

Abstract
Ferroportin (Fpn), a ferrous iron Fe(II) transporter responsible for the entry of iron into plasma, is regulated post-translationally through internalization and degradation following binding of the hormone hepcidin. Cellular iron export is impaired in mice and humans with aceruloplasminemia, an iron overload disease due to mutations in the ferroxidase ceruloplasmin (Cp). In the absence of Cp Fpn is rapidly internalized and degraded. Depletion of extracellular Fe(II) by the yeast ferroxidase Fet3p or iron chelators can maintain cell surface Fpn in the absence of Cp. Iron remains bound to Fpn in the absence of multicopper oxidases. Fpn with bound iron is recognized by a ubiquitin ligase, which ubiquitinates Fpn on lysine 253. Mutation of lysine 253 to alanine prevents ubiquitination and maintains Fpn-iron on cell surface in the absence of ferroxidase activity. The requirement for a ferroxidase to maintain iron transport activity represents a new mechanism of regulating cellular iron export, a new function for Cp and an explanation for brain iron overload in patients with aceruloplasminemia.
Keywords: ceruloplasmin, copper, ferroportin, iron, transport
Introduction
Ferroportin (Fpn) is the only known mammalian cellular iron exporter and is present on most cell types that deliver iron into plasma (Ganz and Nemeth, 2006). Expression of Fpn leads to export of iron and a decrease in the cellular iron storage protein ferritin (Nemeth et al, 2004). Fpn is regulated by several mechanisms, but particularly through its interaction with hepcidin, a peptide produced by the liver in response to iron stores, erythropoiesis, hypoxia and inflammation. Hepcidin binds to Fpn, resulting in its internalization and degradation. The removal of Fpn from the cell surface completes a homeostatic loop in which iron transport is coordinated with iron need. Most hereditary iron overload diseases can be explained by inadequate production of hepcidin in response to iron stores or by mutations in Fpn (Pietrangelo, 2006). Conversely, increased expression of hepcidin as a result of inflammatory mediators leads to the clearance of cell surface Fpn. This decrease in Fpn prevents iron export, leading to a precipitous drop in plasma iron (hypoferremia), which if persistent results in an iron-deficient anemia referred to as the anemia of chronic disease or chronic inflammation. Thus, the presence of cell surface Fpn may be the critical determinant in mammalian iron homeostasis.
In humans and mice, loss of function mutations in the multicopper oxidase ceruloplasmin (Cp) lead to excessive iron accumulation in the liver and in various regions of the brain (Harris, 2003), as well as hypoferremia and iron-limited erythropoiesis. Multicopper oxidases, which catalyze the oxidation of four atoms of Fe(II) to Fe(III), have essential roles in iron metabolism in all eukaryotes. In Saccharomyces cerevisiae, the high-affinity iron transport system consists of the multicopper oxidase Fet3p and the transmembrane iron permease Ftr1p. Both proteins must be synthesized together for either to be targeted to the cell surface (Van Ho et al, 2002). In vertebrates, Cp and hephaestin generate Fe(III), which binds to the plasma protein transferrin, enabling delivery of iron to developing red blood cells and other tissues (Shim and Harris, 2003). Here, we show that an additional function of multicopper oxidases is to stabilize Fpn on the cell surface. Inhibition of Cp activity or synthesis prevents stable expression of Fpn on the surface of glioma cells, astrocytes and macrophages and leads to decreased iron export.
Results
Loss of Cp activity prevents Fpn-mediated iron export
We examined the requirement of Cp for Fpn function in C6 glioma cells, which express a glycosylphosphatidylinositol (GPI)-linked Cp (Patel and David, 1997). Vertebrate genomes contain a single gene for Cp, but cells can synthesize either a secreted form of Cp or a GPI-linked Cp anchored to the external surface of the plasma membrane. Incubation of C6 glioma cells with the copper chelator bathocuproine disulfonate (BCS) prevented expression of Cp on the cell surface and led to a decrease in cellular Cp (Figure 1A). Assay of the media did not show the presence of Cp, indicating that GPI-apoCp is not cleaved from the cell surface. Our results, which do not distinguish between increased degradation and decreased synthesis of GPI-Cp in BCS-treated cells, show that BCS treatment prevents the accumulation of cell surface GPI-Cp.
Figure 1.
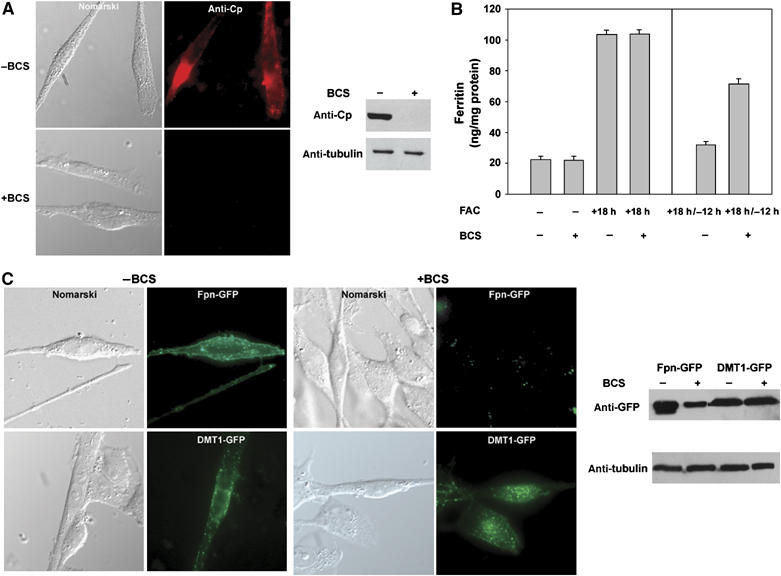
Copper depletion reduces iron export by preventing expression of GPI-Cp and Fpn in C6 glioma cells. (A) Rat glioma C6 cells were incubated in the presence or absence of BCS (250 μM) overnight. The cells were immunostained for Cp (red) and cell extracts were examined for Cp by Western blot analysis. Tubulin was assayed as a loading control. (B) C6 glioma cells were incubated with FAC for 18 h in the presence or absence of BCS (left panel). The cells were washed off FAC and were maintained in the presence or absence of BCS for 12 h. The amount of cell-associated ferritin was determined (right panel). Error bars represent the standard error of the mean of three independent experiments. (C) Cells transfected with Fpn-GFP or DMT1-GFP were incubated in the presence or absence of BCS and analyzed by epifluorescence and Western blot.
