

Abstract
We recently identified p140Cap as a novel adaptor protein, expressed in epithelial-rich tissues and phosphorylated upon cell matrix adhesion and growth factor treatment. Here, we characterise p140Cap as a novel Src-binding protein, which regulates Src activation via C-terminal Src kinase (Csk). p140Cap silencing increases cell spreading, migration rate and Src kinase activity. Accordingly, increased expression of p140Cap activates Csk, leading to inhibition of Src and downstream signalling as well as of cell motility and invasion. Moreover, cell proliferation and ‘in vivo' breast cancer cell growth are strongly impaired by high levels of p140Cap, providing the first evidence that p140Cap is a novel negative regulator of tumour growth.
Keywords: cell signalling, invasion, motility, p140Cap, tumour growth
Introduction
Signalling originated from the integrin family of cell-matrix receptors is central to many physiological and pathological processes such as embryogenesis, inflammatory response, tissue repair and cancer cell progression. Early integrin signalling induces activation of Src family kinases, focal adhesion kinase (FAK) and Rho family GTPases (Damsky and Ilic, 2002; Frame et al, 2002; Miranti and Brugge, 2002; Schwartz and Ginsberg, 2002; Giancotti, 2003; Burridge and Wennerberg, 2004; Cabodi and Defilippi, 2006). Upon integrin-mediated adhesion, Src kinase regulates cell growth, spreading and migration through increased phosphorylation of FAK as well as other key adaptor molecules, like p130Cas (Mitra et al, 2005; Defilippi et al, 2006). As a result of the phosphorylation (Goldberg et al, 2003; Shin et al, 2004), p130Cas recruits other proteins such as Crk and DOCK180 that regulate Rac activation, which is crucial for actin cytoskeleton organisation and cell motility (Chodniewicz and Klemke, 2004). Cells derived from mice deficient in the three members of the Src family (Src/Yes/Fyn), FAK or p130Cas consistently exhibit impaired cell spreading and migration (Ilic et al, 1995, 1998; Klinghoffer et al, 1999). On the other hand, in transformed cells, increased Src activity induces reorganisation of epithelial adhesion systems and the actin cytoskeleton, leading to cell scattering and epithelial-mesenchymal transition (Boyer et al, 1997; Avizienyte et al, 2002).
We recently identified p140Cap as a novel adaptor protein indirectly associated to p130Cas (Di Stefano et al, 2004). p140Cap co-distributes with cortical actin and actin stress fibres, but not with focal adhesions. Interestingly, expression of p140Cap in NIH3T3 and in ECV304 cells delays the onset of cell spreading in the early phases of cell adhesion to fibronectin (FN). Moreover, this protein is tyrosine phosphorylated upon integrin-dependent adhesion or EGF treatment, indicating a potential role as a downstream effector of cell-matrix and growth factor signalling (Di Stefano et al, 2004). p140Cap is expressed in the mammary gland and in breast cancer cells such as MCF7, T47D, MDA-MB-231 and MDA-MB-435. By loss- and gain-of-function approaches, we show here that p140Cap affects breast cancer cell motility, invasion and ‘in vivo' tumour growth and is a novel Src-interacting protein that acts as a negative regulator of Src via activation of Csk.
Results
p140Cap downregulation by shRNA enhances cell migration and integrin-dependent Src kinase activity
Our previous data showed that p140Cap expression affects cell spreading in epithelial cells (Di Stefano et al, 2004). In this work, p140Cap expression was silenced in MCF7 cells by infection with pSuper Retro-p140-2 (p140-2) recombinant retroviruses or by transient transfection with siRNA oligos (siRNA2). Empty pSuper Retro, expressing GFP (Ctr) and scrambled siRNA oligos (siRNA1), were used as negative controls. Stably p140-2 expressing MCF7 cells and MCF7 cells transiently transfected with siRNA oligos showed a 70–85% reduction of p140Cap expression (Figure 1A). These cells were allowed to migrate towards 25 U/ml HGF chemoattractant stimulus. The mean of three experiments (Figure 1B) showed that MCF7 p140-2 cells migrated twofold more relative to Ctr cells, indicating that downregulation of p140Cap expression enhances HGF-stimulated cell migration. Transiently transfected siRNA oligos yielded similar results, confirming that the observed effects were not due to retroviral infection. In addition, MCF7 p140-2 cells transiently transfected with mouse p140Cap full-length complementary DNA to recover protein expression (p140-2/R) (Figure 1A), consistently migrated at a level comparable to control cells.
Figure 1.
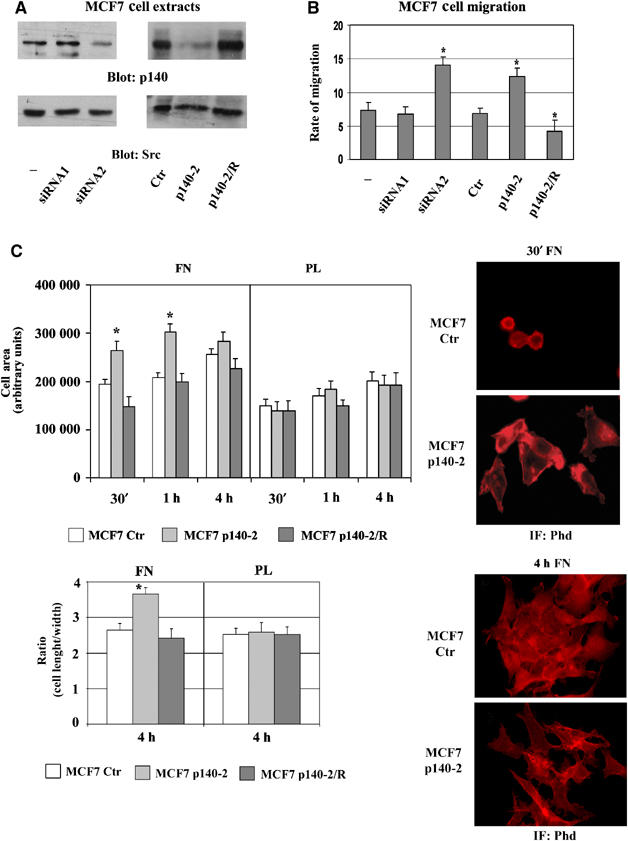
p140Cap silencing enhances cells spreading and migration. (A) p140Cap expression was evaluated by Western blotting on SDS–PAGE in extracts of MCF7 p140-2, MCF7 Ctr, MCF7 cells transiently transfected with control (−), scrambled (siRNA1) or human p140Cap (siRNA2) siRNAs and in MCF7 p140-2/R, transiently transfected with murine pcDNA3.1 Myc-p140Cap to reconstitute p140Cap expression. The same blot was re-probed with antibodies to Src kinase. (B) Cells as in (A) were tested for migration in Transwell assays. Cells were seeded on the upper surface, left to migrate for 9 h in the presence or the absence of HGF (25 U/ml), then fixed, stained and counted. Numbers on the y-axes represent the rate of migration as the ratio between the number of cells migrated in response to the presence and the absence of HGF. (C) MCF7 Ctr, p140-2 and p140-2/R cells were plated on FN and on PL for the indicated times, fixed and stained with phalloidin (Phd). Left upper panel, the histogram represents the mean cell area for each time point in arbitrary units. The area of attached cells was calculated by Metamorph Software in 20 random fields (200 cells) at a magnification of × 60. Left lower panel, the histogram represents the mean ratio between cell length and width as a measure of cell shape at 4 h of adhesion on at least 200 cells. Right panels, representative fields at 30 min (upper) and 4 h (lower) of adhesion are shown at × 60 magnification. The results are representative of six independent experiments (*P<0.05).
