokines and chemokines in the pathogenesis of PD, we studied the temporal profile of their mRNA expression by quantitative real-time PCR in the striatum in the mouse MPTP model. This region of brain is innervated by the dopaminergic neurons that are lost in PD and several lines of evidence suggest that the dopaminergic nerve endings in the striatum are the primary site of pathological changes in PD and animal models of the disorder (Bradbury et al., 1986, Eberling et al., 1997, Nurmi et al., 2001, Rinne et al., 2001). One view is that the nerve endings of SNpc dopaminergic neurons in the striatum are initially damaged or compromised and over time this leads to the retrograde degeneration of the neurons in the SNpc (Burns et al., 1984, Bradbury et al., 1986, Eberling et al., 1997). This is particularly evident for MPTP, which selectively accumulates in striatal dopaminergic nerve endings via uptake through the high affinity dopamine transporter (Chiba et al., 1985, Javitch et al., 1985, Gainetdinov et al., 1997, Bezard et al., 1999). This has rapid metabolic consequences and within 24 hours of MPTP treatment the nerve endings already show morphological changes indicative of degeneration that precede by several days degeneration in the cell soma in the SNpc (Jackson-Lewis et al., 1995). Therefore, events occurring in the striatum in the MPTP model in particular are taken to be representative of early stages of any pathological mechanism involved in the degenerative process.
Two strains of mice, Swiss Webster (SWR) and C57BL/6J, that differ in their sensitivity toward MPTP toxicity (German et al., 1996, Hamre et al., 1999), were used to assess if there were quantitative changes in inflammation-associated gene products related to the pathogenic changes triggered by MPTP. In C57BL/6J mice, acute systemic administration of MPTP results in a loss of dopaminergic neurons in the SNpc, whereas in SWR mice only minimal cell loss is evident (German et al., 1996, Hamre et al., 1999). The reason for this genetically-determined difference is currently unknown, although astrocytes and microglia have been implicated (Smeyne et al., 2001, Smeyne et al., 2005). Here, we show that acute administration of MPTP triggers marked changes in mRNA levels for a number of cytokines and chemokines as well as their receptors. Of the cytokines examined, none demonstrate changes only in the MPTP-sensitive C57BL/6J strain. We have focused on Mcp-1/Clc2 as a potential early mediator of MPTP-induced neurodegeneration and Mip-1α/Ccl3 and Mip-1β/Ccl4 as transcriptional targets for this cytokine. This has been achieved by assessing striatal inflammation-associated gene expression and SNpc neuronal degeneration in wild type and Mcp-1/Ccl2 knockout mice.
Experimental Procedures
Animals, experiments and substances
Female C57BL/6J mice, SWR mice and Mcp-1/Ccl2 homozygous knockout mice (B6.129S4-Ccl2tm1Rol/J) were purchased from Jackson Laboratories (Bar Harbor, Maine). Animals were housed on a 12:12 light:dark schedule with free access to food and water.
At 3–4 months of age animals were administered a single dose (40 mg/kg i.p.) of MPTP-HCl (Sigma-Aldrich, St Louis, Missouri) dissolved in sterile saline. In another set of experiments, the efficacy of MPTP to induce SNpc DA neuron death was evaluated using a well-characterized protocol (20 mg/kg MPTP given i.p. every two hours for a total of 4 injections) (Hamre et al., 1999). All studies were approved by the St. Jude Children’s Research Hospital Animal Care and Use Committee (ACUC) and were conducted in accordance with the National Institute of Health Guide for the Care and Use of Laboratory Animals (NIH Publication No. 80-23, revised 1996).
Dissection of tissue and extraction of total RNA
Animals were sacrificed by cervical dislocation at specific time points and the striatum was rapidly dissected on ice and immediately transferred to dry ice to preserve RNA integrity. Trizol® (Invitrogen, Carlsbad) extraction of total RNA was performed according to manufacturer instructions. Total RNA was quantified by spectrophotometry (UltraSpec 2100 Pro, Amersham) and its integrity established by formaldehyde agarose gel electrophoresis. Samples that appeared degraded were discarded.
Real time PCR and preparation of standards
Total RNA was reverse transcribed using a TaqMan® reverse transcription kit (Applied Biosystems, Foster City, CA) according to manufacturer instructions. Primers and fluorogenic probes for real-time PCR were designed with Primer Express Software version 1.5 for Macintosh (Applied Biosystems, Foster City, CA) and prepared by the Hartwell Center for Bioinformatics and Biotechnology (St Jude Children’s Research Hospital, Memphis TN). The following primer/probe sets were used (underlined nucleotides in the probe sequence indicate the exon-exon boundary): Fn14/Tnfrsf12a (NM_013749) forward 5′-GCC GCC GGA GAG AAA AG-3′, probe 5′-Fam-TTA CTA CCC CCA TAG AGG AGA CTG GTG GAG AG-BHQ -3′, reverse 5′-GCC ACA CCT GGG CAG C-3′; Gadph (NM_001001303) forward 5′-TGG ATC TGA CGT GCC GC-3′, probe 5′-Fam-TGG AGA AAC CTG CCA AGT ATG ATG ACA TCA-BHQ -3′, reverse 5′-TGC CTG CTT CAC CAC CTT C-3′; Tnfα (NM_013693) forward 5′-TCT ATG GCC CAG ACC CTC AC-3′, probe 5′-Fam-CTC AGA TCA TCT TCT CAA AAT TCG AGT GAC AAG C-BHQ-3′, reverse 5′-TTG CTA CGA CGT GGG CTA CA-3′; Mcp-1/Ccl2 (NM_011333) forward 5′-GAG CAT CCA CGT GTT GGC T-3′, probe 5′-Fam-AGC CAG ATG CAG TTA ACG CCC CAC T-BHQ -3′, reverse 5′-TGG TGA ATG AGT AGC AGC AGG T-3′; Tweak/Tnfsf12 (NM_011614) forward 5′-CCT CGC CGA GCT ATT GCA-3′, probe 5′-Fam-CCC ATT ATG AGG TTC ATC CTC GGC CA-BHQ-3′, reverse 5′-TGC TTG TGC TCC ATC CTG TC-3′; Sdf1/Cxcl12 (NM_021704) forward 5′-CCG CGC TCT GCA TCA GT-3′, probe 5′-Fam-ACG GTA AAC CAG TCA GCC TGA GCT ACC G-BHQ -3′, reverse 5′-TCG AAG AAC CGG CAG GGG-3′; Mip-1α/Ccl3 (NM_011337) forward 5′-CAT GAC ACT CTG CAA CCA AGT CTT-3′, probe 5′-Fam-CCA TAT GGA GCT GAC ACC CCG ACT G-BHQ -3′, reverse 5′-TCC GGC TGT AGG AGA AGC A-3′; Mip-1β/Ccl4 (NM_013652) forward 5′-TGC TCG TGG CTG CCT TCT-3′, probe 5′-Fam-CTC CAG GGT TCT CAG CAC CAA TGG G-BHQ -3′, reverse 5′-CAG GAA GTG GGA GGG TCA GA-3′; Il-6 (NM_031168) forward 5′-TGT TCT CTG GGA AAT CGT GGA-3′, probe 5′-Fam-ATG AGA AAA GAG TTG TGC AAT GGC AAT TCT G-BHQ -3′, reverse 5′-AAG TGC ATC ATC GTT GTT CAT ACA-3′; Fractalkine/Cx3cl1 (NM_009142) forward 5′-CCG CGT TCT TCC ATT TGT GT-3′, probe 5′-Fam-CTG CCG GGT CAG CAC CTC GG-BHQ -3′, reverse 5′-GCA CAT GAT TTC GCA TTT CG-3′; Fractalkine receptor/Cx3cr1 (NM_009987) forward 5′-TGG GTG AGT GAC TGG CAC TTC-3′, probe 5′-Fam-CCT TCC CAT CTG CTC AGG ACC TCA CC-BHQ -3′, reverse 5′-CAT ACT CAA AAT TCT TTA GAT CCA GTT CA-3′; Cxcr4 (NM_009911) forward 5′-CAC CAC GGC TGT AGA GCG A-3′, probe 5′-Fam-TGT TGC CAT GGA ACC GAT CAG TGT GAG TAT ATA-BHQ -3′, reverse 5′-CTC CAG AAC CCA CTT CTT CAG AGT-3′. The primer/probe set for Sdf1/Cxcl12 does not discriminate for its three isoforms.
PCR was performed using TaqMan® PCR Core Reagent Kit (Applied Biosystems, Foster City, CA) according to manufacturer guidelines, using an ABI Prism 7900HT (Applied Biosystems, Foster City, CA). PCR conditions were: UNG incubation, 50 °C for 2 minutes; AmpliTaq activation 95 °C for 10 minutes; PCR denaturation step 95 °C for 15 seconds; PCR annealing step 60 °C for 1 minute; 40 cycles of PCR were performed. PCR results were normalized versus GAPDH.
Absolute quantification was performed with standard curves obtained cloning the gene of interest into pcDNA3 plasmid (Invitrogen, Carlsbad, CA) as explained hereafter: a first round of PCR with primers flanking the region of interest was performed with Platinum® Pfx proof reading polymerase (Invitrogen, Carlsbad, CA). The PCR product was gel purified with QIAquick® Gel Extraction Kit (Qiagen, Valencia, CA) and amplified a second time with Platinum® Pfx using the same primers to which sites for restriction enzymes were attached. The product was gel purified, cut and ligated with T4 DNA Ligase (Invitrogen, Carlsbad, California) into pcDNA3. Plasmids were extracted with QIAfilter® Plasmid Maxi Kit (Qiagen, Valencia, California) according to manufacturer instruction and sequenced by the Hartwell Center for Bioinformatics and Biotechnology (St Jude Children’s Research Hospital, Memphis TN) that also provided sequencing primers Sp6 and T7. Only those constructs with proper insert were used as standards. The following is a list of the primers used (restriction enzymes used are reported in parentheses, and their recognized sequence is underlined): Gapdh forward (BamHI)-CGC GGA TCC GCG ATG GTG AAG GTC GGT GTG AA, reverse (EcoRI)-CCG GAA TTC CGG TTC TTA CTC CTT GGA GGC CA; Fn14/Tnfrsf12a forward (BamHI)-CGC GGA TCC GCG GCA ATC ATG GCT CCG GGT TG, reverse (EcoRI)-CCG GAA TTC CGG TGA ATC ACC ACC TCG CCC CA; Tweak/Tnfsf12 forward (Hindi)-CCC AAG CTT GGG ATG GCC GCC CGT CGG AGC CA, reverse (XhoI)-CCG CTC GAG CGG GAG AGC AAG GCC CCT CAG TG; Sdf-1/Cxcl12 forward (EcoRI)-CCG GAA TTC CGG ATG GAC GCC AAG GTC GTC GC, reverse (XhoI)-CCG CTC GAG CGG TAT AAC TGT GCC CAG AGG CCC CAG; Mip-1α/Ccl3 forward (BamHI)-CGC GGA TCC GCG ATG AAG GTC TCC ACC ACT GC, reverse (EcoRI)-CCG GAA TTC CGG AGA CTC TCA GGC ATT CAG TT; Mip-1β/Ccl4 forward (BamHI)-CGC GGA TCC GCG ATG AAG CTC TGC GTG TCT GC, reverse (EcoRI)-CCG GAA TTC CGG GCT GGA GCT GCT CAG TTC AA; Il-6 forward (BamHI)-CGC GGA TCC GCG ATG AAG TTC CTC TCT GCA AG, reverse (EcoRI)-CCG GAA TTC CGG CTA GGT TTG CCG AGT AGA TC; fractalkine/Cx3cl1 forward (KpnI)-CGG GGT ACC CCG ATG GCT CCC TCG CCG CTC GC, reverse (XhoI)-CCG CTC GAG CGG CAC AGG CAG GCA GGC AAG CAG CT; Mcp-1/Ccl2 forward (BamHI)-CGC GGA TCC GCG TCT CTT CCT CCA CCA CCA TG, reverse (EcoRI)-CCG GAA TTC CGG GCA TCA CAG TCC GAG TCA CA; Tnf-α forward (BamHI)-CGC GGA TCC GCG ATG AGC ACA GAA AGC ATG AT, reverse (EcoRI)-CCG GAA TTC CGG CTT CAC AGA GCA ATG ACT CC. Amplicons for Cx3cr1 and Cxcr4 were cloned directly into pCR®II-TOPO® vector (Invitrogen, Carlsbad, CA) using their corresponding primers for real time PCR. Products were gel purified, incubated with Taq polymerase (QIAGEN, Valencia, CA) and dATP at 72 °C for 10 minutes to add overhanging As and ligated. Plasmids were extracted as previously described.
Tyrosine hydroxylase staining and cell counting
Animals were given an overdose of 2,2,2-tribromoethanol and when deeply anesthetized, were transcardially perfused with 0.1 M phosphate-buffered saline (PBS) followed by 4% paraformaldehyde (PFA). Whole brains were removed and postfixed in fresh 4% PFA overnight at 4 °C and embedded in Paraplast Plus ® (Surgipath Medical, Richmond, IL). Serial sagittal sections through the SNpc were cut at 10 μm and collected on poly-ionic glass microscope slides (Superfrost Plus, Fisher). The slides were processed for DA neuron visualization using a polyclonal TH antibody (1:250; Pel-Freeze, Rogers, AR) as previously described (Hamre et al., 1999).
The total number of SNpc neurons was estimated with the optical fractionator method (West et al., 1991) using the Bioquant Nova Image Analysis System (R&M Biometrics, Nashville, TN, USA). In C57BL/6J mice and the B6.129S4-Ccl2tm1Rol/J mice, the average rostral–caudal length of the SNpc was empirically determined to be approximately 1 mm. After randomly choosing the 1st section, every 5th section was sampled throughout the entire anterior to posterior extent of the SNpc (German et al., 1996). On average, 40 sections per SNpc were analyzed. The sampling frame used in the analysis was 5625 square microns and we counted a minimum of 15 frames in each SNpc per section. The SNpc in each section was outlined at low power (2.5X) while SNpc neurons were counted using high magnification (40X). Neurons were counted if they had a TH-positive soma with a clear nucleus; and if they were within the counting frame or touched the line of inclusion. Neurons that touched the line of exclusion were not counted. On occasion, SNpc neurons that were Nissl-positive, but TH-negative were observed. These cells were included in the total cell number count. SNpc dopaminergic neurons were counted separately on both the right and left sides of the midbrain and added together to give a final cell number.