Incubation of C6 glioma cells with iron leads to an increase in ferritin (Figure 1B). Upon incubation in iron-free medium, ferritin levels decrease due to export of iron by Fpn. Incubation of C6 glioma cells with BCS reduced the ability of cells to lower their ferritin levels. The decreased ability to export iron was seen even in BCS-treated cells transfected with plasmids expressing Fpn-GFP (Supplementary Figure 1). Addition of copper to BCS-incubated cells led to loss of ferritin, indicating that BCS-induced copper depletion was responsible for the high levels of ferritin (data not shown). Transfection of C6 glioma cells with a plasmid containing Fpn-GFP led to the presence of cell surface Fpn (Figure 1C), whereas in cells incubated with BCS, there was a dramatic decrease in Fpn-GFP. The effect of BCS was specific for Fpn, as transfection of cells with a plasmid containing Divalent Metal Transporter1 (DMT1-GFP) led to the expression of DMT1-GFP regardless of whether BCS was present or not (Figure 1C). These results show that copper deprivation does not affect transfection frequency or expression from the CMV promoter.
To confirm the requirement of Cp for cell surface localization of Fpn, we examined the effect of silencing GPI-Cp. Transfection of C6 glioma cells with RNAi oligonucleotide pools specific for GPI-Cp led to decreased levels of GPI-Cp (Figure 2A) but did not affect accumulation of ferritin, when cells were incubated with iron (Figure 2B, left panel). Cells silenced for GPI-Cp did not decrease ferritin to the levels seen in control cells, when iron-loaded cells were incubated in iron-free medium (Figure 2B, right panel). When GPI-Cp-silenced cells were transfected with an Fpn-GFP plasmid, there was no expression of Fpn on the cell surface or within cells (Figure 2C), while transfection with a plasmid containing DMT1-GFP lead to abundant expression of endosomal DMT1-GFP, both in GPI-Cp-silenced cells and in cells transfected with nonspecific oligonucleotide pools. C6 glioma cells were isolated from a glial tumor and the cells have been maintained in culture for years. The loss of Cp also affects Fpn expression in primary cells. Astrocytes isolated from wild-type mice have cell surface Fpn, whereas astrocytes isolated from homozygous Cp−/− mice have no detectable Fpn on the cell surface (Figure 2D). Bone marrow macrophages also express GPI-Cp. A similar failure to localize Fpn on the cell surface was seen in BCS-treated bone marrow macrophages transfected with an Fpn-GFP plasmid (Supplementary Figure 2). These results indicate that the absence of Cp leads to the loss of cell surface Fpn in primary as well as cultured cell types. The loss of Fpn due to the absence of Cp is however cell type dependent. HEK293 cells do not express Cp, and in the absence of serum, both endogenous Fpn and transfected Fpn-GFP in HEK293 cells showed a reduced ability to export iron, yet there was no degradation of cell surface Fpn-GFP (Supplementary Figures 3 and 4). These results suggest that the Cp-dependent loss of Fpn may be restricted to cell types that synthesize Cp or GPI-Cp.
Figure 2.
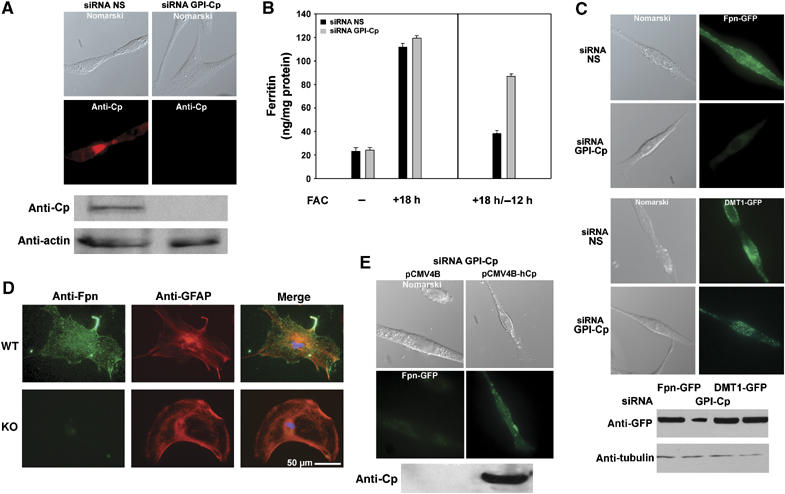
GPI-Cp depletion by siRNA prevents Fpn accumulation. (A) Cells transfected with either control (NS, non-specific) or oligonucleotides specific for rat GPI-Cp were examined for Cp (red) by immunofluorescence and Western blot analysis. Actin was assayed as a loading control. (B) Cells transfected with either control (black bars) or GPI-Cp-specific (gray bars) oligonucleotide pools were incubated with FAC (left panel) and then in the absence of FAC for 12 h (right panel). Ferritin levels were determined. Error bars represent the standard error of the mean of three independent experiments. (C) GPI-Cp-silenced or control cells, transfected with Fpn-GFP or DMT1-GFP, were examined by epifluorescence microscopy and Western blot analysis after 24 h. (D) Astrocytes isolated from Cp+/+ or Cp−/− mice were immunostained for Fpn (green), GFAP (red) and nuclei (DAPI-blue). (E) Cells silenced for GPI-Cp were transfected with Fpn-GFP, a control plasmid (pCMV4B) or a plasmid expressing human secreted Cp (pCMV4B-hCp) and Fpn-GFP and Cp expression examined by fluorescence and Western blot respectively.
To confirm that loss of expression of Fpn was due to the absence of GPI-Cp, C6 glioma cells silenced for GPI-Cp were transfected with a plasmid that contained secreted human Cp under the control of the CMV promoter. This construct was not affected by the silencing oligonucleotide pools, which are specific for rat GPI-Cp. Cells silenced for rat GPI-Cp, transfected with the secreted human Cp, showed cell surface Fpn-GFP (Figure 2E). Transfection of cells with a membrane-bound multicopper oxidase hephaestin also resulted in the accumulation of Fpn-GFP on the surface of GPI-Cp-silenced cells (data not shown). These results show that the loss of cell surface Fpn-GFP can be rescued by multicopper oxidases, although there appears to be little specificity in the multicopper oxidase, as both secreted and membrane-bound multicopper oxidases can restore cell surface Fpn-GFP.