MCF7 Ctr, p140-2 and p140-2/R cells were allowed to adhere and spread on FN and poly-L-Lysine (PL) as negative control, for different times. Morphometric analysis showed that within 1 h of FN adhesion, MCF7 p140-2 cells, but not MCF7 Ctr and p140-2/R, had an increased area with extensions of large membrane protrusions (Figure 1C, upper panels), thus indicating that downregulation of p140Cap accelerates cell spreading in the early phases of cell adhesion. At 4 h, the difference in the extent of spreading was less evident; however, on FN, whereas Ctr cells showed a typical epithelial polygonal morphology, p140-2 cells presented a more elongated fibroblastoid phenotype (Figure 1C, lower panels). This phenotype was not observed in cells plated on PL (Figure 1C). Cell shape was calculated by the mean ratio between cell length and width on at least 200 cells and the variation was statistically significant.
In the early phases of cell-matrix adhesion, cell spreading and migration might depend on Src kinase activation (Yeatman, 2004) and on Rac GTPase (Burridge and Wennerberg, 2004). Indeed, Src kinase activity analysed in MCF7 Ctr cells plated on FN for different times is upregulated within 30 min of adhesion to FN. Interestingly, in p140-2 cells, Src activity follows the same kinetics but is upregulated by four- to sixfold at the examined times (Figure 2A, left and middle panels), thus indicating that downregulation of p140Cap leads to enhanced integrin-dependent Src activation. For what concern Rac GTPase, its activity peaked within 30–60 min of adhesion on FN in Ctr cells and was downregulated at 90 min (Figure 2B, left and middle panels). Interestingly, in p140-2 cells, Rac activity was increased at 60 min and persisted over 90 min of adhesion, thus indicating that p140Cap downregulation induces sustained Rac activation. Both Src and Rac activities were decreased at the control levels in p140-2/R cells (Figure 2A and B, right panels). Therefore, silencing of p140Cap expression enhances the ability of breast cancer cells to respond to the extracellular matrix in terms of cell spreading, increased motility, Src and Rac activation.
Figure 2.
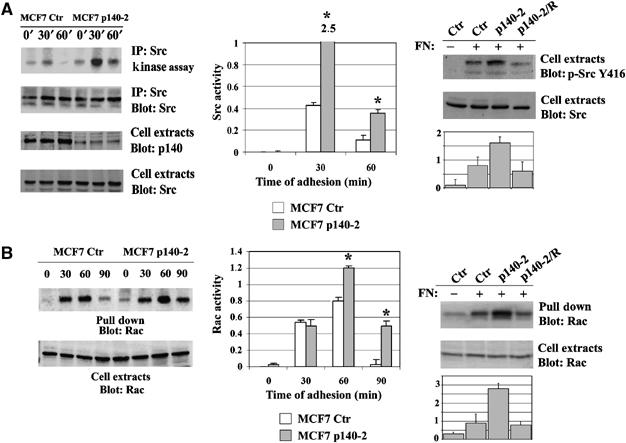
p140Cap silencing enhances Src and Rac activities. (A) Left panel, MCF7 Ctr or p140-2 cells were plated on FN for 30 and 60 min or kept in suspension (0). Right panel, MCF7 Ctr, p140-2 and p140-2/R were plated on FN for 60 min. Src kinase assay was performed as described in Material and Methods using Enolase as Src substrate. The amount of p140Cap and Src in cell extracts was evaluated by Western blot with specific antibodies. (B) Left panel, MCF7 Ctr or p140-2 cells were plated on FN for 30, 60 and 90 min or kept in suspension (0). Right panel, MCF7 Ctr, p140-2 and p140-2/R were plated on FN for 60 min. Activated Rac was pulled down from 1.5 mg of protein extract with the CRIB domain of PAK and detected by Western blot with anti-Rac mAb (upper panel). Total amount of Rac protein in cell extracts is shown in the lower panel. The histograms show the ratio between active and total protein levels in arbitrary units (*P<0.05).
p140Cap overexpression affects cell spreading, migration and invasion of tumour cells
For gain-of-function experiments, two independent populations of stable overexpressing cells (MCF7-p140/P9 and MCF7-p140/P23) were generated by transfecting pcDNA3.1-Myc-p140Cap full-length cDNA (Di Stefano et al, 2004) into MCF7 cells (see Supplementary Figure S1 for level of expression and localisation of the endogenous and exogenous p140Cap).
MCF7-p140/P9, p140/P23 and the control Mock cells, transfected with the empty pcDNA3.1 vector, were allowed to adhere and spread on FN for different times in the absence of serum. The same number of MCF7-p140/P9, p140/P23 and Mock cells adhered to FN and beta1 integrin activation was not affected (Supplementary Figure S2 and data not shown). Morphometric analysis indicated that within 2 h of adhesion, MCF7-p140/P9 and p140/P23 cells exhibited a round morphology and a reduced area (Figure 3A, left panel), compared to Mock cells. Consistent with the RNA interference (RNAi) data, spreading was recovered at 4 h, suggesting that p140Cap interferes only with the early phases of cell spreading. When cells were plated on FN in the presence of serum, whereas the majority of the Mock cells spread on FN within 20 h, MCF7-p140/P9 and p140/P23 cells remained round although completely viable (Figure 3A, right panel, and Supplementary Figures S1C and S2B). The lack of spreading was also associated with the absence of membrane cell protrusions (Figure 3Bd–f). These results indicate that high levels of p140Cap causes a stronger inhibition of cell spreading in the presence of serum, implying that p140Cap is an effector of multiple and additive pathways.
Figure 3.

p140Cap overexpression inhibits cells spreading, migration and invasion. (A) MCF7-Mock, p140/P9 and p140/P23 cells were plated on FN for the indicated times in absence (left panel) or in presence of serum (right panel), fixed and stained with Phd. The histogram represents the mean cell area for each time point in arbitrary units. The area of attached cells was calculated by Metamorph Software in 20 random fields (200 cells) at a magnification of × 60. (B) MCF7-Mock (a–c) and p140/P9 (d–f) cells as in (A) (Right panel) were fixed and stained with Diff Quick kit and photographed at 20X magnification. (C) MCF7-Mock, p140/P9 and p140/P23 cells were tested for migration and invasion in Transwell assays. Cells were left to migrate for 9 h in the presence or the absence of HGF (25 U/ml), then fixed, stained and counted. For invasion, transwells were coated with Matrigel and cells left to invade for 48 h. Numbers on the y-axes represent the fold increase in migration in response to HGF compared to nonstimulated cells. The mean values were calculated on six independent experiments (*P<0.05).
Transwell migration assays showed that, upon HGF stimulus, cells overexpressing p140Cap migrated four times less than MCF7-Mock cells (Figure 3C, upper panels), suggesting that these cells were strongly defective in migration. ‘In vitro' invasion assays into Matrigel-coated Transwell showed that within 48 h of invasion, whereas Mock cells invaded the Matrigel properly in response to the stimulus, MCF7-p140/P9 and p140/P23 cells were significantly impaired in their ability to invade (Figure 3C, lower panel). Hence, p140Cap overexpression affects the ability of cells to move towards a chemoattractant stimulus and to invade extracellular matrix.
p140Cap overexpression compromises integrin-dependent Src activation and downstream signalling
As shown in Figure 4A (left panel), whereas in MCF7-Mock cells Src activity was upregulated within 30 min of adhesion to FN, Src activity was not induced in p140/P9 cells. Src kinase activity was also evaluated by phosphospecific antibodies against the critical tyrosine residue 416 in the Src kinase domain. Upon FN adhesion, tyrosine 416 was phosphorylated only in Mock cells, but not in p140Cap-overexpressing cells (Figure 4A, right panel). Taken together, these results demonstrate that high levels of p140Cap result in inhibition of integrin-dependent Src activation.
Figure 4.

