

Abstract
The HIV-1 central DNA Flap acts as a cis-acting determinant of HIV-1 genome nuclear import. Indeed, DNA-Flap re-insertion within lentiviral-derived gene transfer vectors strongly stimulates gene transfer efficiencies. In this study, we sought to understand the mechanisms by which the central DNA Flap mediates HIV-1 nuclear import. Here, we show that reverse transcription (RT°) occurs within an intact capsid (CA) shell, independently of the routing process towards the nuclear membrane, and that uncoating is not an immediate post-fusion event, but rather occurs at the nuclear pore upon RT° completion. We provide the first observation with ultrastructural resolution of intact intracellular HIV-1 CA shells by scanning electron microscopy. In the absence of central DNA Flap formation, uncoating is impaired and linear DNA remains trapped within an integral CA shell precluding translocation through the nuclear pore. These data show that DNA Flap formation, the very last event of HIV-1 RT°, acts as a viral promoting element for the uncoating of HIV-1 at the nuclear pore.
Keywords: central DNA Flap, HIV-1, nuclear import, uncoating
Introduction
Lentiviruses have the unique ability among retroviruses to integrate and replicate efficiently in nondividing target cells (Gartner et al, 1986; Weinberg et al, 1991). The active nuclear import of their genome through the nuclear membrane is the key to mitosis-independent lentiviral replication.
We have previously shown (Charneau and Clavel, 1991; Charneau et al, 1992, 1994) that HIV-1 and other lentiviruses have a more complex reverse transcription (RT°) strategy than oncoviruses whereby the presence of two additional cis-acting sequences within the lentiviral genome, the central polypurine tract (cPPT) and the central termination sequence (CTS), leads to the formation of a three-stranded DNA structure, the central DNA Flap. Mutations within the cPPT lead to a linear genome lacking the central DNA Flap, and severely impair viral replication (Zennou et al, 2000; Arhel et al, 2006a). Subcellular fractionation together with localization of viral DNA by fluorescence in situ hybridization (FISH) showed that central DNA Flap-defective molecules dock with wild-type kinetics at the nuclear membrane (Zennou et al, 2000). This docking, which involves microtubule- followed by actin-directed movements (Arhel et al, 2006b), is a very rapid process, as the vast majority of the linear DNA is already associated with the nuclear fraction within 6 h post-infection (p.i) or before (Barbosa et al, 1994; Zennou et al, 2000), as is confirmed by real-time imaging of HIV-1 complexes in the cytoplasm of infected cells (McDonald et al, 2002; Arhel et al, 2006b). However, unlike wild-type viral DNA, Flap-defective linear DNA then accumulates at close proximity of the nuclear membrane, indicating a late defect in nuclear import (Zennou et al, 2000). The central DNA Flap therefore acts as a cis-determinant of HIV-1 genome nuclear import, and thus accounting, in part, for the mitosis-independent replication of lentiviruses. More precisely, studies of viral genome localization by in situ hybridization with electron microscopy indicated that this nuclear import defect occurs immediately before viral genome translocation through the nuclear pore and that most Flap defective DNA molecules have not initiated translocation through the nuclear pore (Arhel et al, 2006c).
Consistently with the cis-acting role of the central DNA Flap, its reinsertion in HIV-1-derived gene transfer vectors complements the level of nuclear import from a strong defect to wild-type nuclear import levels, quantitatively indistinguishable from wild-type virus (Zennou et al, 2000). As a result, DNA Flap-containing lentiviral vectors adhere closely to the early steps of wild-type virus infection. Reinsertion of the DNA Flap in HIV-1 vectors strongly stimulates gene transfer efficiencies both in vivo and ex vivo in all tissue and cell types examined (see references in Arhel et al, 2006a), thus making the DNA Flap an essential and widely used component of lentiviral gene transfer vectors. It has been suggested that the role of the central DNA Flap, whereas beneficial in the context of HIV-1-derived vectors, could be nonessential for viral replication in the context of full-length infectious viral genomes (Dvorin et al, 2002; Limon et al, 2002; Marsden and Zack, 2007). However, although mutations in the central DNA Flap do not lead to totally noninfectious viruses, thus pointing to a small fraction of nuclear import that is independent of the DNA Flap, we have recently shown that cPPT mutant viruses show an important nuclear import defect irrespective of the viral strain, cell type or infectivity assay used (Arhel et al, 2006a).
A three-stranded DNA structure acting as a cis-determinant of nuclear import is a novel and intriguing biological phenomenon with no known cellular or viral counterparts. In this work, we unmask the involvement of the central DNA Flap in an unexpected step of the HIV-1 replication cycle: the maturation at the nuclear membrane of RT complexes (RTCs), consisting in a structure proceeding directly from the core of the particle, into pre-integration complexes (PICs) of a size compatible with translocation through the nuclear pore. We further demonstrate that the routing of viral complexes to the nuclear membrane is independent of viral DNA synthesis. This work also revisits the identity of active intracellular HIV-1 replication complexes: direct observation by scanning electron microscopy (SEM) indicates that uncoating, also called decapsidation, does not occur as an immediate post-fusion event, but rather on the cytoplasmic side of the nuclear pore. Maturation of RTC into PIC is impaired in the absence of DNA Flap formation, therefore leading to the trapping of HIV-1 PIC within an integral capsid (CA) shell prohibiting nuclear entry of the HIV-1 genome.
Results
Lack of the central DNA Flap leads to stable perinuclear accumulation of CA proteins
Formation of the central DNA Flap and its role in HIV-1 genome nuclear import constitute a novel and intriguing mechanism. As a result, we can only formulate speculative hypotheses to account for the mechanism implicating the central DNA Flap in HIV-1 genome nuclear import. However, we already know from in vitro work that the central strand displacement leading to the formation of the central DNA Flap requires a high stoichiometry of HIV-1 reverse transcriptase enzyme relative to the viral genome (Charneau et al, 1994), to counteract the tendency of HIV-1 reverse transcriptase to dissociate from its template (Klarmann et al, 1993). It is therefore reasonable to assume that the central strand displacement, which is the last event in the RT° process, occurs within a CA shell that maintains this necessary high stoichiometry of reverse transcriptase enzymes. In fact, HIV-1 capsid size exceeds the maximum diameter of the nuclear pore, implying the requirement for a maturation step before translocation. Concordantly, HIV-1 CA mutants that failed to completely uncoat had impaired nuclear import levels (Dismuke and Aiken, 2006). As we previously hypothesized (Zennou et al, 2000), DNA Flap formation could signal the end of the RT° process and trigger the maturation of RTC into PIC lacking the CA shell. This maturation step was already hinted at by previous work showing a substantial difference in mass between cytoplasmic and nuclear HIV-1 complexes (Karageorgos et al, 1993). According to this hypothesis, the absence of the central DNA Flap would imply that viral DNA remains trapped within an integral CA core, too large to be translocated through the nuclear pore, resulting in an accumulation of CA proteins at the cytoplasmic side of the nuclear membrane.
In a first attempt to test this hypothesis, we examined the intracellular outcome of CA proteins by immunofluorescence confocal microscopy. P4 cells, HeLa-derived HIV-1 infection indicator cells (Charneau et al, 1994, AIDS Reagent Program), were transduced with HIV-1 vectors that include or not the central DNA Flap, fixed at given time points p.i (30 min, 6, 12, 24, 48, and 72 h), and labeled with anti-p24(CA) monoclonal antibodies. CA staining revealed a punctate pattern (Figure 1A) as described previously (Maréchal et al, 2001; McDonald et al, 2002). We found that, at early time points, the CA signal shifts from the plasma membrane to the nuclear membrane where it forms a perinuclear ring as early as 2 h p.i (data not shown and McDonald et al, 2002) and up to 24 h p.i, irrespective of the presence or not of the central DNA Flap (Figure 1A). After this, p24(CA) signal is progressively lost between 24 and 48 h p.i in all cells transduced with vector, including the central DNA Flap (Figure 1A), conceivably as the CA shells disassemble as a necessary event before viral DNA translocation through the nuclear pore. However, in cells transduced with vector lacking the central DNA Flap, the CA signal persists as a perinuclear ring even up to 72 h p.i (Figure 1A). This persistence of CA signal at late time points p.i is reminiscent of the previously reported persistence of DNA Flap-defective genomes at the cytoplasmic face of the nuclear membrane seen by in situ hybridization with electron microscopy (Arhel et al, 2006c).
Figure 1.
