

Abstract
Expression of activated H-Ras induces a unique form of non-apoptotic cell death in human glioblastoma cells and other specific tumor cell lines. The major cytopathological features of this form of death are the accumulation of large phase-lucent, LAMP1-positive, cytoplasmic vacuoles and increased autophagic activity. In this study we sought to determine if induction of cytoplasmic vacuolation a) depends on Ras farnesylation, b) is specific to H-Ras, and c) is mediated by signaling through the major known Ras effector pathways. We find that the unusual effects of activated H-Ras depend on farnesylation and membrane association of the GTPase. Both H-Ras(G12V) and K-Ras4B(G12V) stimulate vacuolation, but activated forms of Cdc42 and RhoA do not. Amino acid substitutions in the Ras effector domain, which are known to selectively impair its interactions with Raf kinase, class-I phosphatidylinositide 3-kinase (PI3K), or Ral nucleotide exchange factors, initially pointed to Raf as a possible mediator of cell vacuolation. However, the MEK inhibitor, PD98059, did not block the induction of vacuoles, and constitutively active Raf-Caax did not mimic the effects of Ras(G12V). Introduction of normal PTEN together with H-Ras(G12V) into U251 glioblastoma cells reduced the PI3K-dependent activation of Akt, but had no effect on vacuolation. Finally, co-expression of H-Ras(G12V) with a dominant-negative form of RalA did not suppress vacuolation. Taken together, the observations indicate that Ras activates non-conventional and perhaps unique effector pathways to induce cytoplasmic vacuolation in glioblastoma cells. Identification of the relevant signaling pathways may uncover specific molecular targets that can be manipulated to activate non-apoptotic cell death in this type of cancer.
Keywords: Ras, Non-Apoptotic Cell Death, Vacuoles, Glioblastoma, Raf, PI 3-Kinase, Farnesylation, Autophagy
1. Introduction
Oncogenic mutations in ras genes occur in approximately 30% of human malignancies [1]. In their active GTP-bound conformation, Ras proteins interact with a variety of downstream effectors that regulate cell proliferation, differentiation, and survival [2,3]. Signals are normally terminated when GTP is hydrolyzed and Ras reverts to the inactive GDP state. The mutant forms of Ras typically found in tumors harbor amino acid substitutions (e.g., G12→V) that reduce their GTPase activity [4,5]. This results in sustained stimulation of Ras pathways that promote cell proliferation and survival [6]. However, in some types of cells activated Ras can trigger cellular senescence [7], apoptosis [8] or non-apoptotic cell death [9]. The most notable example of the latter has been observed in human glioblastoma, gastric carcinoma, and neuroblastoma cells, where introduction of H-Ras(G12V) triggers accumulation of cytoplasmic vacuoles and cell death without caspase activation or DNA fragmentation. [9–11]. This form of cell death was originally described as Type-II programmed cell death [10], a term often used synonymously with autophagic cell death [12,13]. Type-II cell death has received increasing attention as a significant alternative pathway for cell death in cancer [14–18]. However, to date there have been no follow-up studies aimed at defining the signaling pathways through which activated Ras can trigger the initial cytoplasmic vacuolation that ultimately leads to loss of cellular integrity and non-apoptotic death.
In the present study we set out to determine a) if the induction of vacuolation in glioblastoma cells requires H-Ras farnesylation and membrane association, b) if the effect is specific to H-Ras, and c) if vacuolation is linked to activation of the major known Ras effector pathways. We find that the induction of vacuolation requires Ras farnesylation and is sensitive to effector domain mutations, but it does not depend on activation of the Raf, phosphatidylinositide 3-kinase (PI3K), or RalGDS signaling pathways. These observations raise the prospect that Ras may be acting through an atypical or perhaps unique effector pathway to induce vacuolation and non-apoptotic death in glioblastoma cells.
2. Materials and methods
2.1 Cell culture
U251 glioblastoma cells were obtained from the DCT Tumor Repository (National Cancer Institute, Frederick, MD, USA). Cells were maintained at 37°C with 5% CO2 in Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% heat-inactivated fetal bovine serum (FBS). Phase contrast images of the live cells were obtained using an Olympus IX70 inverted microscope equipped with a digital camera, using SPOT imaging software (Diagnostic Instruments Inc., Sterling Heights, MI, USA).
2.2 Generation of expression constructs for nucleofection
H-Ras (G12V) cDNA was purchased from Clontech (Palo Alto, CA, USA). The cDNA was subcloned into a pCMV5 vector that had been modified by PCR to encode either an in-frame myc epitope tag (MEQKLISEEDL) [19] or a FLAG epitope tag (DYKDDDDKG) [20]. Amino acid substitutions (C186S, Y40C, D38E and E37G) were created by PCR modification of the myc-H-Ras(G12V) cDNA using Pfu polymerase (Stratagene, La Jolla, CA, USA) and appropriate oligonucleotide primers. pcDNA3.1 containing the G12V mutant form of 2x myc-tagged K-Ras2(i.e., K-Ras4B) was purchased from the UMR cDNA Resource Center (http://www.cdna.org Rolla, MO, USA). PRK5 vectors encoding myc-Cdc42(G12V) and myc-RhoA(G14V) were provided by A.L. Wilson-Delfosse, Case Western University, Cleveland, OH. Human PTEN in pCMV6-XL5 was purchased from Origene Technologies Inc., Rockville, MD, USA. The pCMV-RafCaax construct was purchased from Clontech. The insert was subcloned into the pCMV5 vector with the myc-tag sequence described above. Human RalA (GenBank accession number NM_005402) was modified by overlap extension PCR to generate the S28N point mutation. The resulting PCR product was subcloned into the aforementioned pCMV5 vector with an in-frame 5’ myc-tag sequence. All of the expression vectors were introduced into the U251 cells by nucleofection, using the Nucleofector II system from Amaxa Biosystems (Koeln, Germany) following the manufacturer’s protocol with Nucleofector Solution T in combination with program T-30. For each reaction, 1–5 μg of plasmid DNA was introduced into 3 × 106 cells that had been harvested in RPMI 1640 medium. Following nucleofection, the cells were plated in 60 mm dishes and analyzed 18–24 hours later.
2.3 Generation of Ras constructs for retroviral expression
Myc-tagged H-Ras constructs, (G12V and S17N) were subcloned into the EcoR1-BamH1 sites of the retroviral expression vector, pFBneo (Stratagene, La Jolla, CA, USA). Retrovirus was produced in HEK293-GPG packaging cells and glioblastoma cells were infected as described previously [21]. Non-infected cells were eliminated by incubation for 2 days post-infection in medium containing 2.0 mg/ml G418.
2.4 Immunofluorescence microscopy
Cells were grown on laminin-coated glass coverslips, washed with Hanks balanced salt solution (HBSS), fixed in ice-cold methanol for 10 min, and blocked with 10% goat serum in PBS. For detection of myc-H-Ras(G12V, C186S) and 2x-myc-K-Ras2(G12V) the procedure was modified slightly to include a preliminary fixation with 3% paraformaldehyde prior to the methanol step. Myc-tagged proteins were detected by incubation for 1 h with an anti-myc monoclonal antibody (Calbiochem, San Diego, CA, USA) diluted in PBS + 10% goat serum, followed by a 1-h incubation with goat anti-mouse IgG conjugated with Alexa Fluor 568 (Invitrogen, Carlsbad, CA, USA). Antibodies and procedures used for immunofluorescence localization of LAMP1 have been described previously [22]. For experiments where myc-tagged RalA was co-expressed with FLAG-tagged Ras, we used an anti-FLAG monoclonal antibody (Sigma, St. Louis, MO, USA) followed by goat anti-mouse IgG-Alexa Fluor 568, combined with rabbit anti-myc IgG conjugated directly to FITC (ICL Labs, Newberg, OR, USA). Photomicrographs were taken with a Nikon Eclipse 800 fluorescent microscope equipped with a digital camera and ImagePro software (Media Cybernetics, Silver Spring, MD, USA).