p140Cap overexpression compromises integrin-dependent Src activation and downstream signalling. MCF7-Mock and p140/P9 were plated on FN or kept in suspension (S) for 30 min and extracted. (A) Left panel, Src kinase assay on Enolase was performed as described in Material and Methods. Right panel, in the same extracts, Src phosphorylation on tyrosine 416 was evaluated using phosphospecific antibodies (Y416). The total amount of Src was evaluated by Western blot. (B) FAK phosphorylation was evaluated in cell extracts using phosphospecific antibodies for the FAK autocatalytic tyrosine 397 (Y397) (Left panel) and FAK tyrosine 925 (Y925) (Right panel). The total amount of FAK was quantified using FAK mAbs. (C) Cell extracts from MCF7-Mock and p140/P9 plated on FN for 30 min were immunoprecipitated with Src antibodies and pre-immune serum as negative control. The amount of FAK co-immunoprecipitated with Src was detected with FAK antibody. The level of immunoprecipitated Src was quantified using Src polyclonal antibodies. (D) Cell extracts as in (A) were immunoprecipitated with anti phosphotyrosine antibodies (P-Tyr) followed by Western blot analysis with p130Cas mAbs. p130Cas was evaluated in cell extracts. (E) Cell extracts as in (A) were analysed for activated Rac as described in Figure 2B. (F) Cell extracts as in (A) were analysed for Akt phosphorylation using phosphospecific antibodies for the Akt Serine 473 Ser(473). The total amount of Akt was quantified using antibodies. The results are representative of three independent experiments. The histograms show the ratio between active or phosphorylated and total protein levels in arbitrary units (*P<0.05).
Upon integrin-mediated adhesion, Src phosphorylates downstream effectors such as FAK and p130Cas (Miranti and Brugge, 2002). To investigate whether p140Cap overexpression affects this signalling, both MCF7-Mock and p140/P9 cells were analysed for phosphorylation of FAK. As depicted in Figure 4B (left panel), p140Cap overexpression did not affect integrin-dependent autophosphorylation on FAK tyrosine 397. In contrast, tyrosine 925, a residue shown to be a specific substrate of Src kinase (Brunton et al, 2005; Mitra et al, 2005), was not phosphorylated in p140/P9 cells (Figure 4B, right panel). Moreover, the binding between FAK and Src is decreased in p140/P9 cells compared to Mock-transfected cells (Figure 4C). These results suggest that high levels of p140Cap do not modify integrin-dependent FAK autophosphorylation but rather, alter its ability to associate with Src, thereby affecting Src-dependent phosphorylation on specific residues. Moreover, integrin-dependent phosphorylation of p130Cas (Figure 4D), as well as its association with Crk (data not shown), were decreased in cells overexpressing p140Cap.
As a consequence of decreased Src/FAK/p130Cas signalling Rac activity could also be affected. Active Rac was not present in p140Cap-overexpressing cells upon adhesion to FN (Figure 4E), indicating that p140Cap overexpression inhibits integrin-dependent Rac activation. In contrast, as shown in Figure 4F, Akt was equally phosphorylated on Ser 473 in MCF7 Mock and p140/P9-overexpressing cells plated on FN, thus suggesting that Src/FAK/p130Cas pathway is a specific target of p140Cap.
p140Cap directly associates with Src kinase
p140Cap has been identified for its ability to indirectly associate with the adaptor protein p130Cas (Di Stefano et al, 2004). As Src kinase is one of the major p130Cas-interacting molecules (Defilippi et al, 2006), we evaluated whether Src might also associate with p140Cap. Src and p140Cap reciprocally co-immunoprecipitated in MCF7 cells (Figure 5A), indicating that the two molecules are associated in a complex. p140Cap presents two proline-rich regions, potentially involved in binding to SH3 domains, as well as phosphorylated tyrosine residues (Figure 5B), which can associate with SH2 domains (Figure 5C, upper panel). GST-Src/SH3 or SH2 domains were used to affinity purify endogenous p140Cap from MCF7 cell extracts. As shown in Figure 5C (lower panel), only the GST-Src/SH3 fusion protein pulled down p140Cap, whereas the GST-Src/SH2 recombinant protein was ineffective. Recombinant GST-Cap3 and -5 fusion proteins (Di Stefano et al, 2004), which contain the first and the second proline-rich regions, respectively, and GST and GST-Cap4 fusion proteins serving as negative controls (Figure 5D, left panel), were incubated with the MBP-Src/SH3 fusion protein in an ‘in vitro' binding assay. Only the GST-Cap5 protein was able to bind to the MBP-Src/SH3 domain (Figure 5D, right panel), indicating that p140Cap and Src directly interact through the binding of the Src SH3 domain with the most carboxy-terminal proline-rich region of p140Cap.
Figure 5.

p140Cap binds directly to Src SH3 domain. (A) MCF7 cell extracts were immunoprecipitated with mAbs to p140Cap or Src. The immunoprecipitates were blotted with Src antibodies. Molecular weights are indicated on the left. (B) MCF7 cell extracts were immunoprecipitated with mAbs to p140Cap or pre-immune serum. The immunoprecipitate were blotted with anti-p140Cap polyclonal antibodies and re-probed with anti-pTyr antibodies. The results are representative of six independent experiments. (C) Upper panel, a schematic representation of full-length p140Cap domains. Lower panel, GST, GST-Src/SH2 and GST-Src/SH3 fusion proteins were used to pull down p140Cap from MCF7 cell extracts. Bound proteins were immunoblotted using p140Cap polyclonal antibodies (left) or stained with Coomassie blue (right) to quantify the loading. The results are representative of two independent experiments. (D) A total of 2 μg of purified p140Cap GST-Cap3, -4 and -5 fusion proteins (left panel) were incubated in ‘in vitro' binding assays with 2 μg of MBP-Src/SH2 or MBP-Src/SH3 proteins. Associated proteins were pulled down with Glutathione–Sepharose, eluted and immunoblotted with anti MBP antibodies. The results are representative of two independent experiments.
p140Cap-dependent Src kinase inhibition is mediated by Csk activation
Csk is a potent negative regulator of Src, due to its ability to phosphorylate the negative regulatory tyrosine 527 on the carboxy-terminal domain of Src (Latour and Veillette, 2001). Densitometric analysis revealed that upon adhesion to FN, phosphorylation of Src tyrosine 527 was increased by 2.5-fold in MCF7-p140/P9 cells in comparison to Mock cells (Figure 6A), suggesting a potential role for p140Cap in regulation of Csk activity.
Figure 6.

p140Cap inhibition of Src kinase activity is dependent on Csk activity. (A) MCF7-Mock and p140/P9 cells were plated on FN for different times or kept in suspension (S). Phosphorylation of Src Tyr-527 was detected by Western blot with specific antibodies. The same blots were re-probed with antibodies to Src. The histogram shows the ratio between Tyr-527 phosphorylation and Src protein levels in arbitrary units. The results are representative of three independent experiments. (B) Left panels, cell extracts of MCF7-Mock and p140/P9 plated on FN for 30 min, were immunoprecipitated with antibodies to Csk or pre-immune rabbit immunoglobulins (PI) and subjected to kinase assay. The amount of immunoprecipitated Csk was evaluated by Western blot with specific antibodies. Right panel, densitometric analysis of Poly-Glu-Tyr signal is reported in arbitrary units. The results are representative of two independent experiments. (C) Mock-MCF7 and p140/P9 cells were infected with GFP or kinase-negative mutant of Csk (Csk-KD) recombinant adenoviruses. Infected cells were plated on FN for 30 min. Src activity was analysed by in vitro kinase assay as shown in Figure 2A. The level of Csk was detected by Western blot. Arrow indicates immunoglobulin. The results are representative of three independent experiments. (D) MCF7-Mock cell extracts were immunoprecipitated with mAbs to p140Cap, Csk or pre-immune serum (PI) as negative control. Immunocomplexes were analysed by Western blotting using antibodies to Src, Csk and p140Cap, respectively. The results are representative of four independent experiments. (E) Cell extracts from HEK293 transfected with p140Cap were immunoprecipitated with mAbs to p140Cap and GAPDH. Upper panel, Western blot was probed with recombinant Csk produced by in vitro transcription/translation followed by anti-Csk antibodies. Middle and lower panels, the blots were re-probed with anti-p140Cap and GAPDH antibodies. L, lysates; MW, molecular weight.
