

Abstract
Benzo[a]pyrene (B[a]P), a known environmental pollutant and tobacco smoke carcinogen, is metabolically activated to highly tumorigenic B[a]P diol epoxide derivatives that predominantly form N2-guanine adducts in cellular DNA. Although nucleotide excision repair (NER) is an important cellular defense mechanism, the molecular basis of recognition of these bulky lesions is poorly understood. In order to investigate the effects of DNA adduct structure on NER, three stereoisomeric and conformationally different B[a]P-N2-dG lesions were site specifically incorporated into identical 135-mer duplexes and their response to purified NER factors was investigated. Using a permanganate footprinting assay, the NER lesion recognition factor XPC/HR23B exhibits, in each case, remarkably different patterns of helix opening that is also markedly distinct in the case of an intra-strand crosslinked cisplatin adduct. The different extents of helix distortions, as well as differences in the overall binding of XPC/HR23B to double-stranded DNA containing either of the three stereoisomeric B[a]P-N2-dG lesions, are correlated with dual incisions catalyzed by a reconstituted incision system of six purified NER factors, and by the full NER apparatus in cell-free nuclear extracts.
Keywords: benzo[a]pyrene, cisplatin, DNA repair, NER, XPC
Introduction
Polycyclic aromatic hydrocarbons (PAH) are ubiquitous environmental, carcinogenic pollutants that are products of fossil fuel combustion (Luch, 2005), and are known cancer causing substances in tobacco smoke (Hecht, 1999). It is well established that PAH compounds become carcinogenic only after they are metabolically activated to mutagenic and tumorigenic intermediates (Conney, 1982). Benzo[a]pyrene (B[a]P), an extensively studied PAH compound (Luch, 2005), is stereospecifically metabolized by cytochrome P450 to highly reactive and stereoisomeric diol epoxides, including the ultimate carcinogenic form of B[a]P, the diol epoxide (+)-7R,8S-dihydrodiol-9S,10R-epoxy-7,8,9,10-tetrahydroB[a]P ((+)-BPDE) (Buening et al, 1978). The latter reacts with the exocyclic amino group of guanines in DNA to form mostly the (+)-trans-, and to a lower extent, the stereoisomeric (+)-cis-B[a]P-N2-dG guanine adducts; the (−)-BPDE enantiomer forms (−)-trans- B[a]P-N2-dG adducts, but also with lower efficiencies (Szeliga and Dipple, 1998). The structures and absolute configurations of these three adducts are depicted in Figure 1A. The major reaction product of (+)-BPDE with DNA found in mammalian cells and tissues is the (+)-trans-B[a]P-N2-dG adduct (Weinstein et al, 1976; Koreeda et al, 1978). This lesion has been implicated in tobacco-related mutations in lung tissues (Pfeifer et al, 2002) and lung cancer (Rojas et al, 1998), as well as other forms of human cancers (Kriek et al, 1998; Luch, 2005).
Figure 1.
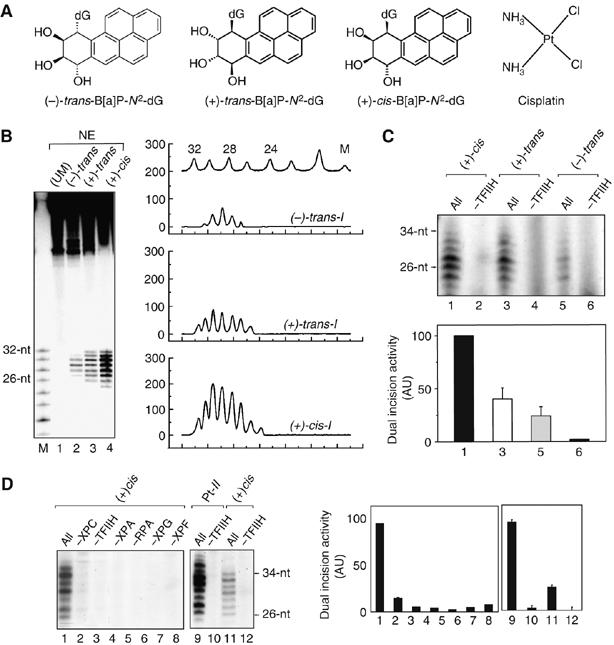
NER of modified 135-mer duplexes. (A) Structures of the the stereoisomeric (+)-cis−, (+)-trans−, and (−)-trans-B[a]P-N2-dG adducts and cisplatin. (B) Autoradiographs of dual excision products: NER of (+)-cis-I, (+)-trans-I, and (−)-trans-I 135-mer duplexes. The duplexes were internally 32P 5′ end labeled at C−6 (Table I). Dual incision products obtained after incubation with NEs for 60 min at 30°C from human HeLa cells (lanes 2–4). Lane 1: unmodified oligonucleotide 135-mer duplex control after treatment with NE. Lane M: oligonucleotide size markers 32, 30, 28, 26, 24, etc. nucleotides long (from top to bottom). Densitometric analysis of lanes M, 2, 3, and 4 (top to bottom). After normalization with respect to the total radioactivity in each lane, quantitative analysis indicates that the ratio of dual excision products of the (+)-cis-I, (+)-trans-I, and (−)-trans-I duplexes is ∼5:1:0.9, with error bars of ±20% using different extracts (eight experiments). (C) Dual incision products of internally labeled (+)-cis-I, (+)-trans-I, and (-)-trans-I duplexes were catalyzed by the RIS of purified NER factors RIS (labeled ‘All', lanes 1, 3, and 5), or RIS minus TFIIH (labeled −TFIIH, lanes 2, 4, and 6) with an XPC/HR23B concentration of 3.9 nM. The experiments were reproduced at least three times. The incision signals were quantified and plotted in a graph (lower graph, (+)-cis-I: black bar; (+)-trans-I: white bar; (−)-trans-I: gray bar)) (D) Comparison of NER of the same concentrations of the (+)-cis-I and Pt-II duplexes by RIS (‘All', lanes 1 and 9, respectively). The omission of any one of the six NER factors in RIS abolishes the dual incisions (lanes 2–8). NER of the Pt-II duplex DNA by RIS (All, lane 9) or RIS without TFIIH (lane 10). The levels of incision of two independent experiments were quantified and plotted in the bar graph.
The removal of such bulky DNA adducts by the nucleotide excision repair (NER) mechanism is an important line of defense against this disease (Reardon and Sancar, 2005; Gillet and Scharer, 2006). Although NER is a highly efficient process that excises an astounding variety of structurally different DNA lesions, the rate of repair of different lesions can vary over several orders of magnitude (Huang and Sancar, 1994; Gunz et al, 1996; Wood, 1999). The structural features that define the efficiencies of recognition of the DNA lesions by NER factors are still not well understood, in spite of the growing number of examples of chemically diverse lesions that are substrates for NER proteins (Gillet and Scharer, 2006). The relative efficiencies of removal of some DNA lesions and not others may play an important role in the etiology of sporadic cancers (Hanawalt et al, 2003), and individual differences in DNA repair capacity are known to constitute important risk factors for smoking-related lung cancers (Wei and Spitz, 1997). A fundamental knowledge of the mechanisms of NER is therefore critically important, especially the relationship between the structures of different DNA lesions, the DNA distortions that they engender, and their recognition by key NER factors. Such knowledge would be highly useful in chemotherapeutic applications, where the design of efficient antitumor agents that resist removal by cellular DNA repair mechanisms is important (Wang and Lippard, 2005).
