

Abstract
The cornerstone of the functionality of almost all motor proteins is the regulation of their activity by binding interactions with their respective substrates. In most cases, the underlying mechanism of this regulation remains unknown. Here, we reveal a novel mechanism used by secretory preproteins to control the catalytic cycle of the helicase ‘DEAD' motor of SecA, the preprotein translocase ATPase. The central feature of this mechanism is a highly conserved salt-bridge, Gate1, that controls the opening/closure of the nucleotide cleft. Gate1 regulates the propagation of binding signal generated at the Preprotein Binding Domain to the nucleotide cleft, thus allowing the physical coupling of preprotein binding and release to the ATPase cycle. This relay mechanism is at play only after SecA has been previously ‘primed' by binding to SecYEG, the transmembrane protein-conducting channel. The Gate1-controlled relay mechanism is essential for protein translocase catalysis and may be common in helicase motors.
Keywords: ATPase, DEAD motor, helicase, protein secretion
Introduction
More than a third of a cellular proteome is secreted across or inserted into biological membranes through an unusually dynamic nanomachine termed ‘preprotein translocase' or ‘translocon' (Veenendaal et al, 2004; Vrontou and Economou, 2004; Osborne et al, 2005). Bacterial translocase comprises the membrane-embedded SecYEG ‘protein-conducting channel' (Veenendaal et al, 2004; Osborne et al, 2005) and the peripheral motor-like ATPase SecA (Vrontou and Economou, 2004).
SecA is a member of the superfamily 2 (SF2) ‘DExH/D' proteins (Koonin and Gorbalenya, 1992; Vrontou and Economou, 2004; Papanikolau et al, 2007). Many of these proteins are nucleic acid helicases (Caruthers and McKay, 2002; Cavanaugh et al, 2006; Cordin et al, 2006). However, other functions such as disassembly of protein–RNA complexes (Jankowsky et al, 2001; Yang and Jankowsky, 2006) and stabilization of RNA conformations (Solem et al, 2006) have been described. Two structurally homologous RecA-like domains lie at the core of these proteins and form the ‘helicase' or ‘DEAD' (‘Asp–Glu–Ala–Asp') motor (Caruthers and McKay, 2002; Cordin et al, 2006). In SecA, the DEAD motor comprises the discontinuous nucleotide binding domain (NBD; aa 1–220 and aa 378–420; Figure 1A) and the intramolecular regulator of ATP hydrolysis (IRA2; aa 421–610). Nucleotides bind to a cleft formed at the NBD/IRA2 interface. The motor harbors the nine conserved motifs of the DEAD RNA helicase subfamily (Vrontou and Economou, 2004; Cordin et al, 2006; Papanikolau et al, 2007). Catalytic Motifs I and II, which correspond to ATPase ‘Walker boxes' A and B, are located on NBD and are juxtaposed to Motifs V and VI located on IRA2. Motif VI is a flexible helix that carries the ‘arginine finger' Arg574 (Sianidis et al, 2001; Chou et al, 2002; Keramisanou et al, 2006).
Figure 1.
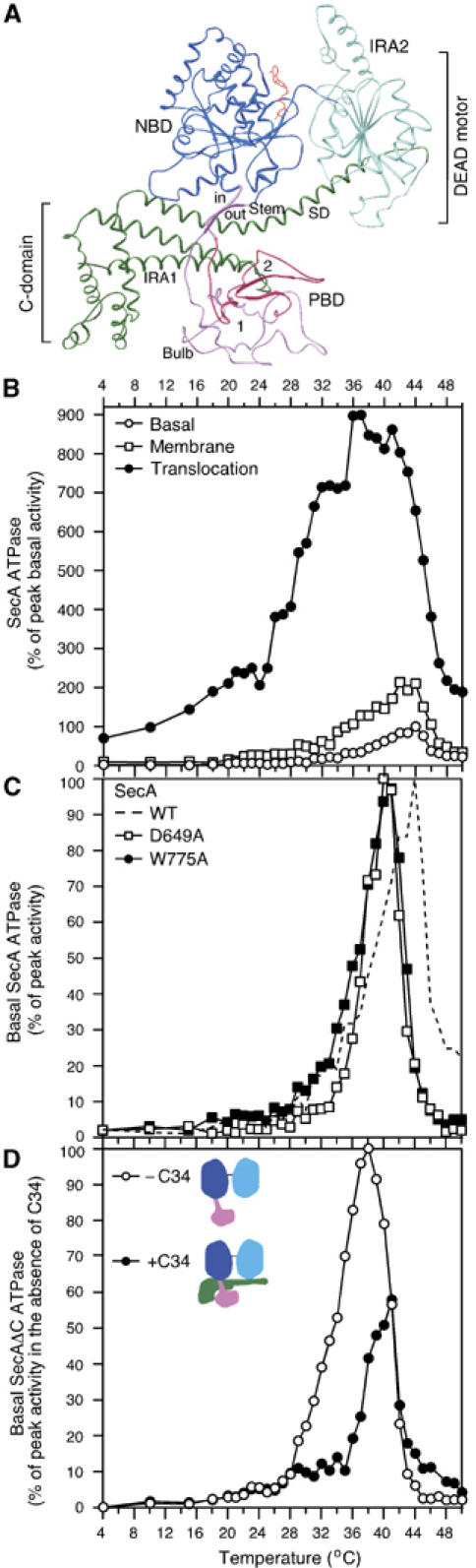
(A) Ribbon representation of ecSecA (Papanikolau et al, 2007). NBD, nucleotide binding domain (blue); IRA2, intramolecular regulator of the ATPase 2 (light blue); PBD, preprotein binding domain (magenta), C-domain (green), ATP (red); SD, scaffold domain. (B) The Kcat values (pmoles Pi/pmol SecA protomer/min) of basal, membrane (urea-treated IMVs; 17 μg protein per milliliter) and translocation (IMVs plus 60 μg/ml proOmpA) ATPase activities of SecA were determined as a function of temperature. The peak Kcat value of basal ATPase was taken as 100% and all other values were expressed as its percentage. Averaged data (n=8) of SecA basal ATPase were used in subsequent graphs. (C) Basal ATPase activities of SecA, SecA(D649A), SecA(W775A) (as in panel B). The peak Kcat value for each ATPase (Supplementary Table 1) was taken as 100% and all other values were expressed as its percentage. (D) Basal ATPase activity of SecAΔC (as in B) in the presence or absence of 10X molar excess of C34 (as indicated). The peak Kcat value for the SecAΔC ATPase (Supplementary Table 1) was taken as 100% and all other values (± C34) were expressed as its percentage. Averaged data (n=12) of SecAΔC were used in subsequent graphs.
Functional specialization of DEAD motors derives from auxiliary ‘specificity domains' or additional subunits (Caruthers and McKay, 2002; Cordin et al, 2006) that can interact with different ligands or protein partners. SecA has two such domains (Figures 1A and 2A): the preprotein binding domain (PBD; aa 221–377) that ‘sprouts out' of a surface-exposed loop of NBD through an antiparallel β-sheet (Stem) and ends in a bilobate ‘Bulb' (Papanikou et al, 2005) and the C-domain (aa 611–901) that controls the DEAD motor ATPase via a long α-helix (scaffold domain, SD) (Vrontou et al, 2004; Keramisanou et al, 2006; Mori and Ito, 2006). During protein translocation, SecA binds to SecYEG (Hartl et al, 1990) and preproteins (Lill et al, 1990), and several of its regions, including the C-domain, insert into the membrane (Economou and Wickner, 1994; Eichler and Wickner, 1997; Jilaveanu and Oliver, 2007). As a result, its ATPase activity is stimulated several fold (Lill et al, 1990). Isolated SecAΔC (aa 1–610), a derivative lacking the C-domain, displays stimulated ATPase and enhanced preprotein binding akin to SecA engaged in translocation (Karamanou et al, 1999; Baud et al, 2002; Vrontou et al, 2004). These events may be linked to cycles of dissociation of the dimeric SecA into monomers (Or et al, 2002; Duong, 2003).
Figure 2.