‘In vitro' kinase assays showed a twofold increase in poly-Glu-Tyr phosphorylation in MCF7-p140/P9 cells relative to control cells (Figure 6B), demonstrating that p140Cap overexpression upregulates Csk activity. Infection with adenoviruses expressing a kinase-defective Csk mutant (Csk-KD) induced a strong activation of Src in MCF7-p140/P9 cells (Figure 6C), indicating that the presence of the kinase-defective Csk counteracts the ability of p140Cap to inhibit Src activity. These data were further supported by the analysis of Src activity in MCF7-Mock and MCF7-p140/P9 cells transiently transfected with Csk siRNA. As depicted in Supplementary Figure S3, silencing of Csk protein led to increased Src phosphorylation in cells overexpressing p140Cap. Taken together, these data show that p140Cap negatively regulates Src kinase through the activation of Csk kinase. Reciprocal immunoprecipitation of p140Cap and Csk from MCF7-Mock cells showed that the two molecules co-immunoprecipitated (Figure 6D), revealing the association of Csk and Src with p140Cap in a macromolecular complex, which could favour the regulation of kinase activities. To investigate the direct interaction between p140Cap and Csk, we performed Far-Western analysis on p140Cap and GAPDH (negative control) immunoprecipitates using recombinant Csk produced by in vitro transcription/translation as probe. As shown in Figure 6E, the Csk antibody detected a band at 140 kDa only in the p140Cap and not in the GAPDH immunoprecipitates, indicating that the Csk protein binds to p140Cap on the filter. Therefore, this experiment demonstrates that the p140Cap and Csk directly interact.
The carboxy-terminal proline-rich region of p140Cap is required for inhibition of c-Src kinase, cell spreading, motility and invasion
To assess the role of the carboxy-terminal proline-rich region PPPPPRR in cell signalling, MCF7 cells were transfected with cDNAs expressing either a large Myc-tagged truncated form of p140Cap (MCF7-p140Delta) or a small deleted mutant (MCF7-p140Pro) lacking amino acids 1000–1048, which include specifically the PPPPPRR sequence (Figure 7A). Co-immunoprecipitation experiments indicated that these mutants failed to bind to Src (Figure 7B), confirming the relevance of the proline-rich sequence in Src binding. By immunofluorescence experiments with anti-Myc antibodies, p140Delta protein was found to localise as the endogenous one with cortical actin (see Supplementary Figure S1C).
Figure 7.

Src-binding domain is essential for p140Cap role in biological processes. (A) Left panel, a schematic representation of full-length p140Cap protein, p140Pro and p140Delta mutants. Right panel, cell extracts of MCF7p140/P9, p140Pro and p140Delta were analysed by Western blot using Myc antibodies. The same filter was re-probed with Src-specific antibodies. (B) Extracts of HEK293 cells transiently transfected with p140FL, p140Pro and p140Delta were immunoprecipitated with Src antibodies. The immunoprecipitate were analysed by Western blot with Myc and Src antibodies. (C) The histogram represents the mean cell area for MCF7-Mock, p140/P9, p140Pro and p140Delta cells plated on FN for the indicated times, calculated as described in Figure 1C. (D) Left panel, the same cells as in (C) were induced to migrate and to invade as described in Figure 3C. (E) Extracts of MCF7-Mock and p140Delta cells plated on FN for 30 min or kept in suspension (S) were tested for in vitro Src kinase assay as shown in Figure 2A (left panel) or for Rac activation as shown in Figure 2B. The results are representative of three independent experiments (*P<0.05).
Morphometric analysis showed that MCF7-p140Delta and p140Pro plated on FN, covered a cell area similar to Mock cells (Figure 7C). Moreover, as shown in Figure 7D, cells expressing the mutated forms of p140Cap migrated and invaded at the same rate as Mock cells.
Following FN adhesion, Src activity in the p140Delta was found comparable to that observed in the Mock-transfected cells (Figure 7E). Similarly, in mutant cells, the level of integrin-mediated active Rac was unaffected (Figure 7F). In addition, MCF7 p140-2 cells transiently transfected with mouse p140Delta cDNA show a comparable rate of Rac activation, migration and adhesion as MCF7 p140-2 cells (see Supplementary Figure S4), indicating that this mutant does not modifiy the phenotype of p140Cap-silenced cells.
Therefore, these data show that the carboxy-terminal domain of p140Cap, containing the Src-binding site, is essential for downregulation of downstream signalling and of cell motility and invasion.
p140Cap overexpression inhibits tumorigenic properties of cancer cells
Different breast cancer cell lines (MCF7, T47D, MDA-MB-231 and MDA-MB-435), on the basis of their phenotype and invasiveness, distribute at the opposite boundaries of a spectrum of differentiation from epithelial to mesenchymal phenotype (Lacroix and Leclercq, 2004). Indeed, MCF7 and T47D, which express markers typical of the luminal epithelial phenotype of breast cells, are weakly migratory and invasive, whereas MDA-MB-231 or 435 have a ‘basal-like' phenotype and are highly invasive ‘in vitro' and ‘in vivo'. p140Cap is expressed as a single faint band in MDA-MB-231 and MDA-MB-435 cell lines, whereas is highly expressed in MCF7 and T47D cells (Figure 8A, upper panel), suggesting a correlation between expression of p140Cap and cell morphological and invasive properties. Interestingly, both MCF7 and MDA-MB-231 are inhibited in cell growth by the use of the specific Src inhibitor SU6656 (Supplementary Figure S5), indicating that Src kinase activity is required for their proliferation. Consistently, MDA-MB-231 p140/P12 and p140/P16 cell populations overexpressing pcDNA3.1-Myc-p140Cap full-length cDNA (Figure 8A, lower panel) were strongly inhibited in migration and invasion compared to Mock cells (Figure 8B). This shows that p140Cap overexpression compromises migratory properties in a highly invasive breast cancer cell line such as MDA-MB-231.
Figure 8.

p140Cap regulates anchorage-independent and in vivo tumour growth. (A) Upper panel, expression of p140Cap was evaluated in extracts of MCF7, T47D, and MDA-MB-231 and MDA-MB-435 breast cancer cells by Western blot with p140Cap antibodies. The blot was re-probed with Src antibodies. Lower panel, MDA-MB-231 cells stably transfected with p140Cap-Myc were analysed by Western blot with anti-Myc tag antibodies. Cell population P12 and P16 were selected for further experiments. (B) MDA-MB-231 Mock, p140/P12 and p140/P16 cells were tested for their ability to migrate for 2 h (upper panel) or to invade Matrigel-coated Transwells for 12 h (lower panel) as described in Figure 3C. The mean values were calculated on five independent experiments (*P<0.05). (C) 107 MDA-MB-231 Mock, p140/P16 and p140Delta cells, transfected with the deleted mutant p140Delta, were injected subcutaneously in SCID mice. Progressively growing neoplastic masses were measured with calipers and the average value recorded as mean tumour diameter. Differences in tumour volume were evaluated with Student's t-test. (D) 105 MDA-MB-231 Mock, p140/P16 or p140Delta cells were allowed to grow in the presence of 10% FCS for 12 days. Every two days, cells were detached and counted. The mean number of cells from three separate experiments is reported on the y-axis. (E) Left panel, 105 MCF7-Mock, p140/P9 and p140Delta cells were allowed to grow for 12 days and counted as in (D) (right upper panel). Extracts of MCF7-Mock and p140/P9 cells grown for 2, 4 and 6 days were analysed by Western blot with cyclin D1 and actin antibodies. Right lower panel, extracts of MCF7-Mock and p140/P9 cells starved for 24 h and treated for 30 min with 20% FCS or left untreated, were analysed for Src kinase assay as described in Figure 2A. The amount of Src in immunoprecipitates was evaluated by Western blot. The results are representative of three independent experiments.