2.5 Western blot analysis
Antibodies used for western blotting were obtained from the following sources: p44/42 MAP Kinase (ERK1/2), phospho-p44/42 MAP Kinase (ERK1/2) (Thr202/Tyr204), phospho-Akt (Ser473), and total Akt (Cell Signaling Technology, Danvers, MA, USA); myc epitope tag (EMD Biosciences, San Diego, CA, USA); Raf-1 and PTEN (Santa Cruz Biotechnology, Santa Cruz, CA, USA). Protein determinations, SDS-PAGE, and western blot analyses were performed as described previously [19]. Chemiluminescent immunoblot signals were quantified using a Kodak 440CF Image Station.
3. Results
3.1 Induction of cytoplasmic vacuolation in human glioblastoma cells by activated H-Ras(G12V) requires Ras farnesylation
In their initial study Chi et al. [10] demonstrated that expression of oncogenic H-Ras(G12V), but not GDP-locked H-Ras(S17N), causes massive accumulation of phase-lucent cytoplasmic vacuoles in U251 glioblastoma cells and gastric carcinoma cells. The vacuolated cells round up, detach from the substrate and disintegrate. This appears to be a form of non-apoptotic programmed cell death, since it occurs without substantial mitochondrial swelling, caspase activation or chromosomal DNA fragmentation [10]. We have observed a similar effect of activated H-Ras in a broad spectrum of human glioblastoma cell lines, but not in other commonly studied transformed cell lines (Overmeyer et al., unpublished).
H-Ras normally undergoes a series of sequential modifications commencing with the attachment of a farnesyl moiety to cys-186, followed by proteolytic removal of the three distal amino acids, and finally, carboxyl-methylation of the terminal cysteine [23]. In H-Ras, palmitylation of two upstream cysteines also occurs. In the absence of farnesylation, H-Ras remains predominantly in the cytosol, and subsequent modifications by membrane-associated enzymes do not occur [24]. Therefore, to evaluate the importance of membrane localization of H-Ras(G12V) for induction of the vacuolar phenotype, we introduced an amino acid substitution that prevents farnesylation; i.e., C186→S. The cysteine mutation eliminated the ability of activated H-Ras to induce phase lucent cytoplasmic vacuoles (Fig. 1a) and the cells continued to proliferate without detaching from the dish (not shown).
Figure 1.
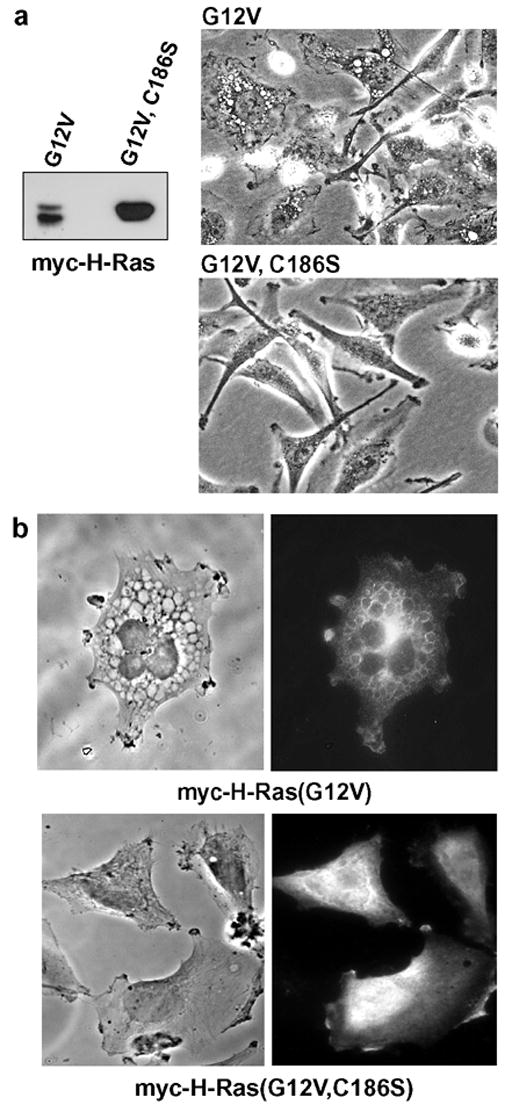
Stimulation of vacuole formation by activated Ras depends on farnesylation. (a) U251 glioblastoma cells were nucleofected with expression vectors encoding the indicated myc-tagged constructs. After 24 hours equal amounts of cellular protein were subjected to western blot analysis using the myc antibody. At the same time, live cells in parallel cultures were examined by phase contrast microscopy (300 × magnification). (b) U251 cells were nucleofected with expression vectors encoding the indicated myc-tagged constructs. After 24 hours the expressed proteins were localized by immunofluorescence microscopy as described in Materials and methods. The left panel of each figure shows the same cells under phase contrast.
Many of the Ras-induced vacuoles contain the lysosomal membrane protein, LAMP1 (ref. 10 and Fig. 4 in this report), suggesting that they arise from lysosomal compartments. However, these structures have not yet been characterized in sufficient detail rule out other origins (e.g., late endosomes, autophagosomes, autolysosomes). Immunofluorescent localization of myc-tagged H-Ras(G12V) revealed a striking association of the GTPase in the membranes surrounding the cytoplasmic vacuoles (Fig. 1b). In contrast to cells expressing H-Ras(G12V), the cells expressing the non-farnesylated myc-H-Ras(G12V, C186S) did not form vacuoles and the Ras protein exhibited a diffuse cytoplasmic distribution (Fig. 1c).
Figure 4.
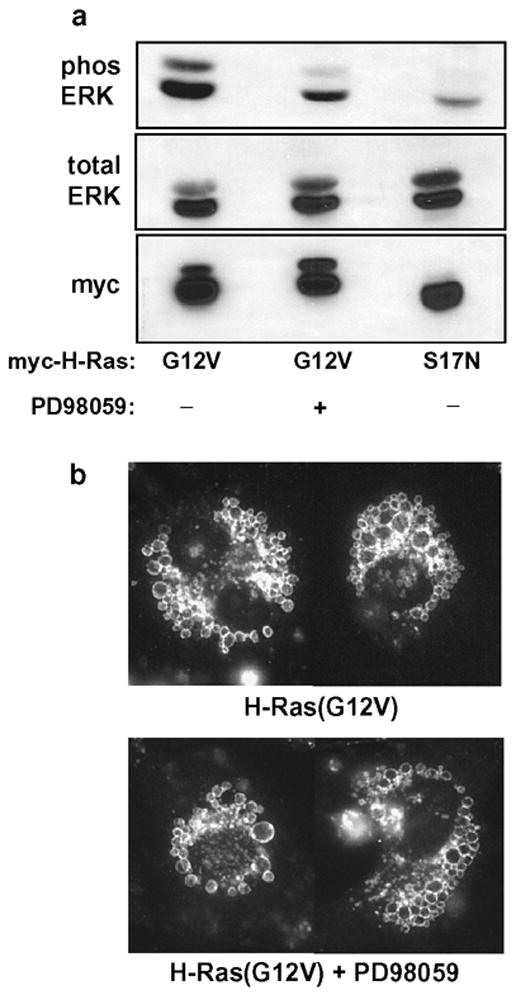
Induction of vacuoles by H-Ras(G12V) does not depend on activation of the ERK pathway. To obtain uniform expression of exogenous Ras, U251 cells were infected with retrovirus encoding myc-H-Ras(G12V) or myc-H-Ras(S17N). At the time that the cells were plated in selection medium (see Methods), the MEK inhibitor, PD98059 (25 μM) or an equivalent volume of DMSO vehicle, was added to the culture medium. Cells were processed for western blot analysis or immunofluorescent detection of LAMP1-positive vacuoles after 42 h in the presence of the inhibitor. (a) Activation of ERK was monitored in equal aliquots of whole cell lysate (normalized for protein) using antibodies to detect phosphorylated ERK1/2 (phos ERK) and total ERK1/2 . (b) Immunofluorescence microscopy to detect LAMP1-positive vacuoles was performed as described in Materials and methods. The panels are composites showing representative cells from the cultures incubated with or without PD98059.