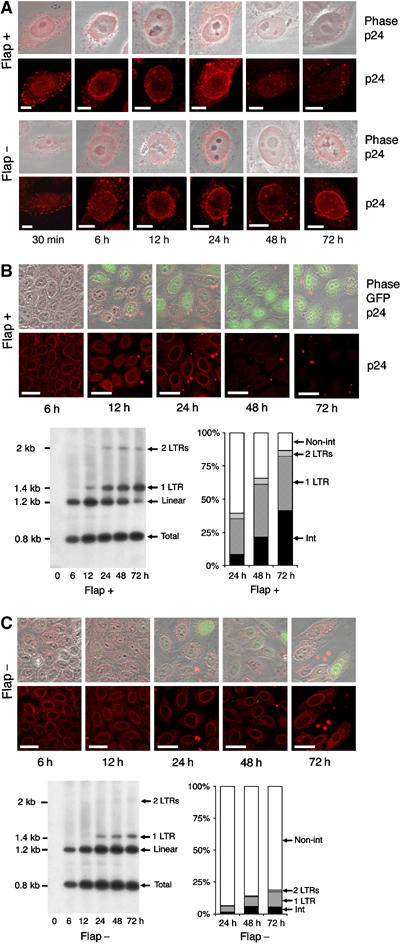
Disappearance of CA proteins at the nuclear membrane correlates in time with nuclear entry of linear vector DNA. (A) Fluorescence confocal microscopy images of incoming CA proteins with time following transduction. P4 cells were transduced with TRIP Flap+ vector, HR Flap− vector. Lower panels show p24 staining and upper panels merge images of p24 staining with phase contrast. The thickness of confocal images is 0.8 μm. Scale bars=3 μm. (B and C) Correlated Southern blotting and immunofluorescence studies. Bottom left-hand panels show intracellular vector DNA profiles following transduction by Southern blot analysis using a vector-specific probe (Zennou et al, 2000). P4 cells were transduced with equal amounts of TRIP-GFP (B) or HR-GFP (C) vector normalized on p24 contents of the supernatants. Bottom right-hand panels show the phosphorimager quantification of intracellular vector DNA forms expressed as percentage of total vector DNA (Zennou et al, 2000). Upper images are the immunofluorescence images (merge of 4 confocal slices) obtained in parallel of the Southern analysis, showing p24 staining and a merge of p24 staining, phase contrast, and GFP expression. Scale bars=10 μm.
Disappearance of HIV-1 CA perinuclear signal correlates with nuclear entry of linear DNA
We hypothesized that the disappearance of the CA perinuclear signal following infection with wild-type virus likely corresponds to the dissociation of CA proteins and uncoating of viral complexes at the nuclear membrane, and that this uncoating event necessarily precedes translocation of the viral genome through the nuclear pore. To test this hypothesis, we sought to determine how the kinetics of loss of CA signal correlate with the nuclear entry of vector DNA in transduced cells. We used a Southern blot and phosphorimaging technique already described (Zennou et al, 2000) in which restriction and probe design allows for precise quantification of each of the viral or vector DNA forms present in infected or transduced cells. Once reverse transcribed in the cytoplasm, viral or vector DNA is imported into the nucleus, where it either integrates or circularizes as one- or two-long terminal repeat (LTR) circles. We have previously shown that after completion of a single cycle of infection, more than 50% of viral DNA from wild-type HIV-1 is processed into integrated provirus, with most of the remaining being nuclear localized circular forms. On the other hand, as expected for a defect in nuclear import, 90% of viral DNA from DNA Flap mutant viruses accumulates as linear nontranslocated DNA molecules (Zennou et al, 2000).
We carried out a precise kinetic analysis of vector DNA processing in parallel with p24(CA) detection by immunofluorescence. We found that total vector DNA accumulates until 12 h p.i and then remains constant irrespective of the presence or not of the central DNA Flap (Figure 1B and C). This indicates that, whereas DNA synthesis begins asynchronously following fusion, with full-length linear DNA detected as early as 4 h p.i (Barbosa et al, 1994; Kim et al, 1989), RT° is only fully completed by 12 h. It also indicates that, contrary to a previous report (Ao et al, 2004), intracellular viral DNA is stable irrespective of the presence or not of the central DNA Flap.
In the case of vector DNA containing the central DNA Flap, linear DNA then slowly disappears between 12 and 72 h, with a concomitant increase in nuclear DNA forms, which represent 39, 66, and 86% of total intracellular vector DNA forms at 24, 48, and 72 h p.i, respectively (Figure 1B). These kinetics of vector DNA nuclear entry closely follow those of disappearance of perinuclear CA signal. In contrast, in the case of vector lacking the central DNA Flap, high levels of linear unintegrated DNA persist up to 72 h p.i, with nuclear vector DNA representing as little as 7, 14, and 19% of total intracellular forms at 24, 48, and 72 h p.i respectively, indicating a clear defect in nuclear import (Figure 1C). The persistence of Flap-defective linear DNA correlates with the stable accumulation of perinuclear CA signal.
Taken together, these results indicate that nuclear import of HIV-1 DNA is closely linked in time with the disappearance of perinuclear CA signal, which itself could be a consequence of an uncoating event immediately before translocation.
Routing of HIV-1 complexes is independent of the reverse transcription process
In a parallel experiment, cells were transduced with a HIV-1 vector including the central DNA Flap in the presence of nevirapine, a non-nucleosidic HIV-1 reverse transcriptase inhibitor, inhibiting the RT° process and thus the formation of the central DNA Flap. Interestingly, this resulted in a stable perinuclear accumulation of CA proteins (Figure 2), quantitatively similar to that obtained after transduction with a vector lacking the central DNA Flap (Figure 1C). These data established that routing of HIV-1 RTCs to the nuclear membrane and viral DNA synthesis are two independent processes. This implies that, contrary to a previous report (Bukrinskaya et al, 1998), the establishment of efficient HIV-1 RT° does not implicate the cytoskeleton.
Figure 2.
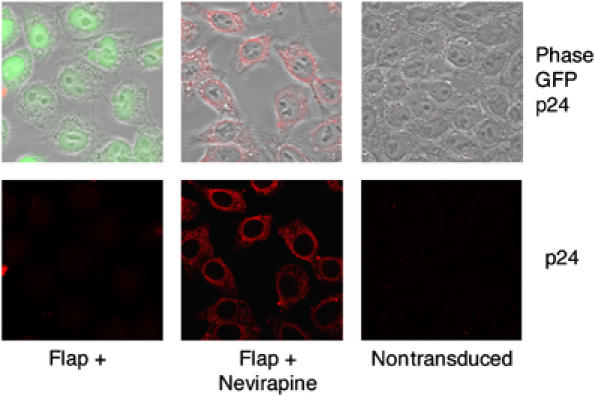
Inhibition of HIV-1 RT° does not perturb routing to the nuclear membrane but results in stable perinuclear accumulation of viral CA. P4 cells were transduced with TRIP-GFP Flap+ vector in the presence or absence of 1 μM nevirapine, or were left nontransduced as control. Immunofluorescence images are a merge of four confocal stacks showing p24(CA) staining at 48 h p.i. Scale bar=5 μm.
The perinuclear CA signal corresponds to intact capsids at close proximity of nuclear pores
To determine the precise nature of the complex corresponding to the perinuclear p24(CA) signal, we used transmission electron microscopy (TEM) to observe cells transduced or infected with DNA Flap-defective vectors or viruses. Having previously established that there is a lack of efficient translocation into the nuclear compartment in the absence of the central DNA Flap (Arhel et al, 2006c), but that viral DNA is systematically associated with the nuclear fraction in cell fractionation experiments (Zennou et al, 2000), we deduced that viral complexes were likely stably docked at the nuclear membrane. In fact, p24(CA) staining revealed some measure of colocalization with nuclear pore complexes (NPC), indicating that CA staining corresponds to NPC-associated viral material (Figure 3A). At late time points p.i, in cells infected with DNA Flap-defective viruses, but not with wild-type virus, we found structures resembling both in shape and size, as described previously in vitro (Benjamin et al, 2005; Briggs et al, 2006), HIV-1 CA shells on the outer nuclear membrane and at close proximity to nuclear pores (Figure 3B). The viral nature of these structures was confirmed by pre-embedding p24(CA) immunogold labeling, which revealed the presence at the nuclear membrane of viral capsids labeled with gold particles (Figure 3B).
Figure 3.