In SCID mice, subcutaneous injection of MDA-MB-231 Mock and MDA-MB-231 p140Delta cells, overexpressing the p140CapDelta mutant (data not shown), gave rise to tumours of 1.4 cm mean diameter within forty days after challenge, whereas injection with MDA-MB-231 p140/P16 cells did not cause tumour growth (Figure 8C), indicating that cells expressing high levels of p140Cap protein do not grow ‘in vivo'. Consistent with a role of p140Cap overexpression in cell proliferation, both MDA-MB-231 and MCF7 cells overexpressing p140Cap were strongly defective in proliferation compared to Mock and p140Delta cells (Figure 8D and E). The defective growth was not due to an increased apoptosis rate (Supplementary Figure S6). Indeed, p140Cap-overexpressing cells were defective in cyclin D1 induction as well as in upregulation of Src activity in response to serum (Figure 8F). Taken together, these data show that p140Cap overexpression strongly inhibits cell growth.
Finally, MCF7 Mock, Ctr and p140Delta grew in anchorage-independent soft agar assays forming a mean of 84 colonies. In contrast, cells overexpressing p140Cap were strongly inhibited (Table I). p140Cap downregulation by shRNA consistently resulted in an increased number of colonies, with a mean value of 122 (Table I). In conclusion, these data show that p140Cap expression regulates breast cancer cell tumorigenic properties.
Table 1.
p140Cap regulates anchorage-independent growth
| Cell type | No. of colonies |
|---|---|
| MCF7-Mock | 82±10.4 |
| MCF7-p140/P9 | 3.4±1.6* |
| MCF7-p140Delta | 86.2±7 |
| MCF7-Ctr | 89.5±6.3 |
| MCF7-p140-2 | 122±5.9* |
| MCF7-Mock, MCF7-p140/P9, MCF7-p140Delta, MCF7 Ctr and MCF7 p140-2 cells were evaluated in soft agar assay for anchorage-independent notes. A total of 5 × 104 cells were plated in six-well dishes coated with agar as described in Material and Methods. After 30 days, colonies visible by the eye were counted in each well. The mean values were calculated in four independent experiments (*P<0.05). | |
Discussion
In this work, we identify p140Cap as an adaptor protein that affects integrin-dependent signalling and suppresses tumorigenic properties of breast cancer cells. Silencing p140Cap by RNAi increases Src kinase activity, leading to enhanced cell migration and anchorage-independent growth. Accordingly, elevated expression of p140Cap activates Csk and leads to inhibition of Src kinase activity, resulting in decreased cell spreading, motility and invasion. Moreover, high levels of p140Cap inhibit cell proliferation and ‘in vivo' tumour growth.
p140Cap binds to Src kinase and regulates its activity through Csk kinase
p140Cap was originally characterised as a protein that indirectly associates to p130Cas, a major Src-binding protein (Defilippi et al, 2006) through its carboxy-terminal domain (Di Stefano et al, 2004). The putative direct binding molecule, however, remained elusive. The carboxy-terminal domain of p140Cap contains a proline-rich sequence (PPPPPRR). This sequence is a perfect match with the class II PxxPx+ consensus motif known to bind to the Src SH3 domain (Kay et al, 2000), suggesting that Src might bind directly to p140Cap. Our data generated from the ‘in vitro' binding assay show that this region is able to directly interact with the Src SH3 domain. Moreover, two deletion mutants of p140Cap lacking the interacting region loss the ability to immunoprecipitate Src, thus confirming that p140Cap directly associates with Src through this sequence.
Our data show that Src kinase activity is dramatically regulated by the levels of expression of p140Cap. In fact, RNAi experiments demonstrate that silencing p140Cap expression is sufficient to upregulate Src kinase activity fivefold over the level of control cells. On the contrary, p140Cap overexpression inhibits Src activation. Moreover, mutant forms of p140Cap lacking the proline-rich domain carboxy-terminal, which includes the Src-interacting region, do not deter Src activity, indicating that Src/p140Cap interaction is required for inhibition of Src kinase. It has been well established that Src kinase activity is negatively controlled by endogenous regulators such as the Csk. This regulator acts by phosphorylating the inhibitory tyrosine 527 in the Src carboxy-terminal domain, thereby allowing the binding of the Src SH2 domain and the acquirement of a close, inactive conformation (Koegl et al, 1995; Yamaguchi and Hendrickson, 1996; Gonfloni et al, 2000). Our data show that high levels of p140Cap overexpression induces higher Csk activity, and increases phosphorylation of Src tyrosine 527, leading to the downregulation of Src activity. It is interesting to note that phosphorylation of Src tyrosine 527 is absent in suspended cells where Src is not active. These data are supported by a recent report by Zhang et al (2004), who shows a similar level of tyrosine 527 phosphorylation upon integrin activation in suspended cells, implying that in this condition even if the inhibitory tyrosine 527 is not phosphorylated and does not block the SH2 domain in a close configuration, an increased activity of tyrosine PTPases on tyrosine 416 might maintain Src inactive.
A Csk kinase-deficient mutant and Csk silencing by siRNA consistently rescue Src kinase activity in p140Cap-overexpressing cells, demonstrating a crucial role of Csk in p140Cap regulation of Src activity. Moreover, by Far Western analysis, our data show that p140Cap associate directly to Csk in a macromolecular complex, thus indicating that p140Cap behaves as a novel binding partner in the cell machinery recruiting Csk and Src and regulating their activity in breast cancer cells.
p140Cap expression regulates cell signalling, migration and invasion in a Src-dependent manner
In the recent years, many proteins have been reported to cooperate with Src in driving downstream signalling either upon cell matrix adhesion or growth factor stimulation (reviewed by Miranti and Brugge, 2002; Parsons, 2003). Src has been reported to bind to the autocatalytic tyrosine 397 of FAK and to phosphorylate regulatory loop tyrosines in the FAK kinase domain, further promoting its catalytic activity. The FAK tyrosine 925 residue has been reported to be a specific Src kinase-dependent site (Brunton et al, 2005). According to the inhibition of integrin-dependent Src kinase activity, p140Cap overexpression does not modify phosphorylation of tyrosine 397, which is mediated by FAK activity, but rather inhibits Src-dependent phosphorylation of tyrosine 925. Moreover, p140Cap overexpression affects the ability of FAK to associate with Src, likely because inactive Src does not expose free SH2 domains able to interact with phosphorylated tyrosines on FAK.
p140Cap-elevated expression also impairs integrin-dependent p130Cas phosphorylation. It is well established that Src-dependent p130Cas phosphorylation leads to the assembly of an Src-p130Cas-Crk signalling complex (Bouton et al, 2001; Defilippi et al, 2006) required for cell migration and invasion through activation of the small GTPase Rac (Etienne-Manneville and Hall, 2002; Burridge and Wennerberg, 2004; Jaffe and Hall, 2005). Consistent with these findings, elevated levels of p140Cap severely impair integrin-dependent Rac GTPase activity (Price et al, 1998), whereas knockdown of p140Cap expression by RNAi induces a sustained Rac activation. As expected for the major role of Rac in actin cytoskeleton dynamics and cell migration, high levels of p140Cap impair spreading on cell matrix and extension of lamellipodia and filopodia, as well as cell migration and invasion. These data are further supported by the RNAi experiments, which decrease the level of p140Cap in MCF7 cells, thereby triggering an increase in cell spreading in the early phases of cell adhesion, and inducing a fibroblastic-like shape and increased motility. In addition, our unpublished data show that silencing of p140Cap increases cell spreading and migration also in normal breast cells such as MCF10 cells, suggesting a more general role for p140Cap in regulation of cell motility (Di Stefano and Defilippi, unpublished data).