Statistical analysis
Statistical analysis was performed with GraphPad Prism® version 4.03 for Windows® (GraphPad Software Inc., San Diego, CA). Each sample from a single animal was analyzed in triplicate and results were expressed as the average of 3 to 5 animals. The temporal profile of each gene was analyzed by one-way ANOVA followed by Bonferroni’s multiple comparisons test to assess statistical significance versus respective control (time zero). Differences between strains were analyzed by two-way ANOVA followed by Bonferroni’s post-test.
Results
MPTP induces rapid changes in Tnf-α and Il-6 mRNA levels
Prior studies have reported increases in Tnf-α and Il-6 using the MPTP model (Kaku et al., 1999, Mandel et al., 2000, Hebert et al., 2003), and mice deficient in Tnf-α receptors or Il-6 have altered responsiveness to MPTP (Bolin et al., 2002, Sriram et al., 2002, Cardenas and Bolin, 2003, Ferger et al., 2004, Leng et al., 2005). Therefore, we examined levels of Tnf-α and Il-6 mRNA in both sensitive and resistant strains of mice. Following administration of MPTP (1 × 40 mg/kg) the levels of Tnf-α mRNA increased dramatically and then declined back to basal levels over a 8–24 hour period (Figure 1A). No statistical difference in the magnitude or time course of these changes were seen between the sensitive (C57BL/6J) and resistant (SWR) strains. Unlike Tnf-α, Il-6 mRNA displayed quantitative differences between the two strains (Figure 1B). Although the time courses of changes were roughly the same in the two strains, absolute mRNA levels of Il-6 mRNA were consistently higher in SWR mice.
Figure 1. Temporal profile of Tnf-α and Il-6 mRNA levels in MPTP-treated SWR and C57BL/6J mice.
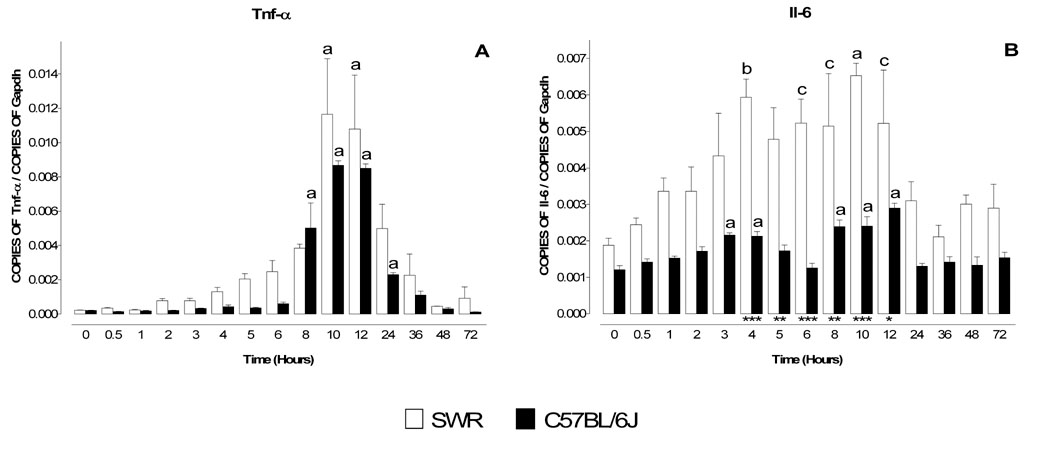
MPTP sensitive (C57BL/6J) and resistant (SWR) mice were injected at time zero with a single dose of MPTP (40 mg/kg) and sacrificed at 0.5, 1, 2, 3, 4, 5, 6, 8, 10, 12, 24, 36, 48 and 72 hr. Levels of mRNA for Tnf-α (A) and Il-6 (B) were evaluated by quantitative RT-PCR. Results are expressed as the number of copies of a specific mRNA (normalized versus the number of copies of Gapdh mRNA) versus time (hours). White bars represent results from SWR mice and black bars those from C57BL/6J mice. Tnf-α mRNA levels are dramatically increased 8–24 hr following MPTP injection, although the time course and the magnitude of the changes are identical in both MPTP-sensitive (C57BL/6J) and MPTP-insensitive (SWR) mice. In contrast the mRNA profile of Il-6 was different in the two strains of mice as levels of Il-6 mRNA were significantly higher in SWR than in C57BL/6J (B). Data are presented as mean ± S.E.M. of three to five animals. Differences between strains were statistically analyzed by two way ANOVA followed by Bonferroni post-test and are indicated with asterisks placed at the bottom of each significantly different point (*** P<0.001, ** P<0.01, * P<0.05). Differences versus control (time point zero) within a single strain were analyzed with one way ANOVA and Bonferroni post test and statistical significances are indicated with letters placed on top of each bar (a P<0.001, b P<0.01, c P<0.05).
Differential expression of chemokine/cytokine ligand and receptor mRNAs in the MPTP model
To gain a better understanding of the molecular components of the inflammatory response in MPTP-sensitive and -resistant strains of mice we assessed expression of mRNA for cytokines and chemokines and their respective receptors by quantitative RT-PCR.
Tweak/Tnfsf12 is a weak inducer of apoptosis belonging to the Tnf-α superfamily that functions by binding to its receptor Fn14/Tnfrsf12a (Wiley et al., 2001, Brown et al., 2006). Levels of Tweak/Tnfsf12 mRNA were similar in untreated C57BL/6J and SWR mice and remained essentially unchanged (C57BL/6J) or declined slightly (SWR) following administration of 1 × 40 mg/kg MPTP (Figure 2A). In striking contrast, levels of mRNA for Fn14/Tnfrsf12a increased substantially in both strains beginning around 3 hours after treatment and declining by 48 hours post treatment (Figure 2B).
Figure 2. Temporal profile of mRNA levels for Tweak/Tnfsf12, Fractalkine/Cx3cl1, Sdf-1/Cxcl12 and their respective receptors Fn14/Tnfrsf12a, Cx3cr1 and Cxcr4 in MPTP-treated SWR and C57BL/6J mice.
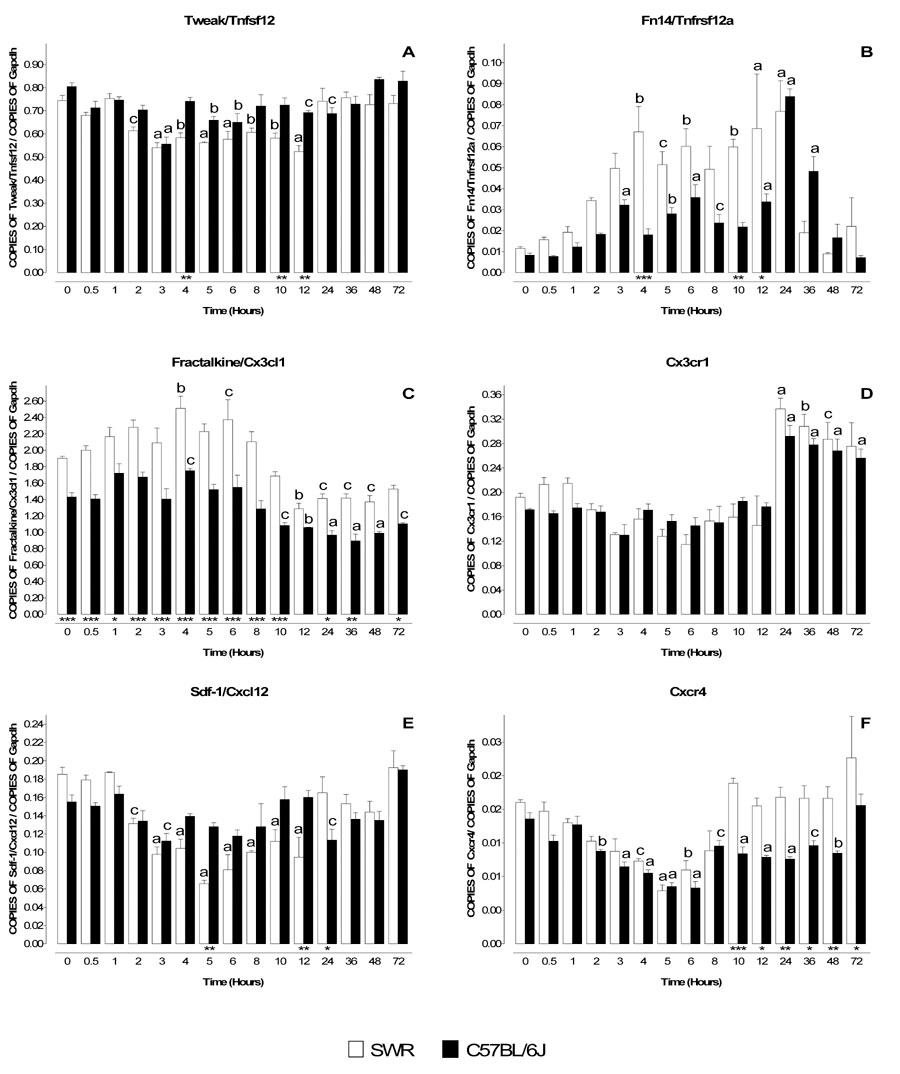
MPTP sensitive (C57BL/6J) and resistant (SWR) mice were injected at time zero with a single dose of MPTP (40 mg/kg) and sacrificed at 0.5, 1, 2, 3, 4, 5, 6, 8, 10, 12, 24, 36, 48 and 72 hr. Levels of mRNA for Tweak/Tnfsf12 (A), Fractalkine/Cx3cl1 (C), Sdf-1/Cxcl12 (E) and their respective receptors Fn14/Tnfrsf12a (B), Cx3cr1 (D) and Cxcr4 (F) was evaluated by quantitative RT-PCR. Results are expressed as the number of copies of a specific mRNA (normalized versus the number of copies of Gapdh mRNA) versus time (hours). White bars represent results from SWR mice and black bars those from C57BL/6J mice. mRNA for Tweak/Tnfsf12, decreases slightly in MPTP-resistant mice (SWR) and are largely unaltered (A) in MPTP-sensitive mice (C57BL/6J). In contrast its receptor Fn14/Tnfrsf12a, increases substantially in both strains of mice (B). Fractalkine/Cx3cl1 and its receptor Cx3cr1 display a reciprocal regulation following MPTP administration, with receptor mRNA being upregulated (D) and ligand mRNA decreasing (C). Fractalkine/Cx3cl1 was the only chemokine tested whose endogenous mRNA levels were statistically different between the two strains (C). The mRNA for Sdf-1/Cxcl12 and its receptor Cxcr4 are coordinately downregulated in both strains of mice (E,F), though levels of Cxcr4 in SWR returns to baseline earlier (10 hr) than in MPTP-sensitive mice (72 hr) (F). Data are presented as mean ± S.E.M. of three to five animals. Differences between strains were statistically analyzed by two way ANOVA followed by Bonferroni post-test and are indicated with asterisks placed at the bottom of each significantly different point (*** P<0.001, ** P<0.01, * P<0.05). Differences versus control (time point zero) within a single strain were analyzed with one way ANOVA and Bonferroni post test and statistical significances are indicated with letters placed on top of each bar (a P<0.001, b P<0.01, c P<0.05).
Fractalkine/Cx3cl1 and its receptor, Cx3cr1 regulate communication between microglia and neurons (Harrison et al., 1998, Nishiyori et al., 1998, Meucci et al., 2000, Hatori et al., 2002) and protect neurons in several model of neurodegeneration (Meucci et al., 2000, Zujovic et al., 2000, Mizuno et al., 2003, Limatola et al., 2005, Cardona et al., 2006). Basal mRNA levels for fractalkine/Cx3cl1 were higher in SWR compared to C57BL/6J mice (Figure 2C). After a single MPTP injection (40 mg/kg), fractalkine/Cx3cl1 mRNA levels declined in both strains beginning at 10 hr and remained depressed up to 72 hours post treatment (Figure 2C). In marked contrast Cx3cr1 exhibited reciprocal regulation and between 24 and 72 hours its mRNA levels were elevated in both strains (Figure 2D).
To establish whether reciprocal regulation was common to all cytokine ligand-receptor pairs in the MPTP model, we assessed expression of Sdf-1/Cxcl12 and its receptor Cxcr4, that to the best of our knowledge have never been studied in the MPTP model. These genes play roles in the development of several brain structures (Lu et al., 2002, Stumm et al., 2003, Vilz et al., 2005), migration of neuronal precursor cells after ischemia (Robin et al., 2006), axonal elongation (Arakawa et al., 2003, Pujol et al., 2005) and neurotransmission (Limatola et al., 2000, Ragozzino et al., 2002, Guyon et al., 2005, Guyon et al., 2006). Unlike the other ligand-receptor pairs, Sdf-1/Cxcl12 and Cxcr4 were coordinately regulated. Sdf-1/Cxcl12 mRNA levels decreased in SWR mice from 2 to 12 hr post-treatment (Fig. 2E). Cxcr4 mRNA levels declined by 2 hours and were maximally reduced between 4 and 6 hours post treatment in both strains (Figs 2E,F). Subsequently, Cxcr4 mRNA levels recovered in SWR mice but remained depressed in C57BL/6J mice for up to 48 hours (Figs 2F).
Strain-dependent regulation of Mip1α/Ccl3 and Mip1β/ccl4 by MPTP
The expression of two other chemokines not previously investigated in the MPTP model was assessed. Mip-1α/Ccl3 and Mip1β/Ccl4 are two structurally related chemokines that are encoded by adjacent genes located on mouse chromosome 11 (Wilson et al., 1990). Mip-1α/Ccl3 and Mip1β/Ccl4 have been implicated in neurodegeneration (Ishizuka et al., 1997, Takami et al., 1997, Simpson et al., 1998, Xia et al., 1998, Xia and Hyman, 1999, Boven et al., 2000, Cowell et al., 2002, Kim et al., 2002, Nishi et al., 2005, Perrin et al., 2005) and their levels are increased in the aged midbrain (Felzien et al., 2001). The mRNA levels for both chemokines were dramatically increased following MPTP administration (Figure 3A and 3B) and displayed pronounced strain-dependent differences. In SWR mice, levels of mRNA for Mip-1α/Ccl3 and Mip1β/Ccl4 were elevated between 4–12 hours and 4–24 hours, respectively. In contrast, in C57BL/6J mice the mRNA levels for Mip-1α/Ccl3 and Mip1β/Ccl4 were elevated for shorter periods, 5–24 hours and 8–12 hours, respectively (Figures 3A,B).