3.2 Activated K-Ras, but not RhoA or Cdc42, can mimic the effects of activated H-Ras
The amino acid sequences of Ras proteins are highly similar throughout their core regions (amino acids 1–170), which contain the nucleotide binding and effector domains [25]. Most of the variation among the Ras proteins occurs in the last 20–24 amino acids, which comprise the COOH-terminal “hypervariable” domain and the farnesylation motif. In the cases of H-Ras and K-Ras4B, differences in the COOH-terminal sequences (Fig. 2a) dictate how the proteins will associate with membranes. For instance, H-Ras associates with membranes via farnesylation and palmitylation, whereas K-Ras4B relies on farnesylation combined with the upstream polylysine domain [26]. The different lipid modifications, combined with variations in amino acids between 167–172, also influence the targeting of H-Ras and K-Ras to separate membrane microdomains [26,27].
Figure 2. H- and K-Ras, but not Rho family GTPases, induce vacuolation.
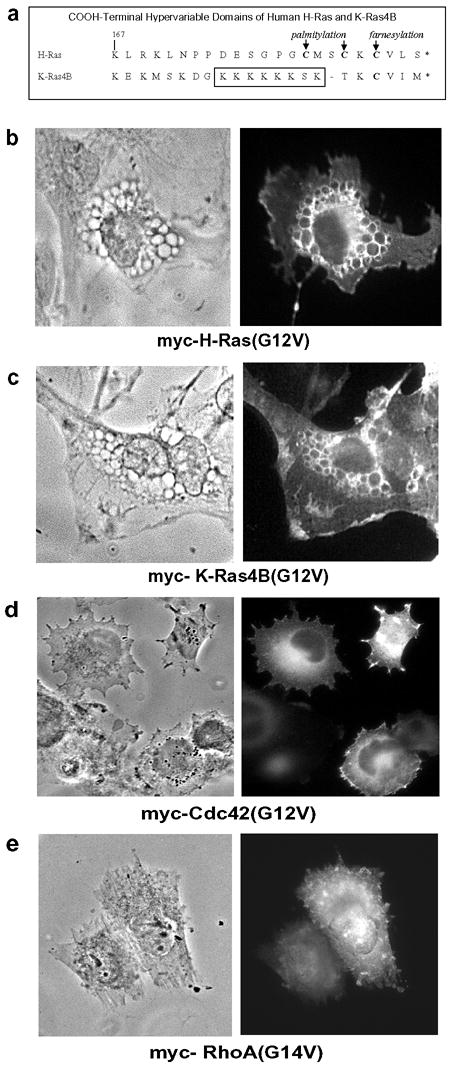
(a) Sequence alignment of the COOH-terminal hypervariable domains of H-Ras and K-Ras4B, showing sites of posttranslational lipidation. (b–e) U251 glioblastoma cells were nucleofected with expression vectors encoding the indicated myc-tagged constructs. After 24 hours the expressed proteins were localized by immunofluorescence microscopy using an antibody against the myc epitope (right panel). The left panel of each figure shows the same field of cells under phase contrast.
In light of these distinctions, we asked whether activated K-Ras4B was also capable of inducing the vacuolar phenotype in U251 cells. As shown in Fig. 2c, cells expressing myc-tagged K-Ras4B(G12V) formed phase-lucent vacuoles that were rimmed with the expressed protein, similar to the observations with H-Ras(G12V) (Fig. 2b). As in the case of H-Ras(S17N), the GDP-locked K-Ras4B(S17N) was ineffective at inducing vacuoles (not shown). Although activated forms of both H-Ras and K-Ras produced similar morphological effects, overexpression of other Ras-related GTPases with activating mutations, Cdc42(G12V) (Fig. 2d) and RhoA(G14V) (2e), did not trigger the formation of cytoplasmic vacuoles.
3.3 Effector domain mutations have differential effects on the ability of H-Ras(G12V) to induce vacuoles
The preceding results suggested that the ability of Ras to activate pathways responsible for causing vacuolation depends on domains common to H-Ras and K-Ras rather than on unique COOH-terminal sequences or membrane localization signals. In light of these findings, we used mutagenesis of the Ras effector domain as an initial experimental strategy to evaluate the relative importance of different Ras effector pathways for induction of the vacuolar phenotype. The effector domain (amino acids 32–40) is one of two major “switch” regions that change conformation when the nucleotide binding pocket of Ras is occupied by GTP versus GDP [28,29]. Specific amino acid substitutions in the effector domain selectively prevent the interaction of activated Ras with its major targets: Raf-1, PI3K p110α, or RalGDS [30–33] (summarized in Fig. 3a). We first tested the D38E substitution, which is permissive for binding to Raf, but not to PI3K or RalGDS. Cells expressing H-Ras(G12V, D38E) exhibited robust vacuolation, reduced only slightly when compared with G12V alone (Fig. 3c). This finding suggested that interactions with PI3K and RalGDS are not critical for vacuolation, and pointed to the potential importance of Raf. Consistent with this idea, introduction of theY40C substitution, which allows interaction with PI3K, but blocks interaction with Raf and RalGDS, markedly reduced the ability of H-Ras(G12V) to induce vacuoles (Fig. 3c). The final mutant tested was H-Ras(G12V, E37G). The E37G substitution is known to prevent Ras interaction with both Raf and PI3K, while preserving the interaction with RalGDS and several other effectors. As shown in Fig. 3b, incorporation of this substitution caused a substantial reduction in the percentage of vacuolated cells. Thus, in summary, the combined results with all three effector mutants were inconsistent with a key role for PI3K or RalGDS and pointed to the Raf pathway as the most likely mediator of Ras-induced vacuolation.
Figure 3.
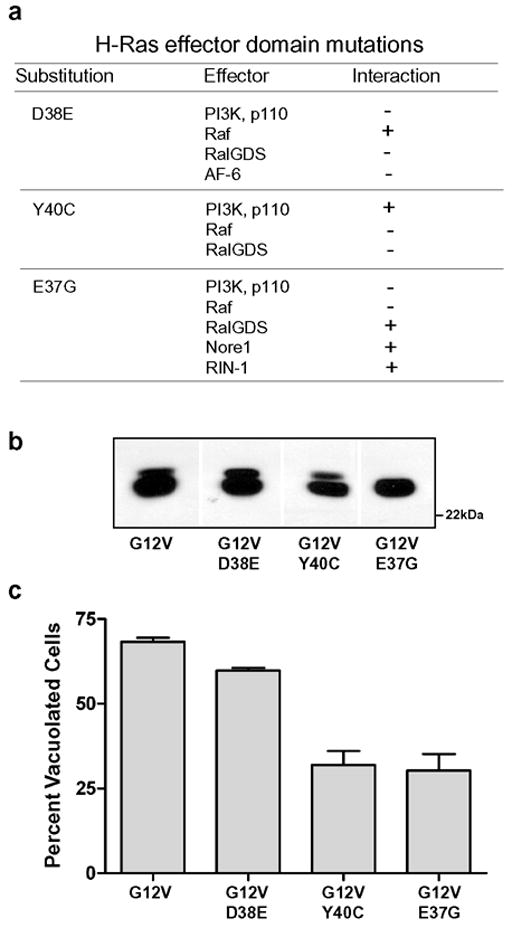
Mutations in the effector domain of H-Ras(G12V) have differential effects on its ability to induce cytoplasmic vacuolation. (a) Summary of Ras effector interactions known to be inhibited (−) or permitted (+) by specific amino acid substitutions, based on information compiled from refs 8,30–33,69,70,72. (b) U251 cells were nucleofected with expression vectors encoding the indicated myc-tagged H-Ras constructs. After 24h, equal aliquots of cell protein were subjected to western blot analysis using the myc antibody (representative blot shown). (c) Parallel cultures were processed for immunofluorescence localization of myc-tagged Ras proteins. Cells staining positive with the myc antibody were photographed and scored for vacuolation. Cells were scored as vacuolated if they contained at least 2 vacuoles ≥ 0.5 μ in diameter and/or >10 smaller vacuoles. At least 65 myc-positive cells were scored to determine the percentage of transfected cells that were vacuolated in each culture. The results for each Ras construct shown in the bar graph are derived from three separate experiments (mean ± S.D.). The decreases in vacuolation observed with G12V+Y40C and G12V+E37G were significant at p<0.01 compared with G12V alone (Student’s t-test).