MCF7 cells express both beta1 and beta4 integrins (data not shown), that in these cells have been implicated in tumour progression and invasion (Wang et al, 2002; Bon et al, 2006). The data presented here indicate that the beta1 signalling induced by plating cells on FN is strongly regulated by levels of p140Cap. Moreover, beta4 integrin-dependent activation of Erk1/2 MAPK is also regulated by p140Cap expression (Supplementary Figure S7), indicating that p140Cap plays a regulatory role both on beta1 and beta4 early signalling. Interestingly, p140Cap does not affect Akt activation both in response to beta1 and beta4 integrins confirming the existence of specific signalling target regulated by p140Cap.
As discussed below, the Src-interacting region in the carboxy-terminal domain of p140Cap is required for inhibition of Src. Cells expressing a truncated form of p140Cap are also capable of triggering integrin-dependent Rac activation, and do not modify their migration and invasion properties. In addition to the Src/p130Cas/Crk pathway, the observation that the activation of Rac is related to that of Src is also supported by recent data showing that Rac GEFs, such as TIAM1 and Vav2, are downstream effectors of Src in epithelial and mesenchymal cells (Servitja et al, 2003).
Taken together, these results indicate that p140Cap is a new upstream regulator of integrin-dependent Src/FAK/p130Cas/Rac signalling in the control of cell motility and invasion (Figure 9).
Figure 9.
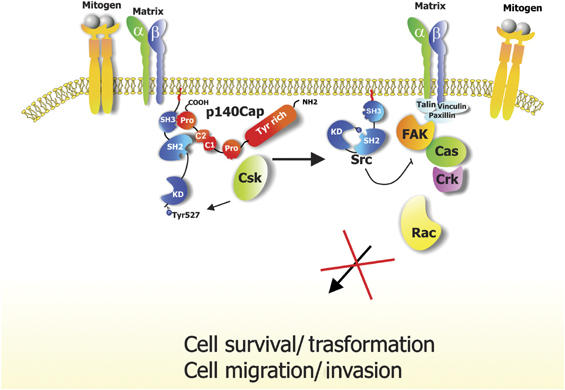
Schematic summary of the signalling pathways affected by p140Cap. Upon cell matrix adhesion or mitogen stimulus, Src is activated and recruits additional transducing molecules such as FAK and p130Cas, leading to cell migration and invasion and regulation of cell growth and transformation. p140Cap participates to these processes by activating Csk, inhibiting Src activity and downstream signalling, thus leading to impaired cell spreading, motility, invasion and growth.
Elevated expression of p140Cap inhibits ‘in vitro' and ‘in vivo' cell growth of tumour cells
Our data also show that elevated expression of p140Cap in both MCF7 and MDA-MB-231 cells inhibits cell growth, indicating that in addition to regulating migratory and invasive phenotypes, p140Cap also profoundly affects cell proliferation. The defect in proliferation is evident in the ‘in vivo' model of tumour formation in SCID mice. Mice injected with MDA-MB-231 cells expressing high levels of p140Cap do not develop tumours within 40 days, whereas mice injected with control cells present visible tumours. Accordingly, overexpressing p140Cap shows a reduced growth rate comparable to one third that of control cells, but do not show obvious signs of cell death by TUNEL assays. Cells overexpressing p140Cap do not form colonies in soft agar, whereas cells in which p140Cap has been downregulated form a significantly increased number of colonies. It is also remarkable that the two cell lines MCF7 and MDA-MB-231, which are characterised by distinct tumorigenic properties (Lacroix and Leclercq, 2004), differ in their level of endogenous p140Cap, which is lower in MDA-MB-231 cells compared to MCF7 cells, suggesting a correlation between low p140Cap expression and increased aggressiveness. Indeed, high levels of p140Cap in MDA-MB-231 cells is sufficient to revert their proliferative and invasive phenotype, ‘in vitro' and ‘in vivo'.
The mechanisms by which p140Cap overexpression blocks cell proliferation might rely on its ability to activate Csk and to inhibit Src kinase. The defect in anchorage-dependent and -independent cell proliferation is recovered in cells expressing a p140Cap mutant lacking the Src-interacting region, implying that the absence of the Src-binding site might be a contributing factor to the defective growth. Indeed, cells overexpressing p140Cap lose the ability to activate Src upon serum stimulation, thus showing that p140Cap might control Src activity not only in response to integrin-mediated adhesion, but also in response to mitogens and growth factors.
Recent data show that Src activation may promote growth during the process of tumorigenesis (Frame, 2002; Yeatman, 2004). In particular, catalytically inactive Src kinase partially inhibits breast cancer cell tumour formation in nude mice, indicating that Src activity is required for this process (Ishizawar et al, 2004; Parsons and Parsons, 2004). Moreover, our data show that the Src kinase inhibitor SU6656 affects both MCF7 and MDA-MB-231 cell growth. Therefore, we believe that our data provide significant insight into a possible role for a p140Cap/Src complex in breast cancer cells.
Taken together, these results indicate that regulation of p140Cap expression represents a novel way to activate Csk and interfere with integrin and growth factor-dependent Src kinase activity and downstream signalling, thereby affecting the tumorigenic properties of cancer cells. Overexpression of the p140Cap adaptor might thus constitute a potential tool to revert cancer cells to a more differentiated, less invasive phenotype.
Materials and methods
Reagents and antibodies
Cell lines, immunofluorescence, immunoprecipitation and Western blotting
Human breast carcinoma MDA-MB-231, MDA-MB-435, MCF7 and T47D cells, obtained from ATTC, were cultured in RPMI 1640 supplemented with 10% FCS and penicillin/streptomycin.
Immunofluorescence was performed as described by Di Stefano et al (2004). Cell images were visualised on a IX70 Olympus microscope and analysed with Metamorph software (Universal Imaging Corporation). For protein analysis, cells were extracted, immunoprecipitated and processed as described by Di Stefano et al (2004).
For far-Western analysis, membranes were blocked for 1 h in TBS, 0.1% Tween-20, 5% BSA and incubated for 3 h with 1 μg/ml of Csk protein. Filters were first decorated with anti-Csk antibody and subsequently with anti-p140Cap and anti-GAPDH antibodies. Csk was produced in vitro using TNT Quick Coupled Transcription/Translation Systems kit (Promega) according to the manufacturer's instructions.
RNA interference
Nonsilencing fluorescence-labelled control, scrambled and human p140Cap-specific siRNAs (AAGCTGTGTCTGTTGAGGCTG) (Xeragon, Quiagen) were transfected in MCF7 cells by Trans Messenger Transfection Reagent (Quiagen) for 72 h as described by the manufacturer. For stable downregulation, the 140Cap-2 sequence specifying for nucleotides 2617–2639 (CGGCAGAGACTGACTTCAA) was inserted into the pSUPER.retro.puro backbone (OligoEngine) (Brummelkamp et al, 2002) after digestion with BglII and HindIII. The resulting construct was sequenced and referred to as pSuper Retro-p140-2. pSuper Retro-p140-2 and pSuper Retro GFP (negative control) retroviruses were produced by co-transfection of 293GP packaging cells (Invitrogen) with pSR vectors and pVSV-G vector (Clontech). Supernatants were harvested over 48–72 h, filtrated and concentrated by ultracentrifugation. Virus titres were assessed by transducing HeLa cells with serial dilutions of viral stocks. Exponentially growing MCF7 cells (1 × 105/ml) were infected with viruses, in the presence of 8 μg/ml polybrene (Sigma) and selected with 1 μg/ml puromycin.