The set of three stereoisomeric (+)-cis-, (+)-trans-, and (−)-trans-B[a]P-N2-dG lesions with base-displaced intercalation, and 5′-directed and 3′-directed minor groove conformations, respectively, are fascinating substrates for studies of structure–function relationships in DNA repair, because of these different stereochemistry-dependent patterns of adduct conformations (Geacintov et al, 1997). Striking correlations between DNA adduct stereochemistry, adduct conformation, and relative susceptibilities to excision by both human (Hess et al, 1997a) and prokaryotic (Zou et al, 1995) repair enzymes have been reported. The hallmark of human NER is the excision of oligonucleotides 24–32 bases long that contain the damaged nucleotides (Huang et al, 1994; Gillet and Scharer, 2006). The dual incisions that generate these fragments require the sequential assembly and concerted action of the six NER factors XPC/HR23B, TFIIH, XPA, RPA, XPG, and XPF/ERCC1. Recent advances in generating the individual purified factors have allowed detailed studies of the sequence in which the different NER factors assemble on the damaged DNA duplexes and bring about these incisions (Riedl et al, 2003; Tapias et al, 2004).
In this work, we have used an interdisciplinary approach to investigate the impact of the different conformations of three stereoisomeric B[a]P-N2-dG lesions on their recognition by purified NER factors. Permanganate footprinting assays were used to study the positioning and patterns of local helix opening by these NER factors in the vicinity of the lesions (Jiang and Gralla, 1995; Evans et al, 1997; Riedl et al, 2003; Tapias et al, 2004). The results are interpreted in terms of the conformational characteristics of the different B[a]P-N2-dG adducts established by NMR methods (Cosman et al, 1992, 1993; de los Santos et al, 1992), and by molecular modeling and molecular dynamic simulation methods. Striking differences are observed in the patterns of strand opening by XPC/HR23B that are correlated with the relatively inefficient DNA repair of the biologically most prevalent and mutagenic (Seo et al, 2000), (+)-trans-B[a]P-N2-dG adduct. New insights are obtained into the factors that govern the recognition and excision of this structurally diverse group of stereoisomeric bulky adducts by the human DNA repair apparatus.
Results
B[a]PDE-modified DNA substrates are repaired by purified NER factors
We constructed 135-mer double-stranded substrates containing single, stereoisomeric (+)-trans-, (−)-trans-, or (+)-cis-B[a]P-N2-dG adducts (G0*) positioned near the centers of the duplexes in identical sequence contexts (Table I). Duplexes containing single intra-strand crosslinked cisplatin lesions (cis-diamminedichloroplatinum (II)) (Riedl et al, 2003; Tapias et al, 2004) (Table I) were used as positive controls.
Table 1.
Double-stranded sequences studied
| Designation of duplexesa | Modified sequences and numbering scheme |
|---|---|
| (+)-trans-I,(−)-trans-I,(+)-cis-I | 5′-…C−9T−8G−7A−6C−5C−4A−3T−2C−1G0*C+1T+2A+3C+4C+5G+6A+7G+8… G0*=(+)−trans-, (−)-trans-, or (+)-cis-B[a]P-N2-dG |
| Pt-II | 5′-C−11T−10C−9T−8T−7C−6T−5T−4C−3T−2G−1*T0G+1* C+2A+3… G−1*T0G+1*=intra-strand Pt crosslink from G−1* to G+1* |
| aBoth sequences were annealed with their natural complementary strands. The complete sequence is provided in Supplementary data. | |
The three 135-mer DNA substrates with the different B[a]P-N2-dG lesions were incubated either in a repair proficient mammalian HeLa cell nuclear extract (NE), or with the reconstituted incision system (RIS) consisting of the six purified recombinant NER factors XPC/HR23B, TFIIH, XPA, RPA, XPG, and XPF/ERCC1. The excision reactions in NE result in the appearance of the characteristic oligonucleotide fragments with lengths in the 26–32 nucleotide range (Figure 1B). The NER efficiencies in NE follow the order (+)-cis >> (+)-trans/(−)-trans, and analogous trends have been reported with whole-cell extracts (Hess et al, 1997a).
The RIS results in incisions of the B[a]PDE-modified duplexes with relative efficiencies that qualitatively resemble those observed in NE, generating a similar order of repair efficiencies : (+)-cis > (+)-trans > (−)-trans (Figure 1C). The RIS incision efficiencies of the (+)-cis-adducts are ∼2.5 times greater than for the (+)-trans-, and ∼5 times better than for the (−)-trans-adducts. The (+)-cis/(−)-trans incision ratio is similar to that observed with NEs from whole cells, while the (+)-cis/(−)-trans ratio is ∼ two times higher in the case of the NE factors. This relative order of repair efficiencies is conserved at different incubation times in time course experiments with the RIS factors (Supplementary data). The dual incision of the B[a]PDE-modified duplexes requires a combination of all of the six NER factors; leaving out any one of the NER factor abolishes the removal of the damaged DNA fragment (Figure 1D). Similar results were observed with the Pt-II duplexes, and a typical incision result obtained with RIS, and RIS–TFIIH are depicted in Figure 1D for comparison. It seems important to note that the Pt-II substrate is a significantly better substrate for excision by the purified RIS system than any of the B[a]P-derived adducts. According to a quantitative densitometry analysis, the Pt-II substrate is repaired four times better than the (+)-cis-I duplex (Figure 1D, lanes 9–12).
Opening of the damaged DNA duplexes by NER factors
We next investigated the impact of the stereoisomeric B[a]P-N2-dG lesions on the interactions of the different NER factors with DNA. The (+)-trans-, (−)-trans-, or (+)-cis-I duplexes were compared with one another and with the Pt-II duplex (Riedl et al, 2003; Tapias et al, 2004), using KMnO4 footprinting methods (Jiang and Gralla, 1995). The KMnO4 footprinting experiments were performed using either (1) the XPC/HR23B heterodimer alone, (2) the Reconstituted Opening System (ROS) comprising all of the factors in RIS minus XPF/ERCC1, (3) ROS without TFIIH, or (4) RIS.
The binding of XPC/HR23B alone to the Pt-II duplex causes significant opening of the duplex at nucleotides G-1, T0, C+2, and A+3 (Figure 2A, lane 2). In the presence of the ROS combination of NER factors, additional opening of the duplex, attributed to TFIIH, at T+2, C−3, T−5, C−6, T−8, and T−10 is observable (lanes 3 and 4). Addition of the endonuclease containing factor XPF/ERCC1 does not produce any additional KMnO4-sensitive bands (lane 5) in the immediate vicinity of the lesion, although an incision occurs more distantly on its 3′-side (not shown in Figure 2A).
Figure 2.