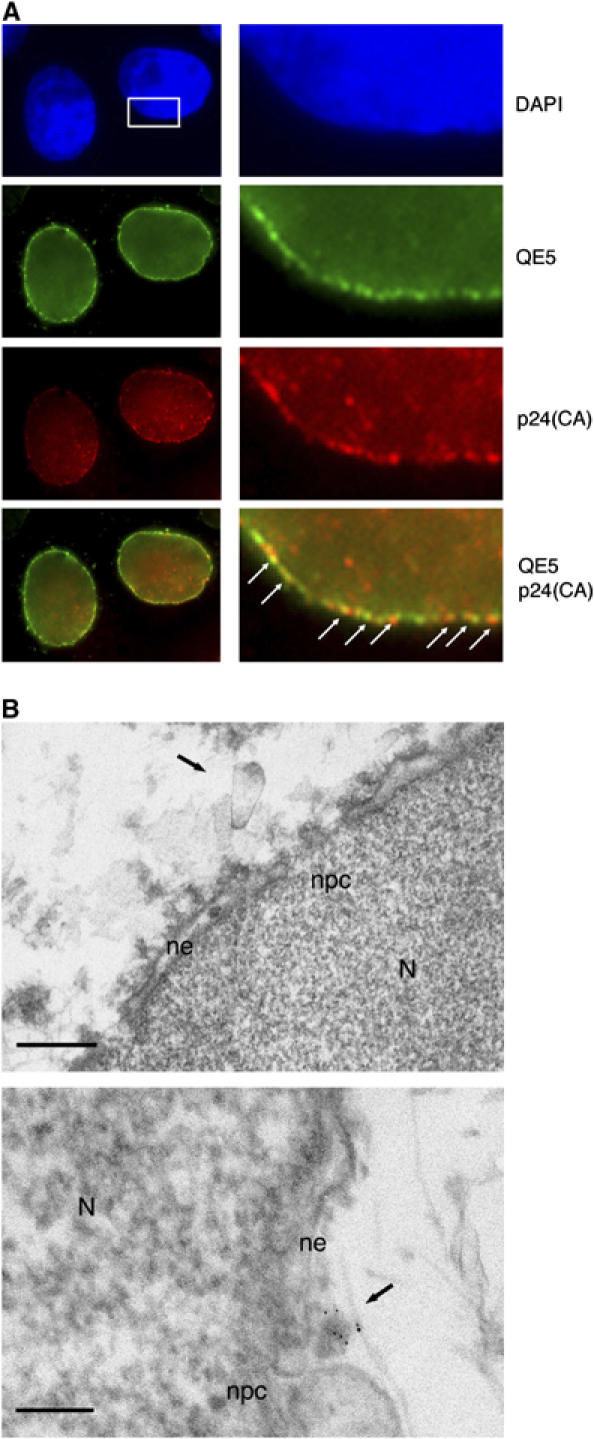
Detection by TEM of integral HIV-1 capsids at the cytoplasmic face of the nuclear pore. (A) Fluorescence confocal images of P4 cells 72 h p.i with an HR-Luc vector. Nuclei are visualized with DAPI staining. Nuclear pores are labeled with mouse monoclonal QE5 antibody (kind gift from Dr U Aebi), which recognizes CAN/Nup214, p62, and p153. CA is labeled using a polyclonal anti-p24 antibody. Right-hand panels are greater magnifications of the boxed area. Arrows point to points of colocalization between p24(CA) and NPCs. Scale bars=3 μm (left-hand panels) and 500 nm (right-hand panels). (B) MT4 cells were infected with cPPT-225T virus and nuclei were isolated 48 h p.i. Upper panel: ultrastructure image showing a capsid-like structure at close proximity of the nuclear membrane and two nuclear pores. Lower panel: immunogold staining with anti-p24(CA) antibodies (gold beads=1 nm) confirms the viral nature of the capsid. N, nucleus; ne, nuclear envelope; npc, nuclear pore complex. Scale bars=100 nm.
As the visualization of intracellular capsids within 60 nm thick TEM slices represents an extremely rare event (no more than 10 observations by ultrastructural analysis), we set out to observe the nuclear surface of transduced cells by SEM. Whole cells, transduced, infected or control, were extracted in situ as described previously (Allen et al, 1998) to expose the outer cytoplasmic face of the nuclear membrane thus rendering it accessible for surface imaging (Figure 4A). This preparation technique fully exposes NPCs, occasionally revealing typical eightfold radial symmetry (Supplementary Figure 1), as well as elements of the cytoskeleton and occasional remnants of the endoplasmic reticulum. In nontransduced control cells, we did not observe any capsid-like structures, nor was there any specific labeling using an anti-p24(CA) antibody; the background level of nonspecific gold labeling was under 1 gold particle/μm2 of nuclear surface (Figure 4B). In contrast, in cells transduced or infected with DNA Flap-defective particles, we readily observed heavily p24(CA)-labeled viral capsids (Figure 4C–F and H), which were clearly distinct from background nonspecific gold labeling (Figure 4B). Viral capsids were typically covered with at least a dozen gold particles which, considering the steric hindrance induced by their large size (10 nm), constitutes high labeling density. Of note, the binding of these 10 nm beads onto 100–125 nm conical capsids occasionally led to loss of typical morphology, which became instead ‘raspberry-like'.
Figure 4.
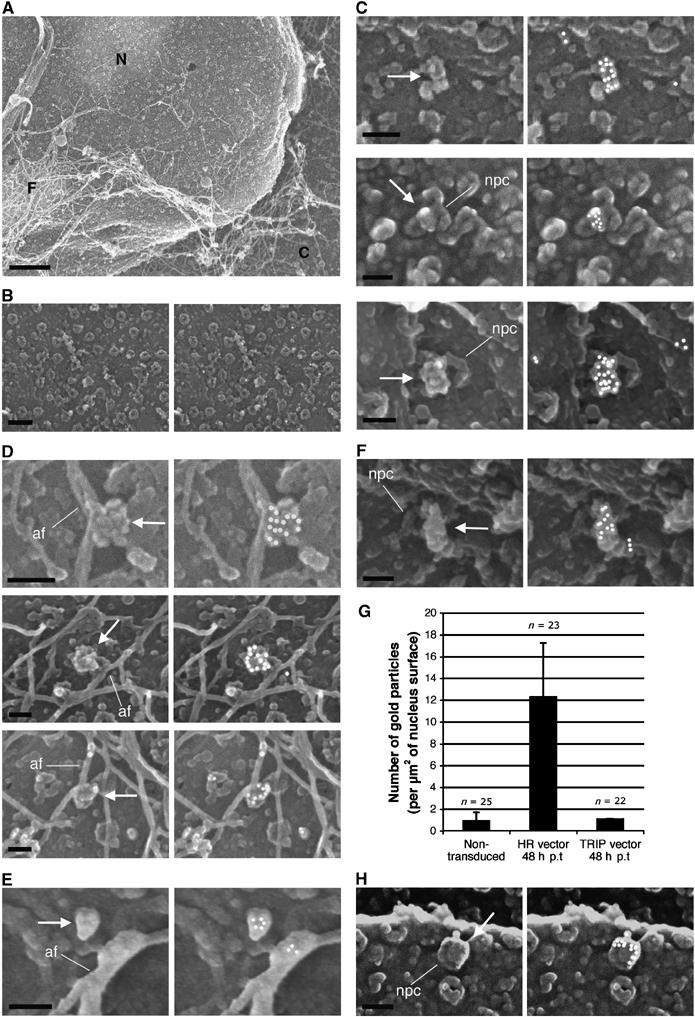
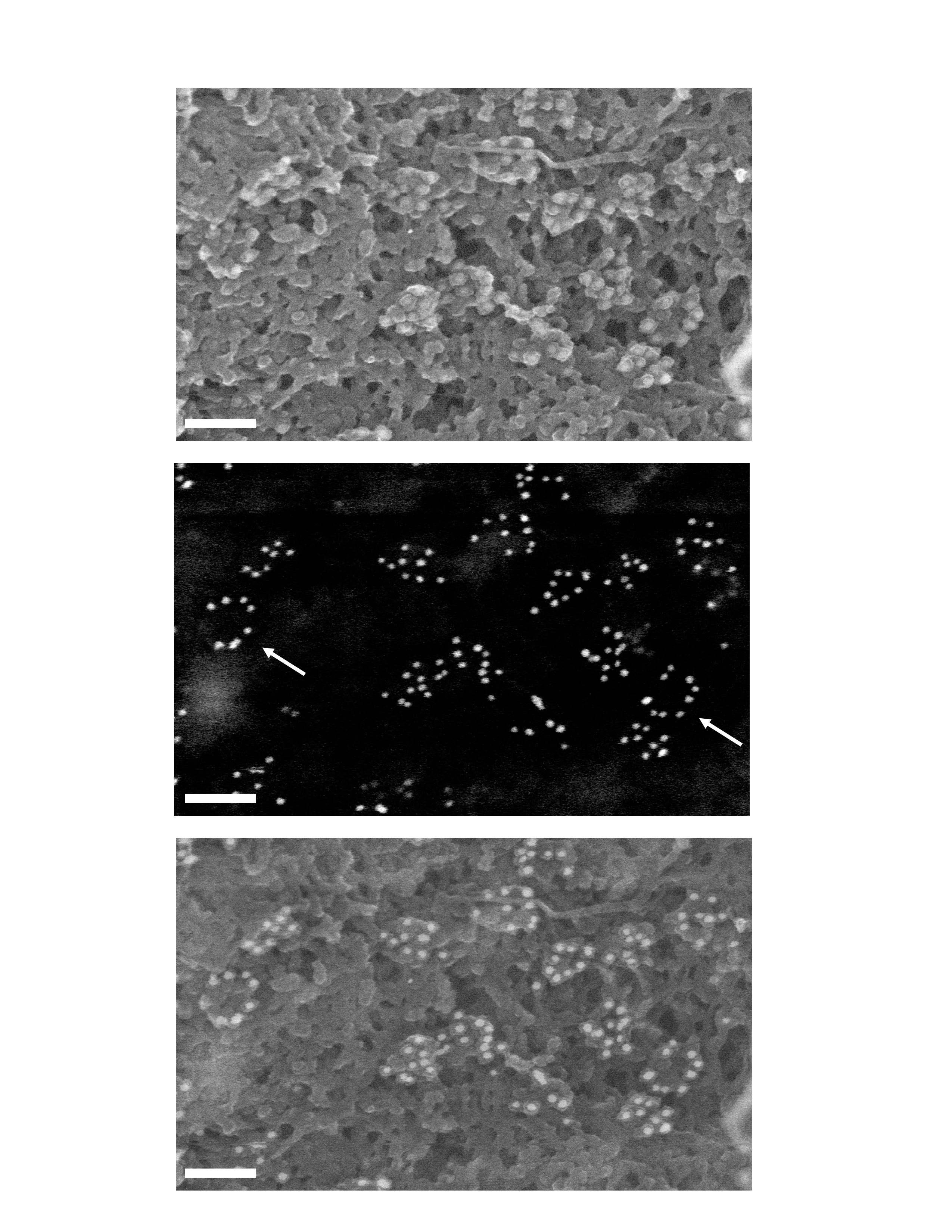
Surface imaging of nuclei by SEM following transduction or infection leads to detection of intact vector and viral capsids at close proximity of nuclear pores. (A) Cytoplasmic extraction of P4 cells in situ exposes nuclear pores and cytoskeletal remnants. C, remnant cytoplasm; N, nucleus; F, cytoskeletal filaments. (B) Control noninfected cells. (C, D) At 48 h p.i with the HR Flap− vector. (E) At 48 h p.i with Flap-defective cPPT-225T mutant virus. (F) At 12 h p.i with the TRIP Flap+ vector. Left panels show secondary electron surface imaging alone, and right panels the backscattered gold signal (anti-p24(CA) labeling) superimposed (Supplementary Figure 2 shows original backscatter images). Arrows point to viral capsids, whereas lines pinpoint key NPC and actin filaments (af). (G) Quantification of p24(CA) staining at the nuclear membrane 48 h p.i with either HR Flap− or TRIP Flap+ vectors, or noninfected. Quantification was carried out on five or more separate nuclei from three independent sample preparations. Capsids were readily detected at late time points p.i in the absence of the central DNA flap, but only at early time points in the presence of the DNA flap. (H) Surface imaging of nuclei by SEM 48 h p.i with LAI-vsv wild-type virus in the presence of nevirapine. Scale bars=1 μm for (A), 200 nm for (B), and 100 nm for all other image panels.