The absence of Cp increases the internalization of Fpn
We determined that Fpn does not accumulate on the cell surface in the absence of multicopper oxidases, because it is rapidly internalized and degraded. This was shown through the use of mutant dynamin. Dynamin is a GTPase required for the internalization of coated pits and caveolae (Damke et al, 1994). Transfection of cells with a wild type dynamin had no effect on the loss of expression of Fpn-FLAG in cells silenced for GPI-Cp (Figure 3A). Expression of a mutant dynamin(K44A) that is unable to hydrolyze GTP leads to an inhibition in membrane internalization. Expression of dynamin(K44A)-GFP in cells silenced for GPI-Cp led to accumulation of Fpn-FLAG on the cell surface. This result demonstrates that the presence of GPI-Cp stabilizes Fpn-FLAG on the cell surface. While GPI-Cp-silenced cells transfected with dynamin(K44A)-GFP had accumulated cell surface Fpn-FLAG, there was no recovery of GPI-Cp (data not shown). This result confirms that Fpn-FLAG does not require simultaneous synthesis of GPI-Cp for Fpn to accumulate at the cell surface. To determine if Fpn is degraded in the lysosome or by the proteasome, cells silenced for GPI-Cp were transfected with an Fpn-GFP containing plasmid. The transfected cells were then incubated in the presence or absence of the lysosome inhibitor chloroquine or the proteosomal inhibitor MG132. Cells incubated with chloroquine showed levels of Fpn similar to non-silenced cells, whereas MG132-treated cells showed decreased amounts of Fpn (Figure 3B). These data indicate that once internalized, Fpn is degraded in the lysosome.
Figure 3.
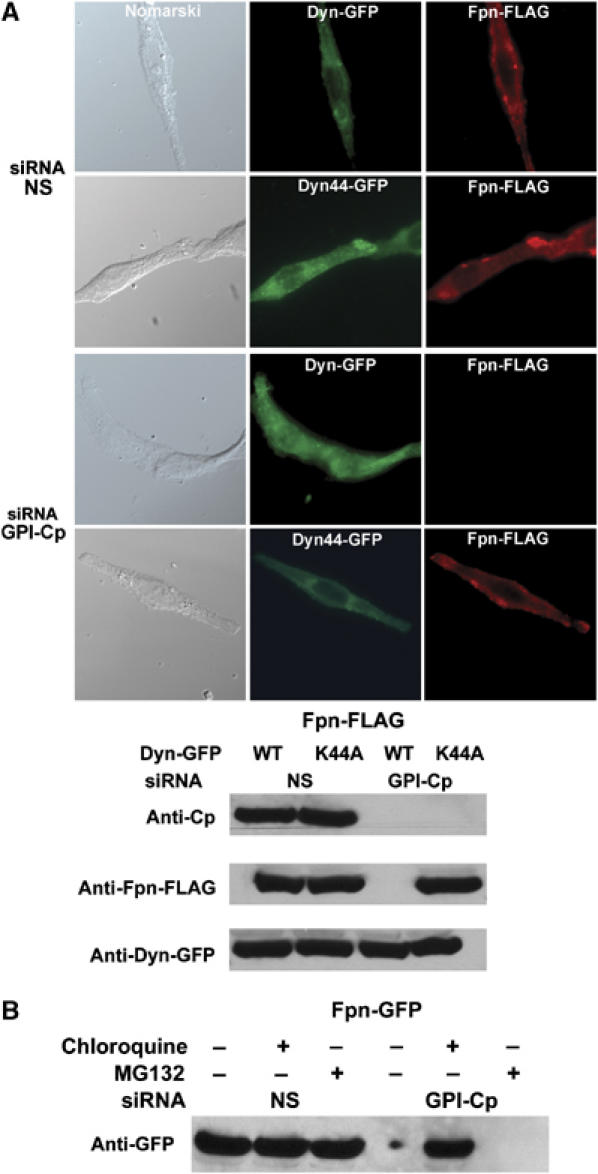
Cell surface Fpn is rapidly internalized in the absence of GPI-Cp. (A) Cells, silenced for GPI-Cp and transfected with a plasmid for Fpn-FLAG, were simultaneously transfected with a plasmid expressing either wild-type dynamin-GFP (Dyn-GFP) or a dominant-negative dynamin (Dyn44-GFP). Cells were examined for Fpn-FLAG (red) and dynamin (green) 24 h post-transfection. (B) Cells silenced for GPI-Cp were transfected with a plasmid expressing Fpn-GFP and 24 h post-transfection incubated with either chloroquine (100 μM) or MG132 (10 μM) for 6 h. Cells were extracted and Fpn-GFP was assayed by Western blot analysis.
Addition of multicopper oxidases to Cp-deficient cells restores cell surface Fpn
Iron transport can be promoted in astrocytes obtained from Cp-deficient mice by the addition of Cp to the medium (Jeong and David, 2003). Addition of exogenous Cp led to expression of Fpn-GFP on the surface of C6 cells that were silenced for GPI-Cp, suggesting that formation of an Fpn–Cp complex in the secretory apparatus is not required for Fpn to accumulate on the cell surface (Figure 4A). An active multicopper oxidase is required for Fpn to accumulate at the cell surface, as addition of human Cp that had been treated with azide to inactivate catalytic activity did not lead to Fpn accumulation (data not shown). Fet3p is a cell surface multicopper oxidase found in fungi, and there is little structural homology between Cp and Fet3p, apart from the amino acids that ligate copper (Askwith et al, 1994). A secreted form of Fet3p, injected into aceruloplasminemic mice, can induce tissue iron release (Harris et al, 2004). Similarly, addition of secreted Fet3p to C6 glioma cells silenced for GPI-Cp led to the accumulation of cell surface Fpn-GFP (Figure 3A).
Figure 4.