Figure 3. Temporal profile of mRNA levels for Mip-1α/Ccl3, Mip-1β/Ccl4 and Mcp-1/Ccl2 in MPTP-treated SWR and C57BL/6J mice.
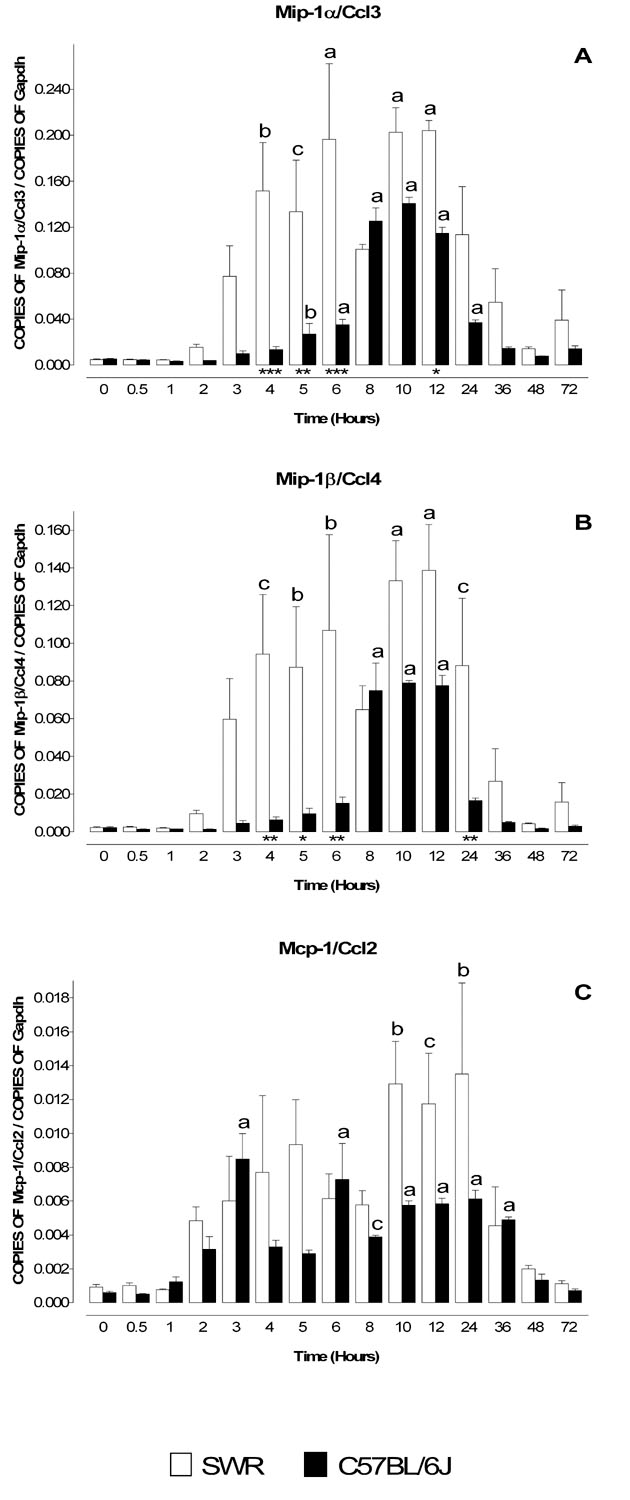
MPTP sensitive (C57BL/6J) and resistant (SWR) mice were injected at time zero with a single dose of MPTP (40 mg/kg) and sacrificed at 0.5, 1, 2, 3, 4, 5, 6, 8, 10, 12, 24, 36, 48 and 72 hr. Levels of mRNA for Mip-1α/Ccl3 (A), Mip-1β/Ccl4 (B) and Mcp-1/Ccl2 (C) were evaluated by quantitative RT-PCR. Results are expressed as the number of copies of a specific mRNA (normalized versus the number of copies of Gapdh mRNA) versus time (hours). White bars represent results from SWR mice and black bars those from C57BL/6J mice. Mip-1α/Ccl3 (A) and Mip-1β/Ccl4 (B) mRNA levels increase following MPTP injection but their temporal profiles differ between the MPTP-sensitive (C57BL/6J) and MPTP-insensitive (SWR) strains of mice. In SWR mice, levels of Mip-1α/Ccl3 and Mip-1β/Ccl4 mRNA are elevated at earlier time points than in C57BL/6J. Mcp-1/Ccl2 is the earliest chemokine to be transcribed after MPTP administration, with mRNA levels peaking at 3 hours. Data are presented as mean ± S.E.M. of three to five animals. Differences between strains were statistically analyzed by two way ANOVA followed by Bonferroni post-test and are indicated with asterisks placed at the bottom of each significantly different point (*** P<0.001, ** P<0.01, * P<0.05). Differences versus control (time point zero) within a single strain were analyzed with one way ANOVA and Bonferroni post test and statistical significances are indicated with letters placed on top of each bar (a P<0.001, b P<0.01, c P<0.05).
Expression of inflammation-associated genes in MPTP-treated Mcp-1 null mice
Mcp-1/Ccl2 is a widely studied chemokine that is expressed in neurons in the striatum and SNpc (Banisadr et al., 2005a, Banisadr et al., 2005b), and it is implicated in neurodegeneration (Bruno et al., 2000, Muessel et al., 2000, Muessel et al., 2002, Eugenin et al., 2003, Kalehua et al., 2004, Perrin et al., 2005). The mRNA expression pattern of Mcp-1/Ccl2 exhibited an initial peak at 3 hours followed by a second peak between 6–36 hours post MPTP treatment in both strains of mice (Fig. 3C).
As increased expression of Mcp-1/Ccl2 mRNA is one of the earliest gene expression events documented in the MPTP model, this chemokine could be a key initiating component of the response to MPTP. Therefore, we investigated the role of Mcp-1/Ccl2 in the MPTP model by comparing mRNA levels of inflammation-associated gene products in Mcp-1/Ccl2-null and wild type mice. To eliminate potential confounding effects from genetic background, both knockout and wild type mice were backcrossed for 10 generations onto the C57BL/6J background.
Animals from both strains were injected with MPTP (1 × 40 mg/kg) and mRNA levels for the respective gene products were assessed at 2, 5, 12, 24 and 72 hours by real time quantitative PCR. No statistically significant differences between the knockout mice and wild type mice could be detected for Tnf-α, Il-6 (Fig 4), Cx3cr1, Tweak/Tnfsf12, Sdf-1/Cxcl12, Fn14/Tnfrsf12a and Cxcr4 (Fig. 5). However, the mRNA responses for Mip-1α/Ccl3 and Mip-1β/Ccl4 at 5 hours were significantly lower in Mcp-1/Ccl2-null mice compared to wild type C57BL/6J mice (Fig. 6). Therefore, Mcp-1/Ccl2 may play a positive role in modulating expression of Mip-1α/Ccl3 and/or Mip-1β/Ccl4 in response to MPTP challenge.
Figure 4. Comparison of mRNA levels for Tnf-α and Il-6 in MPTP-treated wild type and Mcp-1/Ccl2 knockout mice.
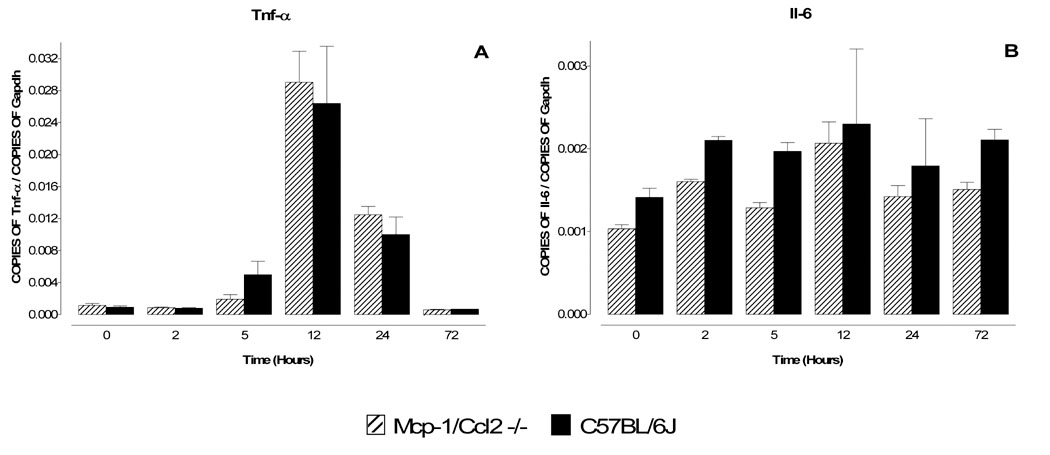
Mcp-1/Ccl2 knockout mice and their respective wild type C57BL/6J controls were injected once with MPTP (40 mg/kg) and sacrificed 2, 5, 12, 24 and 72 hr later. The mRNA levels for Tnf-α (A) and Il-6 (B) were evaluated by quantitative RT-PCR. Results are expressed as the number of copies of a specific mRNA (normalized versus the number of copies of Gapdh mRNA) versus time (hours). Black bars represent results from C57BL/6J mice and hatched bars those from Mcp-1/Ccl2 knockout mice. Neither Tnf-α (A) nor Il-6 (B) mRNA levels differ between the two strains of mice, suggesting that Mcp-1/Ccl2 does not regulate the expression of these two cytokines in the MPTP model. Data presented as mean ± S.E.M. of three to five animals. Differences between strains were statistically analyzed by two way ANOVA followed by Bonferroni post-test and are indicated with asterisks placed at the bottom of each significantly different point (*** P<0.001, ** P<0.01, * P<0.05).
Figure 5. Comparison of mRNA levels for Tweak/Tnfsf12, Fractalkine/Cx3cl1, Sdf-1/Cxcl12, Fn14/Tnfrsf12a, Cx3cr1 and Cxcr4 in MPTP-treated wild type and Mcp-1/Ccl2 knockout mice.
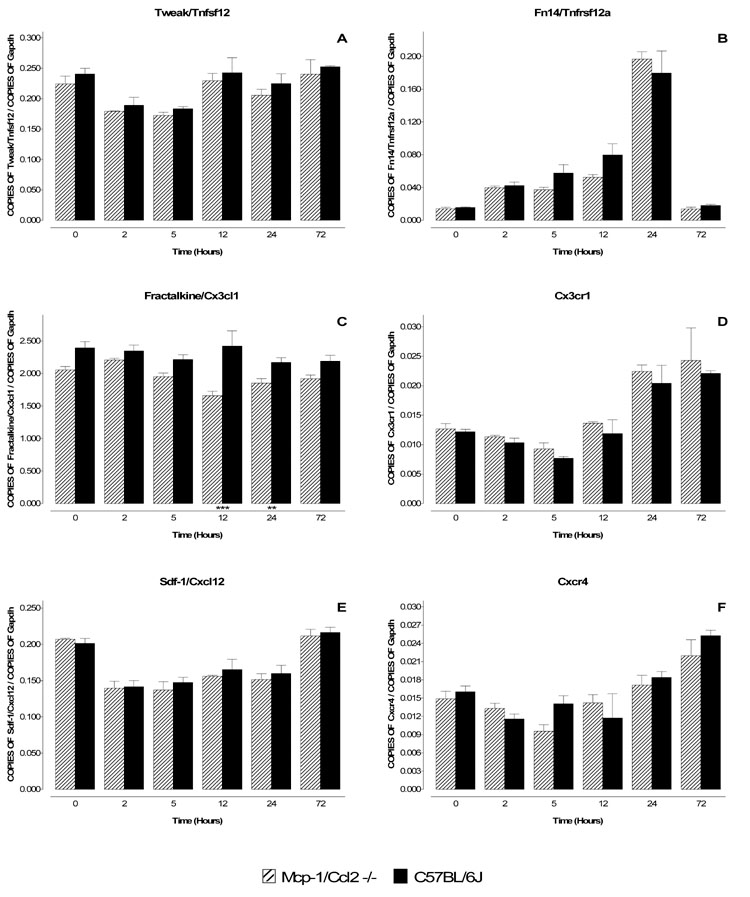
Mcp-1/Ccl2 knockout mice and their respective wild type C57BL/6J controls were injected once with MPTP (40 mg/kg) and sacrificed 2, 5, 12, 24 and 72 hr later. The mRNA levels for Tweak/Tnfsf12 (A), Fractalkine/Cx3cl1 (C), Sdf-1/Cxcl12 (E) and their respective receptors, Fn14/Tnfrsf12a (B), Cx3cr1 (D) and Cxcr4 (F) were evaluated by quantitative RT-PCR. Results are expressed as the number of copies of a specific mRNA (normalized versus the number of copies of Gapdh mRNA) versus time (hours). Black bars represent results from C57BL/6J mice and hatched bars those from Mcp-1/Ccl2 knockout mice. There are no substantial differences in mRNA levels for any of the cytokines/chemokines (A,C,E) and their respective receptors (B,D,F) in the two strains of mice, indicating that Mcp-1/Ccl2 does not regulate their expression in the MPTP model. Data are presented as mean ± S.E.M. of three to five animals. Differences between strains were statistically analyzed by two way ANOVA followed by Bonferroni post-test and are indicated with asterisks placed at the bottom of each significantly different point (*** P<0.001, ** P<0.01, * P<0.05).
Figure 6. Comparison of mRNA levels for Mip-1α/Ccl3 and Mip-1β/Ccl4 in MPTP-treated wild type and Mcp-1/Ccl2 knockout mice.