3.4 Induction of cytoplasmic vacuolation by activated Ras does not depend on activation of Raf signaling pathways
In light of the preceding studies, we used alternative approaches to determine if the stimulation of cytoplasmic vacuoles in U251 cells by H-Ras(G12V) is directly related to its activation of the Raf→MEK→ERK1/2 signaling pathway. A specific MEK inhibitor (PD98059) was included in the medium for 42 h after infection of the cells with retrovirus expressing myc- H-Ras(G12V). The MEK inhibitor suppressed the H-Ras(G12V)-induced phosphorylation of ERK1/2 to a level approaching that detected in the negative control cells expressing myc-H-Ras(S17N) (Fig. 4a). However, the suppression of ERK phosphorylation did not prevent the accumulation of LAMP1-positive vacuoles in the cells expressing H-Ras(G12V) (Fig. 4b).
As a different strategy to specifically address the role of Raf activation in cellular vacuolation, we activated the Raf→MEK→ERK1/2 signaling pathway independently of Ras. This was done by attaching a Caax prenylation motif to the C-terminus of Raf, thereby generating a constitutively active form of Raf that remains associated with the cell membrane independent of Ras [34]. When this construct was expressed in U251 cells, it induced an increase in ERK phosphorylation that was even greater than that induced by H-Ras(G12V) (Fig. 5b). However, the activation of ERK by Raf-Caax did not trigger vacuolation of the glioblastoma cells (Fig. 5c). These results demonstrate that the induction of vacuolation by activated Ras involves effector pathways distinct from the Raf→MEK→ERK1/2 cascade.
Figure 5.
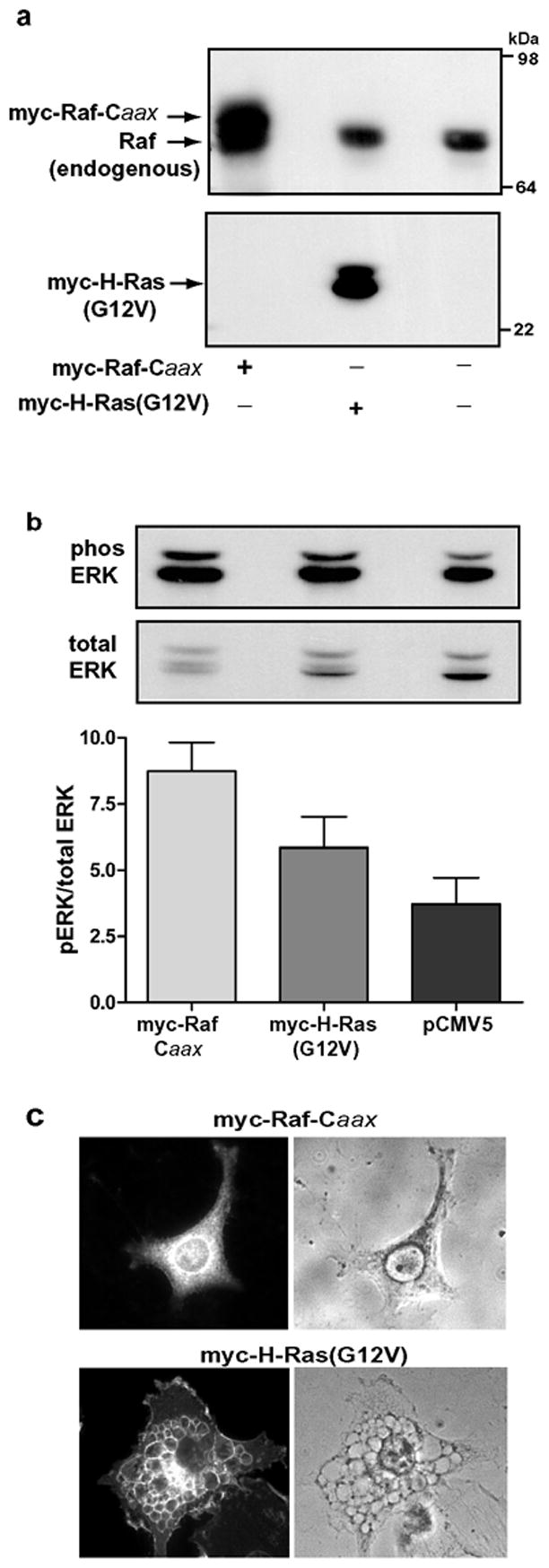
Overexpression of constitutively active Raf-1 does not mimic the effect of activated H-Ras. U251 cells were nucleofected with empty vector or with expression vectors encoding myc-Raf-Caax or myc-H-Ras(G12V). The cells were processed for western blot analysis or immunofluorescence 24 h later. (a) Immunoblot analysis with an antibody against Raf confirms that myc-Raf-Caax (upper band) is overexpressed relative to endogenous Raf. (b) Phospho-ERK and total ERK were determined by immunoblot analysis (representative blot shown). The ratio of phospho-ERK to total ERK was determined by quantifying the immunoblot signals with a Kodak Image station. The results shown in the bar graph are the means (±SD) of separate determinations performed on three cultures. (c) Cells expressing myc-Raf-Caax or myc-H- Ras(G12V) were identified by immunofluorescence using antibody against the myc epitope. The same cells were examined under phase contrast to assess the presence of vacuoles. The nucleofection efficiencies in both sets of cultures were approximately 50%. There were no vacuolated myc-positive cells in the cultures expressing myc-Raf-Caax.
3.5 Class I PI3K is not an essential mediator of vacuolation induced by H-Ras(G12V)
The GTP-bound form of Ras activates the p110 catalytic subunit of class I PI3K [35], resulting in increased production of PI(3,4,5)P3 [36]. Although the results of the effector domain mutagenesis studies (Fig. 3) suggested that interaction with PI3K is not essential for H-Ras(G12V) to trigger cellular vacuolation, we wanted to confirm this by a separate method. The use of PI3K inhibitors like wortmannin was not desirable because they can perturb endocytic trafficking and cause the formation of vacuoles derived from multivesicular endosomes [37]. Instead, we took advantage of the fact that U251 cells, like many other human glioblastoma lines, carry a mutation in the PTEN gene [38]. PTEN normally dephosphorylates the 3’ position of PI(3,4,5)P3 , facilitating turnover of the PI3K product and downregulating signaling to the Akt pathway [39,40]. By introducing normal PTEN into U251 cells expressing H-Ras(G12V) (Fig. 6a), we were able to effectively down-regulate PI3K signaling, measured by Akt phosphorylation.(Fig. 6b). Although coexpression of PTEN with H-Ras(G12V) reduced phosphorylation of Akt by more than 60%, there was no detectable reduction of vacuolation (Fig. 6c).
Figure 6.