At late time points p.i, we readily observed intact capsids at the cytoplasmic face of the nuclear membrane only in the case of vectors lacking the central DNA Flap (Figure 4G). These capsids were predominantly located directly onto NPCs, slightly off-centered relative to the lumen of the pore (Figure 4C). Alternatively, viral capsids were also found associated with filaments of the cytoskeleton adjacent to the nuclear membrane (Figure 4D), which given their diameter (under 20 nm) were identified as actin filaments.
No capsids were observed in cells transduced with vector, including the central DNA Flap at late time points p.i. In these samples, gold labeling was never detected in clusters and corresponded to probable capsid debris that appeared as small structures (under 40 nm in diameter) on the nuclear membrane or directly attached to the NPC, labeled by no more than 2–3 gold beads (Figure 5A), very distinct from the heavily labeled CA structures detected in Flap− samples at the same time points (Figure 5B). At early time points, however, intact capsids were readily detectable in both Flap+ (Figure 4F) and Flap– samples (18.3 and 17.8 gold particles/μm2, respectively on average), with no morphological differences.
Figure 5.
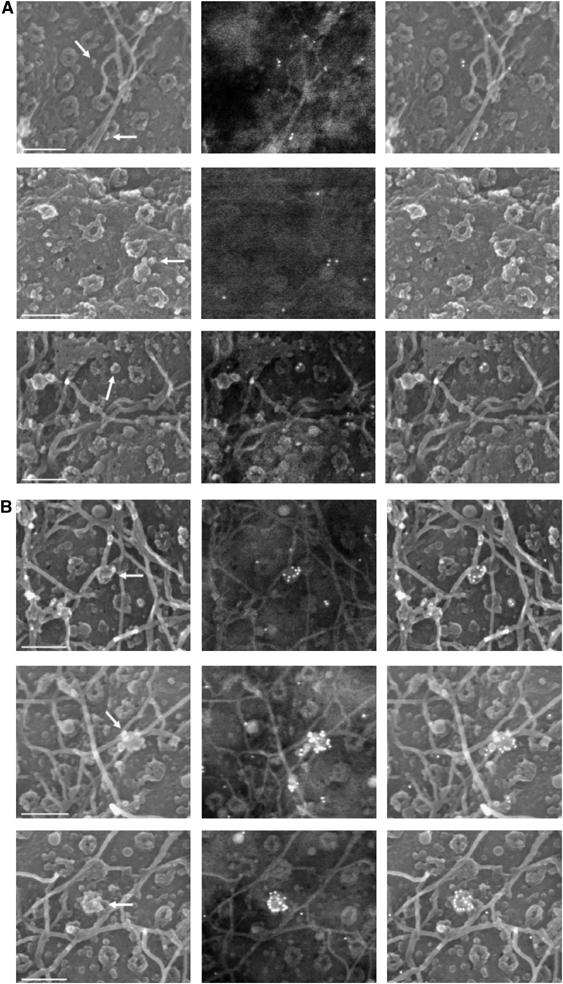
SEM imaging of nuclei at late time points p.i reveals CA debris in Flap+ samples and integral CA shells in Flap− samples. Images show nuclear surfaces in transduced cells 48 h p.i with (A) the TRIP Flap+ vector and (B) the HR flap− vector. The figure shows secondary electron surface imaging alone (left panels), original corresponding backscattered gold signals (anti-p24(CA) labeling, central panels), and the two superimposed (with artificial highlighting of gold signal, right panels). Arrows point to CA debris in (A), and to intact HIV-1 vector capsids in (B). Scale bars=200 nm.
Similarly, we observed no morphological difference between capsids derived from vectors or from viruses. Viral capsids were readily detected at late time points p.i with the DNA Flap-defective virus cPPT-225T (Figure 4E). Moreover, infection with wild-type virus in the presence of nevirapine restores the detection of integral viral capsids at the nuclear membrane (Figure 4H), indicating that integral capsids containing non-reverse-transcribed genomic RNA can also dock at the nuclear pore. It has been suggested that RT° of viral RNA can take place within the host cell nucleus (Bukrinsky et al, 1993). In fact, as docked viral complexes cofractionate with the nuclear fraction, we suggest that RT° can take place at the cytoplasmic face of the nuclear pore and not in the nucleus. As added proof, inhibition of RT° results in a perinuclear accumulation of viral complexes (Figure 2). The abortive intracellular replication HIV-1 complexes in the presence of a reverse transcriptase inhibitor correspond to integral CA shells containing the non-reverse-transcribed genomic RNA dimers.
To obtain images of viral capsids at the nuclear membrane as close as possible to their actual morphology, avoiding possible drawbacks linked to the use of solvents and the dehydration step, and eliminating the added bulk of the 10 nm gold particles attached to the labeled capsids, we observed samples by cryo-SEM rather than the conventional SEM. The surface of the nuclear membrane and structure of nuclear pores appeared very much the same by cryo-SEM as they did with critical point drying (Figure 6), indicating that possible artifacts linked to sample cryo-preparation are limited. Moreover, in cells transduced with vector lacking the central DNA Flap, we identified a number of structures strongly resembling the well-documented (Benjamin et al, 2005; Briggs et al, 2006) conical shape of HIV-1 CA shells, 100–125 nm in length and close to nuclear pores (Figure 6).
Figure 6.

Detection of capsid-like structures at proximity of nuclear pores by cryo SEM. P4 cells were transduced with HR Flap− vector and prepared by cryo-fixation 48 h p.i. (A) Negative control. (B) HR-transduced cells. Preparation of samples by cryo-fixation reveals capsids with typical conical morphology at close proximity of nuclear pores. Arrows indicate capsid-like structures of typical morphology, and lines point to adjacent NPCs. Supplementary Figure 3 provides an outlining of these structures with artificial coloring. Scale bars=100 nm.
Based on all these observations, we conclude that HIV-1 RTCs present at the nuclear membrane contain an integral CA shell that proceeds directly from the core of the incoming viral particle. Consequently, the uncoating step in HIV-1 replication is not an immediate post-fusion event, but rather occurs at the nuclear pore upon completion of viral DNA synthesis. Failure to complete DNA synthesis (as in the presence of nevirapine), or more specifically to generate the central DNA Flap by, central strand displacement at the end of RT°, leads to the stable accumulation of intact capsids at close proximity of nuclear pores.
In vitro DNA Flap formation within vector particles leads to uncoating
To confirm that uncoating occurs upon completion of viral DNA synthesis and formation of the central DNA Flap, we used an endogenous RT (ERT) assay on intact vector particles containing or not the central DNA Flap, and measured uncoating by adapting the previously published fate-of-capsid assay to our ERT reactions (Stremlau et al, 2006). ERT reactions were carried out on nonconcentrated supernatants containing Flap+ or Flap− vector particles (50 to 100 ng p24 antigen per ERT reaction), at 37°C in the presence of dNTPs, EGTA, and 0.01% NP40. Controls included ERT reactions carried out in the absence of dNTPs or in the presence of nevirapine. ERT efficiency was optimized by varying all critical parameters (detergent concentration, temperature, dNTP concentration, addition of EGTA; data not shown) and was systematically monitored by quantitative PCR (qPCR) on late RT° products and early minus strong stop DNA ((−)ssDNA) products. Optimal ERT yielded 50–60% late RT° products relative to (−)ssDNA for both Flap+ and Flap− vectors (Figure 7A, upper panel), whereas ERT reactions carried out in the absence of dNTPs or in the presence of nevirapine yielded near-to-no late RT° products (Figure 7A, upper panel). In parallel to qPCR, ERT products were also analyzed by Southern blotting, and full-length vector DNA was detected for both Flap+ and Flap− samples (3647 and 3457 bp, respectively; Figure 7B), demonstrating RT° completion.
Figure 7.
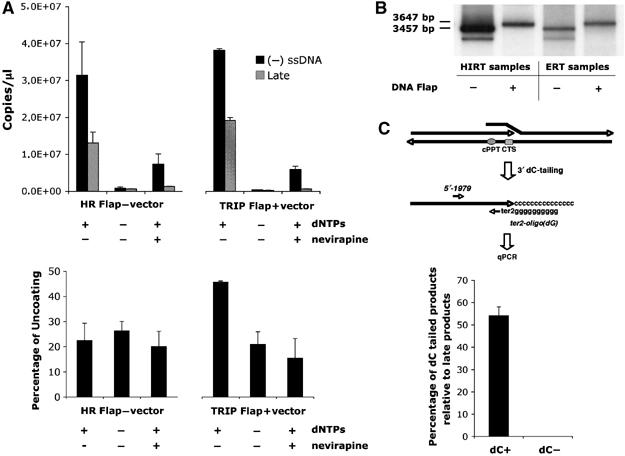
ERT and uncoating assay shows increased vector capsid uncoating upon successful central DNA Flap formation. (A) ERT was carried out on vector supernatants, and qPCR was used to quantify late RT° products and early minus strong stop DNA ((−)ssDNA) products (upper panel). The qPCR quantification was carried out in triplicate and is highly representative of nine independent experiments carried out on three distinct vector productions. ERT reactions were then layered onto sucrose cushion to isolate monomeric CA proteins, indicative of productive uncoating, which were quantified by p24 ELISA assay (lower panel). The p24 ELISA quantification is represented as a percentage of monomeric CA obtained for the corresponding Triton X-100-treated vector samples (% uncoating), and is a mean of three independent experiments carried out on three distinct vector productions, representative of five independent experiments. (B) Southern blotting using a vector specific probe was used to monitor the presence of full-length reverse-transcribed products in the ERT reactions compared with Hirt samples. The radiograph shown is representative of three independent experiments. (C) Formation of the central DNA Flap in Flap+ samples was assessed by dC-tailing of the 3′ (+) strand overlap followed by qPCR using primers specific for the ter2-dCtail region and a 5′ region ∼200 bp upstream (upper panel). The number of copies of dC-tailed amplified products is represented as a percentage of late RT° products (lower panel). The quantification was carried out in triplicate and is highly representative of five independent experiments.
To determine the proportion of late RT° products that are complete reverse-transcribed DNA genomes, with (+) strand overlap in the case of vectors containing the central DNA Flap, we carried out dC-tailing of the 3′ extremity of the (+) strand using terminal transferase. The added 3′poly(dC) tail and the immediately upstream ter2 position in the CTS (Charneau et al, 1994) were then used as complementary template sequence for qPCR amplification using the ter2 oligo(dG) primer and a specific upstream 5′-1979 primer (Figure 7C, upper panel). qPCR reactions carried out on ERT samples without dC-tailing led to no detectable product amplification (Figure 7C, lower panel). Similarly, dC-tailed HR Flap− ERT samples led to no specific amplification product by qPCR (data not shown). However, qPCR reactions on dC-tailed TRIP Flap+ ERT samples led to the amplification of a specific product, which corresponded to 55–65% of late product amplification. We thus ascertained that optimized ERT reactions on Flap+ vectors led to functional DNA Flap formation in about 40% of particles taking into account the yield of endogenous RT.
To determine the degree of uncoating associated with successful RT, we carried out the previously published fate-of-capsid assay on our ERT reactions (Stremlau et al, 2006). Briefly, ERT reactions were layered onto a 50% sucrose cushion and ultracentrifuged at 125 000 g for 2 h at 4°C. After centrifugation, the uppermost 100 μl fraction of the supernatant, which contains monomeric CA (Stremlau et al, 2006), was collected and tested by Western blotting (data not shown) or quantified using a p24 ELISA assay (Figure 7A, lower panel). As a control, ERT reactions were incubated with 0.5% Triton X-100 before layering onto sucrose cushion, a treatment that leads to the complete disassembly of the CA shells. The percentage of uncoating is indicated by the amount of monomeric p24 antigen in the uppermost 100 μl fraction relative to the total amount of p24 measured in the corresponding 0.5% Triton X-100 sample. Flap+ ERT samples were found to lead to 40–45% monomeric CA proteins compared with the Triton X-100 control, whereas Flap− ERT samples systematically only led to 15–30% (Figure 7A, lower panel). Taken together, these results bring an additional argument for a direct role of the central DNA Flap in the uncoating process and further suggests that uncoating does not depend on cellular factors, at least none that would not be already present in the core of the particle.
Integral CA shells contain reverse-transcribed HIV-1 viral DNA
The colocalization of p24(CA) with RTCs in the cytoplasm of infected cells has already been observed using Vpr-green fluorescent protein (GFP)-labeled HIV-1 particles (McDonald et al, 2002). To determine whether integral capsids at the nuclear pore contain viral linear DNA, we carried out in situ hybridization with TEM as we published previously (Arhel et al, 2006c), but without the usual protease treatment to preserve protein ultrastructures. MT4 cells, HTLV-1 transformed human CD4+ T lymphocytes, were infected with DNA Flap-defective cPPT-225 T virus and viral DNA molecules were detected with a virus-specific DNA probe (Zennou et al, 2000). Images reveal barely detectable CA shells around the viral DNA genome, both within the cytoplasm and at the nuclear pore (Figure 8A). Moreover, co-detection of p24(CA) signal together with in situ DNA hybridization reveals colocalization of viral DNA with CA proteins (Figure 8B). We conclude that the integral capsids observed on the cytoplasmic face of the nuclear membrane within infected cells also contain the reverse-transcribed viral DNA genome, indicating that capsids remain associated with the viral genome from entry up to the nuclear pore.
Figure 8.
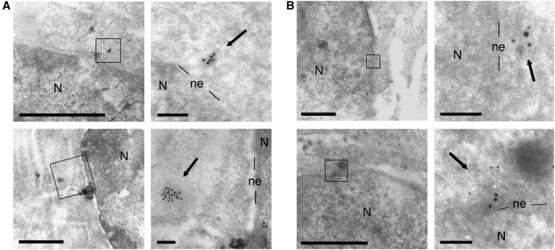
HIV-1 viral genomes lacking the central DNA Flap are trapped within integral capsids at the cytoplasmic face of the nuclear membrane. MT4 lymphocytes were infected with cPPT-225T replicative virus and fixed at the peak of infection. The protocol was carried out without the usual protease treatment to preserve ultrastructures. (A) viral DNA was revealed by in situ hybridization using a virus-specific probe (Zennou et al, 2000; gold beads=10 nm). (B) Co-detection of viral DNA (gold beads=10 nm) with p24(CA) signal by immunogold labeling (gold beads=5 nm). Right-hand images are greater magnifications of the boxed areas in left-hand images. Arrows point to viral genomes. Scale bars=1 μm (left images) and 100 nm (right images). N, nucleus; ne, nuclear envelope.
Discussion
In this work, we provide new insights into the intriguing mechanisms by which the HIV-1 central DNA Flap is implicated in viral genome nuclear import. We knew from previous studies that lack the central DNA Flap results in perinuclear accumulation of viral genomes that do not cross the nuclear membrane, indicating a lack of initiation of translocation through the nuclear pore (Arhel et al, 2006c). RTCs, which are organized as a polymerized CA shell directly issued from the core of the particle, accumulate transiently at close proximity of nuclear pores following infection with wild-type virus, whereas they remain stably blocked at this stage in the case of DNA Flap mutants. DNA Flap formation, the last event in HIV-1 RT°, facilitates RTC maturation into PIC before viral DNA translocation through the nuclear pore. Consequently, the uncoating step in HIV-1 replication is not an immediate post-fusion event, but rather occurs at the nuclear pore upon completion of viral DNA synthesis. Default of nuclear import in Flap-defective viruses and vectors is due to the trapping of viral genomes within intact capsids that do not mature. An updated model for HIV-1 intracellular transport and DNA Flap-dependent nuclear import is provided in Figure 9.
Figure 9.
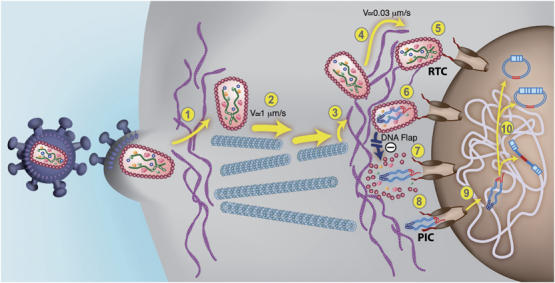
An updated model for HIV-1 intracellular transport and DNA Flap-dependent nuclear import. (1) HIV-1 viruses enter target cells by receptor-mediated fusion and need to cross and/or interact with the actin cortex underlying the plasma membrane for productive infection to take place (Campbell et al, 2004). Post-fusion uncoating does not take place at this stage. (2) HIV-1 RTCs proceeding directly from the core of the particle undergo rapid and saltatory microtubule-directed movement towards the nuclear compartment (McDonald et al, 2002; Arhel et al, 2006b). (3) RTCs undergo transition from microtubules onto actin filaments at close proximity to the nuclear compartment and then (4) slow actin-directed movements towards the nuclear membrane (Arhel et al, 2006b). (5) HIV-1 CAs containing viral genome dock at NPCs. Interaction with cytoplasmic filaments emanating from NPCs is hypothetical. (6) As docking of RTCs at the nuclear pore is more rapid than RT°, we surmise that most of viral DNA synthesis occurs within an integral CA core docked at the NPC. Upon RT° completion and formation of the central DNA Flap, which is consequent upon the final strand displacement event, (7) docked RTCs can undergo DNA-Flap-dependent maturation, leading to PIC formation lacking the CA shell. Complexes that lack DNA Flap formation accumulate within intact HIV-1 CAs that forbid translocation through the nuclear pore. (8) The HIV-1 PIC is translocated through the nuclear pore. The folded structure of the PIC via integrase dimerization is hypothetical (Zennou et al, 2000). (9) The HIV-1 PIC undergoes a poorly characterized diffusive intranuclear transport (Arhel et al, 2006b) and (10) either integrates into host cell chromatin or circularizes into 1- or 2-LTR circles.
HIV-1 reverse transcription occurs within an integral CA core
In this work, we provide definitive evidence, by direct SEM visualization, for the presence of intact HIV-1 capsids in close proximity to the nuclear membrane on actin filaments, and at the nuclear pore frequently slightly off-centered from the lumen of the nuclear pore. Moreover, we show that the reverse-transcribed viral DNA colocalizes with p24(CA) detection (as previously shown by McDonald et al, 2002), which corresponds to intact CA shells, indicating that capsids remain associated with the viral genome from entry up to when the complex reaches the nuclear pore. In some mitosis-dependent retroviral systems, the association of CA proteins with the viral genome in the cytoplasm has already been documented (Bowerman et al, 1989; Saib et al, 1997; Petit et al, 2003). However, in the case of HIV-1, most studies involving isolation and biochemical analysis of intracellular RTCs led to the conclusion that the CA proteins are not associated with the HIV-1 linear DNA genome (Bukrinsky et al, 1993; Fassati and Goff, 2001). We believe that this apparent discrepancy can be resolved by two lines of arguments. First, the HIV-1 CA shell is a fragile p24 assembly (Borroto-Esoda and Boone, 1991; Welker et al, 2000), and therefore, it is not unlikely that the protocol used for isolation of RTCs by density gradient leads to the dissociation of CA proteins from the viral genome. As an example, we found, while optimizing our ERT assay, that HIV-1 capsids disassemble completely after as little as 10 s incubation with 0.1% Triton X-100 or after overnight incubation at 41°C rather than 37°C (data not shown). Second, because transit to the nuclear membrane is very rapid following infection, biochemical analyses of cytoplasmic RTCs are carried out early after infection. At this point, an important fraction of viral particles that entered by endocytosis are on the way to being degraded (Maréchal et al, 1998). We also think that some functional RTCs in the routing process are lost and degraded; it has already been observed that not all Vpr-GFP-labeled HIV-1 complexes reach the nuclear membrane, but that some particles undergo stochastic movement with no significant displacement towards the nucleus (McDonald et al, 2002). In light of our work, these likely correspond to the reported fraction of cytoplasmic RTCs that do not contain detectable p24(CA) (McDonald et al, 2002). Therefore, it is likely that most HIV-1 complexes present within biochemical fractions represent complexes that have either lost their capsids as a result of the isolation protocol, or were damaged or engaged in a pathway of degradation at the time of isolation or observation.
In fact, the very mechanisms of RT° provide compelling arguments that it cannot take place at high dilution of the viral components in the cytoplasm of infected cells, but requires the structural environment of a RTC, where these components are confined to a CA protein assembly. The HIV-1 RTC contains numerous copies of reverse transcriptase polymerase (about 80 to 120 per capsid; Layne et al, 1992). However, due to the important distributive nature of the reverse transcriptase enzyme (Klarmann et al, 1993), and the need to maintain a high stoichiometry of enzyme to viral template for rate limiting steps such as polymerization pauses, strand transfers, and formation of the central DNA Flap (Klarmann et al, 1993; Charneau et al, 1994), it is only logical that in the cytoplasm of infected cells RT° occurs within an integral CA structure. Therefore, although some visually undetectable morphological changes of the capsid may be involved, as suggested by the impaired RT° of viruses with hyperstable capsids (Forshey et al, 2002), it is likely that full uncoating before RT° completion results in nonproductive infection and degradation of the aborted complex. In support of this is the recent demonstration that the HIV-1 capsid is the target of rhTRIM5α restriction, and that rhTRIM5α exerts restriction by accelerating the uncoating of HIV-1 capsids (Stremlau et al, 2006). Moreover, HIV-1 viruses with unstable cores are impaired for RT° in target cells, but not in vitro (Forshey et al, 2002).
Kinetics of docking and reverse transcription
After fusion with the cell membrane, the HIV-1 virus likely interacts with the actin filament network underlying the plasma membrane (Campbell et al, 2004) before interacting with the microtubule network for a rapid (under 2 h) routing to the nuclear membrane (McDonald et al, 2002; Arhel et al, 2006b). Our data showing HIV-1 capsids on actin filaments at the immediate vicinity of the nuclear pore are in direct support of our previous findings that HIV-1 replicative complexes transit from microtubules onto actin filaments before actually reaching the nuclear pore (Arhel et al, 2006b).
A distinctive feature of the early steps in lentiviral biology is that translocation through the nuclear pore imposes a maturation step from RTC (including viral capsid) into PIC. The central strand displacement leading to the formation of the central DNA Flap is the final event in the process of RT° and thus marks RT° completion. Here, we show that in the absence of central DNA Flap, formation of viral DNA remains trapped on the cytoplasmic face of the nuclear membrane within an integral capsid. We therefore unmask a mechanistic relationship between central termination of RT° (formation of the DNA Flap) and maturation of RTC into PIC that involves at least the loss of the CA assembly. The molecular mechanisms underlying DNA Flap-dependent maturation of HIV-1 capsids remain an open question. As we demonstrate that DNA Flap-dependent uncoating can be reconstituted in vitro within purified vector particles, there is likely no direct implication of cell components in the uncoating process unless these are already present in the core of the particle. One possible hypothesis could be that uncoating is the consequence of a major morphological change due to a rearrangement of the RTC following formation of the central DNA Flap, which would no longer be compatible with its containment within a CA shell. The in vitro uncoating assay will provide a valuable tool to further study this intriguing step of HIV-1 replication. A subsequent role for the DNA Flap in the actual translocation process or intranuclear routing, once it is rendered accessible to the nuclear import machinery, can by no means be excluded. Interaction with cellular and/or viral karyophilic shuttling proteins could then be either direct, or via other cellular factors such as the recently identified tRNAs (Zaitseva et al, 2006).
The docking of intact viral capsids to the nuclear pore and uncoating before nuclear translocation are phenomena that are already described for other viral systems (Smith and Helenius, 2004). As mentioned above, it is probable that HIV-1 capsids also dock to the nuclear pore via specific interactions with nucleoporins although an interaction between p24(CA) and components of the nuclear pore has yet to be identified. Unlike previously suggested, it is not likely that either phosphorylated MA, IN or Vpr are implicated in docking of the RTC to the nuclear pore, as these are confined within a CA shell that prevents their access to cellular nuclear import machinery. Contrary to Ad2 and HSV-1, docking of HIV-1 CAs to the nuclear pore is not sufficient to immediately trigger uncoating, as docking precedes translocation by several hours (10 to 20 h), indicating that HIV-1 likely possesses a viral control of successful RT° completion before uncoating and translocation into the nucleus. Our work shows that the decision for uncoating is governed by formation of the central DNA Flap, which marks the end of RT°.
Materials and methods
Antibodies
Primary antibodies against p24(CA) proteins were mouse monoclonal p25a and p25c (clones 11-25 and 90-3-55, Laboratoire d'Ingenierie des Anticorps, Institut Pasteur) for immunofluorescence and TEM; and p25e (clone 157-20) and 183-H12-5C (AIDS Reagent Program) for SEM. Secondary antibodies were Cy3-labeled goat anti-mouse IgG (Amersham Pharmacia) for confocal immunofluorescence microscopy, goat anti-mouse IgG H&L conjugated 10 nm gold (British Biocell International) for SEM, goat anti-mouse IgG- and IgM-conjugated 5 nm colloidal gold solution (Amersham Pharmacia) for TEM.
HIV-1 virus and HIV-1-based vector production and transduction
The conservative cPPT mutant virus, cPPT-225T, is a cPPT-225 mutant (Charneau et al, 1992), containing two additional T-to-C base mutations in the cPPT sequence rendering it more severely replication defective (data not shown). Pseudotyped viruses, which were used to maintain one-round infections, were produced by transient transfection of 293T cells by calcium phosphate co-precipitation with the proviral plasmid deleted in the env gene, pLAI-env and p225T-env, and with a VSV-G envelope expression plasmid (pHCMV-G; Yee et al, 1994).
The HIV-1 vectors HR-GFP, derived from HR'CMVLacZ (Naldini et al, 1996), and TRIP-GFP, which contains the cis-acting sequences required for formation of the central DNA Flap (Zennou et al, 2000), encode the GFP under the control of the CMV promoter to monitor gene transduction. The vector HR-Luc carries the luciferase gene under the control of the CMV promoter, and was used when GFP expression was not desirable. HIV-1 vectors were produced by transient transfection of 293T cells with the vector plasmid, encapsidation plasmid (p8.7), and the VSV envelope expression plasmid as described previously (Naldini et al, 1996). P4 or MT4 cells were transduced or infected with supernatants containing vector or viral particles (1 μg/106 cells) in the presence of 20 μg/ml of DEAE-dextran in the case of P4 cells. Nevirapine (Boehringer Ingelheim) was added at a final concentration of 1 μM.
Southern blotting analysis
The quantitative follow-up of the synthesis, circularization, and integration of HIV-1 intracellular vector DNA was carried out by Southern blotting, following DpnI, EcoNI, AvaII, and XhoI digestion as described previously (Zennou et al, 2000). ERT products were also analyzed by Southern blotting, following DpnI digestion only.
Confocal fluorescence microscopy
Cells were fixed in 4% paraformaldehyde (PFA) in PBS for 10 min and permeabilized in 0.5% Triton X-100 (PBS) for 15 min. Nonspecific binding was blocked by incubation in 0.3% BSA (PBS) for 10 min. Incubation with primary antibodies diluted 1:100 in 0.3% BSA was carried out for 2 h at room temperature, then with Cy3-conjugated antibody diluted 1:1000 for 30 min in the dark. Cells were mounted in Vectashield mounting medium (Vector Laboratories) and viewed using Leica upright confocal microscope (TCS4D). Stacks were consistently taken at equivalent voltage and offset, with optical sections of 0.5 or 0.8 μm. Images shown in Figure 1B-C are a merge of four confocal slices.
Transmission electron microscopy
Samples were prepared according to standard TEM protocols (see Supplementary data for details).
Scanning electron microscopy
Cells were grown on silicon chips (Agar Scientific) and pre-fixed in situ in 1% PFA/0.01% GA for 15 s. Extraction of cytoplasmic contents in situ was carried out in 0.5% Triton X-100 for 30 min to expose the nuclear membrane and pores. Samples were post-fixed in 4% PFA for 20 min and incubated successively in NH4Cl 0.25%, 1% BSA, and 1% BSA/1% NGS (normal goat serum, Nordic Immunology). Cells were incubated with primary anti-p24(CA) antibodies diluted 1:20 in 1% BSA/1% NGS for 2 h, and with secondary gold conjugate diluted 1:25 in 0.1% gelatin for 1 h. Cells were post-fixed in 3% GA for 1 h and in 1% OsO4 (0.1% Sörensen, pH 7.3) for 30 min. After critical point drying, nuclei were fractured as described previously (Allen et al, 1998), coated with chromium, and observed with a field emission in-lens ISI/Topcon DS 130F SEM microscope at 28 kV acceleration voltage. The criteria used to score an HIV-1 capsid were first, its heavy immunogold labeling with anti-CA antibodies, and second, its similar morphology compared to the well-characterized purified HIV-1 cores (Benjamin et al, 2005; Briggs et al, 2006). The same criteria were used in TEM and cryo SEM.
Cryo-SEM
Cells were grown on silicon chips and were pre-fixed and extracted in situ as for SEM. After brief fixation in 3% GA (PBS), the silicon chips were plunged in liquid N2. Samples were prepared using the Gatan cryo transfer unit Alto 2500, coated with Au-Pd, and visualized with a Jeol field emission-SEM JSM 6700F at 5 kV acceleration voltage.
Endogenous reverse transcription
Supernatants containing between 50 and 100 ng p24 of vector particles were mixed with 2 × ERT buffer (100 mM Tris–HCl (pH 8.0), 0.2 mM each dNTPs, 6 mM MgCl2, 6 mM EGTA and 0.02% NP40) and were incubated at 37°C for 16 h. For PCR analysis, reactions were then terminated by the addition of an equal volume of stop buffer (2% SDS, 20 mM EDTA, 200 μg/ml proteinase K), followed by a 4-h incubation at 56°C. As negative controls, reactions were run without dNTPs or in the presence of 5 μM nevirapine.
Analysis of ERT products: qPCR and dC-tailing
After ERT, viral DNA was phenol-chloroform extracted, ethanol precipitated, and resuspended in 35 μl H2O. Dpn I digestion was included to eliminate residual transfection plasmid. Quantitative PCR (qPCR) was performed in triplicate using SYBR Green JumpStart Taq Ready Mix (Sigma) and 300 nM of each primer. qPCR analysis of early ERT products was carried out using M667/AA55 M primers specific for minus-strand strong stop DNA, as described previously (Zhang et al, 1993). Late ERT products were detected using M667/M661 primers, which amplify the RU5-primer binding site 5′ noncoding (5NC) region (Zhang et al, 1993).
For dC-tailing by terminal transferase, ERT products were heat-denatured at 95°C for 5 min, and 3′ dC-tailing was carried out as described previously (Charneau et al, 1994). As a negative control, reactions were run without terminal transferase. Detection of dC-tailed products by qPCR was performed using a ter2-oligo(dG) primer (5′-GGGGGGGGGGAAAATTTTG-3′), complementary to the ter2 site of the CTS and the added 3′ poly(dC) tail, and the 5′-1979 primer (5′- CAGAGACAGATCCATTCGATTAGTG-3′) upstream the cPPT sequence. Results are expressed as percentage of dC-tailed products relative to late RT° products.
Uncoating assay
After ERT, samples were diluted in PBS qsp 2 ml, layered onto a 7 ml 50% sucrose cushion, and ultracentrifuged as described previously (Stremlau et al, 2006). After centrifugation, 100 μl from the uppermost part of the supernatant was collected and tested for p24 content by ELISA assay (PerkinElmer Life Sciences) according to the manufacturer's instructions.
In situ DNA hybridization with TEM
Infected cells were fixed at 48 h p.i. and in situ DNA hybridization was carried out as described previously using a biotinylated double-stranded virus-specific DNA probe (Arhel et al, 2006c).
Supplementary Material
Supplementary Figure 1
{kind=link}
Supplementary Figure 2
{kind=link}
Supplementary Figure 3
{kind=link}
Supplementary Figures Legends
Supplementary Data
Acknowledgments
The 183-H12-5C Monoclonal Antibody (Chesebro et al, 1992) was obtained through the AIDS Reagent Program. This work was supported by grants from the Institut Pasteur, the Agence Nationale de Recherche sur le SIDA (ANRS), the Agence Nationale de la Recherche (ANR) and the Fondation pour la Recherche Médicale (FRM).
References
- Allen TD, Rutherford SA, Bennion GR, Wiese C, Riepert S, Kiseleva E, Goldberg MW (1998) Three-dimensional surface structure analysis of the nucleus. In Nuclear Structure and Function. Berrios M (ed), pp 125–138. San Diego, CA: Academic Press [DOI] [PubMed]
- Ao J, Yao X, Cohen EA (2004) Assessment of the role of the central DNA flap in HIV-1 replication by using a single-cycle replication system. J Virol 78: 3170–3177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arhel NJ, Genovesio G, Kim KA, Miko S, Perret E, Olivo-Marin JC, Shorte S, Charneau P (2006b) Quantitative 4D trascking of cytoplasmic and nuclear HIV-1 complexes. Nat Meth 3: 817–824 [DOI] [PubMed] [Google Scholar]
- Arhel NJ, Munier S, Souque P, Mollier K, Charneau P (2006a) Nuclear import defect of DNA Flap mutant HIV-1 viruses is not dependent on the viral strain or target cell type. J Virol 80: 10262–10269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arhel NJ, Souquere-Besse S, Charneau P (2006c) Wild-type and central DNA Flap defective HIV-1 DNA genomes: intracellular visualization at ultrastructural resolution levels. Retrovirology 3: 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbosa P, Charneau P, Dumey N, Clavel F (1994) Kinetic analysis of HIV-1 early replicative steps in a coculture system. AIDS Res Hum Retroviruses 10: 53–59 [DOI] [PubMed] [Google Scholar]
- Benjamin J, Ganser-Pornillos BK, Tivol WF, Sundquist WI, Jensen GJ (2005) Three-dimensional structure of HIV-1 virus-like particles by electron cryotomography. J Mol Biol 346: 577–588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borroto-Esoda K, Boone LR (1991) Equine infectious anemia virus and HIV DNA synthesis in vitro: characterization of the endogenous reverse transcriptase reaction. J Virol 65: 1952–1959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowerman B, Brown PO, Bishop JM, Varmus HE (1989) A nucleoprotein complex mediates the integration of retroviral DNA. Genes Dev 3: 469–478 [DOI] [PubMed] [Google Scholar]
- Briggs JA, Grunewald K, Glass B, Forster F, Krausslich HG, Fuller SD (2006) The mechanism of HIV-1 core assembly: insights from three-dimensional reconstructions of authentic virions. Structure 14: 15–20 [DOI] [PubMed] [Google Scholar]
- Bukrinskaya A, Brichacek B, Mann A, Stevenson M (1998) Establishment of a functional HIV-1 reverse transcription complex involves the cytoskeleton. J Exp Med 188: 2113–2125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bukrinsky MI, Sharova N, McDonald TL, Pushkarskaya T, Tarpley WG, Stevenson M (1993) Association of integrase, matrix, and reverse transcriptase antigens of HIV-1 with viral nucleic acids following acute infection. Proc Natl Acad Sci USA 90: 6125–6129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell EM, Nunez R, Hope TJ (2004) Disruption of the actin cytoskeleton can complement the ability of Nef to enhance HIV-1 infectivity. J Virol 78: 5745–5755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charneau P, Alizon M, Clavel F (1992) A second origin of DNA plus-strand synthesis is required for optimal HIV replication. J Virol 66: 2814–2820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charneau P, Clavel F (1991) A single-stranded gap in HIV unintegrated linear DNA defined by a central copy of the polypurine tract. J Virol 65: 2415–2421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charneau P, Mirambeau G, Roux P, Paulous S, Buc H, Clavel F (1994) HIV-1 reverse transcription. A termination step at the center of the genome. J Mol Biol 241: 651–662 [DOI] [PubMed] [Google Scholar]
- Chesebro B, Wehrly K, Nishio J, Perryman S (1992) Macrophage-tropic HIV virus isolates from different patients exhibit unusual V3 envelope sequence homogeneity in comparison with T-cell-tropic isolates: definition of critical amino acids involved in cell tropism. J Virol 66: 6547–6554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dismuke DJ, Aiken C (2006) Evidence for a functional link between uncoating of the HIV-1 core and nuclear import of the viral preintegration complex. J Virol 80: 3712–3720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dvorin JD, Bell P, Maul GG, Yamashita M, Emerman M, Malim MH (2002) Reassessment of the roles of integrase and the central DNA flap in HIV-1 nuclear import. J Virol 76: 12087–12096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fassati A, Goff SP (2001) Characterization of intracellular reverse transcription complexes of HIV-1. J Virol 75: 3626–3635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forshey BM, von Schwedler U, Sundquist WI, Aiken C (2002) Formation of a HIV-1 core of optimal stability is crucial for viral replication. J Virol 76: 5667–5677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gartner S, Markovits P, Markovitz DM, Kaplan MH, Gallo RC, Popovic M (1986) The role of mononuclear phagocytes in HTLV-III/LAV infection. Science 233: 215–219 [DOI] [PubMed] [Google Scholar]
- Karageorgos L, Li P, Burrell C (1993) Characterization of HIV replication complexes early after cell-to-cell infection. AIDS Res Hum Retroviruses 9: 817–823 [DOI] [PubMed] [Google Scholar]
- Kim SY, Byrn R, Groopman J, Baltimore D (1989) Temporal aspects of DNA and RNA synthesis during HIV infection: evidence for differential gene expression. J Virol 63: 3708–3713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klarmann GJ, Schauber CA, Preston BD (1993) Template-directed pausing of DNA synthesis by HIV-1 reverse transcriptase during polymerization of HIV-1 sequences in vitro. J Biol Chem 268: 9793–9802 [PubMed] [Google Scholar]
- Layne SP, Merges MJ, Dembo M, Spouge JL, Conley SR, Moore JP, Raina JL, Renz H, Gelderblom HR, Nara PL (1992) Factors underlying spontaneous inactivation and susceptibility to neutralization of HIV. Virology 189: 695–714 [DOI] [PubMed] [Google Scholar]
- Limon A, Nakajima N, Lu R, Ghory HZ, Engelman A (2002) Wild-type levels of nuclear localization and HIV-1 replication in the absence of the central DNA flap. J Virol 76: 12078–12086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maréchal V, Clavel F, Heard JM, Schwartz O (1998) Cytosolic Gag p24 as an index of productive entry of HIV-1. J Virol 72: 2208–2212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maréchal V, Prevost MC, Petit C, Perret E, Heard JM, Schwartz O (2001) HIV-1 entry into macrophages mediated by macropinocytosis. J Virol 75: 11166–11177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsden MD, Zack JA (2007) Human immunodeficiency virus bearing a disrupted central DNA flap is pathogenic in vivo. J Virol 81: 6146–6150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald D, Vodicka MA, Lucero G, Svitkina TM, Borisy GG, Emerman M, Hope TJ (2002) Visualization of the intracellular behavior of HIV in living cells. J Cell Biol 159: 441–452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naldini L, Blomer U, Gallay P, Ory D, Mulligan R, Gage FH, Verma IM, Trono D (1996) In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector. Science 272: 263–267 [DOI] [PubMed] [Google Scholar]
- Petit C, Giron ML, Tobaly-Tapiero J, Bittoun P, Real E, Jacob Y, Tordo N, De The H, Saib A (2003) Targeting of incoming retroviral Gag to the centrosome involves a direct interaction with the dynein light chain 8. J Cell Sci 116: 3433–3442 [DOI] [PubMed] [Google Scholar]
- Saib A, Puvion-Dutilleul F, Schmid M, Peries J, de The H (1997) Nuclear targeting of incoming human foamy virus Gag proteins involves a centriolar step. J Virol 71: 1155–1161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith AE, Helenius A (2004) How viruses enter animal cells. Science 304: 237–242 [DOI] [PubMed] [Google Scholar]
- Stremlau M, Perron M, Lee M, Li Y, Song B, Javanbakht H, Diaz-Griffero F, Anderson DJ, Sundquist WI, Sodroski J (2006) Specific recognition and accelerated uncoating of retroviral capsids by the TRIM5alpha restriction factor. Proc Natl Acad Sci USA 103: 5514–5519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberg JB, Matthews TJ, Cullen BR, Malim MH (1991) Productive HIV-1 infection of nonproliferating human monocytes. J Exp Med 174: 1477–1482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welker R, Hohenberg H, Tessmer U, Huckhagel C, Krausslich HG (2000) Biochemical and structural analysis of isolated mature cores of HIV-1. J Virol 74: 1168–1177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yee JK, Miyanohara A, LaPorte P, Bouic K, Burns JC, Friedmann T (1994) A general method for the generation of high-titer, pantropic retroviral vectors: highly efficient infection of primary hepatocytes. Proc Natl Acad Sci USA 91: 9564–9568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaitseva L, Myers R, Fassati A (2006) tRNAs promote nuclear import of HIV-1 intracellular reverse transcription complexes. PLoS Biol 4: e322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zennou V, Petit C, Guetard D, Nerhbass U, Montagnier L, Charneau P (2000) HIV-1 genome nuclear import is mediated by a central DNA flap. Cell 101: 173–185 [DOI] [PubMed] [Google Scholar]
- Zhang H, Zhang Y, Spicer TP, Abbott LZ, Abbott M, Poiesz BJ (1993) Reverse transcription takes place within extracellular HIV-1 virions: potential biological significance. AIDS Res Hum Retroviruses 9: 1287–1296 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Figures Legends
Supplementary Data