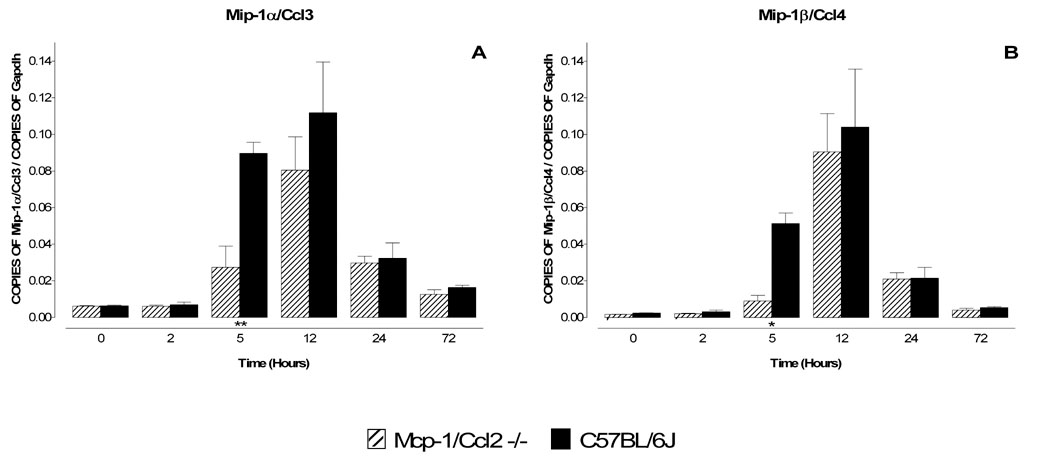
Mcp-1/Ccl2 knockout mice and their respective C57BL/6J wild type controls were injected once with MPTP (40 mg/kg) and sacrificed 2, 5, 12, 24 and 72 hr later. The mRNA levels for Mip-1α/Ccl3 and Mip-1β/Ccl4 were evaluated by quantitative RT-PCR. Results are expressed as the number of copies of a specific mRNA (normalized versus the number of copies of Gapdh mRNA) versus time (hours). Black bars represent results from C57BL/6J mice and hatched bars those from Mcp-1/Ccl2 knockout mice. Genetic ablation of Mcp-1/Ccl2 results in a delayed increase in mRNA encoding both Mip-1α/Ccl3 and Mip-1β/Ccl4 (A,B). Data are presented as mean ± S.E.M. of three to five animals. Differences between strains were statistically analyzed by two way ANOVA followed by Bonferroni post-test and are indicated with asterisks placed at the bottom of each significantly different point (*** P<0.001, ** P<0.01, * P<0.05).
Loss of TH-positive SNpc neurons in MPTP-treated Mcp-1/Ccl2-null mice
To assess the biological significance of Mcp-1/Ccl2 in MPTP toxicity, wild type C57BL/6J mice and Mcp-1/Ccl2-null mice on a C57BL/6J background were treated with MPTP (4 × 20mg/kg) and neuronal loss in the SNpc determined 14 days later by stereological reconstruction (West et al., 1991) of TH-immunostained and Nissl counterstained sections. This paradigm is well characterized and leads to neuronal loss in C57BL/6J mice (Sonsalla and Heikkila, 1986, Smeyne et al., 2005). Stereological counting revealed no statistically significant differences in the number of resident TH-positive neurons in the SNpc of wild type and Mcp-1/Ccl2-null mice (Figure 7). Furthermore, there was the same extent of neuronal loss in both strains of mice following MPTP treatment (Figure 7). Thus, loss of Mcp-1/Ccl2 did not confer resistance to MPTP. It remains to be established whether loss of Mcp-1/Ccl2 on an SWR background can confer MPTP-sensitivity.
Figure 7. Comparison of SNpc cell loss after MPTP treatment in wild type and Mcp-1/Ccl2 knockout mice.
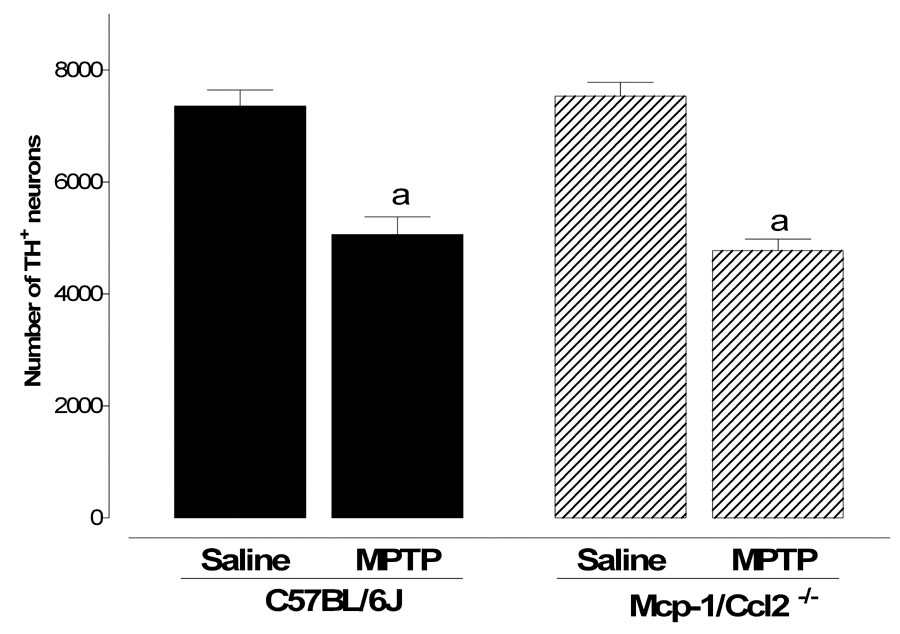
Both Mcp-1/Ccl2 knockout mice and their respective wild type controls were injected with MPTP (20 mg/kg) every two hours for a total of 4 injections and sacrificed 14 days later. The total number of dopaminergic neurons in the SNpc was estimated with the optical fractionator method on sections immunostained for tyrosine hydroxylase. Black bars represent results from C57BL/6J mice and hatched bars those from Mcp-1/Ccl2 knockout mice. Genetic ablation of Mcp-1/Ccl2 does not protect mice from MPTP-induced loss of dopaminergic neurons in the SNpc. Data are presented as mean ± S.E.M. of five animals. Differences were statistically analyzed by two way ANOVA followed by Bonferroni post-test (*** P<0.001, ** P<0.01, * P<0.05).
Discussion
The etiology of PD is thought to involve interplay between environmental agents and genetic risk factors (Poirier et al., 1991, Tsang and Soong, 2003, Allam et al., 2005). A number of inbred strains of mice with differential sensitivity to MPTP have been identified and can be used to identify genetic factors (Hamre et al., 1999, Schwarting et al., 1999, Smeyne et al., 2001, Cook et al., 2003, Liu et al., 2003a, McLaughlin et al., 2006). As inflammatory responses have been associated with PD, genetically determined differences in these responses could contribute to the risk for developing this disease. Here we have measured mRNA levels of various cytokines and chemokines as surrogate markers of inflammation in the striatum of MPTP-sensitive and MPTP-resistant strains of mice. This strategy permits us to establish not only whether inflammatory responses occur in the MPTP model (and if so whether to the same extent in sensitive and resistant strains of mice) but also whether specific inflammatory mediators might be the direct or indirect genetic basis for the strain differences.
MPTP modulates the levels of a number of cytokine and chemokine mRNAs in the striatum. The earliest mRNA changes are coincident with some of the acute effects of MPTP in the striatum, such as dumping and depletion of dopamine in SNpc nerve terminals (Urakami et al., 1988) and may be the direct consequences of these events. Many of the mRNA changes also precede or are coincident with frank morphological manifestations of MPTP-induced damage to dopaminergic nerve endings in the striatum which have been shown to occur within the first 24 hours (Cochiolo et al., 2000) as well as degeneration of TH-positive neurons in the SNpc that occurs maximally between days 4 and 7 post-treatment (Jackson-Lewis et al., 1995, Faherty et al., 2005). Therefore, these mRNA alterations may either contribute to nerve damage and cell loss and/or be a response to these processes. With a few exceptions, the general temporal patterns of mRNA changes triggered by MPTP are similar in both MPTP-sensitive and MPTP-resistant strains of mice. Therefore, despite the fact that SWR mice exhibit minimal loss of neurons compared to C57BL/6J mice, MPTP apparently elicits inflammatory responses in both strains. This opens the possibility that inflammation may contribute to the pathogenic process in both positive and negative fashions.
Tnf-α and Il-6 have been intensively investigated in the MPTP and other neurodegenerative paradigms (Mandel et al., 2000, Nagatsu et al., 2000, Nagatsu and Sawada, 2005). Here, we confirm that striatal Tnf-α levels increase in the MPTP model; however, the temporal profiles and absolute levels for Tnf-α mRNA were statistically indistinguishable in the MPTP-sensitive C57BL/6J and MPTP-insensitive SWR strains of mice. Genetic ablation of either Tnf-α or both its receptors, Tnfrsf1 and Tnfrsf2 is reported to confer protection against MPTP toxicity as measured by attenuation of dopamine depletion in the striatum (Sriram et al., 2002, Ferger et al., 2004, Sriram et al., 2006a, Sriram et al., 2006b), although neither genetic ablation nor pharmacological manipulation of Tnf-α prevents neuronal loss in the SNpc (Rousselet et al., 2002, Ferger et al., 2004, Leng et al., 2005). Therefore, our results are consistent with the view that although Tnf-α may influence dopaminergic terminals in the striatum, it cannot explain genetic resistance to MPTP as measured by loss of TH-positive neurons in the SNpc.
Our results for Il-6 mRNA levels also support prior studies showing that MPTP increases striatal Il-6 levels (Kaku et al., 1999, Hebert et al., 2003). Additionally, we show that MPTP elicits higher levels of Il-6 mRNA in SWR compared to C57BL/6J mice, suggesting that this cytokine may contribute to MPTP resistance. Indeed, Il-6 knockout mice are more sensitive to MPTP toxicity (Bolin et al., 2002), potentially due to reduced MPTP-triggered microgliosis (Cardenas and Bolin, 2003).
Tweak/Tnfsf12 is a proinflammatory member of the Tnf-α family and induces apoptosis in several experimental models by interacting with its receptor Fn14/Tnfrsf12a (Wiley et al., 2001, Nakayama et al., 2003, Yepes et al., 2005). Upon binding its receptor, Tweak/Tnfsf12 activates the NF-κB signaling pathway and modulates transcription of a number of cytokine and proinflammatory genes, including Il-6 and Mcp-1/Ccl2 (Kim et al., 2004, Potrovita et al., 2004) whose expression also change following MPTP treatment. Thus, Tweak/Tnfsf12-Fn14/Tnfrsf12a interactions could contribute to some of the transcriptional changes observed in the MPTP paradigm. As Tweak/Tnfsf12 is expressed by microglia and astrocytes and elicits astrocyte proliferation (Desplat-Jego et al., 2002) it may also contribute to the generation of reactive astrocytes reported in the MPTP model (Francis et al., 1995, Araki et al., 2001, Chen et al., 2002). Although MPTP only had a marginal effect on Tweak/Tnfsf12 expression, it elicited a significant increase in Fn14/Tnfrsf12a mRNA levels. Fn14/Tnfrsf12a is an immediate-early gene, and its induction has been observed following growth factor stimulation (Meighan-Mantha et al., 1999) and a number of insults to the nervous system (Tanabe et al., 2003, Potrovita et al., 2004). Furthermore, both ligand and receptor are markedly upregulated in a focal cerebral ischemia model (Potrovita et al., 2004) and a soluble decoy version of Fn14/Tnfrsf12a reduces infarct volume (Yepes et al., 2005), suggesting that it mediates neurodegenerative phenomena. In this study, there was a trend towards reduced expression of Tweak/Tnfsf12 mRNA at times when the mRNA for Fn14/Tnfrsf12a increased substantially. This finding is intriguing in light of a report that Fn14/Tnfrsf12a regulates neurite outgrowth through a ligand-independent mechanism (Tanabe et al., 2003). It is possible that the greatly increased expression of Fn14/Tnfrsf12a combined with the concomitant reduction of Tweak/Tnfsf12 might represent a mechanism geared to mitigate damage to striatal neurons that are synaptic targets for SNpc dopaminergic nerve endings.
Fractalkine/Cx3cl1 was the only gene studied whose basal level of expression differed significantly between SWR and C57BL/6J mice. This chemokine is protective in several in vitro models of neurodegeneration (Boehme et al., 2000, Meucci et al., 2000, Zujovic et al., 2000, Mizuno et al., 2003, Limatola et al., 2005). Moreover, genetic ablation of either Fractalkine/Cx3cl1 or its receptor Cx3cr1, protects in vivo from MPTP toxicity (Cardona et al., 2006). Thus, despite the fact that Fractalkine/Cx3cl1 expression does not differ qualitatively between the two strains, the higher levels in SWR mice might contribute to their resistance to MPTP. Fractalkine/Cx3cl1 is expressed in vivo on neurons (Harrison et al., 1998) whereas its receptor Cx3cr1 is expressed on microglia (Harrison et al., 1998), suggesting cross-talk between these cell types. Microglia are well-known to participate in neurodegenerative processes (McGeer and McGeer, 1998a, McGeer and McGeer, 1998b, Orr et al., 2002, Gao et al., 2003, Liu and Hong, 2003, Liu et al., 2003b) and their pharmacological inactivation protects from MPTP toxicity (Wu et al., 2002, Liu and Hong, 2003). Therefore, the reciprocal modulation of this chemokine and its receptor by MPTP may influence microglia/neuron interactions (Harrison et al., 1998, Nishiyori et al., 1998).
Sdf-1/Cxcl12 and its receptor Cxcr4 are downregulated in both strains of mice and have been associated with multiple processes in the developing and adult nervous systems. Critically, both ligand and receptor mediate release of Tnf-α and glutamate from astrocytes (Bezzi et al., 2001), implying pro-inflammatory actions. Indeed, downregulation of Cxcr4 has been suggested as the mechanism of action for dexamethasone protection of neurons (Felszeghy et al., 2004). They also play roles in the development of several brain regions (Bagri et al., 2002, Tissir et al., 2004, Luo et al., 2005) and regulate synaptic activity in striatal cholinergic neurons (Banisadr et al., 2002a) and TH positive dopaminergic neurons in the SNpc (Banisadr et al., 2002a, Guyon et al., 2006). Sdf-1/Cxcl12 and Cxcr4 regulate axonal development and pathfinding in the developing CNS (Chalasani et al., 2003, Gilmour et al., 2004, Li et al., 2005, Lieberam et al., 2005, Pujol et al., 2005), therefore, they might play analogous roles in adult brain by contributing to the sprouting of new fibers from the SNpc that occurs after mechanical or chemical (6-OHDA and MPTP) damage in the striatum (Mitsumoto et al., 1998, Liberatore et al., 1999, Finkelstein et al., 2000, Song and Haber, 2000).