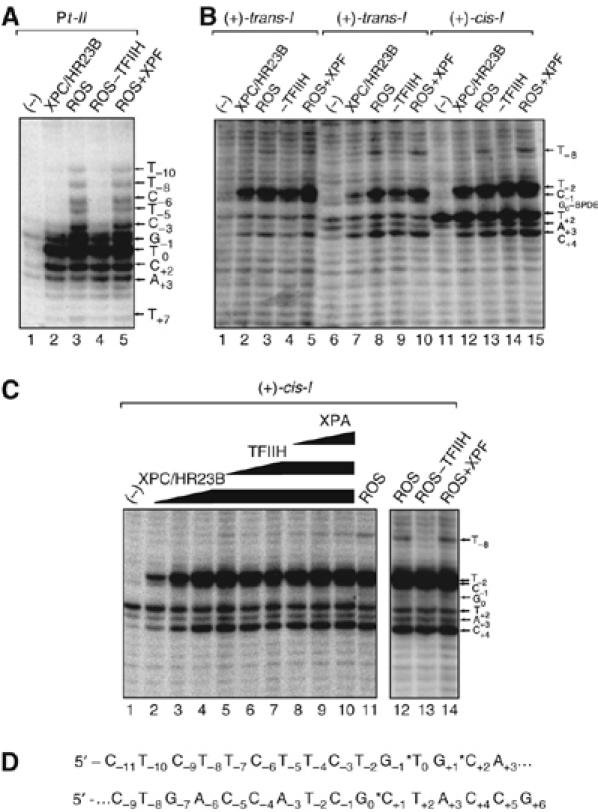
Contribution of XPC/HR23B and the ROS to the opening of the (+)-cis-I, (+)-trans-I, (−)-trans-I, and Pt-II duplexes. (A) KMnO4 footprinting experiments carried out either with (A) the Pt-II duplex or (B) the (+)-trans-I (lanes 1–5), (−)-trans-I (lanes 6–10), or the (+)-cis-I duplexes (lanes 11–15). (ROS: XPC/HR23B, TFIIH, XPA, RPA, and XPG). The 135-mer duplexes (5.7 nM final concentrations) were incubated in solutions with either ROS, ROS–TFIIH, or RIS (labeled ROS+XPF) as indicated at the top of each panel. The bases modified by KMNO4 are indicated by the arrows on the right side of the panels. (C) Effects of increasing amounts of XPC/HR23B (1.3, 2.6, and 5.2 nM final concentrations), followed by the addition of TFIIH, and subsequently XPA, on the opening of the (+)-cis-I duplexes (5.7 nM). Filled squares, (+)-cis-I; open triangles, (+)-trans-I; filled diamonds, (−)-trans-I. (D) Sequence of the Pt-II duplex (top) and the B[a]P-modified duplexes I (bottom). Details are provided in Table I.
The extent of helix opening around the (+)-trans-, (−)-trans-, and (+)-cis stereoisomeric adducts is shown in Figure 2B. The factor XPC/HR23B exhibits the major effect on the opening of the duplexes in the immediate vicinity of each lesion (lanes 2, 7, and 12). The binding of XPC/HR23B causes the appearance of major bands mainly at C+4, C−1 in the two trans-adducts (Figures 2B, 3A, and B) and at T-2 in the case of the (+)-cis-I duplex, and a less intense band is also observed at C+4 (Figure 2B, lanes 12–15). The nucleotide T+2 is permanganate sensitive in the (−)-trans-I and (+)-cis-I duplexes (and to a somewhat lesser extent in the (+)-trans-I duplex, Figure 3B), even before the addition of any repair factors; its intensity does not change significantly in the (−)-trans-I and (+)-cis-I duplexes when XPC/HR23B is added (Figure 2B, lanes 6, 7 and 11, 12). This confirms that the stronger sensitivity of T-2 to permanganate is induced by the binding of XPC/HR23B to the (+)-cis-I duplex rather than to the intrinsic reactivity of thymine with KMnO4. The addition of all the other factors seems to stabilize the open structure, and does not lead to any visible additional changes, except for the appearance of a new band at T-8. This band is attributed to TFIIH, since removing this factor from the ROS leads to disappearance of the helix opening at T-8 (Figure 2B, compare lanes 3, 8, and 13 with lanes 4, 9, and 14). As in the case of the Pt-II substrate, the addition of XPF does not generate any additional KMnO4-sensitives sites. The patterns of opening of the duplexes caused by the ROS are different in the case of the Pt-II and cis/trans-I duplexes, extending over a region of ∼17 and ∼12 base pairs around the lesion sites, respectively.
Figure 3.
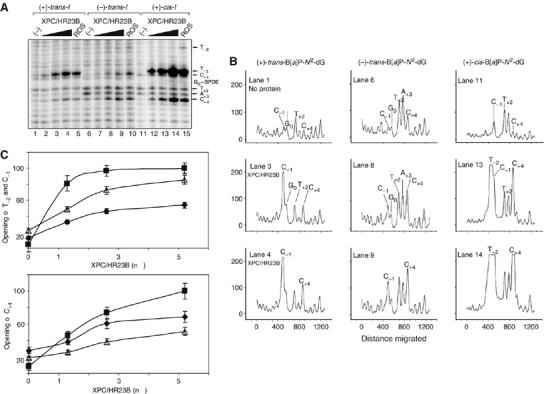
The patterns and extent of opening of the modified duplexes by XPC/HR23B depends on B[a]P-N2-dG adduct stereochemistry. (A) Increasing amounts of XPC/HR23B (1.5, 2.5, and 5.5 nM) were incubated with either the (+)-trans-I (lanes 1–4), (−)-trans-I (lanes 6–9), or the (+)-cis-I duplexes (lanes 11–14); a shorter exposure permitted a better resolution of the cleaved bands. (B) Densitometric analysis of the autoradiogram (panel A) showing the distributions and relative intensities of KMnO4-induced bands at different nucleotides in the vicinity of the B[a]P-N2-dG adducts. The vertical scales are identical in each of the nine panels. (C) The experiment was carried out two times and the signals in T−2 and C−1 (upper graph) or C+4 (lower graph) were quantified and plotted in a graph ((+)-cis-I, black square; (+)-trans-I, open triangle; (−)-trans-I, black diamonds).
In order to obtain further information on the roles of each of the purified NER factors, we studied the effects of adding the different factors sequentially on the opening of the duplex in the vicinity of the (+)-cis-B[a]P-N2-dG lesion (Figure 2C). The addition of increasing amounts of XPC/HR23B gives rise to intense signals when the protein concentration is increased, mainly at nucleotides C+4, T−2, and C−1 (lanes 2–4). However, neither the subsequent addition of TFIIH, XPA (lanes 5–10) nor the entire ROS (lanes 11, and 12), or ROS+XPF (lane 14) led to any further significant changes in the KMnO4—induced cleavage patterns, except for the T−8 band (compare lanes 12–14). It is evident that the factor XPC/HR23B is dominant in opening the duplex in the vicinity of the (+)-cis-B[a]P-N2-dG lesions in a region from T−2 to C+4 that includes the modified guanine.
To better assess the differences in the opening of the duplexes containing each of the three stereoisomeric B[a]P-N2-dG lesions, we compared the effects of increasing the amount of XPC under less saturating conditions of autoradiogram exposure (Figure 3A). Remarkable differences are observed in the intensity patterns of the different bands associated with the interaction of XPC/HR23B with the (+)-trans-I, (−)-trans-I and (+)-cis-I duplexes. The intensities of the different bands as a function of increasing XPC/HR23B concentrations (Figure 3B) and a quantitative analysis of the corresponding extent of helix opening at nucleotides C−1/T−2 and C+4 have been quantified and plotted in (Figure 3C) in order to more easily assess the changes in intensities (on the same scales) at each of the relevant cleavage sites. Increasing the concentrations of XPC/HR23B leads mainly to specific enhancements in the intensities of the T−2, C−1 and the C+4 bands in the (+)-cis-I duplex and the T−2 signals are already saturated at a relative value of 100% at an XPC/HR23B concentration of 2.5 nM (lanes 13 and 14; Figure 3B and C). It is interesting that, in contrast to the (+)-cis-I duplex, the nucleotide T−2 is not sensitive to permanganate in either the (+)-trans-I or the (−)-trans-I duplexes (lanes 1–10). Furthermore, the overall extent of opening of the duplexes is significantly more pronounced in the case of the (+)-cis-I (Figure 3A, lanes 12–15) than in the case of the (+)- and (−)-trans-I duplexes (Figure 3A, lanes 1–10). There is also a difference in the cleavage patterns of the two trans-I duplexes. In the (+)-trans-I duplex, the exposure of the C-1 nucleotide in the XPC/HR23B–DNA complexes is more pronounced than that of the C+4 nucleotide (Figure 3C), with negligible additional changes due to XPC/HR23B at T+2, and A+3 (Figure 3B). In contrast, in the case of the (−)-trans-I duplex, the reverse pattern of relative intensities is observed with the C+4 band that increases more than the C−1 band in intensity as the XPC/HR23B protein concentration is increased (Figure 3B and C).