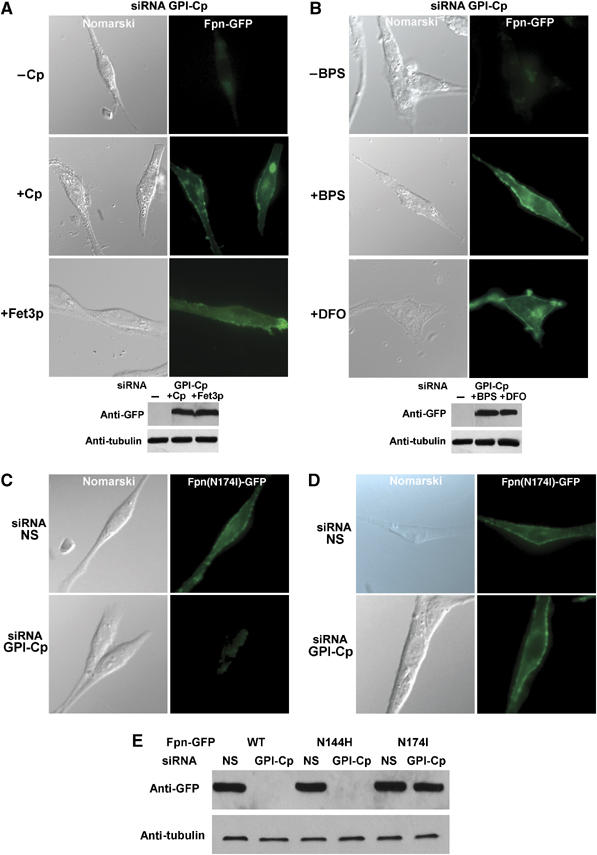
Reduction of extracellular Fe(II) pools inhibits the internalization of Fpn in GPI-Cp-silenced cells. (A) Cells silenced for GPI-Cp were transfected with an Fpn-GFP plasmid. Human secreted Cp (2.0 μM) or secreted Fet3p (2.0 μM) was added to the medium and the cells were examined after 24 h by epifluorescence microscopy and Western blot analysis. (B) Cells, silenced for GPI-Cp and transfected with a plasmid expressing Fpn-GFP, were incubated for 24 h with 600 μM BPS or 600 μM DFO-dextran and examined by fluorescence microscopy and Western blot. (C) Cells transfected with control or GPI-Cp-specific oligonucleotides were transfected with plasmids expressing either transport-competent Fpn(N144H)-GFP or (D) transport-incompetent Fpn(N174I)-GFP and examined by epifluorescence microscopy or (E) Western blot analysis.
Ferroxidases prevent Fpn internalization by lowering extracellular Fe(II)
We next determined how Cp stabilizes Fpn on the cell surface. Previous work showed that GPI-Cp and Fpn can be co-immunoprecipitated (Jeong and David, 2003), suggesting that binding of Cp to Fpn promotes Fpn stability on the cell surface. Co-immunoprecipitation experiments using C6 glioma cells, however, failed to show any evidence of a complex between transfected Fpn-GFP and either human secreted Cp or secreted Fet3p (data not shown). It is possible that multicopper oxidases, by promoting iron oxidation, increase iron transport activity and active transport of iron leads to Fpn stabilization on the cell surface. To determine if multicopper oxidases stabilize cell surface Fpn by reducing extracellular Fe(II) concentration, we incubated GPI-Cp-silenced C6 glioma cells with bathophenanthroline disulfonate (BPS), an impermeable Fe(II) chelator or desferrioxamine (DFO) conjugated to dextran to ensure that DFO is not permeable. While DFO is an Fe(III) chelator, its affinity for iron is so high (1033) that it alters the Fe(II)/Fe(III) equilibrium and will deplete free Fe(II). Addition of either chelator to GPI-Cp-silenced cells led to accumulation of Fpn-GFP on the cell surface (Figure 4B). This result suggests that the ability of multicopper oxidases to lower extracellular ferrous iron stabilizes Fpn on the cell surface.
Hepcidin, a peptide produced by the liver in response to inflammation and iron stores, regulates iron entry into plasma by binding to Fpn and effecting its internalization and degradation (Nemeth et al, 2004). Hepcidin binding to Fpn induces the phosphorylation of either of two adjacent tyrosine residues (Y302,Y303) (De Domenico et al, 2007). Mutation of both resides to phenylalanine prevents hepcidin-mediated internalization. Fpn(Y302-303F)-GFP, however, is rapidly internalized in GPI-Cp-silenced cells (data not shown). This result demonstrates that internalization in the absence of Cp is independent of hepcidin.
Internalization of Fpn in the absence of Cp leads to increased Fpn-associated iron
We identified two Fpn mutations that are not internalized when expressed in HEK293T cells exposed to hepcidin (De Domenico et al, 2005, 2006a, 2006b). Fpn mutant (N174I) is unable to export iron while Fpn mutant (N144H) is transport competent. Fpn(N144 H)-GFP expressed in GPI-Cp silenced C6 glioma cells was not found at the cell surface, whereas Fpn(N174I)-GFP accumulated at the cell surface (Figure 4C–E). Transport-incompetent Fpn(N174I)GFP is stable on the cell surface of GPI-Cp silenced cells while transport-competent Fpn-GFP is not stable. This result suggests that some aspect of the transport process, which is not completed in the absence of Cp activity, is responsible for the loss of Fpn.
We considered the possibility that Cp or an Fe chelator affects iron transport by promoting loss of iron from Fpn. We tested this hypothesis by incubating GPI-Cp-silenced C6 glioma cells with Tf(59Fe)2 and then examining immunoprecipitated Fpn for 59Fe. There was no detectable immunoprecipitation of 59Fe-Fpn from cells transfected with nonspecific oligonucleotide pools (Figure 5A, column 1). This is expected, as the retention time of 59Fe in transport-competent Fpn is likely to be low. There was little 59Fe associated with immunoprecipitated Fpn in GPI-Cp-silenced cells, as there is little Fpn (Figure 5A, column 3). There was, however, appreciable 59Fe-Fpn in GPI-Cp-silenced cells expressing dynamin(K44A)-GFP that traps Fpn on the cell surface (Figure 5A, column 4). No 59Fe was found to be associated with DMT1-GFP in GPI-Cp-silenced cells, indicating that the association of 59Fe with Fpn is not an artifact of immunoprecipitation (data not shown). Addition of Cp or impermeable iron chelators permitted Fpn to accumulate at the cell surface of GPI-Cp-silenced cells (Figure 5A, column 5). Similarly, addition of Cp to 59Fe-labeled GPI-Cp-silenced cells expressing dynamin(K44A)-GFP led to loss of Fpn-associated 59Fe. No 59Fe was found to be associated with transport-incompetent Fpn(174I)-GFP in GPI-Cp silenced cells (Figure 5B).