The only two genes that showed marked quantitative differences between sensitive and resistant strains of mice were Mip-1α/Ccl3 and Mip-1β/Ccl4. To the best of our knowledge this is the first time that these chemokines have been implicated in the MPTP model. Not only were both chemokines induced to higher levels in SWR mice but they also showed earlier expression in the resistant strain than in the MPTP-sensitive strain.
Both Mip-1α/Ccl3 and Mip-1β/Ccl4 are elevated in several neurodegenerative disorders including multiple sclerosis (MS) (Simpson et al., 1998, Boven et al., 2000), cerebral ischemia (Takami et al., 1997, Cowell et al., 2002, Kim et al., 2002, Nishi et al., 2005), Alzheimer’s disease (AD) (Xia et al., 1998, Xia and Hyman, 1999) as well as in experimental models of demyelination (McMahon et al., 2001), experimental allergic encephalopathy (EAE) (Matejuk et al., 2002), experimental autoimmune neuritis (Zou et al., 1999) and Wallerian degeneration (Perrin et al., 2005). Moreover, of particular relevance to PD, their expression is increased in the midbrain of mice during aging (Felzien et al., 2001). Despite the evidence suggesting that Mip-1α/Ccl3 and Mip-1β/Ccl4 are involved in neurodegenerative processes, their precise role is not well established. Experimental models of retrograde degeneration suggest that they are involved in repair processes. For example, antibodies against Mip-1α/Ccl3 and Mip-1β/Ccl4 reduce macrophage activation and myelin clearance after Wallerian degeneration (Perrin et al., 2005). Moreover after olfactory bulbectomy, Mip-1α/Ccl3 and Mip-1β/Ccl4 contribute to regenerative processes in the olfactory epithelium (Getchell et al., 2002, Kwong et al., 2004). By analogy it is possible that these genes could contribute to repair processes in PD and MPTP. Mcp-1/Ccl2 showed the earliest response to MPTP.
Mcp-1/Ccl2 is localized to neuronal cell bodies and filaments in the rat SNpc (Banisadr et al., 2005a) and its unique receptor, Ccr2 is highly expressed on cholinergic neurons in the striatum and TH positive neurons in the SNpc (Banisadr et al., 2002b, Banisadr et al., 2005b). Mcp-1/Ccl2 and its receptor have been implicated in several models of neurodegeneration (Bruno et al., 2000, Muessel et al., 2000, Siebert et al., 2000, Eugenin et al., 2003, Kalehua et al., 2004) including retrograde degeneration (Ghirnikar et al., 2001, Muessel et al., 2002, Perrin et al., 2005). In addition, although controversial, polymorphisms in the Mcp-1/Ccl2 promoter have been linked to sporadic PD (Nishimura et al., 2003, Huerta et al., 2004). We have confirmed that Mcp-1/Ccl2 is induced in striatum after MPTP administration (Sriram et al., 2006b). Moreover, we show that genetic ablation of Mcp-1/Ccl2 significantly delays or attenuates the MPTP-elicited increase of Mip-1α/Ccl3 and Mip1β/Ccl4 mRNA levels; indicating that Mcp-1/Ccl2 contributes to the early transcriptional responses of these two chemokines to MPTP. However, Mcp-1/Ccl2 is clearly not the only factor regulating expression of these two genes, as at later times following MPTP administration mRNA levels of Mip-1α/Ccl3 and Mip1β/Ccl4 are indistinguishable in wild type and knockout mice (Figure 6). It will be important to determine whether the more rapid, Mcp-1/Ccl2-dependent changes in Mip-1α/Ccl3 and Mip1β/Ccl4 mRNA occur in the same cell types as the later, Mcp-1/Ccl2-independent alterations. Since Mip-1α/Ccl3 and Mip1β/Ccl4 are induced to higher levels and with distinct kinetics in SWR mice, differences in Mcp-1/Ccl2 signaling in this strain might contribute to their resistance to MPTP. The absence of Mcp-1/Ccl2 did not significantly protect SNpc TH+ neurons from MPTP toxicity. A more telling experiment will be to assess whether loss of Mcp-1/Ccl2 causes the normally resistant SWR strain to become sensitive. Finally, though the acute model of MPTP (4 X 20 mg/kg) is widely used (Jackson-Lewis et al., 1995, Hamre et al., 1999, Kaku et al., 1999, Schwarting et al., 1999, Araki et al., 2001, Bolin et al., 2002, Rousselet et al., 2002, Cook et al., 2003, Ferger et al., 2004, Faherty et al., 2005, Smeyne et al., 2005), inflammation could potentially have a different role when animals are chronically treated with this neurotoxicant. Indeed, MPTP dosage and its injection schedule are important determinants of the mode of neurodegeneration in the SNpc (Sonsalla and Heikkila, 1986, Jackson-Lewis et al., 1995, Eberhardt and Schulz, 2003, Youdim and Arraf, 2004, Novikova et al., 2006). Therefore, it would be interesting in the future to asses the role of inflammation in subchronic/chronic models of MPTP intoxication.
We have shown here that MPTP modulates the mRNA levels of several cytokine and chemokine genes in the striatum of both resistant (SWR) and sensitive (C57BL/6J) strains of mice. Thus inflammatory responses occur in both strains of mice, independent of their sensitivity to MPTP. Therefore, the genetically determined differential sensitivity to this toxin in SWR and C57BL/6J mice could be related to the intrinsic ability of SNpc neurons to tolerate inflammation rather than in strain-dependent differences in the response itself.
Acknowledgments
The authors would like to thank Ms. Jennifer Parris for her assistance during the preparation of this manuscript and the Hartwell Center for Bioinformatics and Biotechnology for the synthesis of realtime PCR primers and probes and for sequencing. This work was supported in part by the National Institutes of Health Cancer Center CORE Grant CA 21765, the American Lebanese Syrian Associated Charities (ALSAC) and the National Institutes of Health grants R01-ES010772 and R01-NS042828 to J.I.M.
Abbreviations
- DA
dopamine
- Tnf-α
tumor necrosis factor α
- Sdf-1/Cxcl12
stromal derived factor 1/CXC chemokine ligand 12
- Mip-1α/Ccl3
macrophage inflammatory protein 1 alpha/CC chemokine ligand 3
- Mip-1β/Ccl4
macrophage inflammatory protein 1 beta/CC chemokine ligand 4
- Tweak/Tnfsf12
Tnf-like weak inducer of apoptosis/tumor necrosis factor superfamily member 12
- Fn14/Tnfrsf12a
fibroblast growth factor-inducible 14/tumor necrosis factor receptor superfamily member 12a
- TH
tyrosine hydroxylase
- Il-6
interleukin 6
- Cx3cl1
fractalkine
- Cx3cr1
fractalkine receptor
- PD
Parkinson’s disease
- MPTP
1-methyl-4-phenyl-1,2,3,6-tetrahydropyrimidine
- Mcp-1/Ccl2
monocyte chemoattractant protein 1/CC chemokine ligand 2
- PCR
polymerase chain reaction
- SNpc
substantia nigra pars compacta
- 6OHDA
6 hydroxydopamine
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adler MW, Geller EB, Chen X, Rogers TJ. Viewing chemokines as a third major system of communication in the brain. Aaps J. 2005;7:E865–870. doi: 10.1208/aapsj070484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allam MF, Del Castillo AS, Navajas RF. Parkinson’s disease risk factors: genetic, environmental, or both? Neurol Res. 2005;27:206–208. doi: 10.1179/016164105X22057. [DOI] [PubMed] [Google Scholar]
- Arakawa Y, Bito H, Furuyashiki T, Tsuji T, Takemoto-Kimura S, Kimura K, Nozaki K, Hashimoto N, Narumiya S. Control of axon elongation via an SDF-1alpha/Rho/mDia pathway in cultured cerebellar granule neurons. J Cell Biol. 2003;161:381–391. doi: 10.1083/jcb.200210149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araki T, Mikami T, Tanji H, Matsubara M, Imai Y, Mizugaki M, Itoyama Y. Biochemical and immunohistological changes in the brain of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-treated mouse. Eur J Pharm Sci. 2001;12:231–238. doi: 10.1016/s0928-0987(00)00170-6. [DOI] [PubMed] [Google Scholar]
- Bacon KB, Harrison JK. Chemokines and their receptors in neurobiology: perspectives in physiology and homeostasis. J Neuroimmunol. 2000;104:92–97. doi: 10.1016/s0165-5728(99)00266-0. [DOI] [PubMed] [Google Scholar]
- Bagri A, Gurney T, He X, Zou YR, Littman DR, Tessier-Lavigne M, Pleasure SJ. The chemokine SDF1 regulates migration of dentate granule cells. Development. 2002;129:4249–4260. doi: 10.1242/dev.129.18.4249. [DOI] [PubMed] [Google Scholar]
- Bajetto A, Bonavia R, Barbero S, Schettini G. Characterization of chemokines and their receptors in the central nervous system: physiopathological implications. J Neurochem. 2002;82:1311–1329. doi: 10.1046/j.1471-4159.2002.01091.x. [DOI] [PubMed] [Google Scholar]
- Banisadr G, Fontanges P, Haour F, Kitabgi P, Rostene W, Melik Parsadaniantz S. Neuroanatomical distribution of CXCR4 in adult rat brain and its localization in cholinergic and dopaminergic neurons. Eur J Neurosci. 2002a;16:1661–1671. doi: 10.1046/j.1460-9568.2002.02237.x. [DOI] [PubMed] [Google Scholar]
- Banisadr G, Gosselin RD, Mechighel P, Kitabgi P, Rostene W, Parsadaniantz SM. Highly regionalized neuronal expression of monocyte chemoattractant protein-1 (MCP-1/CCL2) in rat brain: Evidence for its colocalization with neurotransmitters and neuropeptides. J Comp Neurol. 2005a;489:275–292. doi: 10.1002/cne.20598. [DOI] [PubMed] [Google Scholar]
- Banisadr G, Gosselin RD, Mechighel P, Rostene W, Kitabgi P, Melik Parsadaniantz S. Constitutive neuronal expression of CCR2 chemokine receptor and its colocalization with neurotransmitters in normal rat brain: functional effect of MCP-1/CCL2 on calcium mobilization in primary cultured neurons. J Comp Neurol. 2005b;492:178–192. doi: 10.1002/cne.20729. [DOI] [PubMed] [Google Scholar]
- Banisadr G, Queraud-Lesaux F, Boutterin MC, Pelaprat D, Zalc B, Rostene W, Haour F, Parsadaniantz SM. Distribution, cellular localization and functional role of CCR2 chemokine receptors in adult rat brain. J Neurochem. 2002b;81:257–269. doi: 10.1046/j.1471-4159.2002.00809.x. [DOI] [PubMed] [Google Scholar]
- Banisadr G, Rostene W, Kitabgi P, Parsadaniantz SM. Chemokines and brain functions. Curr Drug Targets Inflamm Allergy. 2005c;4:387–399. doi: 10.2174/1568010054022097. [DOI] [PubMed] [Google Scholar]
- Barcia C, Fernandez Barreiro A, Poza M, Herrero MT. Parkinson’s disease and inflammatory changes. Neurotox Res. 2003;5:411–418. doi: 10.1007/BF03033170. [DOI] [PubMed] [Google Scholar]
- Bezard E, Gross CE, Fournier MC, Dovero S, Bloch B, Jaber M. Absence of MPTP-induced neuronal death in mice lacking the dopamine transporter. Exp Neurol. 1999;155:268–273. doi: 10.1006/exnr.1998.6995. [DOI] [PubMed] [Google Scholar]
- Bezzi P, Domercq M, Brambilla L, Galli R, Schols D, De Clercq E, Vescovi A, Bagetta G, Kollias G, Meldolesi J, Volterra A. CXCR4-activated astrocyte glutamate release via TNFalpha: amplification by microglia triggers neurotoxicity. Nat Neurosci. 2001;4:702–710. doi: 10.1038/89490. [DOI] [PubMed] [Google Scholar]
- Biber K, de Jong EK, van Weering HR, Boddeke HW. Chemokines and their receptors in central nervous system disease. Curr Drug Targets. 2006;7:29–46. doi: 10.2174/138945006775270196. [DOI] [PubMed] [Google Scholar]
- Boehme SA, Lio FM, Maciejewski-Lenoir D, Bacon KB, Conlon PJ. The chemokine fractalkine inhibits Fas-mediated cell death of brain microglia. J Immunol. 2000;165:397–403. doi: 10.4049/jimmunol.165.1.397. [DOI] [PubMed] [Google Scholar]
- Bolin LM, Strycharska-Orczyk I, Murray R, Langston JW, Di Monte D. Increased vulnerability of dopaminergic neurons in MPTP-lesioned interleukin-6 deficient mice. J Neurochem. 2002;83:167–175. doi: 10.1046/j.1471-4159.2002.01131.x. [DOI] [PubMed] [Google Scholar]
- Boven LA, Montagne L, Nottet HS, De Groot CJ. Macrophage inflammatory protein-1alpha (MIP-1alpha), MIP-1beta, and RANTES mRNA semiquantification and protein expression in active demyelinating multiple sclerosis (MS) lesions. Clin Exp Immunol. 2000;122:257–263. doi: 10.1046/j.1365-2249.2000.01334.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradbury AJ, Costall B, Jenner PG, Kelly ME, Marsden CD, Naylor RJ. MPP+ can disrupt the nigrostriatal dopamine system by acting in the terminal area. Neuropharmacology. 1986;25:939–941. doi: 10.1016/0028-3908(86)90025-0. [DOI] [PubMed] [Google Scholar]