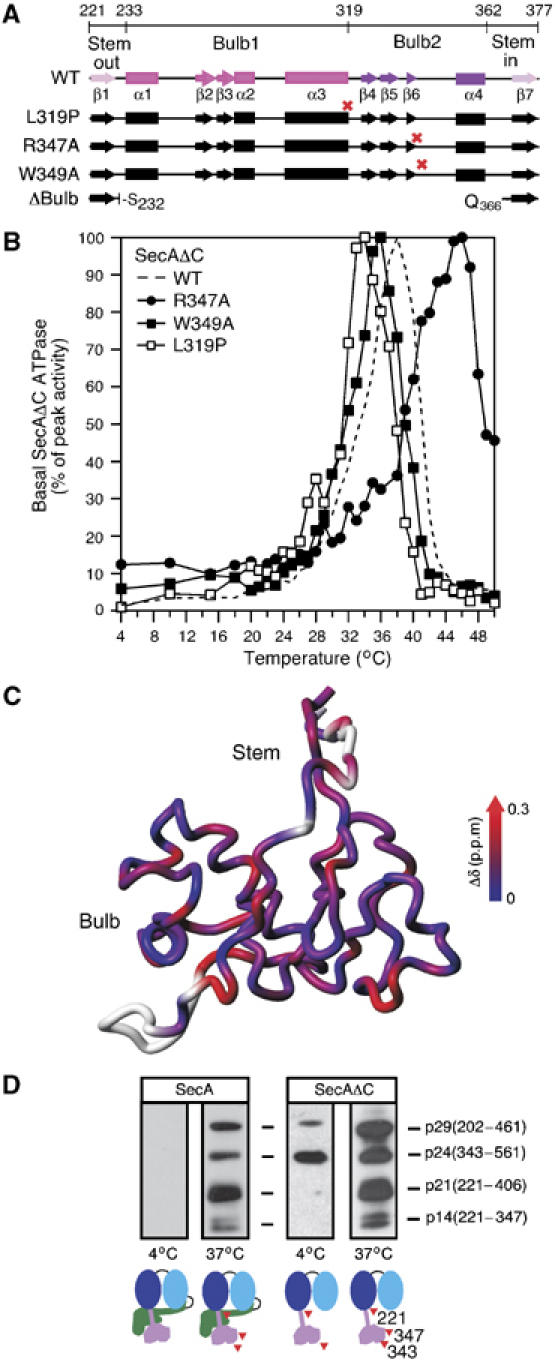
(A) PBD deletions, point mutations (red crosses) and secondary structure are indicated on a region map. β1 and β7 (pink) together form the antiparallel sheet of the Stem. Between them lie the two globular halves of the Bulb (1, magenta and 2, purple). See also Figure 1A. (B) Basal ATPase activities of SecAΔC, SecAΔC(R347A), SecAΔC(W349A) and SecAΔC(L319P) (as in Figure 1D). (C) Chemical shift change (Δδ) as a function of temperature mapped onto the structure of PBD. Higher Δδ values indicate temperature-induced unfolding. (D) SecA or SecAΔC (245 pmol in 100 μl buffer B) were treated (13 min) by limited trypsinolysis at 4°C (0.25 μg/ml) or 37°C (0.025 μg/ml). Trypsin was inactivated with Pefabloc (9 mM). Following SDS–PAGE, polypeptides were immunostained by α-PBD-specific antibody and were identified by N-terminal sequencing.
How the various substrates modulate DEAD motor ATP catalysis and thus control vectorial movement of SecA and other SF2 proteins is a major unresolved question. To address this, we examined the requirements for ATPase activation and the concomitant structural alterations of the DEAD motor in SecA. We demonstrate that SecA undergoes conformational relay cycles between the specificity domains and the DEAD motor. These are modulated by preproteins and controlled by Gate1, a salt-bridge that links the two DEAD motor domains and controls whether the DEAD motor is ‘loose' and nucleotide-free or ‘tight' with a bound nucleotide. This mechanism is a novel essential feature of protein translocase catalysis and may be widespread in helicases.
Results
Preprotein/SecYEG synergy relieves an energetic barrier to SecA ATPase activation
To gain insight into the translocase motor function and identify limiting steps and structural changes, we measured ATP hydrolysis by SecA as a function of temperature (hereafter we will refer to this assay as: thermal ATPase; Figure 1B). Soluble SecA, in the absence of translocation ligands, is barely active below 35°C (basal ATPase (Lill et al, 1990)). This basal ATPase displays an unusually narrow activity window (∼10°C), with an optimum at 43–44°C (Figure 1B). Addition of inverted inner membrane vesicles harboring embedded SecYEG (IMVs) induces some ATPase stimulation (∼2-fold; membrane ATPase) without nevertheless shifting the catalytic optimum temperature significantly. Addition of the model preprotein proOmpA in the absence of IMVs leaves SecA basal thermal ATPase unaffected (data not shown). Strikingly, addition of both IMVs and preprotein not only significantly stimulates ATP hydrolysis (∼9-fold; translocation ATPase) (Lill et al, 1990) but also causes a, previously undocumented, dramatic shift of both the onset and the catalytic optimum to lower temperatures (∼15 and ∼37°C, respectively; Figure 1B compare ‘translocation' with ‘membrane' ATPase). Translocation ATPase correlates directly with ongoing protein translocation (Lill et al, 1990). Notably, during active secretion, preproteins stimulate the ATPase activity of the translocase motor across most of the full spectrum of permissive temperatures for Escherichia coli viability (∼8–48°C).
The thermal ATPase properties of SecA imply that the protein possesses an intrinsic mechanism that imposes an energetic barrier to its catalytic activation. This barrier can be overcome only by the synergy of preprotein association to SecA that is already primed by binding to SecYEG (Hartl et al, 1990). Primed SecA binds to preprotein with a 20- to 30-fold higher affinity (Hartl et al, 1990; Papanikou et al, 2005). Below, we provide evidence that such a mechanism indeed exists and is essential for protein translocation. Using the thermal ATPase as a tool, we identified the elements in SecA that impose this intrinsic mechanism, dissected their individual contributions and established the order of events necessary for translocase catalytic activation.
The intrinsic temperature optimum of the DEAD motor ATPase is up-shifted by the C-domain
DEAD motor/C-domain interaction suppresses futile ATP hydrolysis (Karamanou et al, 1999). To test if the C-domain contributes additionally to the energetic barrier mechanism assumed above, we examined the basal thermal ATPase of SecA(W775A) and SecA(D649A), two SecA mutants with weakened C-domain/DEAD motor interaction (Baud et al, 2002; Vrontou et al, 2004; Keramisanou et al, 2006). Both mutant proteins display a measurable and reproducible (3–4°C) reduction of temperature at which their optimal ATPase is observed. Yet they still maintain substantial thermal control of their basal ATPase (Figure 1C), despite their higher ATP turnover (Supplementary Table 1).
In agreement with these results, SecA truncated of its C-domain (SecAΔC) exhibits a down-shift of its optimal ATPase temperature to 37°C, yet maintains tight thermal regulation of its ATPase, with a narrow activity peak (Figure 1D). These data suggested that a mechanism inherent to SecAΔC maintains the ATPase at 37°C. Apparently, this ATPase activity is further shifted to higher temperatures (i.e. stabilized) when the C-domain is present. To verify this, we compared SecAΔC ATPase in the absence or presence of isolated C-domain polypeptide (C34; Figure 1D) added in trans. Physical reconstitution of the SecAΔC/C-domain complex not only suppresses the ATPase activity of the DEAD motor down to the levels of SecA (Karamanou et al, 1999) but also shifts the ATPase optimum to higher temperatures (Figure 1D).
We conclude that SecAΔC tightly regulates its basal ATPase activity up to ∼37°C through an inherent, unknown mechanism. This property is further enhanced through physical interaction with the C-domain. It is possible that preproteins modulate both the C-domain and the SecAΔC region in order to activate the translocase
PBD controls the ATPase of the DEAD motor
Preproteins bind to PBD, the remaining ‘specificity domain' within SecAΔC. Could PBD affect thermal regulation of the DEAD motor ATPase? To address this, we sought to identify mutants with altered thermal ATPase by screening several point mutants in conserved residues of PBD. The basal ATPase of each mutant was determined as a function of temperature and compared with that of SecAΔC. Three such mutants were identified (Figure 2A and B; Supplementary Table 1): SecAΔC(W349A) (Papanikou et al, 2005), SecAΔC(L319P) and SecAΔC(R347A). Strikingly, activation of the SecAΔC(W349A) and SecAΔC(L319P) ATPase occurred at lower temperatures compared with SecAΔC (∼34–35°C) (we term these ‘early' mutants), whereas that of SecAΔC(R347A) occurred at significantly higher temperature (∼47°C; ‘late' mutant) (Figure 2B; Supplementary Table 1). Remarkably, SecAΔC(ΔBulb) that misses all of the Bulb region (Papanikou et al, 2005), displays a ‘late' thermal ATPase (see below; Figure 4C), suggesting that the presence of a functional Bulb is required for DEAD motor activation.