Figure 5.
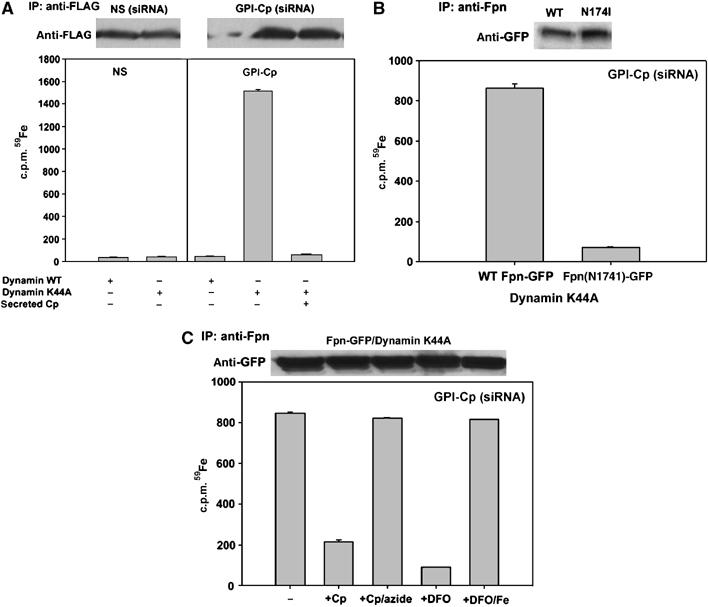
Iron is bound to Fpn in the absence of GPI-Cp. (A) Cells transfected with nonspecific (NS) or GPI-Cp-specific oligonucleotides were then transfected with Fpn-FLAG and dynamin-GFP or dynamin(K44A)-GFP. Twenty-four hours after transfection (72 h post siRNA), cells were incubated for 12 h with Tf (59Fe)2±2.0 μM Cp, washed and Fpn-FLAG was immunoprecipitated using anti-FLAG resin. Fpn-FLAG was eluted with 0.2 mg/ml FLAG peptide, the immunoprecipitates assessed by Western blot and the amount of Fpn bound 59Fe determined. (B) C6 glioma cells silenced for GPI-Cp were transfected with Fpn-GFP or Fpn(174)-GFP and dynamin(K44A)-GFP. Twenty-four hours post-transfection (72 h post GPI-Cp siRNA), cells were treated as in (A), samples immunoprecipitated with rabbit anti-Fpn antibody and protein A/G resin and the amount of immunoprecipitated 59Fe-Fpn determined. (C) Cell lysates from Fpn-GFP/dynamin(K44A)-GFP expressing cells were incubated with Cp, azide-treated Cp, DFO or iron-loaded DFO for 6 h at 4°C. Fpn was immunoprecipitated as in (B) and the amount of 59Fe associated with Fpn-GFP was determined.
We examined if Cp or DFO could promote the loss of iron from 59Fe-Fpn in cell extracts. Addition of Cp or DFO promoted loss of 59Fe from Fpn, but azide-inactivated Cp or iron saturated DFO did not effect the loss of 59Fe. These results suggest that the lack of Cp leads to the retention of iron on Fpn (Figure 5C). These data suggest that iron is not irreversibly bound to Fpn.
Internalization of Fpn in the absence of GPI-Cp results from increased ubiquitination
Transporters are often regulated by their substrates and there is a precedent for transport-incompetent metal transporters not being degraded (Hettema et al, 2004). Conformational changes that occur during transport may signal degradation by exposing buried residues, which may be recognized by a ubiquitin ligase. Addition of ubiquitin targets the protein for lysosomal degradation. We examined whether Fpn-FLAG was ubiquitinated in GPI-Cp-silenced cells expressing dynaminK44A-GFP. Little ubiquitin was found on Fpn-FLAG immunoprecipitated from cells that had been transfected with nonspecific oligonucleotide pools (Figure 6A). The amount of Fpn-FLAG was decreased in GPI-Cp-silenced cells and immunoprecipitated Fpn was ubiquitinated. Fpn was also ubiquitinated in GPI-Cp-silenced cells transfected with dynamin K44A-GFP, which traps Fpn at the cell surface. These results suggest that ubiquitination at the cell surface may lead to Fpn internalization. We previously identified lysine residue K253 in Fpn, which is ubiquitinated following hepcidin-mediated internalization (De Domenico et al, 2007). Mutation of lysine 253 to alanine resulted in an Fpn that was predominantly on the cell surface in GPI-Cp-silenced cells (Figure 6B). (Equivalent immunoprecipitation of Fpn-GFP represents saturation of antibody.) Western analysis confirmed that Fpn(K253A)-GFP was not ubiquitinated or degraded. We speculate that transport-dependent changes in Fpn conformation increase the probability that the K253 will be accessible to an ubiquitin ligase.
Figure 6.