- Brown SA, Hanscom HN, Vu H, Brew SA, Winkles JA. TWEAK binding to the Fn14 cysteine-rich domain depends on charged residues located in both the A1 and D2 modules. Biochem J. 2006;397:297–304. doi: 10.1042/BJ20051362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruno V, Copani A, Besong G, Scoto G, Nicoletti F. Neuroprotective activity of chemokines against N-methyl-D-aspartate or beta-amyloid-induced toxicity in culture. Eur J Pharmacol. 2000;399:117–121. doi: 10.1016/s0014-2999(00)00367-8. [DOI] [PubMed] [Google Scholar]
- Burns RS, Markey SP, Phillips JM, Chiueh CC. The neurotoxicity of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine in the monkey and man. Can J Neurol Sci. 1984;11:166–168. doi: 10.1017/s0317167100046345. [DOI] [PubMed] [Google Scholar]
- Cardenas H, Bolin LM. Compromised reactive microgliosis in MPTP-lesioned IL-6 KO mice. Brain Res. 2003;985:89–97. doi: 10.1016/s0006-8993(03)03172-x. [DOI] [PubMed] [Google Scholar]
- Cardona AE, Pioro EP, Sasse ME, Kostenko V, Cardona SM, Dijkstra IM, Huang D, Kidd G, Dombrowski S, Dutta R, Lee JC, Cook DN, Jung S, Lira SA, Littman DR, Ransohoff RM. Control of microglial neurotoxicity by the fractalkine receptor. Nat Neurosci. 2006;9:917–924. doi: 10.1038/nn1715. [DOI] [PubMed] [Google Scholar]
- Cartier L, Hartley O, Dubois-Dauphin M, Krause KH. Chemokine receptors in the central nervous system: role in brain inflammation and neurodegenerative diseases. Brain Res Brain Res Rev. 2005;48:16–42. doi: 10.1016/j.brainresrev.2004.07.021. [DOI] [PubMed] [Google Scholar]
- Carvey PM, Zhao CH, Hendey B, Lum H, Trachtenberg J, Desai BS, Snyder J, Zhu YG, Ling ZD. 6-Hydroxydopamine-induced alterations in blood-brain barrier permeability. Eur J Neurosci. 2005;22:1158–1168. doi: 10.1111/j.1460-9568.2005.04281.x. [DOI] [PubMed] [Google Scholar]
- Chalasani SH, Sabelko KA, Sunshine MJ, Littman DR, Raper JA. A chemokine, SDF-1, reduces the effectiveness of multiple axonal repellents and is required for normal axon pathfinding. J Neurosci. 2003;23:1360–1371. doi: 10.1523/JNEUROSCI.23-04-01360.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Jacobs E, Schwarzschild MA, McCullough ML, Calle EE, Thun MJ, Ascherio A. Nonsteroidal antiinflammatory drug use and the risk for Parkinson’s disease. Ann Neurol. 2005;58:963–967. doi: 10.1002/ana.20682. [DOI] [PubMed] [Google Scholar]
- Chen H, Zhang SM, Hernan MA, Schwarzschild MA, Willett WC, Colditz GA, Speizer FE, Ascherio A. Nonsteroidal anti-inflammatory drugs and the risk of Parkinson disease. Arch Neurol. 2003;60:1059–1064. doi: 10.1001/archneur.60.8.1059. [DOI] [PubMed] [Google Scholar]
- Chen LW, Wei LC, Qiu Y, Liu HL, Rao ZR, Ju G, Chan YS. Significant up-regulation of nestin protein in the neostriatum of MPTP-treated mice. Are the striatal astrocytes regionally activated after systemic MPTP administration? Brain Res. 2002;925:9–17. doi: 10.1016/s0006-8993(01)03253-x. [DOI] [PubMed] [Google Scholar]
- Chiba K, Trevor AJ, Castagnoli N., Jr Active uptake of MPP+, a metabolite of MPTP, by brain synaptosomes. Biochem Biophys Res Commun. 1985;128:1228–1232. doi: 10.1016/0006-291x(85)91071-x. [DOI] [PubMed] [Google Scholar]
- Cochiolo JA, Ehsanian R, Bruck DK. Acute ultrastructural effects of MPTP on the nigrostriatal pathway of the C57BL/6 adult mouse: evidence of compensatory plasticity in nigrostriatal neurons. J Neurosci Res. 2000;59:126–135. [PubMed] [Google Scholar]
- Cook R, Lu L, Gu J, Williams RW, Smeyne RJ. Identification of a single QTL, Mptp1, for susceptibility to MPTP-induced substantia nigra pars compacta neuron loss in mice. Brain Res Mol Brain Res. 2003;110:279–288. doi: 10.1016/s0169-328x(02)00659-9. [DOI] [PubMed] [Google Scholar]
- Cowell RM, Xu H, Galasso JM, Silverstein FS. Hypoxic-ischemic injury induces macrophage inflammatory protein-1alpha expression in immature rat brain. Stroke. 2002;33:795–801. doi: 10.1161/hs0302.103740. [DOI] [PubMed] [Google Scholar]
- Desplat-Jego S, Varriale S, Creidy R, Terra R, Bernard D, Khrestchatisky M, Izui S, Chicheportiche Y, Boucraut J. TWEAK is expressed by glial cells, induces astrocyte proliferation and increases EAE severity. J Neuroimmunol. 2002;133:116–123. doi: 10.1016/s0165-5728(02)00368-5. [DOI] [PubMed] [Google Scholar]
- Eberhardt O, Schulz JB. Apoptotic mechanisms and antiapoptotic therapy in the MPTP model of Parkinson’s disease. Toxicol Lett. 2003;139:135–151. doi: 10.1016/s0378-4274(02)00428-9. [DOI] [PubMed] [Google Scholar]
- Eberling JL, Bankiewicz KS, Jordan S, VanBrocklin HF, Jagust WJ. PET studies of functional compensation in a primate model of Parkinson’s disease. Neuroreport. 1997;8:2727–2733. doi: 10.1097/00001756-199708180-00017. [DOI] [PubMed] [Google Scholar]
- Eugenin EA, D’Aversa TG, Lopez L, Calderon TM, Berman JW. MCP-1 (CCL2) protects human neurons and astrocytes from NMDA or HIV-tat-induced apoptosis. J Neurochem. 2003;85:1299–1311. doi: 10.1046/j.1471-4159.2003.01775.x. [DOI] [PubMed] [Google Scholar]
- Faherty CJ, Raviie Shepherd K, Herasimtschuk A, Smeyne RJ. Environmental enrichment in adulthood eliminates neuronal death in experimental Parkinsonism. Brain Res Mol Brain Res. 2005;134:170–179. doi: 10.1016/j.molbrainres.2004.08.008. [DOI] [PubMed] [Google Scholar]
- Felszeghy K, Banisadr G, Rostene W, Nyakas C, Haour F. Dexamethasone downregulates chemokine receptor CXCR4 and exerts neuroprotection against hypoxia/ischemia-induced brain injury in neonatal rats. Neuroimmunomodulation. 2004;11:404–413. doi: 10.1159/000080151. [DOI] [PubMed] [Google Scholar]
- Felzien LK, McDonald JT, Gleason SM, Berman NE, Klein RM. Increased chemokine gene expression during aging in the murine brain. Brain Res. 2001;890:137–146. doi: 10.1016/s0006-8993(00)03090-0. [DOI] [PubMed] [Google Scholar]
- Ferger B, Leng A, Mura A, Hengerer B, Feldon J. Genetic ablation of tumor necrosis factor-alpha (TNF-alpha) and pharmacological inhibition of TNF-synthesis attenuates MPTP toxicity in mouse striatum. J Neurochem. 2004;89:822–833. doi: 10.1111/j.1471-4159.2004.02399.x. [DOI] [PubMed] [Google Scholar]
- Ferger B, Teismann P, Earl CD, Kuschinsky K, Oertel WH. Salicylate protects against MPTP-induced impairments in dopaminergic neurotransmission at the striatal and nigral level in mice. Naunyn Schmiedebergs Arch Pharmacol. 1999;360:256–261. doi: 10.1007/s002109900079. [DOI] [PubMed] [Google Scholar]
- Finkelstein DI, Stanic D, Parish CL, Tomas D, Dickson K, Horne MK. Axonal sprouting following lesions of the rat substantia nigra. Neuroscience. 2000;97:99–112. doi: 10.1016/s0306-4522(00)00009-9. [DOI] [PubMed] [Google Scholar]
- Francis JW, Von Visger J, Markelonis GJ, Oh TH. Neuroglial responses to the dopaminergic neurotoxicant 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine in mouse striatum. Neurotoxicol Teratol. 1995;17:7–12. doi: 10.1016/0892-0362(94)00048-i. [DOI] [PubMed] [Google Scholar]
- Gainetdinov RR, Fumagalli F, Jones SR, Caron MG. Dopamine transporter is required for in vivo MPTP neurotoxicity: evidence from mice lacking the transporter. J Neurochem. 1997;69:1322–1325. doi: 10.1046/j.1471-4159.1997.69031322.x. [DOI] [PubMed] [Google Scholar]
- Gao HM, Liu B, Zhang W, Hong JS. Critical role of microglial NADPH oxidase-derived free radicals in the in vitro MPTP model of Parkinson’s disease. Faseb J. 2003;17:1954–1956. doi: 10.1096/fj.03-0109fje. [DOI] [PubMed] [Google Scholar]
- German DC, Nelson EL, Liang CL, Speciale SG, Sinton CM, Sonsalla PK. The neurotoxin MPTP causes degeneration of specific nucleus A8, A9 and A10 dopaminergic neurons in the mouse. Neurodegeneration. 1996;5:299–312. doi: 10.1006/neur.1996.0041. [DOI] [PubMed] [Google Scholar]
- Getchell TV, Subhedar NK, Shah DS, Hackley G, Partin JV, Sen G, Getchell ML. Chemokine regulation of macrophage recruitment into the olfactory epithelium following target ablation: involvement of macrophage inflammatory protein-1alpha and monocyte chemoattractant protein-1. J Neurosci Res. 2002;70:784–793. doi: 10.1002/jnr.10432. [DOI] [PubMed] [Google Scholar]
- Ghirnikar RS, Lee YL, Eng LF. Chemokine antagonist infusion promotes axonal sparing after spinal cord contusion injury in rat. J Neurosci Res. 2001;64:582–589. doi: 10.1002/jnr.1110. [DOI] [PubMed] [Google Scholar]
- Gilmour D, Knaut H, Maischein HM, Nusslein-Volhard C. Towing of sensory axons by their migrating target cells in vivo. Nat Neurosci. 2004;7:491–492. doi: 10.1038/nn1235. [DOI] [PubMed] [Google Scholar]
- Greenfield JG, Bosanquet FD. The brain-stem lesions in Parkinsonism. J Neurol Neurosurg Psychiatry. 1953;16:213–226. doi: 10.1136/jnnp.16.4.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grunblatt E, Mandel S, Youdim MB. MPTP and 6-hydroxydopamine-induced neurodegeneration as models for Parkinson’s disease: neuroprotective strategies. J Neurol. 2000;247(Suppl 2):II95–102. doi: 10.1007/pl00022909. [DOI] [PubMed] [Google Scholar]
- Guyon A, Banisadr G, Rovere C, Cervantes A, Kitabgi P, Melik-Parsadaniantz S, Nahon JL. Complex effects of stromal cell-derived factor-1 alpha on melanin-concentrating hormone neuron excitability. Eur J Neurosci. 2005;21:701–710. doi: 10.1111/j.1460-9568.2005.03890.x. [DOI] [PubMed] [Google Scholar]
- Guyon A, Skrzydelsi D, Rovere C, Rostene W, Parsadaniantz SM, Nahon JL. Stromal cell-derived factor-1alpha modulation of the excitability of rat substantia nigra dopaminergic neurones: presynaptic mechanisms. J Neurochem. 2006;96:1540–1550. doi: 10.1111/j.1471-4159.2006.03659.x. [DOI] [PubMed] [Google Scholar]
- Hakansson A, Westberg L, Nilsson S, Buervenich S, Carmine A, Holmberg B, Sydow O, Olson L, Johnels B, Eriksson E, Nissbrandt H. Interaction of polymorphisms in the genes encoding interleukin-6 and estrogen receptor beta on the susceptibility to Parkinson’s disease. Am J Med Genet B Neuropsychiatr Genet. 2005;133:88–92. doi: 10.1002/ajmg.b.30136. [DOI] [PubMed] [Google Scholar]
- Hamre K, Tharp R, Poon K, Xiong X, Smeyne RJ. Differential strain susceptibility following 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) administration acts in an autosomal dominant fashion: quantitative analysis in seven strains of Mus musculus. Brain Res. 1999;828:91–103. doi: 10.1016/s0006-8993(99)01273-1. [DOI] [PubMed] [Google Scholar]
- Harrison JK, Jiang Y, Chen S, Xia Y, Maciejewski D, McNamara RK, Streit WJ, Salafranca MN, Adhikari S, Thompson DA, Botti P, Bacon KB, Feng L. Role for neuronally derived fractalkine in mediating interactions between neurons and CX3CR1-expressing microglia. Proc Natl Acad Sci U S A. 1998;95:10896–10901. doi: 10.1073/pnas.95.18.10896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassler R. Pathologie der Paralysis agitans und des postenzephalitischen Parkinsonismus. J Psychol Neurol. 1938;48:387–476. [Google Scholar]
- Hatori K, Nagai A, Heisel R, Ryu JK, Kim SU. Fractalkine and fractalkine receptors in human neurons and glial cells. J Neurosci Res. 2002;69:418–426. doi: 10.1002/jnr.10304. [DOI] [PubMed] [Google Scholar]
- Hebert G, Arsaut J, Dantzer R, Demotes-Mainard J. Time-course of the expression of inflammatory cytokines and matrix metalloproteinases in the striatum and mesencephalon of mice injected with 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine, a dopaminergic neurotoxin. Neurosci Lett. 2003;349:191–195. doi: 10.1016/s0304-3940(03)00832-2. [DOI] [PubMed] [Google Scholar]
- Hernan MA, Logroscino G, Garcia Rodriguez LA. Nonsteroidal anti-inflammatory drugs and the incidence of Parkinson disease. Neurology. 2006;66:1097–1099. doi: 10.1212/01.wnl.0000204446.82823.28. [DOI] [PubMed] [Google Scholar]