Figure 4.
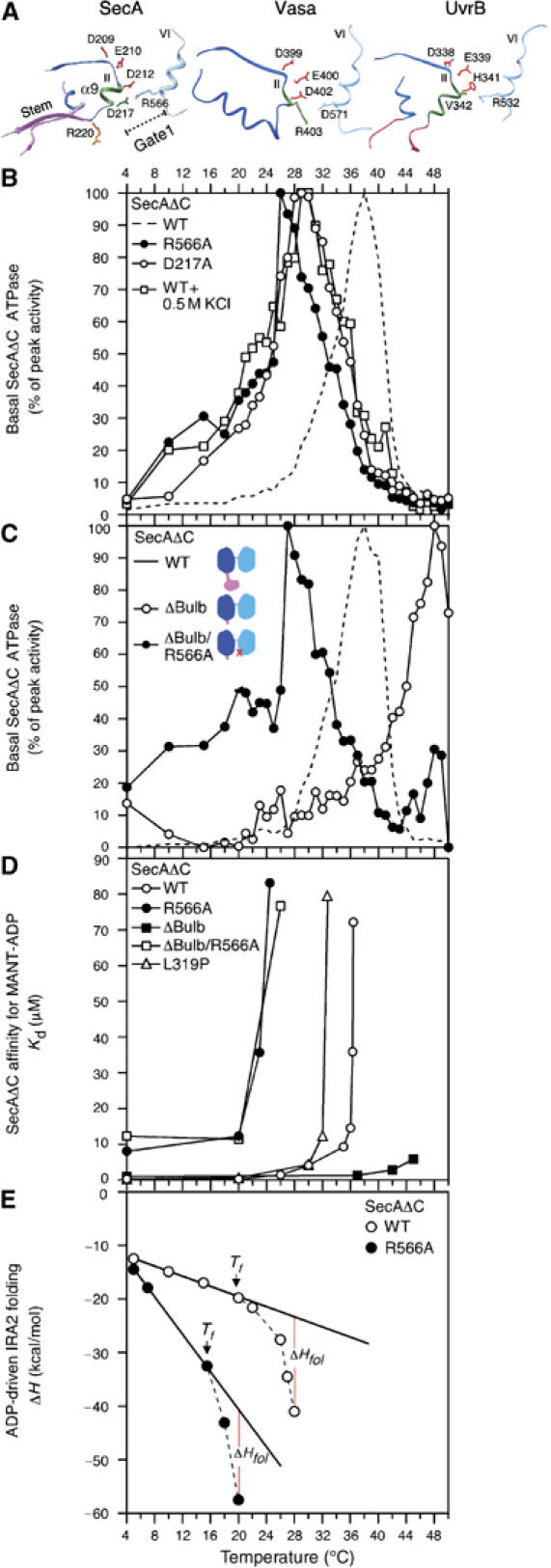
(A) The Gate1 region of ecSecA (Papanikolau et al, 2007) and the corresponding regions in Vasa (Sengoku et al, 2006) and UvrB (Theis et al, 1999). Motif II residue side chains are colored in red. (B) Basal ATPase activities of SecAΔC(R566A), SecAΔC(D217A) and SecAΔC (as in Figure 1D). SecAΔC basal ATPase activity was also determined in the presence of 0.55 M KCl (as indicated). (C) Basal ATPase activities of SecAΔC, SecAΔC(ΔBulb) and SecAΔC(ΔBulb/R566A) (as in Figure 1D). The cartoons (inset) are schematic representations of the three proteins; red cross indicates R566A mutation. (D) Equilibrium dissociation constants (Kd) of SecAΔC and derivatives for the fluorescent analog MANT-ADP plotted as a function of temperature were determined as described (Vrontou et al, 2004). (E) Observed enthalpy change (ΔH) as a function of temperature for the SecAΔC and SecAΔC(R566A) interaction with MgADP (in bufer B with 100 mM KCl). Data were determined using ITC and analyzed as described (Keramisanou et al, 2006).
We concluded that PBD controls the DEAD motor ATPase through a hitherto unknown mechanism. PBD is a necessary positive activator of DEAD motor catalysis.
PBD undergoes temperature-dependent conformational changes
PBD is rooted in NBD (Figure 1A) but is not a physical determinant of the buried nucleotide cleft (Papanikou et al, 2005). Thus, it can regulate the DEAD motor ATPase only through an allosteric mechanism. We posit that this could involve temperature-dependent conformational alterations in PBD.
To test this, we examined the thermal dependence of PBD conformation by recording as a function of temperature NMR heteronuclear single quantum correlation r(1H–15N-HSQC) spectra of SecAΔC/ΔIRA2, a previously reported functional fragment comprising the NBD and PBD domains (Keramisanou et al, 2006) (Figure 2C). The difference in the chemical shift (Δδ) as a function of temperature indicates the degree of conformational changes induced by temperature increase. Several PBD residues in both the Stem and the Bulb change from a structured to an unstructured conformation as temperature increases (i.e. large Δδ; blue to red). Some PBD residues remain unchanged even at 34°C (dark blue). Interestingly, a group of residues including R347, displays prominent resonance broadening suggesting that this region adopts alternate conformational states during thermal transitions.
To test if PBD undergoes temperature-dependent conformational changes also when in the context of SecA (Figure 2D; left) and SecΔC (right), these proteins were treated by limited trypsinolysis at 4°C or 37°C (as indicated) and then immunostained with α-PBD-specific antibodies (Baud et al, 2002). In SecA, at 4°C, no cleavage occurs within PBD. In contrast, at 37°C, the generated peptides indicate that several cleavage sites become exposed. The same peptides are seen with SecAΔC, where some PBD residues are susceptible to tryptic attack already at 4°C. Some of these represent residues that in SecA are directly protected by the C-domain (e.g. R561, R220) (Papanikolau et al, 2007).
We concluded that PBD undergoes extensive intrinsic temperature-dependent conformational changes, which are further regulated by its interaction with the C-domain of SecA.
Preprotein binding to PBD controls the ATPase of the DEAD motor
PBD mutations and temperature affect the DEAD motor ATPase (Figure 2B). Temperature (Figure 2C and D) and preprotein binding (Baud et al, 2002; Papanikou et al, 2005) affect PBD conformation and these changes are sensed by the DEAD motor (Papanikou et al, 2005). It was therefore plausible that preprotein binding to PBD would also affect the thermal ATPase of the DEAD motor. To test this, we used proCH5EE, a model preprotein that binds to SecA in solution with high affinity (Baud et al, 2005; Papanikou et al, 2005). Binding of proCH5EE to PBD causes a down-shift of the thermal ATPase optimum of SecAΔC (Figure 3A) that is also seen with and is characteristic for the ‘early' PBD mutants.
Figure 3.
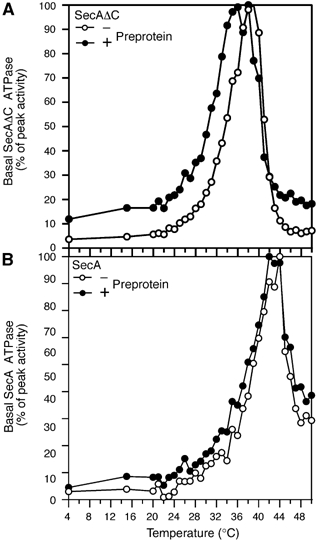
Basal thermal ATPase activities of SecAΔC (A) and SecA (B) in the presence or absence of pCH5EE (‘preprotein'; Baud et al, 2005; Papanikou et al, 2005) (as in Figure 1C).
These data suggested that through binding to PBD (Papanikou et al, 2005), preproteins control ATP hydrolysis in the DEAD motor of SecA.
Control of SecA ATP hydrolysis through a novel ‘gate' in the DEAD motor
How does preprotein binding to PBD affect the DEAD motor ATPase? The PBD Stem is rooted in NBD at the flexible D217–R220 loop (Figure 4A, left). D217 lies at the end of helix α9 (green) that harbors the catalytic helicase Motif II at its other end. In the crystal structures, D217 is proximal to the juxtaposed R566 of IRA2, located at the beginning of Motif VI, and together form a salt-bridge, we term ‘Gate1' (Figure 4A, left). Gate1 ‘bridges' NBD and IRA2 at the bottom (Figure 4A), whereas ATP at the top (Figure 1A) of the cleft. D217 and R566 are also in close proximity in soluble SecA, since cysteinyl substitutions in these two residues become readily oxidized (Supplementary Figure 4A).