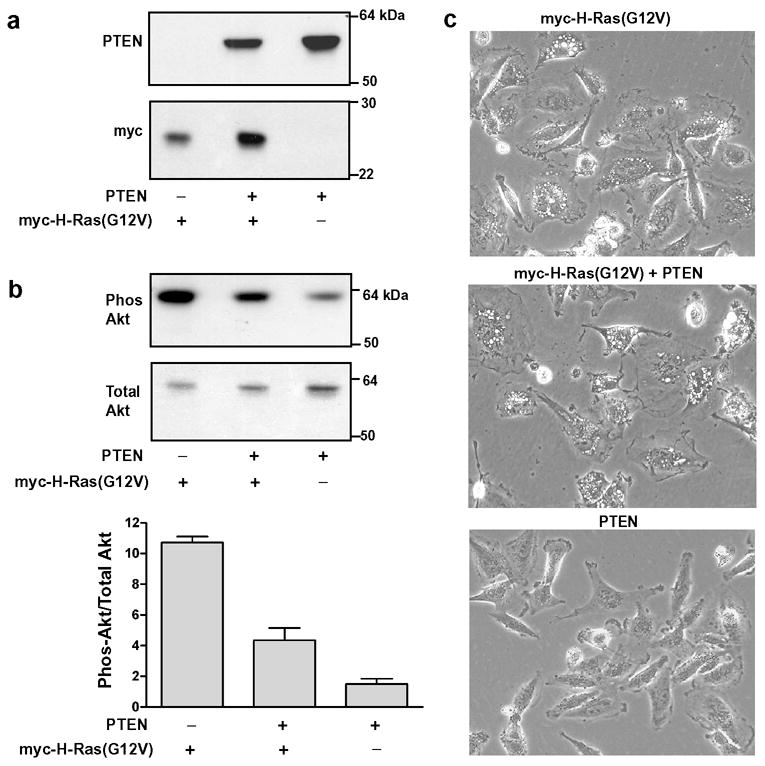
Reduction of the activity of the PI 3K signaling pathway by introduction of PTEN does not prevent the induction of vacuoles by H-Ras(G12V). (a) U251 cells were nucleofected with expression vectors encoding PTEN and/or myc-H-Ras(G12V) as indicated. After 24 h equal aliquots of cell protein were processed for western blot analysis to verify the expression of PTEN and myc-tagged Ras. (b) Phospho-Akt and total Akt were determined by immunoblot analysis (representative blot shown). The bar graph depicts the ratio of phospho-Akt to total Akt (mean ± S.D.) determined by quantifying the immunoblot signals from three separate cultures. The decrease in Akt phosphorylation in PTEN + myc-H-Ras(G12V) was significant at p < 0.05 compared with myc-H-Ras(G12V) alone (Student’s t-test). (c) Phase contrast images of representative fields of cells from the cultures harvested for the blots shown in a & b (200x).
3.6 Ras-induced vacuolation is not blocked by coexpression with dominant-negative RalA
In addition to PI3K and Raf, activated Ras binds and stimulates RalGDS [41,42]. The latter promotes exchange of GTP for GDP on two Ras-related GTPases, RalA and RalB. The Ral GTPases have been implicated in many of the downstream effects of Ras, including cell transformation, activation of transcription factors, and intracellular trafficking [43–45].
Our earlier observation that H-Ras(G12V, D38E) was a strong inducer of vacuolation (Fig. 3) despite its reported inability to interact with RalGDS [46], suggests that activation of RalGDS is not required for the Ras-induced phenotype. To confirm this observation, we coexpressed FLAG-tagged H-Ras(G12V) with myc-tagged RalA (S28N) in U251 cells (Fig. 7). Similar to Ras(S17N), substitution of S28→N in Ral locks the protein in the GDP state and renders it a dominant-negative suppressor of endogenous Ral function [47]. As expected, cells expressing FLAG-Ras(G12V) exhibited typical vacuolation, with localization of the Ras protein to the vacuole membranes. In the cells expressing myc-RalA(S28N) alone, there was no evidence of vacuolation and the expressed protein exhibited a diffuse localization with concentration in the plasma membrane. When FLAG-Ras(G12V) was co-expressed with myc-RalA(S28N), the dominant-negative Ral did not inhibit vacuolation. Interestingly, although activation of the Ral pathway does not appear to be causally related to vacuolation, coexpression of activated H-Ras(G12V) with RalA(S28N) changed the distribution of the latter so that it completely co-localized with H-Ras(G12V) on the vacuole membranes (Fig. 7b). Although we did not explore this further, it would be consistent with a model in which activated H-Ras on the vacuole membrane binds to endogenous RalGDS or other Ral exchange factors, thereby shifting the distribution of the associated GDP-locked RalA(S28N) to this compartment.
Figure 7.
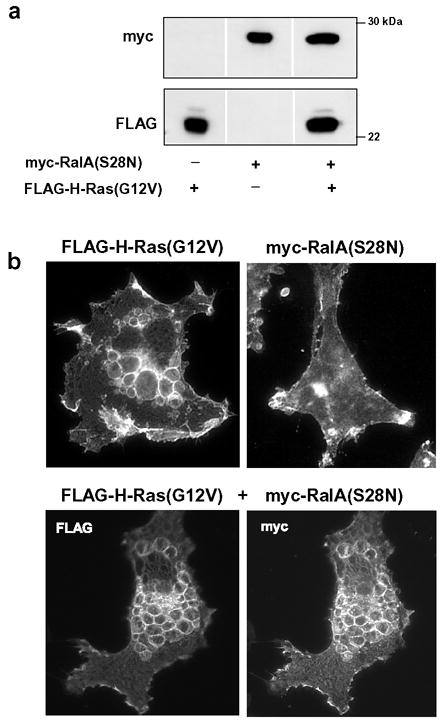
Dominant-negative RalA does not block the induction of vacuolation by activated H-Ras. (a) U251 cells were nucleofected with expression vectors encoding FLAG-H-Ras(G12V), myc-RalA(S28N) or a combination of both. Equal amounts of cell lysate were subjected to western blot analysis to verify expression of the tagged proteins. (b) The upper panels show representative immunofluorescent localization of FLAG-H-Ras(G12V) alone and myc-RalA(S28N) alone. The bottom panels show a typical cell expressing both proteins.
4. Discussion
In the present study we report the first detailed exploration of the molecular signaling pathways underlying a unique form of Type-II (caspase-independent) programmed cell death triggered by expression of activated Ras in glioblastoma [9–10]. The major cytopathological features of this form of cell death are accumulation of phase-lucent, LAMP1-positive, cytoplasmic vacuoles, followed by cell detachment and disintegration.
Our results demonstrate that farnesylation is required for activated H-Ras to stimulate signaling pathways that induce cytoplasmic vacuolation. Farnesylation initially targets Ras proteins to the endoplasmic reticulum, where the Caax motif is further processed by a protease that removes the final three amino acids and a carboxyl methyltransferase that modifies the farnesylated cysteine. H-Ras and K-Ras subsequently take different routes to the plasma membrane [24]. Differences in their hypervariable C-terminal sequences and posttranslational modifications (see Fig. 2a) direct H-Ras to lipid rafts and K-Ras to non-raft regions [26]. We now show that both H-Ras(G12V) and K-Ras4B(G12V) are capable of stimulating cellular vacuolation and localizing to the vacuole membrane. This suggests that the reported difference in H-Ras versus K-Ras membrane subdomain distribution is not a major factor in the signaling mechanism that triggers vacuolation. One possible explanation for this finding is that H-Ras leaves the rafts and joins K-Ras in the bulk plasma membrane when it is activated by GTP [48]. Alternatively, the signals that stimulate vacuolation may emanate from the pool of activated Ras associated with endomembrane compartments. This would be entirely consistent with evidence that Ras signaling can occur in the endoplasmic reticulum, Golgi apparatus and endosomes [49–52].
The amino acid sequences of H-Ras and K-Ras are identical in the domains termed switch-1 (amino acids 32–40) and switch-2 (amino acids 60–76), which undergo GTP-dependent conformational changes [28,53] and determine the interaction of Ras with downstream effectors [54,55]. In contrast, the Rho family members (Cdc42 and RhoA), diverge from Ras in these regions and do not stimulate vacuolation when expressed in their constitutively active forms (Fig. 2). Together with the inability of GDP-locked Ras(S17N) to cause vacuolation and cell death [10], these findings strongly support the conclusion that the vacuolar phenotype is induced through stimulation of specific Ras effector pathways.