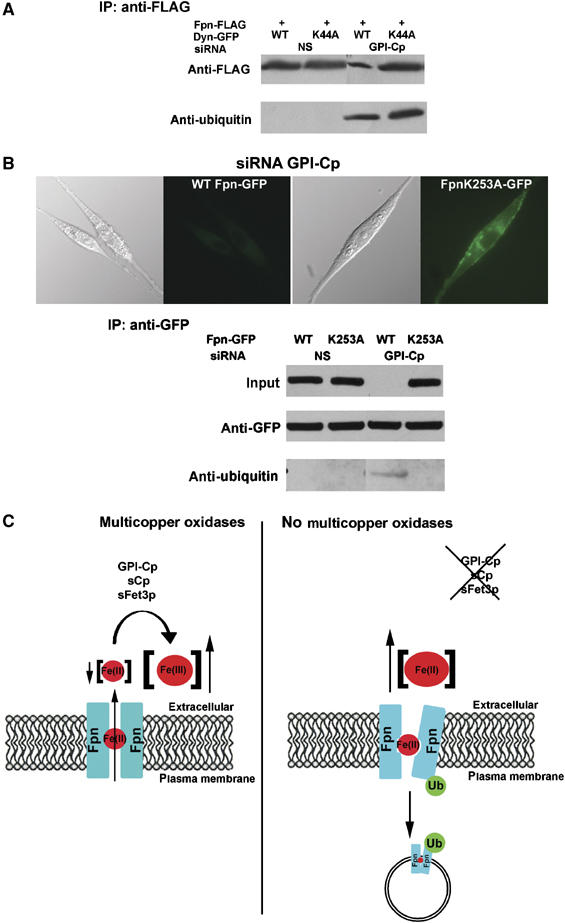
Cell surface Fpn is ubiquitinated in the absence of GI-Cp. (A) Cells transfected with nonspecific (NS) or GPI-Cp-specific oligonucleotides were then transfected with Fpn-FLAG and dynamin-GFP or dynamin(K44A)-GFP. Cells were extracted and immunoprecipitated Fpn-FLAG applied to SDS–PAGE and Western blots probed with either anti-FLAG or anti-ubiquitin antibody. (B) Cells silenced for GPI-Cp were transfected with Fpn-GFP or Fpn(K253A)-GFP. The localization of Fpn-GFP was examined by epifluorescence microscopy. Fpn-GFP was assayed in cell extracts by Western blot analysis and was immunoprecipitated, and the immunoprecipitates were examined for Fpn-GFP or Fpn(K253A)-GFP and the presence of ubiquitin was detected by Western blot. (C) Model for the role of Cp in the cell surface localization of Fpn. Fpn transports Fe(II) from the cytosol across the plasma membrane. Multicopper oxidases ensure low extracellular Fe(II) concentrations by catalyzing the oxidation of Fe(II) to Fe(III). In the absence of Cp, iron may remain in the Fpn channel, affecting Fpn conformation and resulting in Fpn ubiquitination, which leads to Fpn internalization and degradation.
Discussion
Fpn is the only identified iron transporter that exports iron from cells to plasma and as such the presence on cell surface Fpn is the critical feature in regulating plasma iron concentration. In most physiological conditions, the transport of iron by Fpn is regulated by hepcidin, a peptide synthesized by the liver in response to inflammation and hepatic iron load. Hepcidin binds to Fpn and does not inhibit transport activity directly, but rather induces the internalization of Fpn from cell surfaces (Nemeth et al, 2004). Once internalized, Fpn is degraded in the lysosome. The species of iron transported by Fpn is thought to be Fe(II), which is converted to Fe(III) by the multicopper oxidases, ceruloplasmin or hephaestin, which function as ferroxidases. The absence of either multicopper oxidase results in decreased plasma iron and increased iron retention in normally iron exporting cells. Mutations in hephaestin, as seen in the SLA mouse, result primarily in an iron-limited erythropoiesis (Vulpe et al, 1999). Mutations in ceruloplasmin lead to decreased plasma iron and increased iron retention in a variety of cell types (Harris et al, 1998). Patients with aceruloplasminemia present with adult-onset neurological disease, retinal degeneration and diabetes mellitus. Massive iron accumulation and extensive loss of neurons are observed in the basal ganglia. The mechanism leading to cellular iron overload has been unclear. Iron may be exported from cells as Fe(II), and a failure to convert that Fe(II) to Fe(III,) due to the lack of Cp loading iron on for transferring, may lead to Fe(II) being accumulated in cells by non-transferrin transport systems.
In the absence of transferrin, both humans (Hamill et al, 1991) and mice (Bernstein, 1987; Huggenvik et al, 1989) develop a severe anemia due to iron-limited erythropoiesis. Atransferrinemia also leads to severe iron overload in parenchymal tissues such as liver and pancreas. Yet, atransferrinemic humans or mice do not show evidence of neural or cognitive dysfunction as seen in patients with aceruloplasminemia. This observation suggests a function for Cp independent of its ability to load iron onto transferrin. Here, we demonstrate that Cp is responsible for generating an iron gradient, leading to the loss of Fpn-bound iron (Figure 6C). In the absence of Cp, iron bound to Fpn results in Fpn internalization. Thus, removal of Fpn-bound iron is an essential function of Cp, and by extension hephaestin. The conclusion that Cp lowers Fe(II) bound to Fpn is supported by the finding that impermeable iron chelators can lead to the cell surface expression of Fpn in the absence of Cp. This observation indicates that in addition to generating iron bound to transferrin, Cp is required to lower extracellular Fe(II) concentration. Reduced extracellular Fe(II) may create a gradient driving Fpn-mediated iron transport and in the process, remove iron bound to Fpn. In the absence of that gradient, iron bound to Fpn increases the probability that Fpn will be internalized and degraded.
Mutation of residue Fpn K253 prevents ubiquitination and internalization, maintaining Fpn on the cell surface in Cp-negative cells. We hypothesize that an E3-ubiquitin ligase recognizes a conformational state of Fpn resulting from the presence of bound iron. This conformation state might reflect an intermediate in the transport process (Figure 6C). As suggested by Pelham and co-workers, conformational changes in a transporter due to transporter activity might be expected to lead to perturbation of lipid bilayer, or exposure or normally lipid buried amino-acid residues in the aqueous cytosol (Hettema et al, 2004). These altered conformations might be recognized by a ubiquitin ligase, leading to decreased transporter degradation. Under normal transport conditions, the probability of that altered conformation being recognized by the ubiquitin ligase might be low, but would increase with extended transport activity. Continued transport activity would lead to a shorter transporter half-life. Studies with a number of yeast metal transporters have shown that increased transport leads to an increased rate of internalization and degradation (Gitan and Eide, 2000; Hettema et al, 2004; Felice et al, 2005). Internalization and degradation result from the addition of ubiquitin to the transporter. Mutations in the transporter that prevent substrate transport prevent both ubiquitination and degradation.