Does Gate1 participate in thermal regulation of SecAΔC ATPase? To test this, we examined alanyl substitutions of Gate1 residues R566 (Sianidis et al, 2001) and D217 (Figure 4B). Remarkably, both mutants display ‘early' ATPase phenotypes, with optima at 26°C [SecAΔC(R566A)] or 28–29°C [SecAΔC(D217A)]. In agreement with this, salt-bridge weakening by high salt concentrations down-shifts the wild-type (WT) SecAΔC ATPase to ‘early' activation (optimum at 28°C).
Gate1 ‘opening' seems to be coincident with early thermal activation of the DEAD motor ATPase. Gate1 is obviously a critical salt-bridge in the cleft.
PBD regulates DEAD motor ATPase via Gate1
Is PBD-mediated regulation of the DEAD motor ATPase (Figure 2B) exerted through Gate1? If that were the case, artificial ‘opening' of Gate1 by mutation would be sufficient for ‘earlier' activation of SecAΔC(ΔBulb) that displays a ‘late' ATPase (Figure 4C). Indeed, the ATPase of the double mutant SecAΔC(ΔBulb/R566A) (Figure 4C) is activated at similarly low temperatures as that of SecAΔC(R566A) (compare Figure 4C to B). This is a strong indication that PBD association modulates the DEAD motor ATPase via Gate1. We can not currently exclude that PBD might also exert its effect through additional routes.
PBD regulates DEAD motor affinity for nucleotide via Gate1
ADP release from the DEAD motor is the rate-limiting step of ATP hydrolysis (Sianidis et al, 2001; Fak et al, 2004; Keramisanou et al, 2006). Since PBD and Gate1 mutations modulate ATP hydrolysis, it is conceivable that both might also control the affinity of the DEAD motor for ADP.
To test this, we determined the effect of temperature on the equilibrium dissociation constant (Kd) for ADP of SecAΔC and its derivatives. SecAΔC exhibits high affinity for ADP from 4 to 20°C (Kd ∼0.28 μM) (Figure 4D). Above 20°C, SecAΔC gradually looses its affinity for ADP, with an almost complete loss at 36.5°C (Kd>75 μM). Clearly, in SecAΔC, temperature alone transforms its DEAD motor from a High-Affinity (hereafter HAS) to a Low-Affinity (hereafter LAS) state. ‘Early' mutants of either Gate1 (e.g. R566A) or PBD (e.g. L319P) complete the HAS to LAS transformation of SecAΔC at low temperatures (panel D). ‘Late' mutants (e.g. SecAΔC(ΔBulb); panel D) maintain HAS even at 42°C, while introduction of R566A readily transforms the DEAD motor of SecAΔC(ΔBulb/R566A) from HAS to LAS at 26°C.
In conclusion, PBD acting via Gate1 controls the temperature-driven transition of the DEAD motor nucleotide cleft from HAS to LAS. A ‘closed' Gate1 defines HAS; an ‘open' Gate1 defines LAS. Clearly, LAS onset is thermally concurrent with the catalytic activation onset of the DEAD motor ATPase (compare Figure 4Dto B), indicating that the two events are linked. A SecA DEAD motor without specificity domains is necessary and sufficient for ATP binding and hydrolysis, but is defective in efficient transition from HAS to LAS.
Gate1 optimizes ADP-driven IRA2 folding
IRA2 structural elements that face the nucleotide cleft (including Motif VI on which R566 of Gate1 lies; Figure 4A) undergo temperature-driven unfolding antagonized by nucleotide-driven folding (Keramisanou et al, 2006). Do these catalytically essential events (Sianidis et al, 2001; Keramisanou et al, 2006) relate to Gate1-mediated catalytic activation of the DEAD motor ATPase? To test this, the temperature dependence of enthalpy changes (ΔH) caused by nucleotide binding to SecAΔC were measured by isothermal titration calorimetry (ITC) and compared with those of the Gate1 mutant SecAΔC(R566A) (Figure 4E). The ΔH curve for SecAΔC is linear at low temperatures but deviates from linearity at 21°C. This transition (Tf; Figure 4E, vertical arrows) reveals an additional enthalpic component (ΔHfol) resulting from ADP-driven IRA2 folding (Keramisanou et al, 2006). Interestingly, Tf value for ADP binding to the R566A derivative is much lower than for ADP binding to WT SecAΔC, indicating that the nucleotide cleft becomes significantly destabilized as a result of the mutation.
These data demonstrated that the Gate1 salt-bridge stabilizes ADP-driven IRA2 folding.
PBD controls ‘loose/tight' IRA2 conformations via Gate1
Our data indicated that PBD conformational changes are somehow propagated to IRA2 with Gate1 acting as a controller. To further test this conformational ‘cross-talk' hypothesis, we monitored IRA2 conformation in both ‘early' and ‘late' mutants in the presence or absence of ADP. To this end, we used limited trypsinolysis and immunostaining with α-IRA2-specific antibodies (Figure 5A). Undigested SecAΔC and two derivative peptides were monitored: p16, specific for the apoprotein state and p26, specific for the ADP state (Papanikou et al, 2004).
Figure 5.
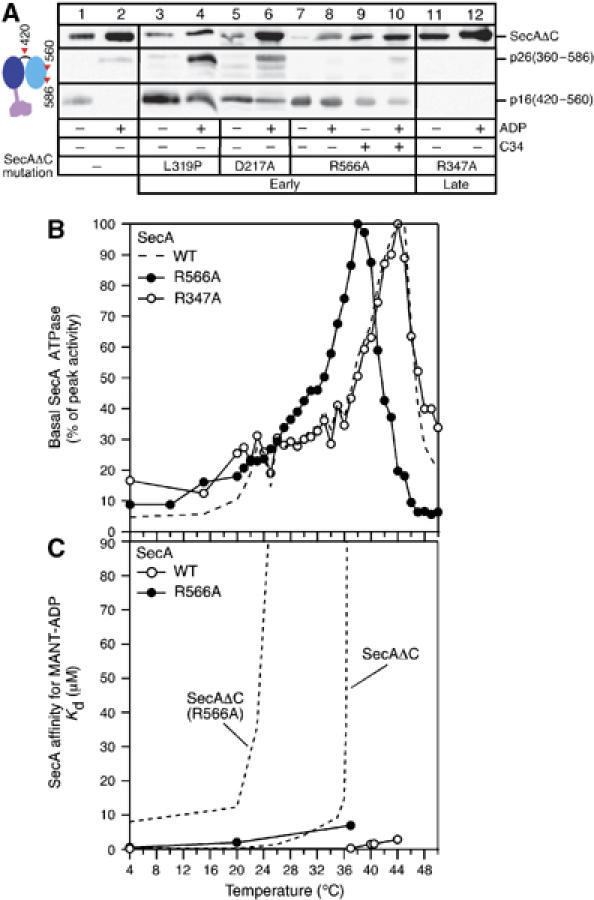
(A) SecAΔC and derivatives (0.9 mg/ml in buffer B) were treated with trypsin (25 μg/ml; 4°C; 13 min) in the absence or presence of ADP (1mM) (as indicated) or a three-fold molar excess of C34 (wherever indicated). Following SDS–PAGE, polypeptides were immunostained by α-IRA2-specific antibody. (B) Basal ATPase activities of SecA, SecA(R566A) and SecA(R347A) (as in Figure 1C). (C) Equilibrium dissociation constants (Kd) of SecA and SecA(R566A) for MANT-ADP plotted as a function of temperature. Two curves (dotted lines) from Figure 4D are included for comparison.
Compared to WT SecAΔC (lane 1), all ‘early' mutants (SecAΔC(L319P), SecAΔC(D217A) and SecAΔC(R566A)) are more protease sensitive and thus the generated amounts of p16 are increased (lanes 3, 5 and 7). ADP binding significantly stabilizes WT SecAΔC; p16 is no longer generated but instead p26 appears (lane 2). This pattern is altered in all ‘early' mutants (lanes 4, 6 and 8). On the other hand, the ‘late' mutant SecAΔC(R347A) is remarkably protease resistant and does not give rise to any detectable amount of either p16 or p26 (lanes 11 and 12).
We conclude that the open/closed states of Gate1 lead to loose/tight IRA2 conformations. These underlie ‘early' and ‘late' activation of the DEAD motor ATPase, respectively. To further test this proposal, the intrinsic fluorescence of IRA2 was measured as a function of temperature (Supplementary Figure 1). Opening/closing of Gate1 can be directly correlated to corresponding changes in the Tmapp of the IRA2 domain. Collectively, the data presented above are best explained with the propagation of conformational changes from PBD through Gate1 to IRA2. We term this the PBD–Gate1–IRA2 conformational relay mechanism.