Binding of XPC/HR23B
Finally, we investigated the efficiency of binding of the purified XPC/HR23B factor to the damaged DNA duplexes containing the (+)-cis-I, (+)-trans-I, or (−)-trans-I B[a]P-N2-dG adducts. The results of electrophoretic mobility shift assay (EMSA) experiments are shown in Figure 4A and B. In Figure 4A, the lowest bands represent the free DNA, while a substantial fraction of the XPC/HR23B–DNA complexes remain in the wells, possibly because of the buffer pH being close to the isoelectric point of the protein complex. A supershifted band is observed upon addition of an XPC mouse antibody (Mab 2H1), as expected for XPC–DNA complexes. Figure 4B represents a plot of the fractions of DNA molecules in complexes with XPC/HR23B, represented by the material indicated by the shifted and supershifted bands (lower and upper arrowheads, respectively) in addition to the material in the wells that is attributed to DNA trapped as protein complexes. These results demonstrate that XPC-HR23B has different affinities for the duplexes with the three stereoisomeric B[a]P-N2-dG lesions. The binding of XPC/HR23B to the (+)/(−)-trans-I duplexes adducts is lower than binding to the (+)-cis-I duplex (Figure 4B).
Figure 4.
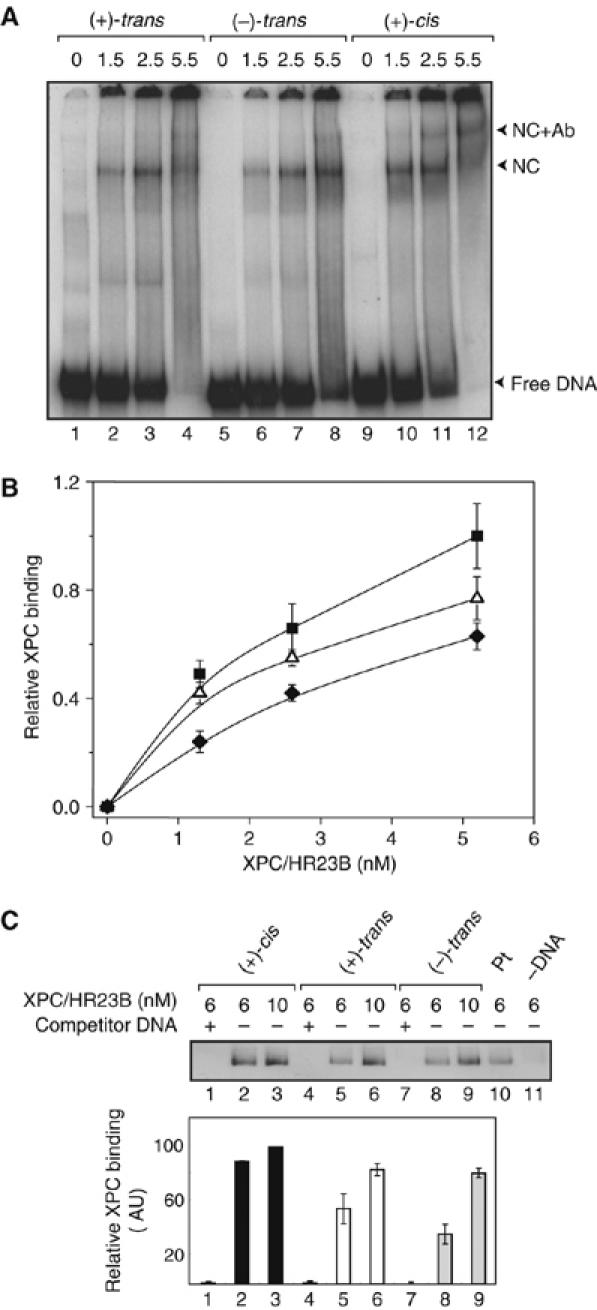
The XPC/HR23B binding to the DNA depends on the B[a]P-N2-dG adduct stereochemistry. (A) An EMSA experiment (4% native polyacrylamide gel) was carried out with 5.7 nM of (+)-trans-I (lanes 1–4), (−)-trans-I (lanes 5–8), or the (+)-cis-I and the indicated amount of XPC/HR23B. The shifts due to the formation of the nucleocomplexes (NC) with XPC/HR23B and supershifts due to XPC/HR23B plus a mouse anti-XPC antibody (MAb 2H1, IGBMC, Illkirch, France) are indicated by the lower and upper arrowheads, respectively, on the right side of the gel. (B) Fractions of DNA molecules bound to XPC/HR23B as a function of concentration of the latter ((+)-cis-I, squares; (+)-trans-I, triangles; (−)-trans-I, diamonds). (C) Binding efficiencies of XPC/HR23B on immobilized (+)-cis-I (lanes 1–3), (+)-trans-I (lanes 4–6), and (−)-trans-I (lanes 7–9) substrates (10 nM final concentration) were analyzed by Western blotting methods. The addition of identical, but unlabeled competitor DNA duplexes (lanes 1, 4, and 7) demonstrates that the binding of XPC/HR23B is specific to the B[a]P-N2-dG lesions in each case. Immobilized platinated DNA (lane 10) and magnetic beads without immobilized DNA (lane 11) were used as controls. The experiments were reproduced three times. The signals were quantified and plotted in the graph ((+)-cis-I, black bar; (+)-trans-I, open bar (−)-trans-I, gray bar).
In order to further support this conclusion, we analyzed the binding of XPC-HR23B by utilizing a magnetic bead/Western blot method (Figure 4C, details in the Materials and methods section). These results confirm that binding is better to the (+)-cis-I than to the (+)-trans-I and to the (−)-trans-I duplexes (Figure 4C) at an XPC/HR23B concentration of 6 nM, but this difference diminishes at a 10 nM protein concentration when the DNA and XPC/HR23B concentrations are close to stoichiometric. The Western blot method is less efficient at lower protein concentrations, but the results are consistent with those obtained by the EMSA method (Figure 4B). In order to ascertain the specificity of the Western blot signals, we also incubated XPC-HR23B with the beads alone, or with a competitor DNA duplex that fully inhibits its binding (Figure 4C, lanes 1, 4, 7, and 11). Interestingly, the NER incision catalyzed by the RIS (Figure 1C) and XPC/HR23B binding can be qualitatively correlated. The quantitative differences in the (+)-cis-I/(+)-trans-I as well as the (+)-cis-I/(−)-trans-I incision and binding ratios can be explained by the involvement of other factors like TFIIH, XPA, etc. that are necessary for carrying out the dual incisions.
Discussion
The recognition of damaged DNA by XPC/HR23B
The recognition by XPC/HR23B of distortions of the DNA double helix, rather than the nature of the lesions, is widely considered to be the initial event in NER (reviewed by Gillet and Scharer (2006)). The initial lesion recognition step has been discussed in terms of a structural distortion of the normal secondary structure of DNA (Szymkowski et al, 1993; Gunz et al, 1996), especially the disruption of Watson–Crick base pairing (Gunz et al, 1996; Hess et al, 1997b; Buschta-Hedayat et al, 1999; Sugasawa et al, 2001), and local flexibility (Isaacs and Spielmann, 2004) associated with bulky lesions. The factor XPC/HR23B is known to bind to DNA that contains bubbles of several mismatched and undamaged DNA bases, but incisions occur only if a chemically modified base is also present (Sugasawa et al, 2001, 2002). Buterin et al (2005) found that an unstacked or flipped out deoxyribonucleotide positioned in the unmodified complementary strand opposite the lesion plays an important role in the recognition and incision of sequences with cis-B[a]P-N2-dG adducts and other lesions. In spite of these important sets of clues, the molecular basis of the role of XPC/HR23B in recognizing damaged DNA sequences is not well understood. Here, we show that the XPC/HR23B factor plays a pivotal initial role in recognizing differences in the conformations of the stereoisomeric B[a]P-N2-dG adducts in identical sequence contexts.