The requirement for an external ferroxidase to maintain surface Fpn is cell type dependent and appears specific to cells that express GPI-Cp. Transfected Fpn is stable in the absence of a ferroxidase in a variety of cultured cells (HeLa, HEK293, FMA3) that do not express GPI-Cp. Fpn is not internalized in the absence of Cp but iron transport activity is reduced. This observation suggests that even in these cell types the ferroxidase activity of Cp promotes iron transport. We speculate that the expression of the specific E3 ligase, which recognizes the altered Fpn conformation, is restricted to those cell types that express GPI-Cp. That there is a specific E3 ligase is suggested by the observation that hepcidin-induced Fpn internalization can occur in all (tested) cultured mammalian cells. Once internalized, Fpn is ubiquitinated on K253 and ubiquitination is required for degradation. Mutation of K253 does not prevent hepcidin-meditated internalization, but prevents the entry of internalized Fpn into the multivesicular body. This result implies that all cells are capable of ubiquitinating Fpn once it is internalized, but that only GPI-Cp producing cells are capable of recognizing the iron-bound form of cell surface Fpn.
The results reported here suggest that Fpn internalization may be regulated by factors other than hepcidin. Fpn levels in astrocytes and glioma cells are particularly affected by the loss of GPI-Cp and this may contribute to the excessive iron overload in the brain seen in aceruloplasminemia. In the absence of endogenous GPI-Cp, external ferroxidase(s) can promote retention of Fpn on cell surface, leading to cellular iron export. This observation suggests that iron chelation therapy for aceruloplasminemia may decrease cellular iron burden by increasing the expression of Fpn.
Materials and methods
Cells and media
Rat C6 glioma cells were maintained in Dulbecco's minimal essential media (DMEM) with 10% fetal bovine serum. Mouse bone marrow macrophages, isolated from mouse femurs, were grown in RPMI 1640 with 20% equine serum for 6 days and adherent cells were further cultured in RPMI 1640 with 20% fetal bovine serum and 30% L cell-conditioned medium. Cells were iron loaded by addition of FAC (10 μM iron) for 18 h.
Transient transfection
All mouse Fpn-GFP constructs (wild type or mutant) or Fpn-FLAG were generated as previously described (De Domenico et al, 2005), and pDynamin-EGFP, pDynaminK44A-EGFP (a generous gift from Dr Matt Mulvey), pDMT1-EGFP-C1 (a generous gift from Dr Hiroshi Gunshin) or pCMV-Tag4b-FLAG-HsecretedCp were transfected using Nucleofector technology (Amaxa, Gaithersburg, MD), according to the manufacturer's instructions.
siRNA transfection
siRNA oligonucleotide pools, nonspecific and rat GPI-Cp specific, were obtained from Dharmacon. C6 cells were transfected using Oligofectamine reagent (Invitrogen), with siRNAs at a final concentration of 100 nM. Eighteen hours post-transfection, cells were trypsinized and plated onto 60-mm plates. Eighteen hours after plating, cells were transfected with pFpn-EGFP-N1, pCMV-Fpn-FLAG, pDynamin-EGFP, pDynaminK44A-EGFP, pDMT1-EGFP-C1 or pCMV-Tag4b-FLAG-HsecretedCp, using the Nucleofector. Cells were grown for 18–24 h and then processed for immunofluorescence microscopy, Western blot analysis or incubated with FAC for 18 h before determining ferritin levels.
Other procedures
Cells were solubilized in 1.0% Triton X-100, 150 mM NaCl, 10 mM EDTA and 10 mM Tris, pH 7.4 with a protease inhibitor cocktail (Roche). Fpn-GFP was immunoprecipitated using rabbit anti-GFP (1:1000, Abcam) and protein A/G Agarose at 4°C overnight. Fpn-FLAG was immunoprecipitated using FLAG beads (Sigma) at 4°C overnight. Total protein concentrations were determined using BCA reagent (Pierce). Protein samples were separated on 4–20% gels (Tris–Glycine) (BioRad) and transferred in Hybond-ECL (Amersham Biosciences). Western analysis was performed using goat anti-Cp antibody (1:1000, Sigma), goat anti-actin (1:1000, Santa Cruz), rabbit anti-GFP (1:10 000, Abcam #ab6556), goat anti-tubulin (1:1000, Gene Tex) or mouse anti-ubiquitin (1:1000, Covance), followed by treatment with peroxidase-conjugated goat anti-rabbit IgG (1:10 000, Jackson ImmunoResearch Labs) and peroxidase-conjugated goat anti-mouse IgG (1:10 000, Jackson ImmunoResearch Labs), or peroxidase-conjugated donkey anti-goat IgG (1:5000, Santa Cruz Biotechnology). Immunofluorescence was performed as described previously (De Domenico et al, 2005), in non-permeabilized cells, using either donkey anti-Cp antibody (1:100, Abcam) or mouse anti-FLAG antibody (1:750, Sigma), followed by treatment with Alexa 594-conjugated goat anti-donkey IgG (1:750, Molecular Probes) or Alexa 594-conjugated goat anti-mouse IgG (1:750, Molecular Probes) as the secondary antibody. Astrocyte immunofluorescence was performed using astrocytes purified from dissociated cell cultures of the neonatal mouse cerebral cortex from Cp−/− and Cp+/+ mice, as described previously (Jeong and David, 2003). Cells were plated on poly-L-lysine-coated round glass coverslips and cultured in DMEM containing 10% fetal bovine serum. Cells were washed in Hank's balanced salt solution (HBSS) and live cultures incubated with anti-FPN1 polyclonal antibody (1:100, Alpha Diagnostics) for 30 min at room temperature. Cells were washed, and then incubated with Alexa-Fluor 488-conjugated goat anti-rabbit IgG (1:200, Molecular Probes) for 30 min. Cells were fixed and permeabilized in acetic acid/ethanol (5:95 (v/v)) at −20°C for 20 min. Cells were then incubated with a monoclonal anti-glial fibrillary acidic protein (GFAP, an astrocyte marker, 1:100, Zymed Laboratories) for 30 min at room temperature and visualized with a rhodamine-conjugated goat anti-rat IgG (1:200, Jackson Immunoresearch). Nuclei were labeled with 100 ng/ml 4′-6-diamidino-2-phenylindole (DAPI, Vector Laboratories) and viewed with a Zeiss Axioskop 2 plus microscope. MG132 and chloroquine (Sigma) were used as described previously (De Domenico et al, 2006a, 2006b). DFO-dextran was a generous gift from Dr Bo Hedland (Biomedical Frontiers). Ferritin analysis was performed as described (De Domenico et al, 2005). Human apo-Tf was iron loaded as described previously (Nemeth et al, 2004). Human ceruloplasmin (Sigma) was inactivated by incubation with 100 μM azide and then extensively dialyzed against PBS before use.