PBD and Gate1-mediated regulation of the DEAD motor ATPase is under C-domain control
How does the PBD–Gate1–IRA2 relay operate in full-length SecA, where the DEAD motor ATPase is under C-domain control (Figure 1C) (Karamanou et al, 1999)? To address this, we examined the effect of Gate1 and/or PBD mutations on the thermal properties of SecA basal ATPase (Figure 5B). The ATPase of SecA(R566A) is significantly stabilized compared with that of SecAΔC(R566A) (optima at 38 and 26°C, respectively). In agreement with this, purified C-domain added in trans significantly stabilizes the ATPase of SecAΔC (Figure 1D) or SecAΔC(R566A) (Supplementary Figure 2A), whereas high salt does not affect the ATPase of SecA (Supplementary Figure 2B) as it does that of SecAΔC (Figure 4B). In contrast, the C-domain destabilizes ‘late' mutants: SecA(R347A) exhibits an optimum at 42°C (Figure 4B), down from 46°C for SecAΔC(R347A) (Figure 2B). In all cases, C-domain-mediated regulation of the DEAD motor thermal ATPase seems hierarchically ‘dominant' over that exerted by either PBD or Gate1.
Nucleotide affinity and catalysis are controlled by IRA2 conformation (Figure 4DE). If the C-domain exerts a dominant role in DEAD motor ATPase regulation, it is anticipated that its presence will affect directly IRA2 conformation. Indeed, supply of C-domain polypeptide to SecAΔC(R566A) in trans in the absence or presence of ADP renders its IRA2 domain significantly protease resistant (Figure 5A; compare lanes 9 and 10 to 7 and 8). Moreover, in the presence of the C-domain, the nucleotide cleft of SecA and SecA(R566A) maintain appreciably high ADP affinity even at high temperatures (Figure 5C; compare SecA to SecAΔC derivatives) and antagonizes proCH5EE-mediated ‘early' activation of DEAD motor ATPase (Figure 3B).
We concluded that the PBD–Gate1–IRA2 relay mechanism is under direct C-domain control and can be modulated by preproteins only when the C-domain acquires the appropriate conformation or becomes detached from the DEAD motor. The C-domain appears to act as a negative regulator of DEAD motor catalysis.
Conformational relay in SecA is essential for protein translocation
If conformational relay between SecA domains via Gate1 is crucial for translocase function, Gate1 mutants should be defective in protein translocation.
SecA Gate1 mutants were tested for their ability to complement a thermo-sensitive BL21secAts strain at 42°C (Mitchell and Oliver, 1993) in vivo (Figure 6A). Growth of SecA(D217A) or SecA(R566A) was seriously compromised. Arginine is irreplaceable at position 566 for in vivo function and cannot be substitued by either a positively charged lysyl or a cysteinyl residue (Supplementary Figure 3). Two more NBD residues, D212 of Motif II and E397, seen in crystal structures to be bound with R566 (Sharma et al, 2003) (Figure 6B), were also examined. SecA(E397A) complemented fully, whereas SecA(D212A) did not (panel A). Clearly, D212 is an essential residue and presumably provides an additional critical salt-bridge between R566 and NBD, and links directly Gate1 with the catalytic DEAD motif. E397 is not an essential residue and might have some contributory role only under certain conditions.
Figure 6.
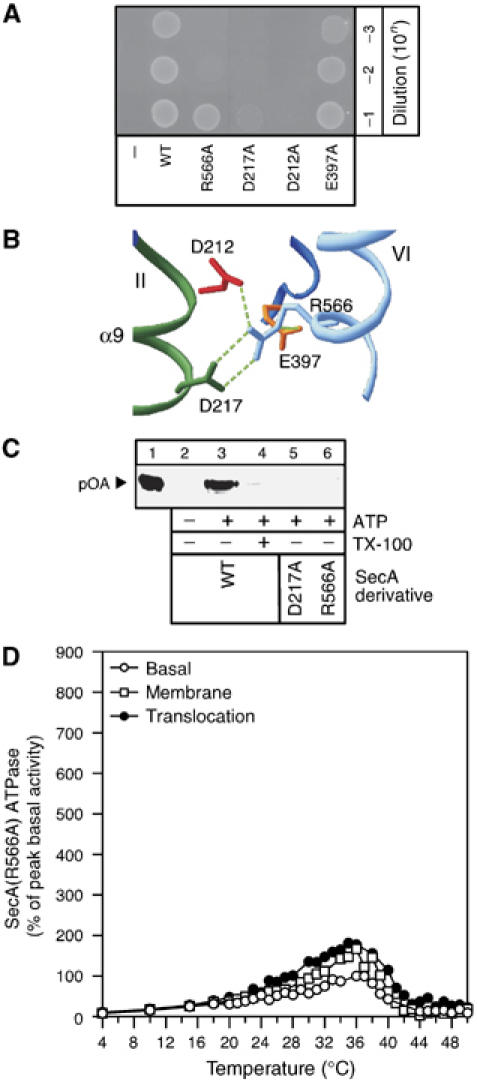
(A) In vivo genetic complementation test of the BL21.19 secAts strain at 42°C (Mitchell and Oliver, 1993) using SecA Gate1 mutants. BL21.19 cultures with pET5 vector alone or its derivatives with cloned secA or secA(R566A) or secA(D217A) or secA(D212A), secA(E397A) genes, grown at 30°C were adjusted to the same density. The indicated dilutions were spotted on LB/ampicillin plates and incubated (20 h; 42°C). (B) Interactions at Gate1 detected by crystallography. NBD residues involved in bonding with R566 of IRA2 in the M. tuberculosis SecA (Sharma et al, 2003). (C) In vitro protein translocation of SecA ‘Gate1' derivatives. ATP-driven in vitro translocation of proOmpA-His (0.1 mg/ml) into SecYEG proteoliposomes (20 μg protein/ml; 100 μl reactions in buffer B, BSA (0.5 mg/ml), 1 mM ATP, 1 mM DTT and SecB (0.4 mg/ml)) catalyzed by SecA or the indicated derivatives. Lane 1: 10% of undigested proOmpA-His input, lane 4: membranes were dissolved with Triton X-100 (1%) before proteinase K (1 mg/ml) addition. Proteins were TCA precipitated (15%), analyzed by SDS–PAGE and immunostained with α-His antibody. (D) Basal, membrane and translocation ATPase activities of SecA(R566A) (as in Figure 1B).
We next examined the ability of purified SecA Gate1 mutants to support ATP-driven protein translocation in vitro (Figure 6C). In contrast to SecA (lane 3), SecA(D217A) and SecA(R566A) (lanes 5 and 6) were unable to catalyze translocation of proOmpA into SecYEG proteoliposomes.
We concluded that PBD–Gate1–IRA2 conformational relay is essential for protein translocation.
Conformational relay in SecA is essential for translocase priming and translocation ATPase
To identify the precise step in the translocation cycle that the Gate1-controlled relay mechanism becomes essential, we first sought to determine whether SecA Gate1 mutants were defective in binding to SecYEG and/or preprotein. However, flotation analysis experiments using IMVs (Papanikou et al, 2005) revealed that binding of SecA(R566A) to SecYEG-containing IMVs was indistinguishable from that of the WT SecA (Supplementary Figure 5, compare panels D and C). Furthermore, the binding of 35S-labelled SecA(R566A) to IMVs was quantitated (Hartl et al, 1990), and was determined to be of comparable strength (Kd=39 nM; Supplementary Figure 5H). Similarly, flotation analysis indicated that binding of proOmpA to SecA(R566A) was indistinguishable to that of SecA (Supplementary Figure 5G and F).
We next quantitated the ‘membrane' and ‘translocation' ATPase activities of Gate1 mutants. The basal ATPase of SecA(R566A) (Figure 6D) or SecA(D217A) (not shown) is only barely stimulated by IMVs (∼0.5-fold) or IMVs plus preproteins (∼1-fold), whereas the corresponding values for WT SecA are 2- to 3.5-fold and ∼9-fold, respectively (Figure 1B; Supplementary Figure 4B, left panel). To test if artificial closure of Gate1 can restore function, the cysteinyl residues of SecA(D217C/R566A) were oxidized. (Supplementary Figure 4B, middle). However, this treatment did not restore membrane or translocation ATPase.