In seeking to define the relevant Ras signaling pathways, we focused initially on the well- known Raf→MEK→ERK1/2 kinase cascade. The first step in the cascade involves the activation of Raf by Ras-GTP [56,57]. Our interest in this pathway was motivated by several reports linking ERK activation to the regulation of macroautophagy, a common feature in Type- II cell death. Specifically, Codogno and colleagues showed that ERK1/2 stimulates autophagosome formation by phosphorylating and activating GAIP (Gα-interacting protein), a GTPase activating protein for Gαi3, [58,59]. Later studies showed that this pathway can be stimulated by Ras [60]. Finally, a separate study in Sertoli cells treated with the carcinogen, lindane, has shown that activation of ERK can cause accumulation of LAMP1-positive vacuoles by interfering with the maturation of autophagosomes into autolysosomes [61]. The results of our studies with the Ras effector domain mutations supported the possible involvement of Raf. For instance, the Y40C and E37G substitutions, which impede interaction with Raf, reduced vacuolation, whereas the D38E mutation, which is permissive for Raf binding, had almost no effect (Fig. 3). However, two follow-up studies indicated that the reduced vacuolation caused by the Y40C and E37G mutations is probably related to interference with effectors other than Raf . First, blocking Raf→ERK signaling with a MEK inhibitor had no effect on Ras-induced vacuolation (Fig. 4). Second, overexpression of a constitutively active form of Raf (Raf-Caax) did not induce vacuoles, despite the fact that it stimulated ERK phosphorylation to an even greater extent than H-Ras(G12V) (Fig. 5)
The ability of Ras-GTP to bind to the p110 catalytic subunit and promote activation of Class-I PI3K is now well established [62]. We initially thought that activation of Class I PI3K by ectopic expression of Ras(G12V) would be an unlikely mechanism to stimulate Type-II cell death in glioblastoma, because the basal activity of the PI3K → Akt pathway is already increased by the absence of PTEN in these cells. In addition, several studies indicate that class-I PI3K is a negative regulator of autophagic activity [63–65]. Our suspicions were borne out by the data from the Ras effector domain studies, where the D38E mutation had little influence on the extent of vacuolation, despite its reported ability to interfere with PI3K interaction. Conversely, introduction of the Y40C mutation into H-Ras(G12V) reduced vacuolation even though this construct is competent to interact with PI 3K. Most importantly, our conclusion that vacuolation is not mediated by stimulation of PI3K was strongly supported by the studies in Fig. 6, where introduction of PTEN into the glioblastoma cells expressing H-Ras(G12V) reduced the activation of Akt by more than 60% without having any effect on vacuolation.
Having ruled out Raf and PI3K as likely mediators of the vacuolar phenotype induced by H-Ras(G12V), we turned our attention to a third major Ras effector, RalGDS. Activation of Ral GTPases has been implicated in the control of endocytic trafficking via the Ral effector, RalBP (RLIP76) [43,44,47]. However, our finding that neither the introduction of the non-permissive D38E effector mutation nor the coexpression of Ras(G12V) with the dominant-negative RalA(S28N) interfered with vacuolation argues against an essential role for pathways regulated by Ral nucleotide exchange factors.
Despite the evidence against Raf, PI3K and RalGDS as key effectors, the reduced vacuolation observed with the Y40C and E37G mutations supports the premise that vacuolation requires activation of specific Ras signaling pathways. Many additional Ras effectors have now been identified [66]. These include the RIN1 nucleotide exchange factor for Rab5 [67], Tiam1 and other exchange factors for Rac [68], Rain, an endomembrane receptor for Ras [69], Nore1, a pro-apoptotic tumor suppressor [8], and AF-6, a mediator of membrane-cytoskeleton interactions [70,71]. Although we have not yet evaluated all of these Ras effector pathways in detail, it is worth noting that the E37G mutation, which reduced vacuolation in our studies, is permissive for Ras binding to RIN1 [72], Nore1 [73] and Rain [69]. Conversely, the D38E mutation, which did not have a major effect on vacuolation, would diminish Ras binding to AF-6 [70]. These preliminary observations raise the intriguing possibility that the unusual cytoplasmic vacuolation induced by activated Ras may be related to interactions with novel effectors found specifically in glioblastoma cells and perhaps other types of cells (neuroblastoma, gastric carcinoma) that respond to Ras in this unique way.
The precise definition of the Ras signaling pathway(s) responsible for induction of the vacuolar phenotype and Type-II cell death could be quite challenging because of the growing list of Ras targets. Further complicating the task, the effects of activated Ras may vary depending on the cell type or species [3,74]. For example, in a mouse glioblastoma model, the introduction of oncogenic K-Ras stimulates gliomagenesis and is essential for tumor maintenance [75,76], contrasting with the death-promoting effects in human glioma cells. Despite these challenges, future studies aimed at pinpointing the Ras signaling pathways responsible for induction of Type-II death could have important clinical implications. For example, migrating glioblastoma cells are generally resistant to apoptosis, due to constitutive activation of one or more pro-survival signaling pathways [77]. By defining the molecular mechanisms for alternative non-apoptotic forms of cell death, it may be possible to identify new targets that can be activated to reduce survival of the malignant cells.
5. Conclusions
Since the original demonstration that ectopic expression of activated H-Ras can induce cytoplasmic vacuolation and Type-II death in glioblastoma cells [10], there have been no systematic studies aimed at defining the molecular mechanisms underlying this unique effect. The results presented in this report represent a significant step forward in narrowing the search for the relevant Ras effectors. We are now able to draw the following conclusions: (1) Cytoplasmic vacuolation can be induced by K-Ras as well as H-Ras, suggesting that common switch regions, rather than unique hypervariable domains or lipid modifications, are the critical factors for stimulation of the relevant signaling pathways. (2) The pathways responsible for triggering vacuolation are specific to Ras; i.e., activated forms of RhoA and Cdc42 do not duplicate this effect. (3) Stimulation of vacuolation depends farnesylation of Ras, implying that the relevant effectors are associated with intracellular membranes. (4) The ability of Ras to induce cytoplasmic vacuolation does not depend on activation of the widely studied Raf→MEK→ERK kinase cascade, the PI3K → Akt pathway, or the RalGDS →Ral pathway. The latter findings underscore the possibility that activated Ras may stimulate unconventional effector pathways to induce non-apoptotic cell death. Although this study does not pinpoint the key effector pathway, it eliminates the most obvious candidates, thereby providing important direction for future studies. In the long term these studies may uncover specific targets that can be manipulated in a therapeutic context to increase the susceptibility of glioblastoma to this alternative form of cell death.