Supplementary Material
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Figure 4
Legend to Supplementary Figures
Acknowledgments
We thank Dr JP Kushner for help in preparing the manuscript and Dr J Gitlin for useful conversations. This work was supported by a grant from the NIH DK079047 to JK and a grant from the Canadian Institutes of Health Research to SD and a Center of Excellence in Hematology Award (NIH CA43014). The financial support of Telethon-Italy GGP06173 to GM is gratefully acknowledged.
References
- Askwith C, Eide D, Van Ho A, Bernard PS, Li L, Davis-Kaplan S, Sipe DM, Kaplan J (1994) The FET3 gene of S. cerevisiae encodes a multicopper oxidase required for ferrous iron uptake. Cell 76: 403–410 [DOI] [PubMed] [Google Scholar]
- Bernstein SE (1987) Hereditary hypotransferrinemia with hemosiderosis, a murine disorder resembling human atransferrinemia. J Lab Clin Med 110: 690–705 [PubMed] [Google Scholar]
- Damke H, Baba T, Warnock DE, Schmid SL (1994) Induction of mutant dynamin specifically blocks endocytic coated vesicle formation. J Cell Biol 127: 915–934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Domenico I, McVey Ward D, Nemeth E, Ganz T, Corradini E, Ferrara F, Musci G, Pietrangelo A, Kaplan J (2006a) Molecular and clinical correlates in iron overload associated with mutations in ferroportin. Haematologica 91: 1092–1095 [PMC free article] [PubMed] [Google Scholar]
- De Domenico I, Vaughn MB, Li L, Bagley D, Musci G, Ward DM, Kaplan J (2006b) Ferroportin-mediated mobilization of ferritin iron precedes ferritin degradation by the proteasome. EMBO J 25: 5396–5404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Domenico I, Ward DM, Nemeth E, Vaughn MB, Musci G, Ganz T, Kaplan J (2005) The molecular basis of ferroportin-linked hemochromatosis. Proc Natl Acad Sci USA 102: 8955–8960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Domenico I, Ward DM, Langelier D, Vaughn MB, Nemeth E, Sundquist WI, Ganz T, Musci G, Kaplan J (2007) The molecular mechanism of hepcidin-mediated ferroportin downregulation. Mol Biol Cell, in press [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felice MR, De Domenico I, Li L, Ward DM, Bartok B, Musci G, Kaplan J (2005) Post-transcriptional regulation of the yeast high affinity iron transport system. J Biol Chem 280: 22181–22190 [DOI] [PubMed] [Google Scholar]
- Ganz T, Nemeth E (2006) Iron imports. IV. Hepcidin and regulation of body iron metabolism. Am J Physiol Gastrointest Liver Physiol 290: G199–G203 [DOI] [PubMed] [Google Scholar]
- Gitan RS, Eide DJ (2000) Zinc-regulated ubiquitin conjugation signals endocytosis of the yeast ZRT1 zinc transporter. Biochem J 346 (Part 2): 329–336 [PMC free article] [PubMed] [Google Scholar]
- Hamill RL, Woods JC, Cook BA (1991) Congenital atransferrinemia. A case report and review of the literature. Am J Clin Pathol 96: 215–218 [DOI] [PubMed] [Google Scholar]
- Harris ZL (2003) Aceruloplasminemia. J Neurol Sci 207: 108–109 [DOI] [PubMed] [Google Scholar]
- Harris ZL, Davis-Kaplan SR, Gitlin JD, Kaplan J (2004) A fungal multicopper oxidase restores iron homeostasis in aceruloplasminemia. Blood 103: 4672–4673 [DOI] [PubMed] [Google Scholar]
- Harris ZL, Klomp LW, Gitlin JD (1998) Aceruloplasminemia: an inherited neurodegenerative disease with impairment of iron homeostasis. Am J Clin Nutr 67: 972S–977S [DOI] [PubMed] [Google Scholar]
- Hettema EH, Valdez-Taubas J, Pelham HR (2004) Bsd2 binds the ubiquitin ligase Rsp5 and mediates the ubiquitination of transmembrane proteins. EMBO J 23: 1279–1288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huggenvik JI, Craven CM, Idzerda RL, Bernstein S, Kaplan J, McKnight GS (1989) A splicing defect in the mouse transferrin gene leads to congenital atransferrinemia. Blood 74: 482–486 [PubMed] [Google Scholar]
- Jeong SY, David S (2003) Glycosylphosphatidylinositol-anchored ceruloplasmin is required for iron efflux from cells in the central nervous system. J Biol Chem 278: 27144–27148 [DOI] [PubMed] [Google Scholar]
- Nemeth E, Tuttle MS, Powelson J, Vaughn MB, Donovan A, Ward DM, Ganz T, Kaplan J (2004) Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science 306: 2090–2093 [DOI] [PubMed] [Google Scholar]
- Patel BN, David S (1997) A novel glycosylphosphatidylinositol-anchored form of ceruloplasmin is expressed by mammalian astrocytes. J Biol Chem 272: 20185–20190 [DOI] [PubMed] [Google Scholar]
- Pietrangelo A (2006) Hereditary hemochromatosis. Biochim Biophys Acta 1763: 700–710 [DOI] [PubMed] [Google Scholar]
- Shim H, Harris ZL (2003) Genetic defects in copper metabolism. J Nutr 133: 1527S–1531S [DOI] [PubMed] [Google Scholar]
- Van Ho A, Ward DM, Kaplan J (2002) Transition metal transport in yeast. Annu Rev Microbiol 56: 237–261 [DOI] [PubMed] [Google Scholar]
- Vulpe CD, Kuo YM, Murphy TL, Cowley L, Askwith C, Libina N, Gitschier J, Anderson GJ (1999) Hephaestin, a ceruloplasmin homologue implicated in intestinal iron transport, is defective in the sla mouse. Nat Genet 21: 195–199 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Figure 4
Legend to Supplementary Figures