Therefore, SecA Gate1 mutants can bind to SecYEG and deliver preproteins to the translocase. Nevertheless, their defective PBD–Gate1–IRA2 conformational relay prevents them from acquiring a functional ‘primed' state and cannot be triggered by preprotein to catalyze multiple ATP turnovers necessary for secretion.
Discussion
The major question of how preproteins control and modulate ATP catalysis in SecA remains elusive. We now reveal a novel preprotein-regulated conformational relay mechanism that controls the DEAD motor of SecA and is essential for SecA-mediated protein translocation (Figure 6A, C and D). These observations were made possible by following thermal catalytic transitions of membrane-bound and soluble SecA, combined with biochemical and biophysical assays. Mutational shifting of catalytic temperature optima allowed dissection of the energetic contributions of SecA domains and ligands.
SecA possesses a complex mechanism that controls the physical interaction between NBD and IRA2, the two DEAD motor domains, thereby regulating the opening/closure of the nucleotide cleft and modulating the ATPase activity of the motor. This mechanism consists of four distinct elements: (a) a DEAD motor that is inherently flexible (Keramisanou et al, 2006) and can acquire ‘tight' and ‘loose' states that correspond to ‘high'- and ‘low'-affinity ADP states, respectively; (b) a Gate1 salt-bridge that controls the physical interaction of the motor domains, thereby modulating both the opening/closure of the cleft and IRA2 folding; (c) a PBD that allosterically regulates DEAD motor conformational states and (d) a C-domain, which through physical contacts, dictates when the above allosteric mechanism becomes activated. SecA activation through the mechanism described here could relate to the endothermic transitions identified by fluorescence (Hunt et al, 2002) and circular dichroism (Sianidis et al, 2001) spectroscopies.
Through intricate salt bridging between D217/D212/R566, Gate1 connects NBD and IRA2 and allows conformational and functional cross-talk between the catalytic Motif II on NBD and Motif VI (Figure 7; blue zigzag) on IRA2. IRA2 regions that face the nucleotide cleft are destabilized at physiological temperature and this causes partial IRA2 detachment from NBD (Keramisanou et al, 2006). Gate1 and nucleotide stabilize IRA2 independently, since disruption of Gate1 destabilizes IRA2 conformation (Figures 4E and 5A; Supplementary Figure 1) and reduces its affinity for NBD (Sianidis et al, 2001), but does not abolish nucleotide-driven folding of IRA2 (Figure 4E). Since Gate1 stabilizes IRA2 even in the absence of nucleotide (Figure 5A), its role may also be critical during parts of the catalytic cycle when the cleft is empty. Through control of IRA2 conformation Gate1 controls ADP release, the rate-limiting step for multiple ATP turnovers (Sianidis et al, 2001; Fak et al, 2004). Gate1 opening results in a remarkable loss of ADP affinity (Figure 4D), leading to activation of SecA ATPase (Figure 4B); in contrast, Gate1 closure stabilizes the ADP state. Mutants with a permanently open Gate1 are functionally inactive both in vivo and in vitro, and do not become additionally stimulated by SecYEG (Figure 6D). The same holds true when Gate is permanently closed (Supplementary Figure 4B). We therefore expect that the Gate1 salt-bridge is not only an essential stability factor but is also essential that Gate1 both opens and closes during catalysis. Linkage of R566A to D212 additionally ensures that these events are linked to ATP hydrolysis. Hence, we expect that several Gate1-controlled HAS to LAS transition cycles lead to the elevated ATPase activity seen during protein translocation (Figure 1B) (Lill et al, 1990).
Figure 7.
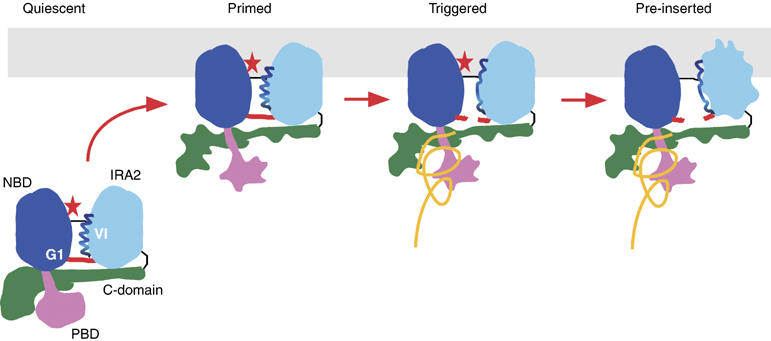
Hypothetical model of preprotein-controlled PBD–Gate1–IRA2 conformational relay that leads to activation of the SecA DEAD motor ATPase (see ‘Discussion' for details). Irregular contours denote conformational changes. Red star, ADP; orange line, preprotein.
The PBD affects IRA2 conformation (Figure 5A; lanes 3–6; Supplementary Figure 1) and controls both DEAD motor nucleotide affinity (Figure 4D) and hydrolysis (Figures 2B and 4C) via Gate1. It is remarkable that a domain with no immediate physical connection (Figure 1A) has such an impact on IRA2 conformation. This was achieved by rooting PBD in NBD via a slender Stem in the immediate vicinity of Gate1 (Figure 4A, left). The Stem is stabilized by its connection to NBD (Papanikou et al, 2005; Keramisanou et al, 2006) and permits approximately 75° Bulb swiveling between a ‘closed' (Hunt et al, 2002) and an ‘open' (Osborne et al, 2004; Papanikolau et al, 2007) state. Presumably, conformational changes in the Bulb, facilitated by temperature (Figure 2C and D) and driven by preprotein binding (Figure 3A) (Chou and Gierasch, 2005; Papanikou et al, 2005; Musial-Siwek et al, 2007), are propagated via the Stem to Gate1. Remarkably, removal of the Bulb keeps the DEAD motor ‘closed' (Figure 4C), indicating that PBD actively manipulates the DEAD motor ‘open' and ‘closed' states. How preprotein binding affects Bulb conformation is currently unknown.
Finally, the PBD–Gate1–IRA2 conformational relay mechanism is under tight C-domain control. The C-domain stabilizes the Motif VI helix (Keramisanou et al, 2006), the conformation of IRA2 (Figure 5A, lanes 9 and 10) (Keramisanou et al, 2006) and PBD (Figure 2C and D) (Vrontou et al, 2004), enhances the affinity of SecA for nucleotide (Figure 5C), delays the ATPase optimum (Figure 5B), suppresses nucleotide turn over (Figure 4B; Supplementary Figure 2 and Supplementary Table 1) and suppresses the effect of PBD mutations (Figure 5B and C) and preprotein-mediated DEAD motor activation (Figure 3B). These data demonstrate that the C-domain is both the central stabilizing element of SecA and an allosteric regulator of catalysis and lies at the top of the regulatory cascade of SecA-domains. This kind of extremely tight regulation of the ATPase of the motor imposes an additional energetic barrier and is not known to occur in other DExD/H proteins. This could result from the subcellular location of SecA function, the only DExD/H protein that acts at and in a membrane environment. Thus, ‘motor' functions of SecA must be physically coupled and coordinated with motions of the membrane-embedded protein-conducting channel. Obviously, activation of SecA during translocation necessitates that the C-domain/DEAD motor/PBD association becomes somehow ‘loosened', as seen with mutations that weaken C-domain/DEAD motor association (Figure 1C) (Karamanou et al, 1999; Vrontou et al, 2004; Keramisanou et al, 2006; Mori and Ito, 2006) and ‘tightened' as seen during C-domain membrane insertion (Economou and Wickner, 1994). The proposed SecA monomerization reaction (Or et al, 2002; Benach et al, 2003; Duong, 2003) may facilitate such C-domain motions. Because of the dramatic effect that preprotein binding has on relieving C-domain suppression, it is possible that preproteins bind also to the C-domain and ‘pry' it away from the ATPase suppressing DEAD motor-bound state. In some of the SecA structures determined to date, the C-domains of the two protomers are in proximity or associate to one another. How these interactions affect the C-domain in its role as a master regulator of the SecA catalytic cycle within each protomer is not currently clear.