Conformations of the stereoisomeric B[a]P-N2-dG adducts
We briefly recall the structural features of the stereoisomeric B[a]P-N2-dG adducts (Figure 5). In the (+)-cis-adduct, hydrogen bonding at the G0*·C basepair is disrupted. The aromatic ring system is inserted into the duplex, thus displacing the modified guanine into the minor groove where it is stacked over the sugar residue of the 5′-flanking C, while the partner cytosine residue is displaced into the major groove (Cosman et al, 1993). The NMR solution structure of the cisplatin-crosslinked adduct shows that Watson–Crick base pairing is also lost at G−1* and T0 (Table I), and that T0 is extruded from the duplex (Teuben et al, 1999), as shown in Figure 5. The flipped out nucleotides in the cisplatin and the (+)-cis-B[a]P-N2-dG adduct are thus a conformational feature shared by the two chemically different lesions. The (+)-trans- and (−)-trans-B[a]P-N2-dG adducts adopt external conformations, with the bulky benzo[a]pyrenyl residues accommodated in the minor groove of B-DNA, aligned either in the 5′-direction of the modified strand in the (+)-trans-adduct (Cosman et al, 1992), or in the 3′-direction in the case of the (−)-trans-adduct (de los Santos et al, 1992). The Watson–Crick hydrogen bonding is maintained at all base pairs in the two duplexes, although at the modified G0*·C basepair hydrogen bonding is weakened (Geacintov et al, 1997).
Figure 5.
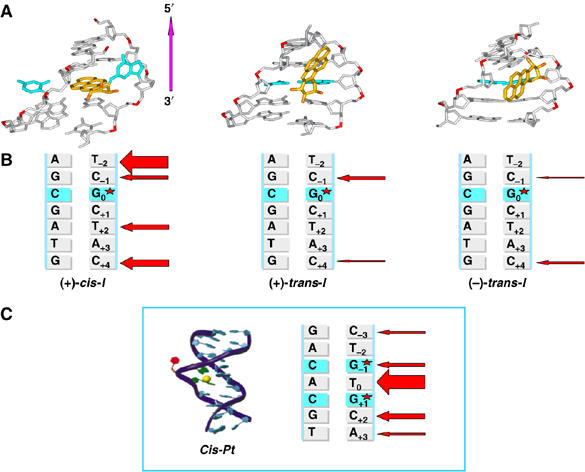
Conformations of the three stereoisomeric B[a]P-N2-dG adducts. (A) View of the central 5-mers of (+)-cis-I′·IC, (+)-trans-I′·IC, and (−)-trans-I′·IC duplexes looking into the minor groove. The structures shown are the best representatives of the ensembles derived from the last 1.5 ns of each molecular dynamic simulation (see Materials and methods and Supplementary data). The B[a]P moiety is colored gold, except for the oxygen atoms, which are orange. The modified guanine and its partner cytosine are cyan. The rest of the DNA duplexes are gray, except for the phosphorus atoms, which are red. Hydrogens and pendant phosphate oxygen atoms are not displayed for clarity. (B) Representations of permanganate-sensitive sites in complexes of XPC/HR23B with (+)-cis-I, (+)-trans-I, and (−)-trans-I duplexes. The thickness of individual red arrows indicates approximately the relative degrees of strand opening at the indicated sites (from Figure 2A and B). (C) Structure of an intrastrand crosslinked cisplatin lesion in a CTG*TG*TC sequence context (adapted and reprinted with permission from Teuben, JM, Bauer, C, Wang, AH and Reedijk, J (1999) Biochemistry, 38, 12305. Copyright (1999) American Chemical Society.
Patterns of helix opening by XPC/HR23B and dual incisions
The patterns of basepair opening depend distinctly on the adduct structure and conformation. In the Pt-II duplex, the strongest opening by XPC/HR23B binding is observed in all but one nucleotide in the region from G−1 to A+3, a region comprising five nucleotides that includes the crosslinked Pt residue. However, the size of this opened region is seven nucleotides long in the (+)-cis-I duplex and the permanganate sensitivity due to XPC/HR23B binding is most pronounced at the terminal nucleotides T−2 and C+4. In the case of the two minor groove (+)- and (−)-trans-I duplexes, the size of the opened region in the duplex is six nucleotides long; in both cases, helix opening is observed at the C−1 and C+4 nucleotides, but with opposite relative intensities (Figure 3A and B).
The higher efficiencies of dual incisions of the Pt-II and (+)-cis-I duplexes relative to those of the two trans-adducts are correlated with flipped out nucleotides in each case (Figure 5). In the Pt-II duplex, it is the T between the two modified guanines that is extruded out of the helix (Teuben et al, 1999), while in the cis-I duplex it is the C in the unmodified strand opposite the lesion that is flipped out. In the latter case, NER is abolished when this C is absent (Buterin et al, 2005). The dual incisions of the two duplexes by the RIS (Figure 1C) of the entire NER apparatus (Figure 1B; Hess et al, 1997a) are significantly lower in the two minor groove trans-I duplexes. The opening of the helix at C-1 in the (+)-trans-I duplex is more pronounced than the opening of the (−)-trans-I duplex at the same site and at C+4 on the other, or 3′-side of G0* (Figure 3B and C). These observations suggest that the (+)-trans-adduct distorts the duplex on the 5′-side of G0*, while the (−)-trans-B[a]P-N2-dG adduct is more distorting on the 3′-side, consistent with the orientations of the B[a]P residues (Figure 5). The levels of helix opening of the different lesions described here decreases in the order Pt-II>(+)-cis-I>>(+)-trans-I>(−)-trans-I (Figures 2A and B, 3A and B) that approximately parallel the relative efficiencies of dual incision efficiencies of the B[a]PDE-modified duplexes catalyzed by the reconstituted complement of repair factors (Figure 1C).
XPC/HR23B helix opening and structural features of B[a]P-N2-dG adducts
The known physical characteristics and NMR solution structures of these stereoisomeric lesions in double-stranded DNA, coupled with an analysis of their structural features by molecular modeling and molecular dynamic simulation methods, provide further insights into our observations. The conformational features of the stereoisomeric B[a]P-N2-dG adducts have been extensively studied in the 11-mer duplex I′·IC′ (Cosman et al, 1992, 1993; de los Santos et al, 1992; Yan et al, 2003) that is identical in sequence to the central portion of the 135-mer duplexes I:
All three B[a]P-N2-dG adducts destabilize the 11-mer I′·IC′ duplexes to different extents (Geacintov et al, 1997) and the melting points of the duplexes I′·IC′, Tm, decrease in the order unmodified I′·IC′ > (+)-cis- > (−)-trans- > (+)-trans-B[a]P-modified I′·IC′. The smaller extent of destabilization caused by the (+)-cis-B[a]P-N2-dG adducts is attributed to carcinogen–base stacking interactions that counteract the loss of Watson–Crick hydrogen bonding interactions in this base-displaced intercalated structure. The differences in the thermodynamic properties and Tm values of the (+)-trans- and (−)-trans-I′·IC′ duplexes have been analyzed in detail by calorimetric methods and are 54 and 58°C, respectively (Marky et al, 1996). EMSAs using native polyacrylamide gels indicate that duplexes with the (+)-trans-adducts are more bent (Xu et al, 1995) and the hydrophobic B[a]P residues are more exposed to the aqueous solvent environment than those with the stereoisomeric (−)-trans-adducts (Marky et al, 1996). Molecular modeling and computational studies indicate that in the (−)-trans-adduct, the aromatic B[a]P residue is partially accommodated within the natural contours of the minor groove on the 3′-side of G0*, without significant distortions of the DNA helix. In contrast, in the (+)-trans-adduct on the 5′-side of G0*, the right-handed helical twist causes steric crowding in the minor groove that is relieved by a local distortion and bending that results in a greater solvent exposure of the B[a]P residue (Yan et al, 2003), thus accounting for the observed lower stabilities of the duplexes with (+)-trans- relative to those with (−)-trans-B[a]P-N2-dG adducts (Marky et al, 1996).