- Huerta C, Alvarez V, Mata IF, Coto E, Ribacoba R, Martinez C, Blazquez M, Guisasola LM, Salvador C, Lahoz CH, Pena J. Chemokines (RANTES and MCP-1) and chemokine-receptors (CCR2 and CCR5) gene polymorphisms in Alzheimer’s and Parkinson’s disease. Neurosci Lett. 2004;370:151–154. doi: 10.1016/j.neulet.2004.08.016. [DOI] [PubMed] [Google Scholar]
- Ishizuka K, Igata-Yi R, Kimura T, Hieshima K, Kukita T, Kin Y, Misumi Y, Yamamoto M, Nomiyama H, Miura R, Takamatsu J, Katsuragi S, Miyakawa T. Expression and distribution of CC chemokine macrophage inflammatory protein-1 alpha/LD78 in the human brain. Neuroreport. 1997;8:1215–1218. doi: 10.1097/00001756-199703240-00031. [DOI] [PubMed] [Google Scholar]
- Jackson-Lewis V, Jakowec M, Burke RE, Przedborski S. Time course and morphology of dopaminergic neuronal death caused by the neurotoxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. Neurodegeneration. 1995;4:257–269. doi: 10.1016/1055-8330(95)90015-2. [DOI] [PubMed] [Google Scholar]
- Javitch JA, D’Amato RJ, Strittmatter SM, Snyder SH. Parkinsonism-inducing neurotoxin, N-methyl-4-phenyl-1,2,3,6 -tetrahydropyridine: uptake of the metabolite N-methyl-4-phenylpyridine by dopamine neurons explains selective toxicity. Proc Natl Acad Sci U S A. 1985;82:2173–2177. doi: 10.1073/pnas.82.7.2173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaku K, Shikimi T, Kamisaki Y, Shinozuka K, Ishino H, Okunishi H, Takaori S. Elevation of striatal interleukin-6 and serum corticosterone contents in MPTP-treated mice. Clin Exp Pharmacol Physiol. 1999;26:680–683. doi: 10.1046/j.1440-1681.1999.03113.x. [DOI] [PubMed] [Google Scholar]
- Kalehua AN, Nagel JE, Whelchel LM, Gides JJ, Pyle RS, Smith RJ, Kusiak JW, Taub DD. Monocyte chemoattractant protein-1 and macrophage inflammatory protein-2 are involved in both excitotoxin-induced neurodegeneration and regeneration. Exp Cell Res. 2004;297:197–211. doi: 10.1016/j.yexcr.2004.02.031. [DOI] [PubMed] [Google Scholar]
- Kim SH, Kang YJ, Kim WJ, Woo DK, Lee Y, Kim DI, Park YB, Kwon BS, Park JE, Lee WH. TWEAK can induce pro-inflammatory cytokines and matrix metalloproteinase-9 in macrophages. Circ J. 2004;68:396–399. doi: 10.1253/circj.68.396. [DOI] [PubMed] [Google Scholar]
- Kim YD, Sohn NW, Kang C, Soh Y. DNA array reveals altered gene expression in response to focal cerebral ischemia. Brain Res Bull. 2002;58:491–498. doi: 10.1016/s0361-9230(02)00823-7. [DOI] [PubMed] [Google Scholar]
- Kurkowska-Jastrzebska I, Wronska A, Kohutnicka M, Czlonkowski A, Czlonkowska A. The inflammatory reaction following 1-methyl-4-phenyl-1,2,3, 6-tetrahydropyridine intoxication in mouse. Exp Neurol. 1999;156:50–61. doi: 10.1006/exnr.1998.6993. [DOI] [PubMed] [Google Scholar]
- Kwong K, Vaishnav RA, Liu Y, Subhedar N, Stromberg AJ, Getchell ML, Getchell TV. Target ablation-induced regulation of macrophage recruitment into the olfactory epithelium of Mip-1alpha−/− mice and restoration of function by exogenous MIP-1alpha. Physiol Genomics. 2004;20:73–86. doi: 10.1152/physiolgenomics.00187.2004. [DOI] [PubMed] [Google Scholar]
- Laing KJ, Secombes CJ. Chemokines. Dev Comp Immunol. 2004;28:443–460. doi: 10.1016/j.dci.2003.09.006. [DOI] [PubMed] [Google Scholar]
- Leng A, Mura A, Feldon J, Ferger B. Tumor necrosis factor-alpha receptor ablation in a chronic MPTP mouse model of Parkinson’s disease. Neurosci Lett. 2005;375:107–111. doi: 10.1016/j.neulet.2004.10.077. [DOI] [PubMed] [Google Scholar]
- Li Q, Shirabe K, Thisse C, Thisse B, Okamoto H, Masai I, Kuwada JY. Chemokine signaling guides axons within the retina in zebrafish. J Neurosci. 2005;25:1711–1717. doi: 10.1523/JNEUROSCI.4393-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberatore GT, Finkelstein DI, Wong JY, Horne MK, Porritt MJ, Donnan GA, Howells DW. Sprouting of dopaminergic axons after striatal injury: confirmation by markers not dependent on dopamine metabolism. Exp Neurol. 1999;159:565–573. doi: 10.1006/exnr.1999.7152. [DOI] [PubMed] [Google Scholar]
- Lieberam I, Agalliu D, Nagasawa T, Ericson J, Jessell TM. A Cxcl12-CXCR4 chemokine signaling pathway defines the initial trajectory of mammalian motor axons. Neuron. 2005;47:667–679. doi: 10.1016/j.neuron.2005.08.011. [DOI] [PubMed] [Google Scholar]
- Limatola C, Giovannelli A, Maggi L, Ragozzino D, Castellani L, Ciotti MT, Vacca F, Mercanti D, Santoni A, Eusebi F. SDF-1alpha-mediated modulation of synaptic transmission in rat cerebellum. Eur J Neurosci. 2000;12:2497–2504. doi: 10.1046/j.1460-9568.2000.00139.x. [DOI] [PubMed] [Google Scholar]
- Limatola C, Lauro C, Catalano M, Ciotti MT, Bertollini C, Di Angelantonio S, Ragozzino D, Eusebi F. Chemokine CX3CL1 protects rat hippocampal neurons against glutamate-mediated excitotoxicity. J Neuroimmunol. 2005;166:19–28. doi: 10.1016/j.jneuroim.2005.03.023. [DOI] [PubMed] [Google Scholar]
- Liu B, Hong JS. Role of microglia in inflammation-mediated neurodegenerative diseases: mechanisms and strategies for therapeutic intervention. J Pharmacol Exp Ther. 2003;304:1–7. doi: 10.1124/jpet.102.035048. [DOI] [PubMed] [Google Scholar]
- Liu L, Hsu SS, Kalia SK, Lozano AM. Injury and strain-dependent dopaminergic neuronal degeneration in the substantia nigra of mice after axotomy or MPTP. Brain Res. 2003a;994:243–252. doi: 10.1016/j.brainres.2003.09.066. [DOI] [PubMed] [Google Scholar]
- Liu Y, Qin L, Li G, Zhang W, An L, Liu B, Hong JS. Dextromethorphan protects dopaminergic neurons against inflammation-mediated degeneration through inhibition of microglial activation. J Pharmacol Exp Ther. 2003b;305:212–218. doi: 10.1124/jpet.102.043166. [DOI] [PubMed] [Google Scholar]
- Lu M, Grove EA, Miller RJ. Abnormal development of the hippocampal dentate gyrus in mice lacking the CXCR4 chemokine receptor. Proc Natl Acad Sci U S A. 2002;99:7090–7095. doi: 10.1073/pnas.092013799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo Y, Cai J, Xue H, Miura T, Rao MS. Functional SDF1 alpha/CXCR4 signaling in the developing spinal cord. J Neurochem. 2005;93:452–462. doi: 10.1111/j.1471-4159.2005.03049.x. [DOI] [PubMed] [Google Scholar]
- Maharaj DS, Saravanan KS, Maharaj H, Mohanakumar KP, Daya S. Acetaminophen and aspirin inhibit superoxide anion generation and lipid peroxidation, and protect against 1-methyl-4-phenyl pyridinium-induced dopaminergic neurotoxicity in rats. Neurochem Int. 2004;44:355–360. doi: 10.1016/s0197-0186(03)00170-0. [DOI] [PubMed] [Google Scholar]
- Mandel S, Grunblatt E, Youdim M. cDNA microarray to study gene expression of dopaminergic neurodegeneration and neuroprotection in MPTP and 6-hydroxydopamine models: implications for idiopathic Parkinson’s disease. J Neural Transm Suppl. 2000:117–124. doi: 10.1007/978-3-7091-6301-6_7. [DOI] [PubMed] [Google Scholar]
- Marchetti B, Abbracchio MP. To be or not to be (inflamed)--is that the question in anti-inflammatory drug therapy of neurodegenerative disorders? Trends Pharmacol Sci. 2005;26:517–525. doi: 10.1016/j.tips.2005.08.007. [DOI] [PubMed] [Google Scholar]
- Matejuk A, Dwyer J, Ito A, Bruender Z, Vandenbark AA, Offner H. Effects of cytokine deficiency on chemokine expression in CNS of mice with EAE. J Neurosci Res. 2002;67:680–688. doi: 10.1002/jnr.10156. [DOI] [PubMed] [Google Scholar]
- McGeer EG, McGeer PL. The importance of inflammatory mechanisms in Alzheimer disease. Exp Gerontol. 1998a;33:371–378. doi: 10.1016/s0531-5565(98)00013-8. [DOI] [PubMed] [Google Scholar]
- McGeer PL, McGeer EG. Glial cell reactions in neurodegenerative diseases: pathophysiology and therapeutic interventions. Alzheimer Dis Assoc Disord. 1998b;12(Suppl 2):S1–6. [PubMed] [Google Scholar]
- McLaughlin P, Zhou Y, Ma T, Liu J, Zhang W, Hong JS, Kovacs M, Zhang J. Proteomic analysis of microglial contribution to mouse strain-dependent dopaminergic neurotoxicity. Glia. 2006;53:567–582. doi: 10.1002/glia.20294. [DOI] [PubMed] [Google Scholar]
- McMahon EJ, Cook DN, Suzuki K, Matsushima GK. Absence of macrophage-inflammatory protein-1alpha delays central nervous system demyelination in the presence of an intact blood-brain barrier. J Immunol. 2001;167:2964–2971. doi: 10.4049/jimmunol.167.5.2964. [DOI] [PubMed] [Google Scholar]
- Meighan-Mantha RL, Hsu DK, Guo Y, Brown SA, Feng SL, Peifley KA, Alberts GF, Copeland NG, Gilbert DJ, Jenkins NA, Richards CM, Winkles JA. The mitogen-inducible Fn14 gene encodes a type I transmembrane protein that modulates fibroblast adhesion and migration. J Biol Chem. 1999;274:33166–33176. doi: 10.1074/jbc.274.46.33166. [DOI] [PubMed] [Google Scholar]
- Meucci O, Fatatis A, Simen AA, Miller RJ. Expression of CX3CR1 chemokine receptors on neurons and their role in neuronal survival. Proc Natl Acad Sci U S A. 2000;97:8075–8080. doi: 10.1073/pnas.090017497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minami M, Katayama T, Satoh M. Brain Cytokines and Chemokines: Roles in Ischemic Injury and Pain. J Pharmacol Sci. 2006 doi: 10.1254/jphs.crj06005x. [DOI] [PubMed] [Google Scholar]
- Mitsumoto Y, Watanabe A, Mori A, Koga N. Spontaneous regeneration of nigrostriatal dopaminergic neurons in MPTP-treated C57BL/6 mice. Biochem Biophys Res Commun. 1998;248:660–663. doi: 10.1006/bbrc.1998.8986. [DOI] [PubMed] [Google Scholar]
- Mizuno T, Kawanokuchi J, Numata K, Suzumura A. Production and neuroprotective functions of fractalkine in the central nervous system. Brain Res. 2003;979:65–70. doi: 10.1016/s0006-8993(03)02867-1. [DOI] [PubMed] [Google Scholar]
- Muessel MJ, Berman NE, Klein RM. Early and specific expression of monocyte chemoattractant protein-1 in the thalamus induced by cortical injury. Brain Res. 2000;870:211–221. doi: 10.1016/s0006-8993(00)02450-1. [DOI] [PubMed] [Google Scholar]
- Muessel MJ, Klein RM, Wilson AM, Berman NE. Ablation of the chemokine monocyte chemoattractant protein-1 delays retrograde neuronal degeneration, attenuates microglial activation, and alters expression of cell death molecules. Brain Res Mol Brain Res. 2002;103:12–27. doi: 10.1016/s0169-328x(02)00158-4. [DOI] [PubMed] [Google Scholar]
- Murphy PM, Baggiolini M, Charo IF, Hebert CA, Horuk R, Matsushima K, Miller LH, Oppenheim JJ, Power CA. International union of pharmacology. XXII. Nomenclature for chemokine receptors. Pharmacol Rev. 2000;52:145–176. [PubMed] [Google Scholar]
- Nagatsu T, Mogi M, Ichinose H, Togari A. Cytokines in Parkinson’s disease. J Neural Transm Suppl. 2000:143–151. [PubMed] [Google Scholar]
- Nagatsu T, Sawada M. Inflammatory process in Parkinson’s disease: role for cytokines. Curr Pharm Des. 2005;11:999–1016. doi: 10.2174/1381612053381620. [DOI] [PubMed] [Google Scholar]
- Nakayama M, Ishidoh K, Kojima Y, Harada N, Kominami E, Okumura K, Yagita H. Fibroblast growth factor-inducible 14 mediates multiple pathways of TWEAK-induced cell death. J Immunol. 2003;170:341–348. doi: 10.4049/jimmunol.170.1.341. [DOI] [PubMed] [Google Scholar]
- Nishi T, Maier CM, Hayashi T, Saito A, Chan PH. Superoxide dismutase 1 overexpression reduces MCP-1 and MIP-1 alpha expression after transient focal cerebral ischemia. J Cereb Blood Flow Metab. 2005;25:1312–1324. doi: 10.1038/sj.jcbfm.9600124. [DOI] [PubMed] [Google Scholar]
- Nishimura M, Kuno S, Mizuta I, Ohta M, Maruyama H, Kaji R, Kawakami H. Influence of monocyte chemoattractant protein 1 gene polymorphism on age at onset of sporadic Parkinson’s disease. Mov Disord. 2003;18:953–955. doi: 10.1002/mds.10462. [DOI] [PubMed] [Google Scholar]
- Nishimura M, Mizuta I, Mizuta E, Yamasaki S, Ohta M, Kuno S. Influence of interleukin-1beta gene polymorphisms on age-at-onset of sporadic Parkinson’s disease. Neurosci Lett. 2000;284:73–76. doi: 10.1016/s0304-3940(00)00991-5. [DOI] [PubMed] [Google Scholar]