Full SecA activation occurs only during protein translocation by the synergy of preprotein and SecYEG. Our data allow the molecular basis of this activation to be rationalized for the first time, since they reveal the individual roles of the SecA domains and their interaction with preprotein. In addition, biochemical removal and re-introduction of individual domains, and the isolation of mutations that freeze or activate catalytic steps allowed us to determine the order of catalytic events: the C-domain must be first detached from the DEAD before all other steps can proceed, PBD must change its conformation before Gate1 can open, the Gate1 salt-bridge must break before IRA2 can become detached, IRA2 must become detached before ADP can be released. These and previous results lead us to propose a six-step working model for protein translocation (Figure 7 depicts the first four): quiescent: cytoplasmic SecA:ADP exists in an inactive state (Sianidis et al, 2001; Fak et al, 2004); priming: binding of SecA:ADP to SecYEG forms a primed translocase holoenzyme. This entails C-domain conformational changes and perhaps partial detachment (Vrontou et al, 2004; Mori and Ito, 2006), allowing limited loosening of Motif IV helix and of Gate1–IRA2, modest membrane ATPase activity and high-affinity preprotein binding (Hartl et al, 1990). SecYEG binds to both the DEAD motor and the C-domain (Economou and Wickner, 1994; Eichler and Wickner, 1997; Jilaveanu and Oliver, 2007). However, the molecular basis of how this interaction contributes to SecA activation remains unknown and will require high-resolution structural knowledge of the SecA-SecYEG interface; triggering: preprotein binding to PBD and perhaps directly to the C-domain fully relieves C-domain suppression, ‘opens' Gate1 and leads to IRA2 detachment; pre-insertion: detached IRA2 renders the DEAD motor ‘loose' and this leads to ADP release from the cleft. This affects PBD conformation with its bound preprotein (Papanikou et al, 2005; Keramisanou et al, 2006); ATP stroke: ATP binding to the empty DEAD motor reverses the previous conformational events, facilitates deeper membrane penetration of SecA (Economou and Wickner, 1994) and drives a short preprotein segment into the membrane (Schiebel et al, 1991); reloading: ATP gets hydrolyzed and the preprotein is partially released (Schiebel et al, 1991). SecA returns to the stable ADP state and the C-domain reassociates tightly to the DEAD motor. Re binding of the succeeding preprotein segment to PBD triggers a new catalytic round.
Based on these data and the previously detailed structural and biochemical similarities, we anticipate that the conformational relay mechanism described here provides a common mechano-chemical conversion strategy underlying catalysis of SecA and other DExD/H proteins. Specificity domains are found inserted in locations similar to that of PBD (e.g. DNA interacting domains in UvrB (Theis et al, 1999) (Figure 4A, right) and RecG (Singleton et al, 2001)), or elsewhere in the DEAD motor (e.g. PcrA), or even be independent polypeptides binding to the motor (Oberer et al, 2005). Helices similar to α9 of SecA (Figures 4A and 6B) are conserved in other SF2 RNA (e.g. Vasa (Sengoku et al, 2006)) and DNA (e.g. UvrB (Theis et al, 1999; Truglio et al, 2006)) helicases (Figure 3A, middle and right). Many helicases form contacts between essential residues at or near regions corresponding to Gate1 (Schmidt et al, 1991; Caruthers and McKay, 2002). Additional bridging of the two DEAD motor domains may be provided by the nucleic acid itself binding to both domains (Velankar et al, 1999; Sengoku et al, 2006) and acting as a molecular staple. In all cases, even though the specificity domains and the substrates differ, the common aim for a molecular tit-for-tat is achieved: substrates via specificity domains and/or direct binding to the DEAD motor (Velankar et al, 1999; Sengoku et al, 2006) and nucleotides modulate the conformational ensemble of the universal DEAD motor. In return, this modulates, in poorly understood ATP-driven steps, the conformation, tertiary organization and topology of the substrate be it unfolded secretory polypeptides, nucleoprotein complexes or nucleic acid. Toward the goal of understanding DExD/H protein–substrate interaction and the role of specificity domains, it is essential to extend structural insight from short oligonucleotides (Velankar et al, 1999; Sengoku et al, 2006) and peptides (Chou and Gierasch, 2005; Musial-Siwek et al, 2007) to longer substrates.
Materials and methods
Genetic manipulations
All constructs are as described in the Supplementary data or are described previously (Jarosik and Oliver, 1991; Karamanou et al, 1999; Sianidis et al, 2001; Baud et al, 2002; Vrontou et al, 2004; Papanikou et al, 2005; Keramisanou et al, 2006).
Thermal ATPase
All assays were performed as described (Lill et al, 1990; Karamanou et al, 1999) in buffer B (50 mM Tris–HCl pH 8.0; 50 mM KCl; 5 mM MgCl2) supplemented with 0.5 mg/ml BSA; 1 mM ATP and 1.5 mM DTT (unless specified otherwise). For membrane ATPase, IMVs (protein content 17 μg/ml) or SecYEG-proteoliposomes (100 μg/ml) were also added. For translocation ATPase, proOmpA (60 μg/ml) was further added.
Miscellaneous
ITC was used to measure changes in enthalpy (ΔH), entropy (ΔS), and free energy (ΔG) of nucleotide binding to SecAΔC, as described (Keramisanou et al, 2006). 1H–15N HSQC spectra of SecADC/DIRA2 were recorded at 14, 22, 26, 32 and 36°C. Fluorescence measurements and limited trypsinolysis were as described (Vrontou et al, 2004). Radioactivity was quantitated by phosphorimaging (Storm 840; GE Healthcare). Binding and kinetic data were analyzed using Prism (GraphPad) or Origin (MicroCal), and structures were visualized with SwissPDBViewer.
Chemicals/biochemicals
Proteases, inhibitors and nucleotides were from Roche; MANT-ADP was from Molecular Probes and all other chemicals were from Sigma. DNA enzymes were from Minotech; oligonucleotides were from the IMBB genomics facility and dNTPs were from Promega. 35S-labelled methionine (1000 Ci/mmol) and chromatography materials (except Ni2+ affinity; Qiagen) were from Amersham. Proteins were purified as described (Karamanou et al, 1999) and were properly folded as determined by CD spectroscopy (Sianidis et al, 2001).
Supplementary Material
Supplementary Information
Acknowledgments
We are grateful to B Pozidis and M Koukaki for materials and assays, A Kuhn for comments, I Skorta for preliminary experiments. This work was supported by grants from the European Union (QLK3-CT-2000-00082 and LSHG-CT-2005-037586 to AE), the Greek General Secretariat of Research and the European Regional Development Fund (01AKMON46 and PENED03ED623 to AE), the US National Institutes of Health grant GM73854 (to CGK), a Scientist Development Grant by American Heart Association (to CGK) and a Rutgers Busch grant (to CGK). GG is a Onassis Foundation predoctoral fellow.