These structural features of the two trans-adducts manifest themselves in terms of different patterns of minor groove widening, relative to unmodified DNA (Figure 6A); analogous changes caused by the (+)-cis-B[a]P-N2-dG adducts are also shown for comparison. These values were calculated by molecular dynamic simulation methods and represent the ensemble averages derived from a 3 ns trajectory (Supplementary data). Consistent with their orientations, the widening of the minor groove caused by the (+)-trans-adduct is up to ∼ 2 Å greater on the 5′-side than the widening caused by the (−)-trans-adduct on the 3′-side, in keeping with the greater structural distortions caused by the (+)-trans lesions (Marky et al, 1996). These structural features are correlated with the differences in the modes of interaction of XPC/HR23B. Although the C−1 and C+4 sites of recognition are the same in these two duplexes, the extent of helix opening in the (−)-trans-I duplex is greater at C+4 on the 3′-side of G0* occupied by the B[a]P residue than at C−1. In the (+)-trans-I duplex, the greater extent of opening at C−1 coincides with the alignment of the B[a]P residue on the 5′-side of G0*.
Figure 6.
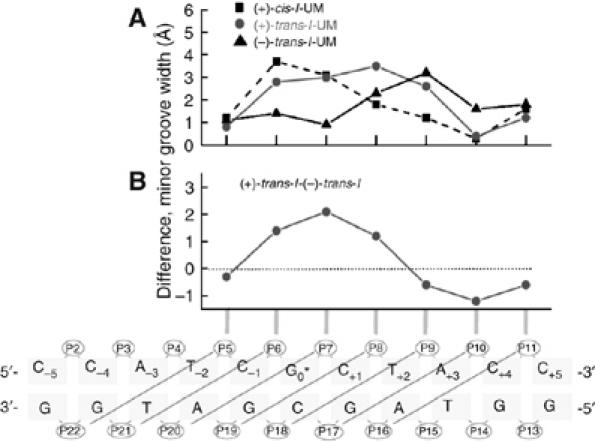
(A) Differences in the width of the minor grooves between the stereoisomeric (+)-cis-, (+)-trans-, and (−)-trans-I′·IC duplexes and the unmodified (UM) I′·I′C 11-mer duplexes in units of Å. The minor groove widths are the ensemble average calculated from individual time points of a 3 ns molecular dynamic simulation. (B) Differences in average minor groove widths between the (+)-trans-I′·I′C and (−)-trans-I′·I′C duplexes that correlate with the 5′- and 3′-orientations of the B[a]P residues in the minor groove relative to G0*. The schematic representation below panel B defines the minor groove widths (diagonal lines). The distances are defined by the phosphorus-to-phosphorus distances, less 5.8 Å to account for the phosphate group diameter in the groove. The absolute values of the minor groove widths in the unmodified I′·I′C duplexes are position dependent and vary from ∼4 to 8 Å (details are provided in Supplementary data).
In the case of the (+)-cis-I duplex, the patterns of minor groove opening are distinct from those of the two trans-adducts, with comparable levels of minor groove opening only at P6–P21 and P7–P20 (Figure 6A). In contrast to the trans-adducts, the extent of helix opening is particularly pronounced at T−2 on the 5′-side of the lesion that has the largest minor groove width, probably due to the nearby displaced modified guanine residue (Figure 3B).
Taken together, any particular DNA structural perturbation caused by bulky lesions such as loss in quality of hydrogen bonding and/or base stacking, will also affect other DNA structural characteristics (Yan et al, 2003). Such deviations from the normal values of the DNA structural parameters may individually and/or collectively serve as signals for the recruitment of NER factors to sites of damaged DNA and influence the efficiencies of dual incisions. We have proposed that such multipartite recognition mechanisms govern the efficiencies of NER of bulky DNA lesions (Geacintov et al, 2002).
Conclusions
The correlations reported here between XPC/HR23B binding, patterns of helix opening, and dual incision efficiencies catalyzed by the RIS of six DNA repair factors suggests an early role of XPC/HR23B in the discrimination between the stereoisomeric B[a]P-N2-dG lesions. Enhanced strand opening is facilitated by a local weakening of Watson–Crick hydrogen bonding and base–base stacking interactions that result in local thermal destabilization of the duplexes caused by the bulky lesions. In turn, helix opening may facilitate the flipping or extrusion of a base from the duplex, a mechanism that has been implicated in human (Buterin et al, 2005) and prokaryotic (Malta et al, 2006) NER. In the presence of the entire NER apparatus, the recruitment and/or the optimal positioning of the subsequent repair factors may also depend on the structural properties and alignment of the XPC/HR23B factor at the damaged DNA site. Finally, the low efficiency of recognition by NER factors of the (+)-trans-B[a]P-N2-dG adduct found in the DNA from lung tissues of smokers (Hecht, 1999; Pfeifer et al, 2002) by XPC/HR23B may contribute to its mutagenic and tumorigenic potentials.
Materials and methods
Substrates and NER assays
The B[a]PDE-modified 11-mer 5-CCATCG*TACC (I′) with (+)-trans-, (−)-trans-, and (+)-cis-B[a]P-N2-dG adduct stereochemistry (G*) were prepared as described (Geacintov et al, 1991). These 11-mers were 32P end labeled and incorporated into 135-mer oligonucleotides (Table I) by standard ligation and annealing methods (Hess et al, 1997a). These internally labeled sequences were then annealed with their complementary strands to form the 135-mer duplexes for NER assays in NEs. The NER factors were produced and purified as previously described (Riedl et al, 2003). The methods used to produce XPC by a baculovirus method is summarized in Supplementary data. The RIS dual incision reactions were carried out in a buffer solution (25 μl) containing 50 mM Tris–HCl pH 7.6, 50 mM KCl, 2.5 mM MgCl2, 0.5 mM DTT, 0.5 mM EDTA, 10% glycerol, 0.02% NP40, and 2 mM ATP. We incubated 100 ng of the DNA with XPC-HR23B (20 ng), TFIIH HAP fraction 5 (50 ng), XPA (30 ng), RPA (200 ng), XPG (50 ng), and XPF/ERCC1 (10 ng), for 40 min at 30°C. The incision products were radioactively labeled after incubation by the incorporation of 32P-labeled 2′-deoxynucleotide triphosphates catalyzed by a polymerase (Laine et al, 2006). The DNA was then purified with a phenol–chloroform extraction before precipitation with ethanol. The labeled excision products were separated on a denaturing 14% polyacrylamide gel and visualized by autoradiography. The NEs were prepared from human HeLa cells as described by Wood et al (1995).
KMnO4 assays
The KMnO4 substrates were obtained by labeling 20 pmol of the 135-mer duplexes at the 3′-end of the damaged strands. The DNA was digested by the restriction enzyme XmaI for 2 h and then incubated 30 min with 15 U of the Klenow fragment, 3 μl α-32P-labeled dCTP (3.3 pmol/μl, 10 μCi/μl), and 1 μl of 5 mM of each dGTP, dTTP, and dATP in 50 μl. The reaction was then stopped with 50 μl of T10E1 and the DNA probe was further recovered after electrophoresis using an 8% non-denaturing gel. The KMnO4 oxidation and cleavage experiments were performed following the protocols that were previously described (Tapias et al, 2004).