DNA constructs, transfection and analysis of stable overexpressing clones
Myc-tagged p140CapDelta mutant was prepared by cloning the first 2448 bp of full-length cDNA into pcDNA3-Myc plasmid. Myc-tagged p140CapPro was prepared by digestion and ligation of full-length cDNA with StyI, deleting bp 3022-3152. Full-length (Di Stefano et al, 2004) and mutant cDNAs were transfected in MCF7 and MDA-MB-231, respectively, using Lipofectamine 2000 (Invitrogen). For stable selection 800 μ/ml of G418 (Gibco) was used. Transiently transfection in HEK293 cells was performed by conventional calcium phosphate precipitation.
Starved MCF7-Mock and p140/P9 cells were infected with recombinant adenoviruses expressing Csk kinase-defective mutant (CskKD) (gift of David Schlaepfer) at a multiplicity of infection of 100 and further incubated for 48 h in complete medium.
Pull-down experiments
pGEX-Src/SH2 and pGEX-Src/SH3 constructs were a kind gift of Dr Sarah Courtneidge (Grand Rapids, MI). Src/SH3 and Src/SH2 fragments were amplified by PCR and cloned into the EcoRI-HindIII restriction sites of the pMALc2 vector to produce MBP fusion proteins. pGEX cDNA fragments coding for distinct p140Cap regions are described elsewhere (Di Stefano et al, 2004). Recombinant GST and MBP fusion proteins were produced in Escherichia coli and used in pull-down assays as described previously (Di Stefano et al, 2004). In vitro binding assays were performed by incubating 2 μg of MBP-Src/SH2 or MBP-Src/SH3 with 2 μg of each purified GST-fusion protein in GST-fish buffer. The complexes were pulled down with Glutathione–Sepharose beads for 30 min to 4°C, eluted in Laemli buffer, run on 8% SDS–PAGE and immunoblotted with anti-MBP antibodies.
Rho GTPases, Src and Csk in vitro activity assay
Cells were washed twice on ice with 1 mM Na3VO4 in PBS and then lysed in RIPA buffer (50 mM Tris (pH7.5), 150 mM NaCl, 1% Triton X-100, 1% Na Deoxycolate, 0.1% SDS). For Rac pull-down, glutathione-coupled Sepharose 4B beads bound to recombinant GST–PAK CRIB domain fusion proteins were incubated with cell extracts at 4°C for 30 min, eluted in Laemmli buffer and analysed for the presence of Rac1 by Western blot.
The c-Src kinase assays were performed as described by (Cabodi et al (2000, 2004) (for detailed description see Supplementary data).
The Csk kinase assay was performed as described for Src kinase assay, immunoprecipitating 200 or 400 μg cell extracts and adding in the reaction 10 μg of Poly Glu-Tyr substrate. Kinase activity was detected by autoradiography and calculated by densitometry analysis using Quantity One software (BIO RAD).
Cell adhesion, spreading, migration and invasion assay
Cell adhesion assays were performed as described previously (Moro et al, 2002). For cell spreading, cells were fixed and stained with Diff-Quik kit (Dade Behering).
For migration assays, Transwell chambers were coated with 10 μg/ml FN. A total of 5 × 104 cells were seeded on the upper side of the filters and incubated in 0.1% BSA RPMI in the presence of 25 U/ml HGF (Sigma) in the bottom wells of the chambers. Cells migrating to the lower side were fixed and stained with Diff-Quick kit. For invasion assays, the upper sides of the filters were coated with 100 μl of Matrigel matrix basement diluted 1:3 in RPMI. Cells were left to invade for 24 h for MDA-MB-231 and 48 h for MCF7 and stained as described above. Cells were counted as described above, and analysed with a × 20 and × 40 objective using Metamorph software.
Cell proliferation and soft agar assay
A total of 105 cells were seeded on 6 cm tissue culture dishes and left to proliferate for 12 days in the presence of medium supplemented with 10% FCS. Every 2 days, cells were detached and manually counted in Burker chambers on triplicate dishes. For the clonogenic assay, 5 × 105 cells were seeded in 0.35% agar on the top of a base layer containing 0.7% agar into six-well plates. After 5 weeks, colonies were counted under a phase-contrast microscope. All of the experiments were performed in triplicate.
In vivo tumour growth
Five-week-old female SCID mice (C.B-17TM/IcrCrl-scidBR) were purchased from Charles River Laboratories (Calco, Italy) and treated in accordance with the European Community guidelines.
The mice were challenged subcutaneously in the left inguinal region with 107 MD-MB-231 Mock, p140/P16 and p140Delta cells suspended in 200 μl of RPMI and 150 μl Matrigel. The incidence and growth of tumours were evaluated twice weekly by measuring with calipers for 40 days the two perpendicular diameters.
Statistical analysis
The results are representative of at least three independent experiments performed in triplicate and are expressed as the means±s.e.m. Statistical analysis of the data was performed using a Student's t-test.
Supplementary Material
Supplementary Information
Acknowledgments
We thank D Schlaepfer (La Jolla, CA) for the gift of Csk-KD adenovirus, SA Courtneidge (Grand Rapids, MI) for the Src SH2 and SH3 domains and R Cojoca for technical assistance. This work was supported by grants of the AIRC and MIUR (PRIN, fondi ex-60% and FIRB 2001), Special project ‘Oncology', Compagnia San Paolo/FIRM, Torino, Italy and Progetti Regione Piemonte.
References
- Avizienyte E, Wyke AW, Jones RJ, McLean GW, Westhoff MA, Brunton VG, Frame MC (2002) Src-induced de-regulation of E-cadherin in colon cancer cells requires integrin signalling. Nat Cell Biol 4: 632–638 [DOI] [PubMed] [Google Scholar]
- Bon G, Folgiero V, Bossi G, Felicioni L, Marchetti A, Sacchi A, Falcioni R (2006) Loss of beta4 integrin subunit reduces the tumorigenicity of MCF7 mammary cells and causes apoptosis upon hormone deprivation. Clin Cancer Res 12: 3280–3287 [DOI] [PubMed] [Google Scholar]
- Bouton AH, Riggins RB, Bruce-Staskal PJ (2001) Functions of the adapter protein Cas: signal convergence and the determination of cellular responses. Oncogene 20: 6448–6458 [DOI] [PubMed] [Google Scholar]
- Boyer B, Roche S, Denoyelle M, Thiery JP (1997) Src and Ras are involved in separate pathways in epithelial cell scattering. EMBO J 16: 5904–5913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brummelkamp TR, Bernards R, Agami R (2002) A system for stable expression of short interfering RNAs in mammalian cells. Science 296: 550–553 [DOI] [PubMed] [Google Scholar]
- Brunton VG, Avizienyte E, Fincham VJ, Serrels B, Metcalf CA III, Sawyer TK, Frame MC (2005) Identification of Src-specific phosphorylation site on focal adhesion kinase: dissection of the role of Src SH2 and catalytic functions and their consequences for tumor cell behavior. Cancer Res 65: 1335–1342 [DOI] [PubMed] [Google Scholar]
- Burridge K, Wennerberg K (2004) Rho and Rac take center stage. Cell 116: 167–179 [DOI] [PubMed] [Google Scholar]
- Cabodi S, Calautti E, Talora C, Kuroki T, Stein PL, Dotto GP (2000) A PKC-eta/Fyn-dependent pathway leading to keratinocyte growth arrest and differentiation. Mol Cell 6: 1121–1129 [DOI] [PubMed] [Google Scholar]