References
- Baud C, Karamanou S, Sianidis G, Vrontou E, Politou AS, Economou A (2002) Allosteric communication between signal peptides and the SecA protein DEAD motor ATPase domain. J Biol Chem 277: 13724–13731 [DOI] [PubMed] [Google Scholar]
- Baud C, Papanikou E, Karamanou S, Sianidis G, Kuhn A, Economou A (2005) Purification of a functional mature region from a SecA-dependent preprotein. Protein Expr Purif 40: 336–339 [DOI] [PubMed] [Google Scholar]
- Benach J, Chou YT, Fak JJ, Itkin A, Nicolae DD, Smith PC, Wittrock G, Floyd DL, Golsaz CM, Gierasch LM, Hunt JF (2003) Phospholipid-induced monomerization and signal-peptide-induced oligomerization of SecA. J Biol Chem 278: 3628–3638 [DOI] [PubMed] [Google Scholar]
- Caruthers JM, McKay DB (2002) Helicase structure and mechanism. Curr Opin Struct Biol 12: 123–133 [DOI] [PubMed] [Google Scholar]
- Cavanaugh LF, Palmer AG III, Gierasch LM, Hunt JF (2006) Disorder breathes life into a DEAD motor. Nat Struct Mol Biol 13: 566–569 [DOI] [PubMed] [Google Scholar]
- Chou YT, Gierasch LM (2005) The conformation of a signal peptide bound by Escherichia coli preprotein translocase SecA. J Biol Chem 280: 32753–32760 [DOI] [PubMed] [Google Scholar]
- Chou YT, Swain JF, Gierasch LM (2002) Functionally significant mobile regions of Escherichia coli SecA ATPase identified by NMR. J Biol Chem 277: 50985–50990 [DOI] [PubMed] [Google Scholar]
- Cordin O, Banroques J, Tanner NK, Linder P (2006) The DEAD-box protein family of RNA helicases. Gene 367: 17–37 [DOI] [PubMed] [Google Scholar]
- Duong F (2003) Binding, activation and dissociation of the dimeric SecA ATPase at the dimeric SecYEG translocase. EMBO J 22: 4375–4384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Economou A, Wickner W (1994) SecA promotes preprotein translocation by undergoing ATP-driven cycles of membrane insertion and deinsertion. Cell 78: 835–843 [DOI] [PubMed] [Google Scholar]
- Eichler J, Wickner W (1997) Both an N-terminal 65-kDa domain and a C-terminal 30-kDa domain of SecA cycle into the membrane at SecYEG during translocation. Proc Natl Acad Sci USA 94: 5574–5581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fak JJ, Itkin A, Ciobanu DD, Lin EC, Song XJ, Chou YT, Gierasch LM, Hunt JF (2004) Nucleotide exchange from the high-affinity ATP-binding site in SecA is the rate-limiting step in the ATPase cycle of the soluble enzyme and occurs through a specialized conformational state. Biochemistry 43: 7307–7327 [DOI] [PubMed] [Google Scholar]
- Hartl FU, Lecker S, Schiebel E, Hendrick JP, Wickner W (1990) The binding cascade of SecB to SecA to SecY/E mediates preprotein targeting to the E. coli plasma membrane. Cell 63: 269–279 [DOI] [PubMed] [Google Scholar]
- Hunt JF, Weinkauf S, Henry L, Fak JJ, McNicholas P, Oliver DB, Deisenhofer J (2002) Nucleotide control of interdomain interactions in the conformational reaction cycle of SecA. Science 297: 2018–2026 [DOI] [PubMed] [Google Scholar]
- Jankowsky E, Gross CH, Shuman S, Pyle AM (2001) Active disruption of an RNA–protein interaction by a DExH/D RNA helicase. Science 291: 121–125 [DOI] [PubMed] [Google Scholar]
- Jarosik GP, Oliver DB (1991) Isolation and analysis of dominant secA mutations in Escherichia coli. J Bacteriol 173: 860–868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jilaveanu LB, Oliver DB (2007) In vivo membrane topology of Escherichia coli SecA ATPase reveals extensive periplasmic exposure of multiple functionally important domains clustering on one face of SecA. J Biol Chem 282: 4661–4668 [DOI] [PubMed] [Google Scholar]
- Karamanou S, Vrontou E, Sianidis G, Baud C, Roos T, Kuhn A, Politou AS, Economou A (1999) A molecular switch in SecA protein couples ATP hydrolysis to protein translocation. Mol Microbiol 34: 1133–1145 [DOI] [PubMed] [Google Scholar]
- Keramisanou D, Biris N, Gelis I, Sianidis G, Karamanou S, Economou A, Kalodimos CG (2006) Disorder–order folding transitions underlie catalysis in the helicase motor of SecA. Nat Struct Mol Biol 13: 594–602 [DOI] [PubMed] [Google Scholar]
- Koonin EV, Gorbalenya AE (1992) Autogenous translation regulation by Escherichia coli ATPase SecA may be mediated by an intrinsic RNA helicase activity of this protein. FEBS Lett 298: 6–8 [DOI] [PubMed] [Google Scholar]
- Lill R, Dowhan W, Wickner W (1990) The ATPase activity of SecA is regulated by acidic phospholipids, SecY, and the leader and mature domains of precursor proteins. Cell 60: 271–280 [DOI] [PubMed] [Google Scholar]
- Mitchell C, Oliver D (1993) Two distinct ATP-binding domains are needed to promote protein export by Escherichia coli SecA ATPase. Mol Microbiol 10: 483–497 [DOI] [PubMed] [Google Scholar]
- Mori H, Ito K (2006) The long alpha-helix of SecA is important for the ATPase coupling of translocation. J Biol Chem 281: 36249–36256 [DOI] [PubMed] [Google Scholar]
- Musial-Siwek M, Rusch SL, Kendall DA (2007) Selective photoaffinity labeling identifies the signal peptide binding domain on SecA. J Mol Biol 365: 637–648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberer M, Marintchev A, Wagner G (2005) Structural basis for the enhancement of eIF4A helicase activity by eIF4G. Genes Dev 19: 2212–2223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Or E, Navon A, Rapoport T (2002) Dissociation of the dimeric SecA ATPase during protein translocation across the bacterial membrane. EMBO J 21: 4470–4479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osborne AR, Clemons WM Jr, Rapoport TA (2004) A large conformational change of the translocation ATPase SecA. Proc Natl Acad Sci USA 101: 10937–10942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osborne AR, Rapoport TA, van den Berg B (2005) Protein translocation by the Sec61/SecY channel. Annu Rev Cell Dev Biol 21: 529–550 [DOI] [PubMed] [Google Scholar]
- Papanikolau Y, Papadovasilaki M, Ravelli RB, McCarthy AA, Cusack S, Economou A, Petratos K (2007) Structure of dimeric SecA, the Escherichia coli preprotein translocase motor. J Mol Biol 366: 1545–1557 [DOI] [PubMed] [Google Scholar]
- Papanikou E, Karamanou S, Baud C, Frank M, Sianidis G, Keramisanou D, Kalodimos CG, Kuhn A, Economou A (2005) Identification of the preprotein binding domain of SecA. J Biol Chem 280: 43209–43217 [DOI] [PubMed] [Google Scholar]
- Papanikou E, Karamanou S, Baud C, Sianidis G, Frank M, Economou A (2004) Helicase Motif III in SecA is essential for coupling preprotein binding to translocation ATPase. EMBO Rep 5: 807–811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiebel E, Driessen AJ, Hartl FU, Wickner W (1991) Delta mu H+ and ATP function at different steps of the catalytic cycle of preprotein translocase. Cell 64: 927–939 [DOI] [PubMed] [Google Scholar]
- Schmidt MG, Dolan KM, Oliver DB (1991) Regulation of Escherichia coli secA mRNA translation by a secretion-responsive element. J Bacteriol 173: 6605–6611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sengoku T, Nureki O, Nakamura A, Kobayashi S, Yokoyama S (2006) Structural basis for RNA unwinding by the DEAD-box protein Drosophila Vasa. Cell 125: 287–300 [DOI] [PubMed] [Google Scholar]
- Sharma V, Arockiasamy A, Ronning DR, Savva CG, Holzenburg A, Braunstein M, Jacobs WR Jr, Sacchettini JC (2003) Crystal structure of Mycobacterium tuberculosis SecA, a preprotein translocating ATPase. Proc Natl Acad Sci USA 100: 2243–2248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sianidis G, Karamanou S, Vrontou E, Boulias K, Repanas K, Kyrpides N, Politou AS, Economou A (2001) Cross-talk between catalytic and regulatory elements in a DEAD motor domain is essential for SecA function. EMBO J 20: 961–970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singleton MR, Scaife S, Wigley DB (2001) Structural analysis of DNA replication fork reversal by RecG. Cell 107: 79–89 [DOI] [PubMed] [Google Scholar]
- Solem A, Zingler N, Pyle AM (2006) A DEAD protein that activates intron self-splicing without unwinding RNA. Mol Cell 24: 611–617 [DOI] [PubMed] [Google Scholar]
- Theis K, Chen PJ, Skorvaga M, Van Houten B, Kisker C (1999) Crystal structure of UvrB, a DNA helicase adapted for nucleotide excision repair. EMBO J 18: 6899–6907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truglio JJ, Karakas E, Rhau B, Wang H, DellaVecchia MJ, Van Houten B, Kisker C (2006) Structural basis for DNA recognition and processing by UvrB. Nat Struct Mol Biol 13: 360–364 [DOI] [PubMed] [Google Scholar]
- Veenendaal AK, van der Does C, Driessen AJ (2004) The protein-conducting channel SecYEG. Biochim Biophys Acta 1694: 81–95 [DOI] [PubMed] [Google Scholar]
- Velankar SS, Soultanas P, Dillingham MS, Subramanya HS, Wigley DB (1999) Crystal structures of complexes of PcrA DNA helicase with a DNA substrate indicate an inchworm mechanism. Cell 97: 75–84 [DOI] [PubMed] [Google Scholar]
- Vrontou E, Economou A (2004) Structure and function of SecA, the preprotein translocase nanomotor. Biochim Biophys Acta 1694: 67–80 [DOI] [PubMed] [Google Scholar]
- Vrontou E, Karamanou S, Baud C, Sianidis G, Economou A (2004) Global co-ordination of protein translocation by the SecA IRA1 switch. J Biol Chem 279: 22490–22497 [DOI] [PubMed] [Google Scholar]
- Yang Q, Jankowsky E (2006) The DEAD-box protein Ded1 unwinds RNA duplexes by a mode distinct from translocating helicases. Nat Struct Mol Biol 13: 981–986 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information