Binding assays
The preparation of the substrates was similar as for the KMNO4 reactions, except that the 135-mer duplexes were digested by the restriction enzyme BamHI, and that during the filling reaction we used 3 μl of 100 mM biotin dUTP instead of 5 mM cold dTTP in 50 μl. The binding of the substrate to magnetic beads was performed as described (Laine et al, 2006). The immobilized substrate was then incubated with the indicated amount of XPC-HR23B for 15 min at 30°C in 50 mM Tris–HCl, pH 7.6, 50 mM KCl, 2.5 mM MgCl2, 0.5 mM DTT, 0.5 mM EDTA, 10% glycerol, 0.02% NP40, and 2 mM ATP. The DNA was further washed twice with the same buffer and then with the SDS–PAGE loading buffer. After 5 min at 100°C, the samples were loaded onto a 10% SDS–polyacrylamide gel and the presence of XPC on the DNA was analyzed by Western blot methods using an antibody against XPC (Riedl et al, 2003).
Modeling and molecular dynamic simulations
The structural analyses were based on an ensemble derived from a 3 ns MD simulation for the (+)-cis-I′, (+)-trans-I′, and (−)-trans-I′ in the duplex form with the complementary strand, and the unmodified control duplexes using the NMR solution structures (Cosman et al, 1992, 1993; de los Santos et al, 1992) as initial models (Yan et al, 2003). We employed the AMBER8 simulation package, the Cornell et al (1995) force field, with the parm99 parameter set (Cheatham et al, 1999), together with added force field parameters developed for the three adducts. These trajectories reproduced well the NMR derived interproton distances, as described in Yan et al (2003). Details are provided in Supplementary data.
Supplementary Material
Supplementary data
Acknowledgments
We thank B Bernardes for fruitful discussions This work was supported by La Ligue contre le Cancer (équipe labellisée, No EL2004), l'Association de la Recherche sur le Cancer, the Agence Nationale de la Recherche (ANR-05-MRAR-005-01), and an EEC grant (DNA Repair/LSHG-CT-2005-512113) (J-ME), and by Grant CA 099194 from the National Cancer Institute, National Institutes of Health to NEG and CA 28038 to SB. VM is sponsored by a fellowship INSERM/Région Alsace.
References
- Buening MK, Wislocki PG, Levin W, Yagi H, Thakker DR, Akagi H, Koreeda M, Jerina DM, Conney AH (1978) Tumorigenicity of the optical enantiomers of the diastereomeric benzo[a]pyrene 7,8-diol-9,10-epoxides in newborn mice: exceptional activity of (+)-7beta,8alpha-dihydroxy-9alpha,10alpha-epoxy-7,8,9,10-tetrahydrobenzo[a ]pyrene. Proc Natl Acad Sci USA 75: 5358–5361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buschta-Hedayat N, Buterin T, Hess MT, Missura M, Naegeli H (1999) Recognition of nonhybridizing base pairs during nucleotide excision repair of DNA. Proc Natl Acad Sci USA 96: 6090–6095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buterin T, Meyer C, Giese B, Naegeli H (2005) DNA quality control by conformational readout on the undamaged strand of the double helix. Chem Biol 12: 913–922 [DOI] [PubMed] [Google Scholar]
- Cheatham TE, Cieplak P, Kollman PA (1999) A modified version of the Cornell et al force field with improved sugar pucker phases and helical repeat. J Biomol Struct Dyn 16: 845–862 [DOI] [PubMed] [Google Scholar]
- Conney AH (1982) Induction of microsomal enzymes by foreign chemicals and carcinogenesis by polycyclic aromatic hydrocarbons. Cancer Res 42: 4875–4917 [PubMed] [Google Scholar]
- Cornell WD, Cieplak P, Bayly CI, Gould IR, Merz KM, Ferguson DM, Spellmeyer DC, Fox T, Caldwell JW, Kollman PA (1995) A 2nd generation force-field for the simulation of proteins, nucleic-acids, and organic-molecules. J Am Chem Soc 117: 5179–5197 [Google Scholar]
- Cosman M, de los Santos C, Fiala R, Hingerty BE, Ibanez V, Luna E, Harvey R, Geacintov NE, Broyde S, Patel DJ (1993) Solution conformation of the (+)-cis-anti-[BP]dG adduct in a DNA duplex: intercalation of the covalently attached benzo[a]pyrenyl ring into the helix and displacement of the modified deoxyguanosine. Biochemistry 32: 4145–4155 [DOI] [PubMed] [Google Scholar]
- Cosman M, de los Santos C, Fiala R, Hingerty BE, Singh SB, Ibanez V, Margulis LA, Live D, Geacintov NE, Broyde S, Patel DJ (1992) Solution conformation of the major adduct between the carcinogen (+)-anti-benzo[a]pyrene diol epoxide and DNA. Proc Natl Acad Sci USA 89: 1914–1918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de los Santos C, Cosman M, Hingerty BE, Ibanez V, Margulis LA, Geacintov NE, Broyde S, Patel DJ (1992) Influence of benzo[a]pyrene diol epoxide chirality on solution conformations of DNA covalent adducts: the (−)-trans-anti-[BP]G.C adduct structure and comparison with the (+)-trans-anti-[BP]G.C enantiomer. Biochemistry 31: 5245–5252 [DOI] [PubMed] [Google Scholar]
- Evans E, Moggs JG, Hwang JR, Egly JM, Wood RD (1997) Mechanism of open complex and dual incision formation by human nucleotide excision repair factors. EMBO J 16: 6559–6573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geacintov NE, Broyde S, Buterin T, Naegeli H, Wu M, Yan S, Patel DJ (2002) Thermodynamic and structural factors in the removal of bulky DNA adducts by the nucleotide excision repair machinery. Biopolymers 65: 202–210 [DOI] [PubMed] [Google Scholar]
- Geacintov NE, Cosman M, Hingerty BE, Amin S, Broyde S, Patel DJ (1997) NMR solution structures of stereoisometric covalent polycyclic aromatic carcinogen-DNA adduct: principles, patterns, and diversity. Chem Res Toxicol 10: 111–146 [DOI] [PubMed] [Google Scholar]
- Geacintov NE, Cosman M, Mao B, Alfano A, Ibanez V, Harvey RG (1991) Spectroscopic characteristics and site I/site II classification of cis and trans benzo[a]pyrene diolepoxide enantiomer-guanosine adducts in oligonucleotides and polynucleotides. Carcinogenesis 12: 2099–2108 [DOI] [PubMed] [Google Scholar]
- Gillet LC, Scharer OD (2006) Molecular mechanisms of mammalian global genome nucleotide excision repair. Chem Rev 106: 253–276 [DOI] [PubMed] [Google Scholar]
- Gunz D, Hess MT, Naegeli H (1996) Recognition of DNA adducts by human nucleotide excision repair. Evidence for a thermodynamic probing mechanism. J Biol Chem 271: 25089–25098 [DOI] [PubMed] [Google Scholar]
- Hanawalt PC, Ford JM, Lloyd DR (2003) Functional characterization of global genomic DNA repair and its implications for cancer. Mutat Res 544: 107–114 [DOI] [PubMed] [Google Scholar]
- Hecht SS (1999) Tobacco smoke carcinogens and lung cancer. J Natl Cancer Inst 91: 1194–1210 [DOI] [PubMed] [Google Scholar]