- Cabodi S, Defilippi P (2006) The essence of integrin signal transduction. In Integrins and Development, Danen E (ed), pp. 49–73. Georgetown, TX, USA: Landes Bioscience, Intelligence Unit Series [Google Scholar]
- Cabodi S, Moro L, Baj G, Smeriglio M, Di Stefano P, Gippone S, Surico N, Silengo L, Turco E, Tarone G, Defilippi P (2004) p130Cas interacts with estrogen receptor alpha and modulates non-genomic estrogen signaling in breast cancer cells. J Cell Sci 117: 1603–1611 [DOI] [PubMed] [Google Scholar]
- Chodniewicz D, Klemke RL (2004) Regulation of integrin-mediated cellular responses through assembly of a CAS/Crk scaffold. Biochim Biophys Acta 1692: 63–76 [DOI] [PubMed] [Google Scholar]
- Damsky CH, Ilic D (2002) Integrin signaling: it's where the action is. Curr Opin Cell Biol 14: 594–602 [DOI] [PubMed] [Google Scholar]
- Defilippi P, Di Stefano P, Cabodi S (2006) p130Cas: a versatile scaffold in signaling networks. Trends Cell Biol 16: 257–263 [DOI] [PubMed] [Google Scholar]
- Di Stefano P, Cabodi S, Boeri Erba E, Margaria V, Bergatto E, Giuffrida MG, Silengo L, Tarone G, Turco E, Defilippi P (2004) P130Cas-associated protein (p140Cap) as a new tyrosine-phosphorylated protein involved in cell spreading. Mol Biol Cell 15: 787–800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etienne-Manneville S, Hall A (2002) Rho GTPases in cell biology. Nature 420: 629–635 [DOI] [PubMed] [Google Scholar]
- Frame MC (2002) Src in cancer: deregulation and consequences for cell behaviour. Biochim Biophys Acta 1602: 114–130 [DOI] [PubMed] [Google Scholar]
- Frame MC, Fincham VJ, Carragher NO, Wyke JA (2002) v-Src's hold over actin and cell adhesions. Nat Rev Mol Cell Biol 3: 233–245 [DOI] [PubMed] [Google Scholar]
- Giancotti FG (2003) A structural view of integrin activation and signaling. Dev Cell 4: 149–151 [DOI] [PubMed] [Google Scholar]
- Goldberg GS, Alexander DB, Pellicena P, Zhang ZY, Tsuda H, Miller WT (2003) Src phosphorylates Cas on tyrosine 253 to promote migration of transformed cells. J Biol Chem 278: 46533–46540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonfloni S, Weijland A, Kretzschmar J, Superti-Furga G (2000) Crosstalk between the catalytic and regulatory domains allows bidirectional regulation of Src. Nat Struct Biol 7: 281–286 [DOI] [PubMed] [Google Scholar]
- Ilic D, Almeida EA, Schlaepfer DD, Dazin P, Aizawa S, Damsky CH (1998) Extracellular matrix survival signals transduced by focal adhesion kinase suppress p53-mediated apoptosis. J Cell Biol 143: 547–560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilic D, Furuta Y, Kanazawa S, Takeda N, Sobue K, Nakatsuji N, Nomura S, Fujimoto J, Okada M, Yamamoto T (1995) Reduced cell motility and enhanced focal adhesion contact formation in cells from FAK-deficient mice. Nature 377: 539–544 [DOI] [PubMed] [Google Scholar]
- Ishizawar RC, Tice DA, Karaoli T, Parsons SJ (2004) The C terminus of c-Src inhibits breast tumor cell growth by a kinase-independent mechanism. J Biol Chem 279: 23773–23781 [DOI] [PubMed] [Google Scholar]
- Jaffe AB, Hall A (2005) Rho GTPases: biochemistry and biology. Annu Rev Cell Dev Biol 21: 247–269 [DOI] [PubMed] [Google Scholar]
- Kay BK, Williamson MP, Sudol M (2000) The importance of being proline: the interaction of proline-rich motifs in signaling proteins with their cognate domains. FASEB J 14: 231–241 [PubMed] [Google Scholar]
- Klinghoffer RA, Sachsenmaier C, Cooper JA, Soriano P (1999) Src family kinases are required for integrin but not PDGFR signal transduction. EMBO J 18: 2459–2471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koegl M, Courtneidge SA, Superti-Furga G (1995) Structural requirements for the efficient regulation of the Src protein tyrosine kinase by Csk. Oncogene 11: 2317–2329 [PubMed] [Google Scholar]
- Lacroix M, Leclercq G (2004) Relevance of breast cancer cell lines as models for breast tumours: an update. Breast Cancer Res Treat 83: 249–289 [DOI] [PubMed] [Google Scholar]
- Latour S, Veillette A (2001) Proximal protein tyrosine kinases in immunoreceptor signaling. Curr Opin Immunol 13: 299–306 [DOI] [PubMed] [Google Scholar]
- Miranti CK, Brugge JS (2002) Sensing the environment: a historical perspective on integrin signal transduction. Nat Cell Biol 4: E83–E90 [DOI] [PubMed] [Google Scholar]
- Mitra SK, Hanson DA, Schlaepfer DD (2005) Focal adhesion kinase: in command and control of cell motility. Nat Rev Mol Cell Biol 6: 56–68 [DOI] [PubMed] [Google Scholar]
- Moro L, Dolce L, Cabodi S, Bergatto E, Erba EB, Smeriglio M, Turco E, Retta SF, Giuffrida MG, Venturino M, Godovac-Zimmermann J, Conti A, Schaefer E, Beguinot L, Tacchetti C, Gaggini P, Silengo L, Tarone G, Defilippi P (2002) Integrin-induced epidermal growth factor (EGF) receptor activation requires c-Src and p130Cas and leads to phosphorylation of specific EGF receptor tyrosines. J Biol Chem 277: 9405–9414 [DOI] [PubMed] [Google Scholar]
- Parsons JT (2003) Focal adhesion kinase: the first ten years. J Cell Sci 116: 1409–1416 [DOI] [PubMed] [Google Scholar]
- Parsons SJ, Parsons JT (2004) Src family kinases, key regulators of signal transduction. Oncogene 23: 7906–7909 [DOI] [PubMed] [Google Scholar]
- Price LS, Leng J, Schwartz MA, Bokoch GM (1998) Activation of Rac and Cdc42 by integrins mediates cell spreading. Mol Biol Cell 9: 1863–1871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz MA, Ginsberg MH (2002) Networks and crosstalk: integrin signalling spreads. Nat Cell Biol 4: E65–E68 [DOI] [PubMed] [Google Scholar]
- Servitja JM, Marinissen MJ, Sodhi A, Bustelo XR, Gutkind JS (2003) Rac1 function is required for Src-induced transformation. Evidence of a role for Tiam1 and Vav2 in Rac activation by Src. J Biol Chem 278: 34339–34346 [DOI] [PubMed] [Google Scholar]
- Shin NY, Dise RS, Schneider-Mergener J, Ritchie MD, Kilkenny DM, Hanks SK (2004) Subsets of the major tyrosine phosphorylation sites in Crk-associated substrate (CAS) are sufficient to promote cell migration. J Biol Chem 279: 38331–38337 [DOI] [PubMed] [Google Scholar]
- Wang F, Hansen RK, Radisky D, Yoneda T, Barcellos-Hoff MH, Petersen OW, Turley EA, Bissell MJ (2002) Phenotypic reversion or death of cancer cells by altering signaling path ways in three-dimensional contexts. J Natl Cancer Inst 94: 1494–1503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi H, Hendrickson WA (1996) Structural basis for activation of human lymphocyte kinase Lck upon tyrosine phosphorylation. Nature 384: 484–489 [DOI] [PubMed] [Google Scholar]
- Yeatman TJ (2004) A renaissance for SRC. Nat Rev Cancer 4: 470–480 [DOI] [PubMed] [Google Scholar]
- Zhang SQ, Yang W, Kontaridis MI, Bivona TG, Wen G, Araki T, Luo J, Thompson JA, Schraven BL, Philips MR, Neel BG (2004) Shp2 regulates SRC family kinase activity and Ras/Erk activation by controlling Csk recruitment. Mol Cell 13: 341–355 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information