- Nishiyori A, Minami M, Ohtani Y, Takami S, Yamamoto J, Kawaguchi N, Kume T, Akaike A, Satoh M. Localization of fractalkine and CX3CR1 mRNAs in rat brain: does fractalkine play a role in signaling from neuron to microglia? FEBS Lett. 1998;429:167–172. doi: 10.1016/s0014-5793(98)00583-3. [DOI] [PubMed] [Google Scholar]
- Novikova L, Garris BL, Garris DR, Lau YS. Early signs of neuronal apoptosis in the substantia nigra pars compacta of the progressive neurodegenerative mouse 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine/probenecid model of Parkinson’s disease. Neuroscience. 2006;140:67–76. doi: 10.1016/j.neuroscience.2006.02.007. [DOI] [PubMed] [Google Scholar]
- Nurmi E, Ruottinen HM, Bergman J, Haaparanta M, Solin O, Sonninen P, Rinne JO. Rate of progression in Parkinson’s disease: a 6-[18F]fluoro-L-dopa PET study. Mov Disord. 2001;16:608–615. doi: 10.1002/mds.1139. [DOI] [PubMed] [Google Scholar]
- Orr CF, Rowe DB, Halliday GM. An inflammatory review of Parkinson’s disease. Prog Neurobiol. 2002;68:325–340. doi: 10.1016/s0301-0082(02)00127-2. [DOI] [PubMed] [Google Scholar]
- Parkinson J. An essay on the shaking palsy. 1817. J Neuropsychiatry Clin Neurosci. 1817;14:223–236; discussion 222. doi: 10.1176/jnp.14.2.223. [DOI] [PubMed] [Google Scholar]
- Perrin FE, Lacroix S, Aviles-Trigueros M, David S. Involvement of monocyte chemoattractant protein-1, macrophage inflammatory protein-1alpha and interleukin-1beta in Wallerian degeneration. Brain. 2005;128:854–866. doi: 10.1093/brain/awh407. [DOI] [PubMed] [Google Scholar]
- Poirier J, Kogan S, Gauthier S. Environment, genetics and idiopathic Parkinson’s disease. Can J Neurol Sci. 1991;18:70–76. doi: 10.1017/s0317167100031334. [DOI] [PubMed] [Google Scholar]
- Potrovita I, Zhang W, Burkly L, Hahm K, Lincecum J, Wang MZ, Maurer MH, Rossner M, Schneider A, Schwaninger M. Tumor necrosis factor-like weak inducer of apoptosis-induced neurodegeneration. J Neurosci. 2004;24:8237–8244. doi: 10.1523/JNEUROSCI.1089-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pujol F, Kitabgi P, Boudin H. The chemokine SDF-1 differentially regulates axonal elongation and branching in hippocampal neurons. J Cell Sci. 2005;118:1071–1080. doi: 10.1242/jcs.01694. [DOI] [PubMed] [Google Scholar]
- Ragozzino D, Renzi M, Giovannelli A, Eusebi F. Stimulation of chemokine CXC receptor 4 induces synaptic depression of evoked parallel fibers inputs onto Purkinje neurons in mouse cerebellum. J Neuroimmunol. 2002;127:30–36. doi: 10.1016/s0165-5728(02)00093-0. [DOI] [PubMed] [Google Scholar]
- Rinne OJ, Nurmi E, Ruottinen HM, Bergman J, Eskola O, Solin O. [(18)F]FDOPA and [(18)F]CFT are both sensitive PET markers to detect presynaptic dopaminergic hypofunction in early Parkinson’s disease. Synapse. 2001;40:193–200. doi: 10.1002/syn.1042. [DOI] [PubMed] [Google Scholar]
- Robin AM, Zhang ZG, Wang L, Zhang RL, Katakowski M, Zhang L, Wang Y, Zhang C, Chopp M. Stromal cell-derived factor 1alpha mediates neural progenitor cell motility after focal cerebral ischemia. J Cereb Blood Flow Metab. 2006;26:125–134. doi: 10.1038/sj.jcbfm.9600172. [DOI] [PubMed] [Google Scholar]
- Ross OA, O’Neill C, Rea IM, Lynch T, Gosal D, Wallace A, Curran MD, Middleton D, Gibson JM. Functional promoter region polymorphism of the proinflammatory chemokine IL-8 gene associates with Parkinson’s disease in the Irish. Hum Immunol. 2004;65:340–346. doi: 10.1016/j.humimm.2004.01.015. [DOI] [PubMed] [Google Scholar]
- Rousselet E, Callebert J, Parain K, Joubert C, Hunot S, Hartmann A, Jacque C, Perez-Diaz F, Cohen-Salmon C, Launay JM, Hirsch EC. Role of TNF-alpha receptors in mice intoxicated with the parkinsonian toxin MPTP. Exp Neurol. 2002;177:183–192. doi: 10.1006/exnr.2002.7960. [DOI] [PubMed] [Google Scholar]
- Sairam K, Saravanan KS, Banerjee R, Mohanakumar KP. Non-steroidal anti-inflammatory drug sodium salicylate, but not diclofenac or celecoxib, protects against 1-methyl-4-phenyl pyridinium-induced dopaminergic neurotoxicity in rats. Brain Res. 2003;966:245–252. doi: 10.1016/s0006-8993(02)04174-4. [DOI] [PubMed] [Google Scholar]
- Schiess M. Nonsteroidal anti-inflammatory drugs protect against Parkinson neurodegeneration: can an NSAID a day keep Parkinson disease away? Arch Neurol. 2003;60:1043–1044. doi: 10.1001/archneur.60.8.1043. [DOI] [PubMed] [Google Scholar]
- Schulte T, Schols L, Muller T, Woitalla D, Berger K, Kruger R. Polymorphisms in the interleukin-1 alpha and beta genes and the risk for Parkinson’s disease. Neurosci Lett. 2002;326:70–72. doi: 10.1016/s0304-3940(02)00301-4. [DOI] [PubMed] [Google Scholar]
- Schwarting RK, Sedelis M, Hofele K, Auburger GW, Huston JP. Strain-dependent recovery of open-field behavior and striatal dopamine deficiency in the mouse MPTP model of Parkinson’s disease. Neurotox Res. 1999;1:41–56. doi: 10.1007/BF03033338. [DOI] [PubMed] [Google Scholar]
- Siebert H, Sachse A, Kuziel WA, Maeda N, Bruck W. The chemokine receptor CCR2 is involved in macrophage recruitment to the injured peripheral nervous system. J Neuroimmunol. 2000;110:177–185. doi: 10.1016/s0165-5728(00)00343-x. [DOI] [PubMed] [Google Scholar]
- Simpson JE, Newcombe J, Cuzner ML, Woodroofe MN. Expression of monocyte chemoattractant protein-1 and other beta-chemokines by resident glia and inflammatory cells in multiple sclerosis lesions. J Neuroimmunol. 1998;84:238–249. doi: 10.1016/s0165-5728(97)00208-7. [DOI] [PubMed] [Google Scholar]
- Smeyne M, Goloubeva O, Smeyne RJ. Strain-dependent susceptibility to MPTP and MPP(+)-induced parkinsonism is determined by glia. Glia. 2001;34:73–80. [PubMed] [Google Scholar]
- Smeyne M, Jiao Y, Shepherd KR, Smeyne RJ. Glia cell number modulates sensitivity to MPTP in mice. Glia. 2005;52:144–152. doi: 10.1002/glia.20233. [DOI] [PubMed] [Google Scholar]
- Song DD, Haber SN. Striatal responses to partial dopaminergic lesion: evidence for compensatory sprouting. J Neurosci. 2000;20:5102–5114. doi: 10.1523/JNEUROSCI.20-13-05102.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonsalla PK, Heikkila RE. The influence of dose and dosing interval on MPTP-induced dopaminergic neurotoxicity in mice. Eur J Pharmacol. 1986;129:339–345. doi: 10.1016/0014-2999(86)90444-9. [DOI] [PubMed] [Google Scholar]
- Sriram K, Matheson JM, Benkovic SA, Miller DB, Luster MI, O’Callaghan JP. Mice deficient in TNF receptors are protected against dopaminergic neurotoxicity: implications for Parkinson’s disease. Faseb J. 2002;16:1474–1476. doi: 10.1096/fj.02-0216fje. [DOI] [PubMed] [Google Scholar]
- Sriram K, Matheson JM, Benkovic SA, Miller DB, Luster MI, O’Callaghan JP. Deficiency of TNF receptors suppresses microglial activation and alters the susceptibility of brain regions to MPTP-induced neurotoxicity: role of TNF-alpha. Faseb J. 2006a;20:670–682. doi: 10.1096/fj.05-5106com. [DOI] [PubMed] [Google Scholar]
- Sriram K, Miller DB, O’Callaghan JP. Minocycline attenuates microglial activation but fails to mitigate striatal dopaminergic neurotoxicity: role of tumor necrosis factor-alpha. J Neurochem. 2006b;96:706–718. doi: 10.1111/j.1471-4159.2005.03566.x. [DOI] [PubMed] [Google Scholar]
- Stumm RK, Zhou C, Ara T, Lazarini F, Dubois-Dalcq M, Nagasawa T, Hollt V, Schulz S. CXCR4 regulates interneuron migration in the developing neocortex. J Neurosci. 2003;23:5123–5130. doi: 10.1523/JNEUROSCI.23-12-05123.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takami S, Nishikawa H, Minami M, Nishiyori A, Sato M, Akaike A, Satoh M. Induction of macrophage inflammatory protein MIP-1alpha mRNA on glial cells after focal cerebral ischemia in the rat. Neurosci Lett. 1997;227:173–176. doi: 10.1016/s0304-3940(97)00338-8. [DOI] [PubMed] [Google Scholar]
- Tanabe K, Bonilla I, Winkles JA, Strittmatter SM. Fibroblast growth factor-inducible-14 is induced in axotomized neurons and promotes neurite outgrowth. J Neurosci. 2003;23:9675–9686. doi: 10.1523/JNEUROSCI.23-29-09675.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tissir F, Wang CE, Goffinet AM. Expression of the chemokine receptor Cxcr4 mRNA during mouse brain development. Brain Res Dev Brain Res. 2004;149:63–71. doi: 10.1016/j.devbrainres.2004.01.002. [DOI] [PubMed] [Google Scholar]
- Trétiakoff, C. Contribution à l’étude de l’anatomie pathologique du locus niger) University of Paris; 1919. [Google Scholar]
- Tsang F, Soong TW. Interactions between environmental and genetic factors in the pathophysiology of Parkinson’s disease. IUBMB Life. 2003;55:323–327. doi: 10.1080/1521654031000153058. [DOI] [PubMed] [Google Scholar]
- Urakami K, Masaki N, Shimoda K, Nishikawa S, Takahashi K. Increase of striatal dopamine turnover by stress in MPTP-treated mice. Clin Neuropharmacol. 1988;11:360–368. doi: 10.1097/00002826-198808000-00004. [DOI] [PubMed] [Google Scholar]
- Vilz TO, Moepps B, Engele J, Molly S, Littman DR, Schilling K. The SDF-1/CXCR4 pathway and the development of the cerebellar system. Eur J Neurosci. 2005;22:1831–1839. doi: 10.1111/j.1460-9568.2005.04378.x. [DOI] [PubMed] [Google Scholar]
- West MJ, Slomianka L, Gundersen HJ. Unbiased stereological estimation of the total number of neurons in thesubdivisions of the rat hippocampus using the optical fractionator. Anat Rec. 1991;231:482–497. doi: 10.1002/ar.1092310411. [DOI] [PubMed] [Google Scholar]
- Wiley SR, Cassiano L, Lofton T, Davis-Smith T, Winkles JA, Lindner V, Liu H, Daniel TO, Smith CA, Fanslow WC. A novel TNF receptor family member binds TWEAK and is implicated in angiogenesis. Immunity. 2001;15:837–846. doi: 10.1016/s1074-7613(01)00232-1. [DOI] [PubMed] [Google Scholar]
- Wilson SD, Billings PR, D’Eustachio P, Fournier RE, Geissler E, Lalley PA, Burd PR, Housman DE, Taylor BA, Dorf ME. Clustering of cytokine genes on mouse chromosome 11. J Exp Med. 1990;171:1301–1314. doi: 10.1084/jem.171.4.1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu DC, Jackson-Lewis V, Vila M, Tieu K, Teismann P, Vadseth C, Choi DK, Ischiropoulos H, Przedborski S. Blockade of microglial activation is neuroprotective in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model of Parkinson disease. J Neurosci. 2002;22:1763–1771. doi: 10.1523/JNEUROSCI.22-05-01763.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia MQ, Hyman BT. Chemokines/chemokine receptors in the central nervous system and Alzheimer’s disease. J Neurovirol. 1999;5:32–41. doi: 10.3109/13550289909029743. [DOI] [PubMed] [Google Scholar]
- Xia MQ, Qin SX, Wu LJ, Mackay CR, Hyman BT. Immunohistochemical study of the beta-chemokine receptors CCR3 and CCR5 and their ligands in normal and Alzheimer’s disease brains. Am J Pathol. 1998;153:31–37. doi: 10.1016/s0002-9440(10)65542-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yepes M, Brown SA, Moore EG, Smith EP, Lawrence DA, Winkles JA. A soluble Fn14-Fc decoy receptor reduces infarct volume in a murine model of cerebral ischemia. Am J Pathol. 2005;166:511–520. doi: 10.1016/S0002-9440(10)62273-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youdim MB, Arraf Z. Prevention of MPTP (N-methyl-4-phenyl-1,2,3,6-tetrahydropyridine) dopaminergic neurotoxicity in mice by chronic lithium: involvements of Bcl-2 and Bax. Neuropharmacology. 2004;46:1130–1140. doi: 10.1016/j.neuropharm.2004.02.005. [DOI] [PubMed] [Google Scholar]
- Zou LP, Pelidou SH, Abbas N, Deretzi G, Mix E, Schaltzbeerg M, Winblad B, Zhu J. Dynamics of production of MIP-1alpha, MCP-1 and MIP-2 and potential role of neutralization of these chemokines in the regulation of immune responses during experimental autoimmune neuritis in Lewis rats. J Neuroimmunol. 1999;98:168–175. doi: 10.1016/s0165-5728(99)00100-9. [DOI] [PubMed] [Google Scholar]
- Zujovic V, Benavides J, Vige X, Carter C, Taupin V. Fractalkine modulates TNF-alpha secretion and neurotoxicity induced by microglial activation. Glia. 2000;29:305–315. [PubMed] [Google Scholar]