- Hess MT, Gunz D, Luneva N, Geacintov NE, Naegeli H (1997a) Base pair conformation-dependent excision of benzo[a]pyrene diol epoxide-guanine adducts by human nucleotide excision repair enzymes. Mol Cell Biol 17: 7069–7076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess MT, Schwitter U, Petretta M, Giese B, Naegeli H (1997b) Bipartite substrate discrimination by human nucleotide excision repair. Proc Natl Acad Sci USA 94: 6664–6669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang JC, Hsu DS, Kazantsev A, Sancar A (1994) Substrate spectrum of human excinuclease: repair of abasic sites, methylated bases, mismatches, and bulky adducts. Proc Natl Acad Sci USA 91: 12213–12217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang JC, Sancar A (1994) Determination of minimum substrate size for human excinuclease. J Biol Chem 269: 19034–19040 [PubMed] [Google Scholar]
- Isaacs RJ, Spielmann HP (2004) A model for initial DNA lesion recognition by NER and MMR based on local conformational flexibility. DNA Repair (Amst) 3: 455–464 [DOI] [PubMed] [Google Scholar]
- Jiang Y, Gralla JD (1995) Nucleotide requirements for activated RNA polymerase II open complex formation in vitro. J Biol Chem 270: 1277–1281 [DOI] [PubMed] [Google Scholar]
- Koreeda M, Moore PD, Wislocki PG, Levin W, Yagi H, Jerina DM (1978) Binding of benzo[a]pyrene 7,8-diol-9,10-epoxides to DNA, RNA, and protein of mouse skin occurs with high stereoselectivity. Science 199: 778–781 [DOI] [PubMed] [Google Scholar]
- Kriek E, Rojas M, Alexandrov K, Bartsch H (1998) Polycyclic aromatic hydrocarbon-DNA adducts in humans: relevance as biomarkers for exposure and cancer risk. Mutat Res 400: 215–231 [DOI] [PubMed] [Google Scholar]
- Laine JP, Mocquet V, Egly JM (2006) TFIIH enzymatic activities in transcription and nucleotide excision repair. Methods Enzymol 408: 246–263 [DOI] [PubMed] [Google Scholar]
- Luch A (2005) Nature and nurture—lessons from chemical carcinogenesis. Nat Rev Cancer 5: 113–125 [DOI] [PubMed] [Google Scholar]
- Malta E, Moolenaar GF, Goosen N (2006) Base flipping in nucleotide excision repair. J Biol Chem 281: 2184–2194 [DOI] [PubMed] [Google Scholar]
- Marky LA, Rentzeperis D, Luneva NP, Cosman M, Geacintov NE, Kupke DW (1996) Differential hydration thermodynamics of stereoisomeric DNA-benzo[a]pyrene adducts derived from diol epoxide enantiomers with different tumorigenic potentials. J Am Chem Soc 118: 3804–3810 [Google Scholar]
- Pfeifer GP, Denissenko MF, Olivier M, Tretyakova N, Hecht SS, Hainaut P (2002) Tobacco smoke carcinogens, DNA damage and p53 mutations in smoking-associated cancers. Oncogene 21: 7435–7451 [DOI] [PubMed] [Google Scholar]
- Reardon JT, Sancar A (2005) Nucleotide excision repair. Prog Nucleic Acid Res Mol Biol 79: 183–235 [DOI] [PubMed] [Google Scholar]
- Riedl T, Hanaoka F, Egly JM (2003) The comings and goings of nucleotide excision repair factors on damaged DNA. EMBO J 22: 5293–5303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojas M, Alexandrov K, Cascorbi I, Brockmoller J, Likhachev A, Pozharisski K, Bouvier G, Auburtin G, Mayer L, Kopp-Schneider A, Roots I, Bartsch H (1998) High benzo[a]pyrene diol-epoxide DNA adduct levels in lung and blood cells from individuals with combined CYP1A1 MspI/Msp-GSTM1*0/*0 genotypes. Pharmacogenetics 8: 109–118 [PubMed] [Google Scholar]
- Seo KY, Jelinsky SA, Loechler EL (2000) Factors that influence the mutagenic patterns of DNA adducts from chemical carcinogens. Mutat Res 463: 215–246 [DOI] [PubMed] [Google Scholar]
- Sugasawa K, Okamoto T, Shimizu Y, Masutani C, Iwai S, Hanaoka F (2001) A multistep damage recognition mechanism for global genomic nucleotide excision repair. Genes Dev 15: 507–521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugasawa K, Shimizu Y, Iwai S, Hanaoka F (2002) A molecular mechanism for DNA damage recognition by the xeroderma pigmentosum group C protein complex. DNA Repair (Amst) 1: 95–107 [DOI] [PubMed] [Google Scholar]
- Szeliga J, Dipple A (1998) DNA adduct formation by polycyclic aromatic hydrocarbon dihydrodiol epoxides. Chem Res Toxicol 11: 1–11 [DOI] [PubMed] [Google Scholar]
- Szymkowski DE, Lawrence CW, Wood RD (1993) Repair by human cell extracts of single (6–4) and cyclobutane thymine–thymine photoproducts in DNA. Proc Natl Acad Sci USA 90: 9823–9827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tapias A, Auriol J, Forget D, Enzlin JH, Scharer OD, Coin F, Coulombe B, Egly JM (2004) Ordered conformational changes in damaged DNA induced by nucleotide excision repair factors. J Biol Chem 279: 19074–19083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teuben JM, Bauer C, Wang AH, Reedijk J (1999) Solution structure of a DNA duplex containing a cis-diammineplatinum(II) 1,3-d(GTG) intrastrand cross-link, a major adduct in cells treated with the anticancer drug carboplatin. Biochemistry 38: 12305–12312 [DOI] [PubMed] [Google Scholar]
- Wang D, Lippard SJ (2005) Cellular processing of platinum anticancer drugs. Nat Rev Drug Discov 4: 307–320 [DOI] [PubMed] [Google Scholar]
- Wei Q, Spitz MR (1997) The role of DNA repair capacity in susceptibility to lung cancer: a review. Cancer Metastasis Rev 16: 295–307 [DOI] [PubMed] [Google Scholar]
- Weinstein IB, Jeffrey AM, Jennette KW, Blobstein SH, Harvey RG, Harris C, Autrup H, Kasai H, Nakanishi K (1976) Benzo(a)pyrene diol epoxides as intermediates in nucleic acid binding in vitro and in vivo. Science 193: 592–595 [DOI] [PubMed] [Google Scholar]
- Wood RD (1999) DNA damage recognition during nucleotide excision repair in mammalian cells. Biochimie 81: 39–44 [DOI] [PubMed] [Google Scholar]
- Wood RD, Biggerstaff M, Shivji M (1995) Detection and measurement of nucleotide excision repair synthesis by mammalian cell extracts in vitro. Methods Enzymol 7: 163–175 [Google Scholar]
- Xu R, Mao B, Xu J, Li B, Birke S, Swenberg CE, Geacintov NE (1995) Stereochemistry-dependent bending in oligonucleotide duplexes induced by site-specific covalent benzo[a]pyrene diol epoxide-guanine lesions. Nucleic Acids Res 23: 2314–2319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan S, Wu M, Patel DJ, Geacintov NE, Broyde S (2003) Simulating structural and thermodynamic properties of carcinogen-damaged DNA. Biophys J 84: 2137–2148. erratum: Biophys J (2007) 92: 697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou Y, Liu TM, Geacintov NE, Van Houten B (1995) Interaction of the UvrABC nuclease system with a DNA duplex containing a single stereoisomer of dG-(+)- or dG-(−)-anti-BPDE. Biochemistry 34: 13582–13593 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary data