

Abstract
Under normal physiological conditions, synaptic vesicle endocytosis is regulated by phosphorylation and Ca2+-dependent dephosphorylation of endocytic proteins such as amphiphysin and dynamin. To investigate the regulatory mechanisms that may occur under the conditions of excessive presynaptic Ca2+ influx observed preceding neural hyperexcitation, we examined hippocampal slices following high-potassium or high-frequency electrical stimulation (HFS). In both cases, three truncated forms of amphiphysin I resulted from cleavage by the protease calpain. In vitro, the binding of truncated amphiphysin I to dynamin I and copolymerization into rings with dynamin I were inhibited, but its interaction with liposomes was not affected. Moreover, overexpression of the truncated form of amphiphysin I inhibited endocytosis of transferrin and synaptic vesicles. Inhibiting calpain prevented HFS-induced depression of presynaptic transmission. Finally, calpain-dependent amphiphysin I cleavage attenuated kainate-induced seizures. These results suggest that calpain-dependent cleavage of amphiphysin I inhibits synaptic vesicle endocytosis during neural hyperexcitation and demonstrate a novel post-translational regulation of endocytosis.
Keywords: amphiphysin, calpain, endocytosis, hyperexcitation, seizure
Introduction
Clathrin-mediated endocytosis is the major mechanism for maintaining the synaptic vesicle recycling pool for synaptic transmission in small central synapses during repetitive high-frequency electrical stimulation (HFS) or even physiological stimulation (Granseth et al, 2006). Clathrin-mediated endocytosis consists of four steps: nucleation, invagination, fission and uncoating. Many endocytosis-related proteins are recruited to form complexes on the retrieved pits at each step after calcium-dependent calcineurin activity is activated by neural excitation. Amphiphysin I, a major dynamin-binding partner localized on the collar of retrieved vesicles, plays a key role in clathrin-mediated endocytosis of synaptic vesicles (Wigge and McMahon, 1998; Zhang and Zelhof, 2002; Evergren et al, 2004). Amphiphysin I mediates invagination and fission of synaptic vesicles, senses and facilitates membrane curvature via its BAR domain (Zhang and Zelhof, 2002; Peter et al, 2004) and stimulates the GTPase activity of dynamin in the presence of lipid membrane (Yoshida et al, 2004; Yoshida and Takei, 2005). The function of both amphiphysin I and dynamin is known to be regulated by phosphorylation and Ca2+-dependent dephosphorylation (Slepnev et al, 1998; Cousin and Robinson, 2001; Tan et al, 2003; Tomizawa et al, 2003) under physiological conditions. Under resting conditions, endocytosis proteins such as amphiphysin I and dynamin are phosphorylated by Cdk5 (cyclin-dependent kinase 5). Once neural excitability is transferred to the nerve terminal, calcium influx to the presynaptic terminal occurs through VDCCs and activates phosphatase 2B calcineurin, which dephosphorylates the endocytic proteins. These proteins then form a complex and induce clathrin-mediated endocytosis.
Massive amount of Ca2+ enter presynaptic terminals during the neural hyperexcitation observed in neurodegenerative disorders such as ischemia/anoxia, epilepsy, stroke, trauma and Alzheimer's disease. Previous studies have shown that hyperexcitation, such as repetitive HFS or high potassium stimulation, at presynapses induces short-term depression at postsynapses in hippocampal neurons (Stevens and Wesseling, 1999; Sara et al, 2002). This short-term depression is thought to be a neuroprotective mechanism for protecting neurons from hyperexcitability. However, the mechanism of the short-term depression after Ca2+ influx is unclear. Previous studies have shown that synaptic vesicle endocytosis, which contributes to maintaining synaptic transmission, is inhibited during synaptic depression (Sun et al, 2002; Wu, 2004). These results suggest that inhibition of clathrin-mediated endocytosis may be involved in the short-term depression at mammalian synapses after excessive presynaptic Ca2+ influx.
Calcium-dependent calpain activity is increased by massive calcium influx, and promotes the degradation of key cytoskeletal and membrane proteins or cleavage of many other important proteins such as calcineurin in neuroexcitotoxicity (Wu et al, 2004; Czogalla and Sikorski, 2005), which has been shown to be related to various neurodegenerative disorders. Moreover, several presynaptic proteins such as spectrin (Saido et al, 1993), PKC (Sessoms et al, 1992) and p35 (Lee et al, 2000), a Cdk5 activator, are substrates of calpain. The function of these proteins may be affected by calpain cleavage. In the present study, we found that the important endocytosis protein amphiphysin I is cleaved by calpain during high potassium stimulation, repetitive high-frequency stimulation and kainate (KA)-induced seizures. Calpain-cleaved amphiphysin I lost its function of binding with dynamin in vitro, and overexpression of the truncated form of amphiphysin I in COS-7 cells and neurons inhibited clathrin-mediated endocytosis. Inhibition of clathrin-mediated endocytosis of synaptic vesicles by calpain-dependent amphiphysin I cleavage is involved in repetitive HFS-induced synaptic depression and KA-induced seizures.
Results
High K+ induces multiple amphiphysin I cleavages by calpain in mouse hippocampal slices
We first investigated whether amphiphysin I was cleaved under conditions of excessive Ca2+ influx into nerve terminals (80 mM KCl) or under neurotoxic conditions (500 μM L-glutamate) in the hippocampal slices. In both cases, three major truncated products of amphiphysin I were detected by Western blotting analysis using anti-amphiphysin I antibodies recognizing the N terminus (Figure 1A). The antibodies reacted with 52, 47 and 43-kDa proteins on SDS–PAGE gels. The induction of these truncations was greater with high potassium (high K+) stimulation than with glutamate stimulation (Figure 1A). Moreover, a weak fourth band of amphiphysin I was sometimes detected in the hippocampal slices given high K+ stimulation (arrow in Figure 1A). These results suggest that excessive Ca2+ influx in presynaptic terminus may induce the cleavage of amphiphysin I. The Ca2+-dependent cysteine protease calpain is involved in various neurological processes, and an increase in calpain activity is related to the pathophysiology of neurodegenerative disorders such as neuroexcitotoxicity and ischemia (Rami, 2003; Wu et al, 2004). We therefore examined whether calpain is involved in the cleavage of amphiphysin I after high K+ stimulation. Figure 1B shows the time-dependent cleavages of amphiphysin I and α-spectrin, a physiological substrate of calpain, in hippocampal slices after high K+ stimulation. Amphiphysin I was rapidly cleaved with obvious truncations observed 30 s after the stimulation (Figure 1B), and 41% of amphiphysin I was cleaved 20 min after the stimulation. α-Spectrin was also rapidly cleaved after high K+ stimulation and was more extensively cleaved than amphiphysin I after 20 min of stimulation (Figure 1B). Subcellular fractionation showed that two isoforms of calpain, μ- and m-calpain, existed in the membrane and cytosol fractions of purified synaptosomes (LP2 and LS2 fractions in Figure 1C). Two potent calpain inhibitors, ALLM and ALLN, inhibited the high K+-induced truncations of amphiphysin I in the hippocampal slices (Figure 1D). Moreover, m-calpain cleaved recombinant amphiphysin I in vitro, yielding three truncated forms with molecular masses equal to those of the truncated forms observed in hippocampal slices after high K+ stimulation (Figure 1E). A high concentration (40 nM) of m-calpain almost completely cleaved amphiphysin I to the 43-kDa form, and 52-kDa amphiphysin I was not observed (Figure 1E). We also examined the effect of μ-calpain on amphiphysin I cleavage in vitro (Supplementary Figure 1). μ-Calpain cleaved amphiphysin I to the same three major truncated forms as m-calpain, and in addition to one new truncated form between the 52 and 47-kDa forms. These results suggest that amphiphysin I may be cleaved by calpain during neural hyperexcitation.
Figure 1.
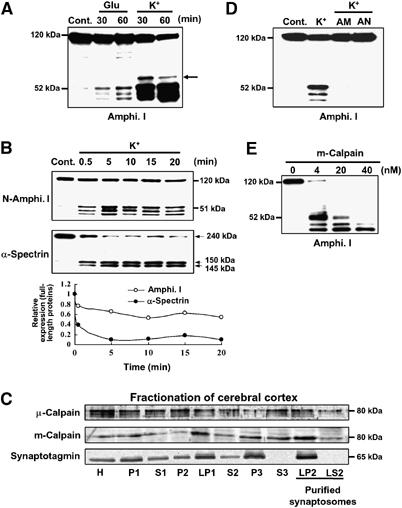
High K+ induces multiple amphiphysin I (Amphi. I) cleavages by calpain in mouse hippocampal slices. (A) Western blotting analysis of hippocampal slices stimulated with L-glutamate or high K+ and then allowed to rest for the indicated times. Probing with anti-amphiphysin I antibody recognizing the N terminus demonstrated the presence of three truncated forms of amphiphysin I. Arrow, fourth truncated form of amphiphysin I. (B) Time-dependent cleavages of amphiphysin I and α-spectrin after high K+ stimulation of hippocampal slices for the indicated times. The lowest panel shows the quantitative analysis of the expression changes of FL amphiphysin I and α-spectrin after high K+ stimulation. The expression levels of both proteins of the control (time=0 min) were set at 1. (C) Subcellular fractionation of μ- and m-calpain from cerebral cortex as shown by probing with anti-μ- and m-calpain antibodies. Anti-synaptotagmin antibodies were used to monitor synaptosomal fractions. H, total homogenate; P2, crude synaptosomal pellet; LP2, membrane fraction of purified synaptosomes; LS2, cytosol fraction of purified synaptosomes. (D) Anti-amphiphysin I immunoblotting showing that calpain inhibitors, ALLM (AM) or ALLN (AN), blocked high K+-induced amphiphysin I cleavage. (E) Anti-amphiphysin I immunoblotting showing the in vitro cleavage of recombinant amphiphysin I (0.5 μg/μl) by various concentrations of recombinant m-calpain.
Identification of the cleavage sites of amphiphysin I by calpain
To determine whether calpain cleaved the N- or C terminus of amphiphysin I, two different anti-amphiphysin I antibodies (designated antibody I and II), which recognized the N- and the C terminus, respectively, were used to detect the truncations in vitro (Figure 2A). Antibody I detected three truncated forms (Figure 2A). In contrast, antibody II detected a truncated form of amphiphysin I with molecular mass of 76 kDa. CBB staining showed three truncations of amphiphysin I and the molecular masses of the truncations on SDS–PAGE agreed with those of amphiphysin I truncations detected with antibody I. However, obvious truncations of amphiphysin I corresponding to the 76-kDa form was not observed on CBB-stained gels. Moreover, antibody II did not detect any truncated forms in the hippocampal slices after high K+ stimulation (Figure 2B). These results suggest that the truncated forms of amphiphysin I are N-terminal fragments, and C-terminal fragments may be degraded after high K+ stimulation. The precise cleavage sites of amphiphysin I were identified by mass spectrometric analysis (MS). Interestingly, MS showed that the protein was cleaved at nine sites of the C terminus (positions 333, 377, 392, 454, 478, 527, 531, 593 and 609) (Figure 2C and Supplementary Figures 2 and 3). Figure 2C shows a comparison of the molecular weights of the truncated forms of amphiphysin I cleaved in vitro by calpain and the recombinant proteins of each truncated form (1–333, 1–377 and 1–392) identified by MS. The molecular weights of the cleaved forms of amphiphysin I agreed with those of the recombinant proteins. These results suggest that positions 333, 377 and 392 correspond to the truncation sites after high K+ stimulation among the cleavage sites identified by MS.
Figure 2.
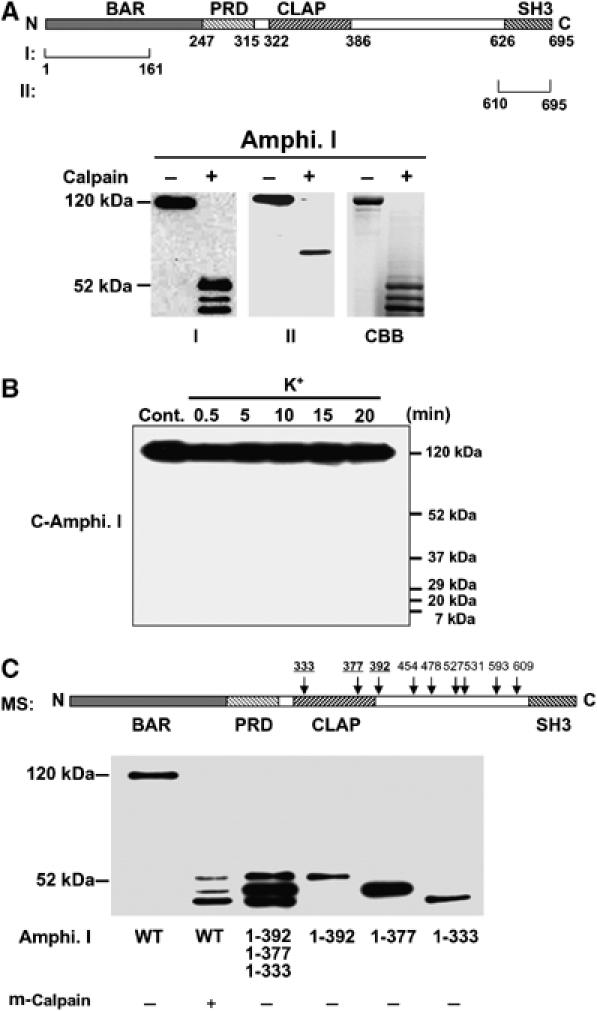
Sites of cleavage of amphiphysin I by calpain. (A) Upper panel, schematic diagram of human amphiphysin I and the recognition sites of each anti-amphiphysin I antibody (recognizing the N terminus (I) or C terminus (II)). Lower panel, Western blotting showing the m-calpain (7 nM)-induced truncation products of recombinant amphiphysin I recognized by anti-amphiphysin I antibodies I and II. CBB staining showing amphiphysin I was mainly cleaved to three fragments corresponding to the truncated products detected by antibody I. (B) No truncated form of amphiphysin I was detected by antibody II in high K+-treated hippocampal slices. (C) Three major truncations of amphiphysin I. Upper panel, scheme of the cleavage sites by m-calpain identified by MS analysis. Lower panel, comparison of the molecular weights of amphiphysin I cleaved in vitro by m-calpain with those of the recombinant proteins of each truncation form identified by MS.
Interaction of amphiphysin I with liposomes and dynamin I inhibits the cleavage by calpain
Amphiphysin I is associated with synaptic plasma membrane through the BAR domain (Zhang and Zelhof, 2002; Peter et al, 2004). This phenomenon can be reconstituted as a tubulation of liposomes in a cell-free system (Figure 3A). Moreover, the protein binds dynamin I via the SH3 domain and coassembles with dynamin I into rings either in solution or on lipid tubules (Figure 3A). We examined whether the association of amphiphysin I with liposomes and/or dynamin I affected the truncations by calpain. Amphiphysin alone was completely cleaved to three truncated forms by calpain (Figure 3B, lane 5). In contrast, amphiphysin I bound to liposomes was resistant to cleavage by calpain regardless of the binding with dynamin I (Figure 3B, lanes 2 and 3). Interaction with dynamin I inhibited the production of two of the truncated forms of amphiphysin I with molecular masses of 47 and 43 kDa (Figure 3B, lane 6). On the other hand, dynamin I was not cleaved by calpain under various conditions (Figure 3C). To investigate whether liposomes influenced calpain activity, calpain cleavage of calcineurin, a good substrate of calpain (Wu et al, 2004), was examined in the presence of liposomes. Calpain cleaved calcineurin as efficiently in the presence of liposomes as in their absence, suggesting that liposomes have no effect on calpain activity (Figure 3D). These results suggest that the association of amphiphysin I with synaptic membranes and dynamin may be crucial for the regulation of its cleavage by calpain. The interaction of amphiphysin I with dynamin I is strictly regulated by phosphorylation and dephosphorylation (Tomizawa et al, 2003; Liang et al, 2007). Phosphorylation of amphiphysin I by Cdk5 inhibits the interaction with endocytic proteins, whereas calcineurin-dependent dephosphorylation of amphiphysin I induces this interaction (Tomizawa et al, 2003). It was next examined whether calpain-dependent cleavage of amphiphysin I was regulated by phosphorylation and dephosphorylation. A phospho-mimic mutant of amphiphysin I (5D) in which five sites (S261, S272, S276, S285 and T310) phosphorylated by Cdk5 were replaced with Asp was cleaved by m-calpain the same way as dephospho-amphiphysin I in vitro (Figure 3E). However, preincubation with FK506, a potent calcineurin inhibitor, increased the levels of high K+-stimulated amphiphysin I cleavages in hippocampal slices compared with roscovitine, a potent Cdk5 inhibitor (Figure 3F). These results suggest that phosphorylation of amphiphysin I does not directly influence the cleavage of amphiphysin I by calpain, but that the phosphorylation inhibits the formation of a complex with endocytic proteins and synaptic vesicle membranes, resulting in the induction of cleavage of amphiphysin I by calpain during hyperexcitation.
Figure 3.
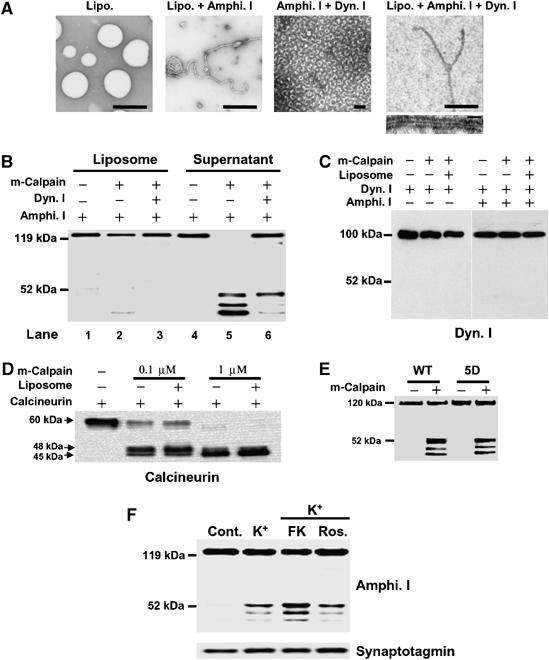
Interaction of amphiphysin I (Amphi. I) with liposomes and dynamin I (Dyn. I) inhibits the cleavage by calpain. (A) Morphological analysis of the interaction of amphiphysin I with liposomes and dynamin I at the electron microscopic level. Amphiphysin I recruits dynamin I onto liposomes, forming collars on tubulated liposomes. Bars in upper panels, 2 μm, bar in lower panel, 100 nm. (B) Effects of the interaction of amphiphysin I with liposomes and dynamin I on its cleavage by calpain are shown by anti-amphiphysin I immunoblotting of liposome and liposome-free fractions of the incubation mixture. (C) Anti-dynamin I immunoblotting shows that dynamin I was not cleaved by calpain in the presence or absence of either liposomes or amphiphysin I. (D) The effect of liposomes on calpain activity was examined by incubating purified calcineurin in the presence or absence of liposomes. Calcineurin cleavage by calpain was examined by Western blotting with anti-calcineurin antibodies. (E) Comparison of calpain-dependent cleavage between WT amphiphysin I and its phospho-mimic mutant form in vitro. For the mutant, Cdk5 phosphorylation sites (S261, S272, S276, S285 and T310) of amphiphysin I were replaced with Asp (5D). Both types of recombinant amphiphysin I were incubated with 2 nM m-calpain for 30 min. (F) The effect of phosphorylation of amphiphysin I on its cleavage by calpain was examined in hippocampal slices treated with either FK506 (FK), an inhibitor of calcineurin, or roscovitine (Ros.), a Cdk5 inhibitor. Hippocampal slices were preincubated with and without 50 μM FK or 10 μM Ros. for 30 min at 30°C and were then treated with 80 mM KCl for 15 min. Homogenized slices were analyzed by immunoblotting with anti-amphiphysin I and anti-synaptotagmin antibodies.
Amphiphysin I cleaved by calpain and a truncated form (1–392) are capable of tubulation with liposomes, but do not form ring structures with dynamin I
To investigate whether calpain-dependent cleavage of amphiphysin I affects its function, the interactions of amphiphysin I with liposomes and dynamin I were examined in vitro (Figure 4). Liposomes formed tubules with cleaved amphiphysin I (Figure 4Ac) just as they did with full-length (FL) amphiphysin I (Figure 4Aa). Recombinant protein corresponding to one of the truncated forms (1–392) also underwent tubule formation with liposomes (Figure 4Ad). These results suggest that amphiphysin I cleaved by calpain may be capable of binding to synaptic membranes. On the other hand, amphiphysin I cleaved by calpain (Figure 4Ag) or the recombinant protein corresponding to the truncated form (Figure 4Ah) did not form ring structures with dynamin I, whereas FL amphiphysin I did form small rings (Figure 4Ae and f). Moreover, when incubated with liposomes and dynamin I, the truncated form failed to form the thick helical collars on tubules (Figure 4Aj), which are formed with FL amphiphysin I (Figure 4Ai). These results indicate that calpain-dependent truncation of amphiphysin I inhibits the interaction with dynamin I. Since both dynamin ring structures and thick helical collars on tubules are required for the formation of endocytic synaptic vesicles, our findings suggest that both of these processes are inhibited following calpain cleavage of amphiphysin I during hyperexcitation.
Figure 4.
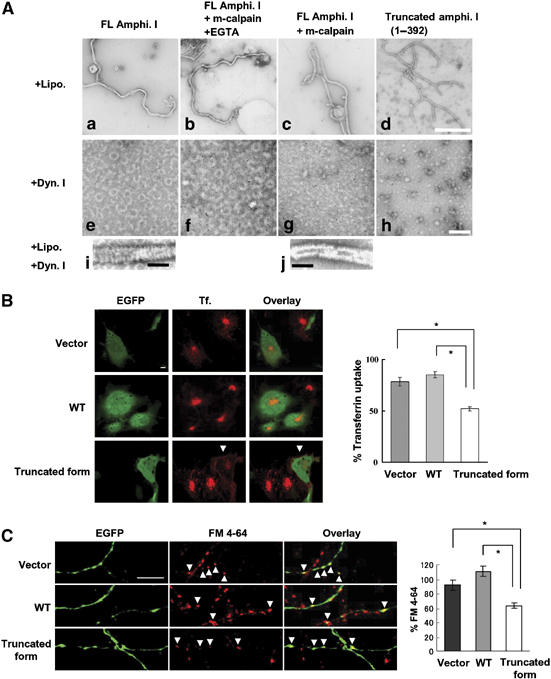
Amphiphysin I cleaved by calpain and a truncated form (1–392) are capable of tubulation with liposomes, but do not form ring structures with dynamin I (A: a–j), and inhibitory effect of calpain-dependent truncated form of amphiphysin I on transferrin uptake in Cos-7 cells (B) and FM 4–64 labeling in primary culture of hippocampal neurons (C). (A: a–d) The effect of truncation on the ability of amphiphysin I to form tubules with liposomes. As a chelator of Ca2+, EGTA inhibits calpain activity. (A: e–h) The effect of both forms of amphiphysin I on the formation of ring structures with dynamin I. (A: i, j) Comparison of helical collars on the tubules of FL and truncated amphiphysin I when incubated with dynamin I plus liposomes. Bars in A: d and h, 500 nm; bars in A: i and j, 100 nm. (B) Confocal laser scanning micrographs of: left panel, EGFP expression on COS-7 cells transfected with pIRES2-EGFP vector only (Vector), the vector containing wild-type amphiphysin I cDNA (WT) and the vector containing truncated amphiphysin I (1–392) cDNA (truncated form); middle panel, transferrin (Tf.) uptake in similarly transfected cells exposed to transferrin 24 h later; and, right panel, showing the overlay of EGFP and transferrin uptake. Transferrin uptake was inhibited in cells transfected with truncated amphiphysin I (arrows); bar, 10 μm. For quantification of the results, transferrin uptake was calculated as the mean fluorescence density for 315–660 cells under each condition and the mean values (±s.e.m.) were expressed as a percentage of normal transferrin uptake in untransfected cells. *P<0.01. (C) Fluorescence imaging micrographs of: left panel, EGFP expression on presynapses of neurons transfected with vector, WT amphiphysin I and the truncated form (amphi. 1–392); middle panel, subtracted images of FM 4–64 (loading minus unloading); right panel showing overlay of EGFP expression and FM 4–64 labeling. Bar, 10 μm. FM 4–64 labeling was inhibited in boutons transfected with truncated amphiphysin I (arrows). Right graph shows the results of quantitative analysis. FM 4–64 labeling was calculated as the integral fluorescence intensity for 47–55 boutons under each condition and the mean values (±s.e.m.) were expressed as a percentage of normal FM 4–64 labeling in untransfected boutons. *P<0.01.
We further investigated whether calpain-dependent cleavage of amphiphysin I influenced clathrin-mediated endocytosis in cells. Wild-type (WT) amphiphysin I and one of its truncated forms (1–392) were overexpressed in Cos-7 cells, and transferrin uptake in the cells was examined (Figure 4B). Transferrin uptake in the truncated amphiphysin I-overexpressing cells was inhibited by 61% compared to that in WT amphiphysin I-overexpressing cells (Figure 4B). Moreover, the results were confirmed in rat hippocampal neurons. We investigated whether calpain-dependent cleavage of amphiphysin I affected presynaptic vesicle endocytosis in rat hippocampal neurons. FM 4–64 labeling in the truncated amphiphysin I-overexpressing boutons was inhibited by 57% compared to that in WT amphiphysin I-overexpressing boutons. These results indicate that calpain-dependent cleavage of amphiphysin I inhibits clathrin-mediated synaptic vesicle endocytosis.
Amphiphysin I cleavage by calpain inhibits excessive synaptic transmission in hippocampal slices and in vivo
Repetitive, high-frequency electrical nerve stimulation (e.g., 10 Hz) causes short-term synaptic depression in hippocampal neurons and neuromuscular junctions (Figure 5C and Wu and Betz, 1998) mainly because synaptic vesicles are depleted (Wang and Kaczmarek, 1998). Clathrin-mediated endocytosis is inhibited during frequency-dependent depression, resulting in the depletion of the recycling pool of synaptic vesicles (Wu and Betz, 1998; Wu, 2004). These data along with the present results led us to hypothesize that amphiphysin I is cleaved by calpain during HFS, and that the cleavage causes high frequency-induced depression of synaptic transmission through the inhibition of clathrin-mediated endocytosis. To test this hypothesis, we examined whether calpain-dependent cleavage of amphiphysin I was involved in the depression of the field EPSP (fEPSP) at the synapses from Schaffer collaterals onto CA1 pyramidal cells in hippocampal slices. HFS (10 Hz) induced amphiphysin I cleavage, whereas low-frequency stimulation (LFS, 0.03 Hz) produced no cleavage (Figure 5A). Blocking calpain with ALLM inhibited the HFS-induced cleavages. Thus, calpain cleaves amphiphysin I during repetitive HFS in hippocampal slices. Perfusion of ALLM for 30 min had no effect on fEPSP slope when slices were stimulated with LFS (Figure 5B). In contrast, ALLM inhibited HFS-induced fEPSP depression (Figure 5C and D). To clarify whether the effect of calpain inhibition on the fEPSP depression is exerted in the presynapse or in the postsynapse, the effect of ALLM on paired-pulse facilitation (PPF) was examined in hippocampal slices. PPF is a transient form of presynaptic plasticity, in which the second of two closely spaced stimuli elicits enhanced transmitter release, because of residual calcium ions in the presynaptic terminal after the first stimulus (Zucker, 1989). PPF at 100 and 150 ms interpulse intervals was decreased after HFS (10 Hz) compared with that after LFS (Figure 5E). In the ALLM-treated slices, in contrast, PPF after HFS was not decreased compared with that after LFS (Figure 5E). These results suggest that cleavages of amphiphysin I by calpain may partially account for the HFS-induced depression of synaptic transmission through the inhibition of clathrin-mediated endocytosis of synaptic vesicles.
Figure 5.
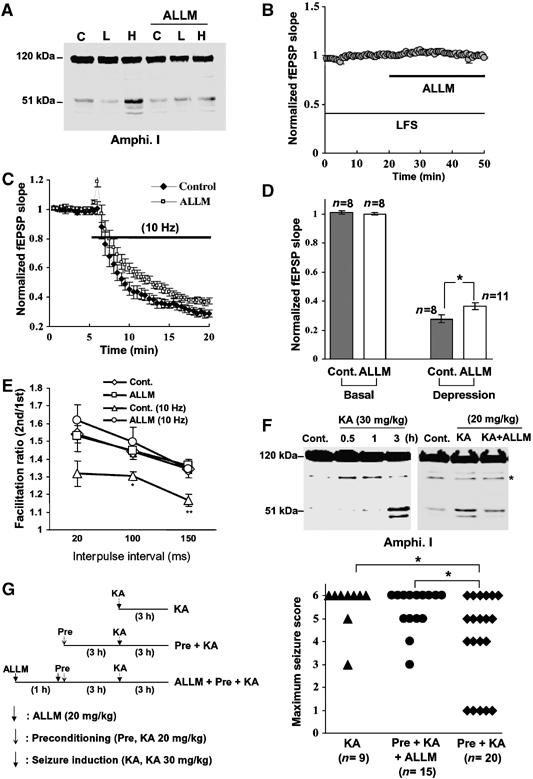
Amphiphysin I cleavage by calpain inhibits excessive synaptic transmission in hippocampal slices and in vivo. (A) Anti-amphiphysin I immunoblot showing the effects of either no electrical stimulation (C), LFS (L, 0.03 Hz) or HFS (H, 10 Hz) on calpain-dependent amphiphysin I cleavage in mouse hippocampal slices, without or with ALLM preincubation. (B) ALLM inhibition of calpain had no effect on fEPSP slope during LFS. (n=7) (C) Effect of calpain inhibition (ALLM) on fEPSP depression induced by HFS. Control, n=8; ALLM, n=11. (D) Comparison of fEPSP slopes at 15 min after LFS and HFS in the ALLM-perfused slices and controls. *P<0.05. (E) PPF in hippocampal slices following LFS is reduced following HFS, an effect that is blocked by inhibiting calpain with ALLM. ALLM had no effect on PPF following LFS. The data represent the ratios of the second EPSP slopes versus the first fEPSP slopes separated by the given intervals. As a control, hippocampal slices were incubated with DMSO instead of ALLM. *P<0.05, **P<0.01 compared with Cont. n=10–15. (F) Anti-amphiphysin I immunoblot showing the ability of KA injections to induce calpain-dependent amphiphysin I cleavage in the hippocampus of FVB/NJ mice. Preincubation with ALLM inhibited the KA-induced cleavage of amphiphysin I. *Nonspecific band. (G) Preconditioning with low-dose KA prevented KA-induced development of seizures in the FVB/NJ mice and ALLM inhibited the preconditioning effect. Left panel, schematic time schedule of the animal experiments. Right panel, the maximum seizure score in mice of each group. Seizure scores were monitored for 3 h after KA (30 mg/kg) injection as described in ‘Methods'. *P<0.05.
Excessive synaptic activity is the primary cause of seizures and seizure-induced brain damage, and many drugs inhibiting the neurotransmitter release machinery in epileptogenic areas are thought to be effective anti-ictal agents (Okada et al, 2002; Costantin et al, 2005). Intraperitoneal injection of KA in FVB/NJ mice induces limbic motor seizures originating in the hippocampus (Lothman and Collins, 1981; Ben-Ari, 1985). Finally, we investigated whether amphiphysin I was cleaved by calpain during hyperexcitation in the hippocampus of KA-injected FVB/NJ mice. Obvious cleavage of amphiphysin I was observed 3 h after 30 and 20 mg/kg KA injection, and administration of ALLM inhibited the KA-induced amphiphysin I cleavage, suggesting that calpain cleaves amphiphysin I during hyperexcitation in vivo (Figure 5F). To clarify the physiological function of amphiphysin I cleavage during neural hyperexcitation, the effect of amphiphysin I cleavage on KA-induced seizure development was examined. KA injection (30 mg/kg) induced seizure behavior in FBV/NJ mice (Figure 5G). The maximum seizure score was stage 6 in almost all KA-injected mice (75%) (Figure 5G). Preconditioning with low-dose KA injection (20 mg/kg) induced amphiphysin I cleavages in the mice (Figure 5F). Preconditioning 3 h before KA injection (30 mg/kg) reduced the KA-induced seizure development (Figure 5G), so that only 30% of the mice developed stage 6 seizures. Moreover, administration of ALLM attenuated the preconditioning effect, so that 60% of the mice developed stage 6 seizures (Figure 5G). These results suggest that amphiphysin I may be cleaved by calpain during neural hyperexcitation in vivo and the cleavage may inhibit excessive neurotransmitter release.
Discussion
In the present study, we found a novel posttranslational regulation of an endocytic protein, amphiphysin I, during neural hyperexcitation. Phosphorylation and dephosphorylation of endocytic proteins are well-known post-translational regulatory mechanisms involved in clathrin-mediated endocytosis in the normal physiological state (Cousin and Robinson, 2001). In contrast, the present results showed that amphiphysin I was not cleaved by calpain under normal physiological conditions. The protein was cleaved during neural hyperexcitation and its cleavage was regulated by its interaction with other endocytic proteins or the cell membrane. We showed that amphiphysin I is cleaved by calpain during high potassium-induced hyperexcitation in rat hippocampal slices, and the cleavage blocked the interaction of amphiphysin I with dynamin I, a key molecule in the fission of clathrin-coated buds from presynaptic membrane through its GTPase activity, in vitro. In addition, the N-terminal fragments of amphiphysin I still retained the ability to bind to liposome membranes after the calpain proteolysis, suggesting a dominant-negative effect of amphiphysin I cleavage by calpain on clathrin-mediated synaptic vesicle endocytosis during hyperexcitation. This was proven by the inhibition of clathrin-mediated endocytosis of transferrin and synaptic vesicles by overexpression of the amphiphysin I truncation product containing amino acids 1–392 in Cos-7 cells and hippocampal neurons. The cleavage of amphiphysin I by calpain is an irreversible process, in contrast to phosphorylation and dephosphorylation. Therefore, the dysfunction of amphiphysin I and its dominant-negative effect resulting from the cleavage are maintained for a long period, resulting in long-lasting inhibition of clathrin-mediated endocytosis and synaptic transmission.
The function of cleavage of amphiphysin I by calpain during neural hyperexcitation may be to inhibit neural hyperexcitability and to protect neurons from neural damage and neuronal cell death. Although the role of synaptic vesicle endocytosis is thought to be to maintain the recycling synaptic vesicle pool for exocytosis during repetitive high-frequency nerve stimulation, previous studies have shown that endocytosis is inhibited during high-frequency stimulation-induced depression (Sun et al, 2002; Wu, 2004). These results suggest that the synaptic depression induced by some presynaptic mechanisms may be important for neuronal auto-protection against neural hyperexcitation, and inhibition of synaptic vesicle endocytosis may play a part in the mechanism. The present study showed that cleavage of amphiphysin I by calpain was induced by HFS in hippocampal slices, and ALLM inhibited the HFS-induced fEPSP depression and blocked the depression-induced inhibition of PPF. We further confirmed this phenomenon in vivo. Preconditioning with low-dose KA induced calpain-related amphiphysin I cleavage and reduced the development of seizures in FVB/NJ mice. However, administration of ALLM attenuated the preconditioning effect. Although in vivo studies did not directly show that calpain regulated assembly of the fission complex of clathrin-mediated synaptic vesicles during hyperexcitation, these results suggest that hyperexcitation-induced amphiphysin I cleavage may play a role in fEPSP depression, which is thought to be a neuroprotective mechanism. Taken together, both the in vivo and in vitro studies showed that hyperexcitation-induced amphiphysin I cleavage plays a role in protecting neurons from neuroexcitotoxicity during hyperexcitation.
There is an SH3 domain in the C terminus of amphiphysin I. The SH3 domain is a potent inhibitor of synaptic vesicle endocytosis (Shupliakov et al, 1997). We first speculated that the calpain cleavage-dependent C-terminal fragment of amphiphysin I was involved in the inhibition of synaptic vesicle endocytosis during hyperexcitation. However, antibody II did not detect any C-terminal fragments of amphiphysin I. Moreover, MS identified nine sites in the C terminus of amphiphysin I as the cleavage sites targeted by calpain. These results suggest that amphiphysin I may undergo multiple cleavages at the C terminus by calpain, and that the C-terminal fragments are rapidly degraded after the cleavage. Finally, the three N-terminal fragments remain and inhibit synaptic vesicle endocytosis through the inhibition of the interaction with synaptic membranes and dynamin.
Both m- and μ-calpain were expressed in the presynaptic terminus. In vitro cleavage studies showed that m-calpain cleaved amphiphysin I to three fragments corresponding to the truncations observed in high K+-treated hippocampal slices. In contrast, μ-calpain cleaved the protein to four fragments. These results suggest that m-calpain may be involved in amphiphysin I cleavage during neural hyperexcitation.
A number of previous studies showed that changes in calpain activity are related to the pathophysiology of neurodegenerative disorders, such as neuroexcitotoxicity, cerebral ischemia and traumatic brain injury (Posmantur et al, 1996; Rami, 2003; Wu et al, 2004). The elevation of intracellular Ca2+ levels during the course of injuries and diseases of the central nervous system causes overactivation of calpain, promoting degradation of key cytoskeletal and membrane proteins, and leading to neuronal cell death (Czogalla and Sikorski, 2005). To date, the key proteins shown to be cleaved by calpain are localized in the postsynapse. Although the reason for the difference in calpain function presynapse versus postsynapse during neural hyperexcitation remains unclear, specific inhibition of calpain in the postsynapse may exert a potent neuroprotective effect.
Materials and methods
Western blotting analysis
Monoclonal anti-amphiphysin I antibodies (I and II) were provided by P De Camilli (Yale University, New Haven, CT). Monoclonal anti-amphiphysin I antibody recognizing the N terminus was purchased from Lab Frontier (LF-MA0047, Seoul, Korea). Polyclonal anti-μ-calpain antibody was from Merck (San Diego, CA), monoclonal anti-m-calpain antibody was from Sigma-Aldrich (Clone 107–82, St Louis, MO), polyclonal anti-calcineurin A antibody was from StressGen Biotech (SPA-610, Victoria, BC, Canada), the polyclonal anti-α-spectrin II antibody was from Santa Cruz Biotechnology (Sc-7465, Santa Cruz, CA); the monoclonal anti-synaptotagmin I antibody was from BD Transduction Laboratories (S39520, San Jose, CA) and polyclonal anti-dynamin I antibody was from Santa Cruz Biotechnology (Sc-6402). Western blotting analysis was performed as described previously (Tomizawa et al, 2002). After incubation with the appropriate secondary antibody conjugated with horseradish peroxidase (Sigma-Aldrich), positive bands were visualized using an enhanced chemiluminescence detection system (Amersham Biosciences, Pittsburgh, PA).
Preparation of hippocampal slices and electric stimulation
Hippocampal slices was prepared as described previously (Tomizawa et al, 2002). Briefly, the hippocampi of male C57BL/6 mice aged 7–8 weeks were dissected, and 300–350 μm transverse slices were prepared. For high K+ stimulation, after equilibration for more than 2 h at room temperature in oxygenated (95% O2, 5% CO2) ACSF solution containing (in mM): 125 NaCl, 26 NaHCO4, 11 glucose, 2.5 KCl, 1.25 NaH2PO4, 2 CaCl2, 1.3 MgSO4, slices were treated with 80 mM potassium solution containing (in mM): 47.5 NaCl, 26 NaHCO4, 11 glucose, 80 KCl, 1.25 NaH2PO4, 2 CaCl2, 1.3 MgSO4 at 32°C. For glutamate stimulation, the slices were incubated with ACSF in the presence of 500 μM L-glutamate (Sigma-Aldrich) for 15 min. The slices were then washed with ACSF and were further incubated for 15 or 45 min. After the reaction, the slices were sonicated in 1% SDS and then boiled for 3 min before Western blot analysis. For calpain inhibition studies, slices were preincubated in either 50 μM ALLM (AM) or ALLN (AN) for 30 min and then stimulated with high K+ in the presence of ALLM or ALLN for 15 min. A Panasonic MED 64 System (Alpha MED Sciences, Osaka, Japan) was used for electrical stimulation. After equilibration for more than 2 h at room temperature, the slices were transferred to the MED probe and fixed with a slice anchor. The slices were stimulated along the Schaffer collaterals and field excitatory postsynaptic potentials (fEPSPs) were recorded in CA1. The intensity of the stimulation was adjusted to produce an fEPSP with a slope of 30–40% of the maximum. For Western blotting analysis, the CA1 region was cut out of the slices and sonicated in 1% SDS buffer.
Preparation of recombinant amphiphysin I and its truncated constructs, and purification of dynamin I
The cDNAs encoding FL human amphiphysin I and its truncation constructs corresponding to amino-acid sequences 1–333, 1–377 and 1–392 were prepared by PCR amplification using specific primers. FL and truncated forms of amphiphysin I cDNAs were subcloned into pGEX-6P vector as BamHI–EcoRI fragments. The expression of GST-fusion proteins was induced by 0.1 mM isopropyl-1-thio-D-galactopyranoside at 25°C for 12 h in LB medium supplemented with 100 μg/ml ampicillin at A600=0.8. The purification of GST-fusion proteins was performed as described previously (Yoshida et al, 2004), and the cleavage of GST with PreScission protease was carried out according to the manufacturer's instructions. Finally, the protein was purified on a Mono Q column equilibrated in 20 mM Tris–HCl (pH 7.7) and 0.2 M NaCl. The protein solution was stored at −80°C, and thawed at 37°C before use.
Dynamin I was purified from bovine brain essentially as described previously (Liu et al, 1994).
Preparation of synaptosomes
Synaptic fractions were prepared from 7-week-old Wistar rat brains as described previously (Tomizawa et al, 2002). Expression of calpain I (μ-) and II (m-) in each fraction was analyzed by Western blotting analysis using specific antibodies.
Preparation of liposomes
Unilamellar liposomes in 0.3 M sucrose (1 mg/ml) were prepared as described previously (Takei et al, 1999).
In vitro proteolysis of amphiphysin I by calpain
Recombinant amphiphysin I (0.5 μg/μl) was incubated with 4–40 nM recombinant m-calpain (Merck) in reaction buffer (50 μl) containing 40 mM Tris–HCl (pH 7.5), 12 mM CaCl2 at 25°C for 30 min. The reaction was stopped by the addition of SDS–PAGE sample buffer, and the reaction mixture was then separated by electrophoresis on a 10% SDS–PAGE gel. The cleavage of amphiphysin I was analyzed by Western blotting with monoclonal anti-amphiphysin I antibody recognizing the N terminus. To investigate the effect of amphiphysin I interaction with liposome or dynamin I on calpain-dependent amphiphysin I cleavage, 0.3 μg/μl liposome or dynamin I was incubated with 0.3 μg/μl amphiphysin I in 20 μl of cytosolic buffer containing 25 mM HEPES–KOH (pH 7.2), 25 mM KCl, 2.5 mM magnesium acetate, 100 mM potassium L-glutamate at 37°C for 15 min, and 7 nM calpain and 12 mM CaCl2 were then added and the mixture was further incubated for 30 min at 25°C. The reaction mixture was then centrifuged at 20 600 g for 10 min, and the proteins in the pellet (liposome fraction) and the supernatant (liposome-free fraction) were analyzed by Western blotting with anti-N-terminal amphiphysin I and anti-dynamin I antibodies.
Mass spectrometry
Mass spectrometry was performed as described previously (Wu et al, 2004). Briefly, standard peptides, angiotensin III and oxidized insulin B chain, were obtained from Sigma-Aldrich. For MALDI-TOF/MS analysis, 2,5-dihydroxybenzoic acid (Wako, Osaka, Japan) was used as a matrix. The water used for all experiments was purified using a MilliQ UV plus water purification system (Millipore, Bedford, MA). Sequencing-grade unmodified trypsin, Glu-C and Asp-N were obtained from Roche Diagnostics (Mannheim, Germany), Lys-C was from Wako (Osaka, Japan) and n-octyl glucoside was obtained from Dojin (Kumamoto, Japan). MALDI-TOF/MS spectra were obtained using a Voyager linear DE or oMALDI-QSTAR pulsar I instrument (Applied Biosystems, Foster City, CA) operated in delayed-extraction mode. The spectra were calibrated using internal standards, angiotensin III and oxidized insulin B chain.
Electron microscopy
Amphiphysin I, liposomes, dynamin I or truncation constructs of amphiphysin I were mixed under the same conditions described in Methods for ‘In vitro proteolysis of amphiphysin I'. For negative staining, samples were treated as described previously (Yoshida et al, 2004) and then observed with a Hitachi H-7100 transmission electron microscope (Tokyo, Japan) at the Central Research Laboratory at Okayama University Medical School.
Transferrin uptake assays
Cos-7 cells were preincubated for 30–60 min in serum-free DMEM (Invitrogen, Carlsbad, CA) at 37°C and then incubated in 25 μg/ml of Alexa Fluor® 546-conjugated transferrin (Invitrogen) for 30 min. The cells were washed in PBS (without Ca2+ and Mg2+) buffer and then transferrin uptake was monitored under a confocal laser microscope (FluoView FV300, Olympus, Tokyo, Japan). To quantify the results, signals were estimated in 21–33 randomly chosen fields each with 15–20 cells with EGFP overexpression from five separate experiments.
Hippocampal neuron culture and FM 4–64 labeling
Hippocampal neuron culture was performed as described previously (Tomizawa et al, 2003). For transfection of amphiphysin I cDNAs, FL and truncated forms of amphiphysin I cDNAs were subcloned into IRES2-EGFP vector (BD Biosciences). Neuronal transfection of various plasmids including WT amphiphysin I, IRES2-EGFP vector and a truncated form of amphiphysin I (1–392) was carried out using the Nucleofector® system (Amaxa Biosystems, Gaithersburg, MD) according to the manufacturer's instructions.
FM-dye imaging was performed as described previously (Tomizawa et al, 2003). Briefly, to load FM 4–64 dye into synaptic vesicles, field stimulation at 20 Hz, 30 s, duration 1.0 ms and intensity 20 V was delivered to the culture through a parallel platinum–iridium electrode immersed into the perfusion chamber. FM 4–64 dye (10 μM, Invitrogen) was present in the perfusion solution from 40 s before stimulation to 40 s after stimulation. After FM 4–64 loading, the culture was rinsed with dye-free perfusion solution for 10 min and fluorescence imaging was performed. An unloading stimulation (10 Hz, 1 min) was delivered to the culture in the dye-free solution to unload the previously loaded dye.
Optical imaging experiments were performed with a Zeiss Axiovert 200 inverted microscope equipped with a CCD camera (Hamamatsu Photonics, Hamamatsu, Japan). Fluorescence images were acquired and analyzed with AquaCosmos software and processed with Photoshop CS and NIH image J software. Images obtained after unloading were used for background subtraction. The intensity of FM 4–64 fluorescence was quantified by measuring the integral FM 4–64 fluorescence intensity of active boutons on subtracted images (loading minus unloading). To compare the results, FM 4–64 fluorescence signals were estimated in 47–55 boutons with EGFP overexpression from five separate experiments.
Animal experiments
Male FVB/NJ mice (25.0–30.0 g, 9 weeks old) were housed individually on a 12-h light/dark schedule with free access to food and water. KA (TOCRIS, Ellisville, MO) and N-acetyl-Leu-Leu-methional (ALLM; Merck) were administered by intraperitoneal injection. For preconditioning, the mice were injected with 20 mg/kg KA 3 h before 30 mg/kg KA injection. ALLM (20 mg/kg) was injected 1 h before the preconditioning and once more simultaneously at preconditioning. Mice were continuously monitored and seizures were scored for 3 h according to a previously defined seizure rating scale (Racine, 1972): stage 0, normal; stage 1, immobility; stage 2, forelimb and/or tail extension, rigid posture; stage 3, repetitive movements, head bobbing; stage 4, rearing and falling; stage 5, continuous rearing and falling; stage 6, severe tonic–clonic seizures (jumping). For Western blotting of calpain-dependent amphiphysin I cleavages, hippocampi were obtained from KA-injected mice at each time.
Statistical analysis
Values are reported as the mean±s.e.m. Data were analyzed using Student's t-test to compare two conditions and one-way ANOVA or the Kruskal–Wallis test to compare multiple conditions; P<0.05 was considered to be significant.
Supplementary Material
Supplementary Figure 1
{kind=link}
Supplementary Figure 2
Supplementary Figure 3
Supplementary Figure Legends
Acknowledgments
We thank P De Camilli for providing amphiphysin I antibodies. This work was supported by the Research Program on Development of Innovative Technology from the Japan Science and Technology Agency (JST), by a grant from the New Energy and Industrial Technology Development Organization (NEDO) and by a Grant-in-Aid for Scientific Research on Priority Areas ‘Membrane Traffic' from the Ministry of Education, Science, Sports and Culture of Japan.
Competing interests statement The authors declare that they have no competing financial interests.
References
- Ben-Ari Y (1985) Limbic seizure and brain damage produced by kainic acid: mechanism and relevance to human temporal lobe epilepsy. Neuroscience 14: 375–403 [DOI] [PubMed] [Google Scholar]
- Costantin L, Bozzi Y, Richichi C, Viegi A, Antonucci F, Funicello M, Gobbi M, Mennini T, Rossetto O, Montecucco C, Maffei L, Vezzani A, Caleo M (2005) Antiepileptic effects of botulinum neurotoxin E. J Neurosci 25: 1943–1951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cousin MA, Robinson PJ (2001) The dephosphins: dephosphorylation by calcineurin triggers synaptic vesicle endocytosis. Trends Neurosci 24: 659–665 [DOI] [PubMed] [Google Scholar]
- Czogalla A, Sikorski AF (2005) Spectrin and calpain: a ‘target' and a ‘sniper' in the pathology of neuronal cells. Cell Mol Life Sci 62: 1913–1924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evergren E, Marcucci M, Tomilin N, Low P, Slepnev V, Andersson F, Gad H, Brodin L, De Camilli P, Shupliakov O (2004) Amphiphysin is a component of clathrin coats formed during synaptic vesicle recycling at the lamprey giant synapse. Traffic 5: 514–528 [DOI] [PubMed] [Google Scholar]
- Granseth B, Odermatt B, Royle SJ, Lagnado L (2006) Clathrin-mediated endocytosis is the dominant mechanism of vesicle retrieval at hippocampal synapses. Neuron 51: 773–786 [DOI] [PubMed] [Google Scholar]
- Lee MS, Kwon YT, Li M, Peng J, Friedlander RM, Tsai LH (2000) Neurotoxicity induces cleavage of p35 to p25 by calpain. Nature 405: 360–364 [DOI] [PubMed] [Google Scholar]
- Liang S, Wei FY, Wu YM, Tanabe K, Abe T, Oda Y, Yoshida Y, Yamada H, Matsui H, Tomizawa K, Takei K (2007) Major Cdk5-dependent phosphorylation sites of amphiphysin 1 are implicated in the regulation of the membrane binding and endocytosis. J Neurochem, in press [DOI] [PubMed] [Google Scholar]
- Liu JP, Powell KA, Sudhof TC, Robinson PJ (1994) Dynamin I is a Ca2+-sensitive phospholipid-binding protein with very high affinity for protein kinase C. J Biol Chem 269: 21043–21050 [PubMed] [Google Scholar]
- Lothman EW, Collins RC (1981) Kainic acid induced limbic seizures: metabolic, behavioral, electroencephalographic and neuropathological correlates. Brain Res 218: 299–318 [DOI] [PubMed] [Google Scholar]
- Okada M, Zhu G, Yoshida S, Kanai K, Hirose S, Kaneko S (2002) Exocytosis mechanism as a new targeting site for mechanisms of action of antiepileptic drugs. Life Sci 72: 465–473 [DOI] [PubMed] [Google Scholar]
- Peter BJ, Kent HM, Mills IG, Vallis Y, Butler PJ, Evans PR, McMahon HT (2004) BAR domains as sensors of membrane curvature: the amphiphysin BAR structure. Science 303: 495–499 [DOI] [PubMed] [Google Scholar]
- Posmantur RM, Kampfl A, Liu SJ, Heck K, Taft WC, Clifton GL, Hayes RL (1996) Cytoskeletal derangements of cortical neuronal processes three hours after traumatic brain injury in rats: an immunofluorescence study. J Neuropathol Exp Neurol 55: 68–80 [DOI] [PubMed] [Google Scholar]
- Racine RJ (1972) Modification of seizure activity by electrical stimulation. II. Motor seizure. Electroencephalogr Clin Neurophysiol 32: 281–294 [DOI] [PubMed] [Google Scholar]
- Rami A (2003) Ischemic neuronal death in the rat hippocampus: the calpain–calpastatin–caspase hypothesis. Neurobiol Dis 13: 75–88 [DOI] [PubMed] [Google Scholar]
- Saido TC, Yokota M, Nagao S, Yamaura I, Tani E, Tsuchiya T, Suzuki K, Kawashima S (1993) Spatial resolution of fodrin proteolysis in postischemic brain. J Biol Chem 268: 25239–25243 [PubMed] [Google Scholar]
- Sara Y, Mozhayeva MG, Liu X, Kavalali ET (2002) Fast vesicle recycling supports neurotransmission during sustained stimulation at hippocampal synapses. J Neurosci 22: 1608–1617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sessoms JS, Chen SJ, Chetkovich DM, Powell CM, Roberson ED, Sweatt JD, Klann E (1992) Ca2+-induced persistent protein kinase C activation in rat hippocampal homogenates. Second Messengers Phosphoproteins 14: 109–126 [PubMed] [Google Scholar]
- Shupliakov O, Low P, Grabs D, Gad H, Chen H, David C, Takei K, De Camili P, Brodin L (1997) Synaptic vesicle endocytosis impaired by disruption of dynamin-SH3 domain interactions. Science 276: 259–263 [DOI] [PubMed] [Google Scholar]
- Slepnev VI, Ochoa GC, Butler MH, Grabs D, De Camilli P (1998) Role of phosphorylation in regulation of the assembly of endocytic coat complexes. Science 281: 821–824 [DOI] [PubMed] [Google Scholar]
- Stevens CF, Wesseling JF (1999) Identification of a novel process limiting the rate of synaptic vesicle cycling at hippocampal synapses. Neuron 24: 1017–1028 [DOI] [PubMed] [Google Scholar]
- Sun JY, Wu XS, Wu LG (2002) Single and multiple vesicle fusion induce different rates of endocytosis at a central synapse. Nature 417: 555–559 [DOI] [PubMed] [Google Scholar]
- Takei K, Slepnev VI, Haucke V, De Camilli P (1999) Functional partnership between amphiphysin and dynamin in clathrin-mediated endocytosis. Nat Cell Biol 1: 33–39 [DOI] [PubMed] [Google Scholar]
- Tan TC, Valova VA, Malladi CS, Graham ME, Berven LA, Jupp OJ, Hansra G, McClure SJ, Sarcevic B, Boadle RA, Larsen MR, Cousin MA, Robinson PJ (2003) Cdk5 is essential for synaptic vesicle endocytosis. Nat Cell Biol 5: 701–710 [DOI] [PubMed] [Google Scholar]
- Tomizawa K, Ohta J, Matsushita M, Moriwaki A, Li ST, Takei K, Matsui H (2002) Cdk5/p35 regulates neurotransmitter release through phosphorylation and downregulation of P/Q-type voltage-dependent calcium channel activity. J Neurosci 22: 2590–2597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomizawa KS, Sunada S, Lu YF, Oda Y, Kinuta M, Ohshima T, Saito T, Wei FY, Matsushita M, Li ST, Tsutsui K, Hisanaga S, Mikoshiba K, Takei K, Matsui H (2003) Cophosphorylation of amphiphysin I and dynamin I by Cdk5 regulates clathrin-mediated endocytosis of synaptic vesicles. J Cell Biol 163: 813–824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang LY, Kaczmarek LK (1998) High-frequency firing helps replenish the readily releasable pool of synaptic vesicles. Nature 394: 384–388 [DOI] [PubMed] [Google Scholar]
- Wigge P, McMahon HT (1998) The amphiphysin family of proteins and their role in endocytosis at the synapse. Trends Neurosci 21: 339–344 [DOI] [PubMed] [Google Scholar]
- Wu HY, Tomizawa K, Oda Y, Wei FY, Lu YF, Matsushita M, Li ST, Moriwaki A, Matsui H (2004) Critical role of calpain-mediated cleavage of calcineurin in excitotoxic neurodegeneration. J Biol Chem 279: 4929–4940 [DOI] [PubMed] [Google Scholar]
- Wu LG (2004) Kinetic regulation of vesicle endocytosis at synapses. Trends Neurosci 27: 548–554 [DOI] [PubMed] [Google Scholar]
- Wu LG, Betz WJ (1998) Kinetics of synaptic depression and vesicle recycling after tetanic stimulation of frog motor nerve terminals. Biophys J 74: 3003–3009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida Y, Kinuta M, Abe T, Liang S, Araki K, Cremona O, Di Paolo G, Moriyama Y, Yasuda T, De Camilli P, Takei K (2004) The stimulatory action of amphiphysin on dynamin function is dependent on lipid bilayer curvature. EMBO J 23: 3483–3491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida Y, Takei K (2005) Stimulation of dynamin GTPase activity by amphiphysin. Methods Enzymol 404: 528–537 [DOI] [PubMed] [Google Scholar]
- Zhang B, Zelhof AC (2002) Amphiphysins: raising the BAR for synaptic vesicle recycling and membrane dynamics. Bin-amphiphysin-Rvsp. Traffic 3: 452–460 [DOI] [PubMed] [Google Scholar]
- Zucker RS (1989) Short-term synaptic plasticity. Annu Rev Neurosci 12: 13–31 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Figure Legends