Acknowledgments
This work was supported by grants to W.A.M. from the National Cancer Institute (RO1 CA34569) and the Charlotte Geyer Foundation. We are grateful to Amy Wilson-Delfosse, Ph.D. for providing the Cdc42 and RhoA expression vectors.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bos JL. ras oncogenes in human cancer: a review. Cancer Res. 1989;49:4682–4689. [PubMed] [Google Scholar]
- 2.Campbell SL, Khosravi-Far R, Rossman KL, Clark GJ, Der CJ. Increasing complexity of Ras signaling. Oncogene. 1998;17:1395–1413. doi: 10.1038/sj.onc.1202174. [DOI] [PubMed] [Google Scholar]
- 3.Shields JM, Pruitt K, McFall A, Shaub A, Der CJ. Understanding Ras: 'it ain't over 'til it's over'. Trends Cell Biol. 2000;10:147–154. doi: 10.1016/s0962-8924(00)01740-2. [DOI] [PubMed] [Google Scholar]
- 4.Der CJ, Finkel T, Cooper GM. Biological and biochemical properties of human rasH genes mutated at codon 61. Cell. 1986;44:167–176. doi: 10.1016/0092-8674(86)90495-2. [DOI] [PubMed] [Google Scholar]
- 5.Lowy DR, Willumsen BM. Function and regulation of Ras. Annu Rev Biochem. 1993;62:851–891. doi: 10.1146/annurev.bi.62.070193.004223. [DOI] [PubMed] [Google Scholar]
- 6.Cox AD, Der CJ. The dark side of Ras: regulation of apoptosis. Oncogene. 2003;22:8999–9006. doi: 10.1038/sj.onc.1207111. [DOI] [PubMed] [Google Scholar]
- 7.Denoyelle C, Abou-Rjaily G, Bezrookove V, Verhaegen M, Johnson TM, Fullen DR, Pointer JN, Gruber SB, Su LD, Nikiforov MA, Kaufman RJ, Bastian BC, Soengas MS. Anti-oncogenic role of the endoplasmic reticulum differentially activated by mutations in the MAPK pathway. Nat Cell Biol. 2006 doi: 10.1038/ncb1471. [Sept 10, Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 8.Vos MD, Martinez A, Ellis CA, Vallecorsa T, Clark GJ. The pro-apoptotic Ras effector Nore1 may serve as a Ras-regulated tumor suppressor in the lung. J Biol Chem. 2003;278:21938–21943. doi: 10.1074/jbc.M211019200. [DOI] [PubMed] [Google Scholar]
- 9.Kitanaka C, Kuchino Y. Caspase-independent programmed cell death with necrotic morphology. Cell Death Differ. 1999;6:508–515. doi: 10.1038/sj.cdd.4400526. [DOI] [PubMed] [Google Scholar]
- 10.Chi S, Kitanaka C, Noguchi K, Mochizuki T, Nagashima Y, Shirouzu M, Fujita H, Yoshida M, Chen W, Asai A, Himeno M, Yokoyama S, Kuchino Y. Oncogenic Ras triggers cell suicide through the activation of a caspase-independent cell death program in human cancer cells. Oncogene. 1999;18:2281–2290. doi: 10.1038/sj.onc.1202538. [DOI] [PubMed] [Google Scholar]
- 11.Kitanaka C, Kato K, Ijiri R, Sakurada K, Tomiyama A, Noguchi K, Nagashima Y, Nakagawara A, Momoi T, Toyoda Y, Kigasawa H, Nishi T, Shirouzu M, Yokoyama S, Tanaka Y, Kuchino Y. Increased Ras expression and caspase-independent neuroblastoma cell death: possible mechanism of spontaneous neuroblastoma regression. J Natl Cancer Inst. 2002;94:358–368. doi: 10.1093/jnci/94.5.358. [DOI] [PubMed] [Google Scholar]
- 12.Zakeri Z, Bursch W, Tenniswood M, Lockshin RA. Cell death:programmed, apoptosis, necrosis or other? Cell Death Differentiat. 1995;2:87–96. [PubMed] [Google Scholar]
- 13.Lockshin RA, Zakeri Z. Apoptosis, autophagy, and more. Int J Biochem Cell Biol. 2004;36:2405–2419. doi: 10.1016/j.biocel.2004.04.011. [DOI] [PubMed] [Google Scholar]
- 14.Bursch W, Ellinger E, Kienzl H, Torok L, Pandey S, Sikorska M, Walker R, Hermann RS. Active cell death induced by the anti-estrogens tamoxifin and ICI 164 384 in human mammary carcioma cells (MCF-7) in culture: the role of autophagy. Carcinogenesis. 1996;17:1595–1607. doi: 10.1093/carcin/17.8.1595. [DOI] [PubMed] [Google Scholar]
- 15.Paglin S, Hollister T, Delohery T, Hackett N, McMahill M, Sphicas E, Domingo D, Yahalom J. A novel response of cancer cells to radiation involves autophagy and formation of acidic vesicles. Cancer Res. 2001;61:439–444. [PubMed] [Google Scholar]
- 16.Kanzawa T, Kondo Y, Ito H, Kondo S, Germano I. Induction of autophagic cell death in malignant glioma cells by arsenic trioxide. Cancer Res. 2003;63:2103–2108. [PubMed] [Google Scholar]
- 17.Kanzawa T, Germano IM, Komata T, Ito H, Kondo Y, Kondo S. Role of autophagy in temozolomide-induced cytotoxicity for malignant glioma cells. Cell Death Differ. 2004;11:448–457. doi: 10.1038/sj.cdd.4401359. [DOI] [PubMed] [Google Scholar]
- 18.Gozuacik D, Kimchi A. Autophagy as a cell death and tumor suppressor mechanism. Oncogene. 2004;23:2891–2906. doi: 10.1038/sj.onc.1207521. [DOI] [PubMed] [Google Scholar]
- 19.Erdman RA, Shellenberger KE, Overmeyer JH, Maltese WA. Rab24 is an atypical member of the Rab GTPase family. Deficient GTPase activity, GDP dissociation inhibitor interaction, and prenylation of Rab24 expressed in cultured cells. J Biol Chem. 2000;275:3848–3856. doi: 10.1074/jbc.275.6.3848. [DOI] [PubMed] [Google Scholar]
- 20.Wilson AL, Erdman RA, Maltese WA. Association of Rab1B with GDP-dissociation inhibitor (GDI) is required for recycling but not initial membrane targeting of the Rab protein. J Biol Chem. 1996;271:10932–10940. doi: 10.1074/jbc.271.18.10932. [DOI] [PubMed] [Google Scholar]
- 21.Zeng X, Overmeyer JH, Maltese WA. Functional specificity of the mammalian Beclin-Vps34 PI 3-kinase complex in macroautophagy versus endocytosis and lysosomal enzyme trafficking. J Cell Sci. 2006;119:259–270. doi: 10.1242/jcs.02735. [DOI] [PubMed] [Google Scholar]
- 22.Johnson EE, Overmeyer JH, Gunning WT, Maltese WA. Gene silencing reveals a specific function of hVps34 phosphatidylinositol 3-kinase in late versus early endosomes. J Cell Sci. 2006;119:1219–1232. doi: 10.1242/jcs.02833. [DOI] [PubMed] [Google Scholar]
- 23.Newman CMH, Magee AI. Posttranslational processing of the ras superfamily of small GTP- binding proteins. Biochim Biophys Acta. 1993;1155:79–96. doi: 10.1016/0304-419x(93)90023-6. [DOI] [PubMed] [Google Scholar]
- 24.Choy E, Chiu VK, Silletti J, Feoktistov M, Morimoto T, Michaelson D, Ivanov IE, Philips MR. Endomembrane trafficking of ras: the CAAX motif targets proteins to the ER and Golgi. Cell. 1999;98:69–80. doi: 10.1016/S0092-8674(00)80607-8. [DOI] [PubMed] [Google Scholar]
- 25.Valencia A, Chardin P, Wittinghofer A, Sander C. The ras protein family: Evolutionary tree and role of conserved amino acids. Biochemistry. 1991;30:4637–4648. doi: 10.1021/bi00233a001. [DOI] [PubMed] [Google Scholar]
- 26.Prior IA, Muncke C, Parton RG, Hancock JF. Direct visualization of Ras proteins in spatially distinct cell surface microdomains. J Cell Biol. 2003;160:165–170. doi: 10.1083/jcb.200209091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jaumot M, Yan J, Clyde-Smith J, Sluimer J, Hancock JF. The Linker domain of the Ha-Ras hypervariable region regulates interactions with exchange factors, Raf-1 and Phosphoinositide 3-Kinase. J Biol Chem. 2002;277:272–278. doi: 10.1074/jbc.M108423200. [DOI] [PubMed] [Google Scholar]
- 28.Krengel U, Schlichting I, Scherer A, Schumann R, Frech M, John J, Kabsch W, Pai EF, Wittinghofer A. Three-dimensional structures of H-ras p21 mutants: Molecular basis for their inability to function as signal switch molecules. Cell. 1990;62:539–548. doi: 10.1016/0092-8674(90)90018-a. [DOI] [PubMed] [Google Scholar]
- 29.Quilliam LA, Hisaka MM, Zhong S, Lowry A, Mosteller RD, Han J, Drugan JK, Broek D, Campbell SL, Der CJ. Involvement of the switch 2 domain of Ras in its interaction with guanine nucleotide exchange factors. J Biol Chem. 1996;271:11076–11082. doi: 10.1074/jbc.271.19.11076. [DOI] [PubMed] [Google Scholar]
- 30.White MA, Nicolette C, Minden A, Polverino A, Van Aelst L, Karin M, Wigler MH. Multiple Ras functions can contribute to mammalian cell transformation. Cell. 1995;80:533–541. doi: 10.1016/0092-8674(95)90507-3. [DOI] [PubMed] [Google Scholar]
- 31.Khosravi-Far R, White MA, Westwick JK, Solski PA, Chrzanowska-Wodnicka M, Van Aelst L, Wigler MH, Der CJ. Oncogenic Ras activation of Raf/mitogen-activated protein kinase-independent pathways is sufficient to cause tumorigenic transformation. Mol Cell Biol. 1996;16:3923–3933. doi: 10.1128/mcb.16.7.3923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rodriguez-Viciana P, Warne PH, Khwaja A, Marte BM, Pappin D, Das P, Waterfield MD, Ridley A, Downward J. Role of phosphoinositide 3-OH kinase in cell transformation and control of the actin cytoskeleton by Ras. Cell. 1997;89:457–467. doi: 10.1016/s0092-8674(00)80226-3. [DOI] [PubMed] [Google Scholar]
- 33.Joneson T, White MA, Wigler MH, Bar-Sagi D. Stimulation of membrane ruffling and MAP kinase activation by distinct effectors of RAS. Science. 1996;271:810–812. doi: 10.1126/science.271.5250.810. [DOI] [PubMed] [Google Scholar]
- 34.Leevers SJ, Paterson HF, Marshall CJ. Requirement for Ras in Raf activation is overcome by targeting Raf to the plasma membrane. Nature. 1994;369:411–414. doi: 10.1038/369411a0. [DOI] [PubMed] [Google Scholar]
- 35.Rodriguez-Viciana P, Warne PH, Vanhaesebroeck B, Waterfield MD, Downward J. Activation of phosphoinositide 3-kinase by interaction with Ras and by point mutation. EMBO J. 1996;15:2442–2451. [PMC free article] [PubMed] [Google Scholar]
- 36.Yan J, Roy S, Apolloni A, Lane A, Hancock JF. Ras isoforms vary in their ability to activate Raf-1 and phosphoinositide 3-kinase. J Biol Chem. 1998;273:24052–24056. doi: 10.1074/jbc.273.37.24052. [DOI] [PubMed] [Google Scholar]
- 37.Reaves BJ, Bright NA, Mullock BM, Luzio JP. The effect of wortmannin on the localisation of lysosomal type I integral membrane glycoproteins suggests a role for phosphoinositide 3-kinase activity in regulating membrane traffic late in the endocytic pathway. J Cell Sci. 1996;109(Pt 4):749–762. doi: 10.1242/jcs.109.4.749. [DOI] [PubMed] [Google Scholar]
- 38.Ishii N, Maier D, Merlo A, Tada M, Sawamura Y, Diserens AC, Van Meir EG. Frequnt co-alterations of TP53, p16/CDKN2A, p14arf, PTEN tumor suppressor genes in human glioma cell lines. Brain Pathol. 1999;9:469–479. doi: 10.1111/j.1750-3639.1999.tb00536.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Maehama T, Dixon JE. The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate. J Biol Chem. 1998;273:13375–13378. doi: 10.1074/jbc.273.22.13375. [DOI] [PubMed] [Google Scholar]
- 40.Wu X, Senechal K, Neshat MS, Whang YE, Sawyers CL. The PTEN/MMAC1 tumor suppressor phosphatase functions as a negative regulator of the phosphoinositide 3-kinase/Akt pathway. Proc Natl Acad Sci USA. 1998;95:15587–15591. doi: 10.1073/pnas.95.26.15587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Matsubara K, Kishida S, Matsuura Y, Kitayama H, Noda M, Kikuchi A. Plasma membrane recruitment of RalGDS is critical for Ras-dependent Ral activation. Oncogene. 1999;18:1303–1312. doi: 10.1038/sj.onc.1202425. [DOI] [PubMed] [Google Scholar]
- 42.Quilliam LA, Rebhun JF, Castro AF. A growing family of guanine nucleotide exchange factors is responsible for activation of Ras-family GTPases. Prog Nucleic Acid Res Mol Biol. 2002;71:391–444. doi: 10.1016/s0079-6603(02)71047-7. [DOI] [PubMed] [Google Scholar]
- 43.Jullien-Flores V, Mahe Y, Mirey G, Leprince C, Meunier-Bisceuil B, Sorkin A, Camonis JH. RLIP76, an effector of the GTPase Ral, interacts with the AP2 complex: involvement of the Ral pathway in receptor endocytosis. J Cell Sci. 2000;113(Pt 16):2837–2844. doi: 10.1242/jcs.113.16.2837. [DOI] [PubMed] [Google Scholar]
- 44.Feig LA. Ral-GTPases: approaching their 15 minutes of fame. Trends Cell Biol. 2003;13:419–425. doi: 10.1016/s0962-8924(03)00152-1. [DOI] [PubMed] [Google Scholar]
- 45.Moskalenko S, Tong C, Rosse C, Mirey G, Formstecher E, Daviet L, Camonis J, White MA. Ral GTPases regulate exocyst assembly through dual subunit interactions. J Biol Chem. 2003;278:51743–51748. doi: 10.1074/jbc.M308702200. [DOI] [PubMed] [Google Scholar]
- 46.Rodriguez-Viciana P, Warne PH, Khwaja A, Marte BM, Pappin D, Das P, Waterfield MD, Ridley A, Downward J. Role of phosphoinositide 3-OH kinase in cell transformation and control of the actin cytoskeleton by Ras. Cell. 1997;89:457–467. doi: 10.1016/s0092-8674(00)80226-3. [DOI] [PubMed] [Google Scholar]
- 47.Nakashima S, Morinaka K, Koyama S, Ikeda M, Kishida M, Okawa K, Iwamatsu A, Kishida S, Kikuchi A. Small G protein Ral and its downstream molecules regulate endocytosis of EGF and insulin receptors. EMBO J. 1999;18:3629–3642. doi: 10.1093/emboj/18.13.3629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Prior IA, Harding A, Yan J, Sluimer J, Parton RG, Hancock JF. GTP-dependent segregation of H-ras from lipid rafts is required for biological activity. Nat Cell Biol. 2001;3:368–375. doi: 10.1038/35070050. [DOI] [PubMed] [Google Scholar]
- 49.Chiu VK, Bivona T, Hach A, Sajous JB, Silletti J, Wiener H, Johnson RL, Cox AD, Philips MR. Ras signalling on the endoplasmic reticulum and the Golgi. Nat Cell Biol. 2002;4:343–350. doi: 10.1038/ncb783. [DOI] [PubMed] [Google Scholar]
- 50.Bivona TG, Philips MR. Ras pathway signaling on endomembranes. Curr Opin Cell Biol. 2003;15:136–142. doi: 10.1016/s0955-0674(03)00016-4. [DOI] [PubMed] [Google Scholar]
- 51.Hancock JF. Ras proteins: different signals from different locations. Nat Rev Mol Cell Biol. 2003;4:373–384. doi: 10.1038/nrm1105. [DOI] [PubMed] [Google Scholar]
- 52.Arozarena I, Matallanas D, Berciano MT, Sanz-Moreno V, Calvo F, Munoz MT, Egea G, Lafarga M, Crespo P. Activation of H-Ras in the endoplasmic reticulum by the RasGRF family guanine nucleotide exchange factors. Mol Cell Biol. 2004;24:1516–1530. doi: 10.1128/MCB.24.4.1516-1530.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pai EF, Krengel U, Petsko GA, Goody RS, Kansch W, Wittinghofer A. Refined crystal structure of the triphosphate conformation of H- ras p21 at 1.35A resolution: implications for the mechanism of GTP hydrolysis. EMBO J. 1990;9:2351–2359. doi: 10.1002/j.1460-2075.1990.tb07409.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Polakis P, McCormick F. Structural requirements for the interaction of p21ras with GAP, exchange factors, and its biological effector target. J Biol Chem. 1993;268:9157–9160. [PubMed] [Google Scholar]
- 55.Moodie SA, Paris M, Villafranca E, Kirshmeier P, Willumsen BM, Wolfman A. Different structural requirements within the switchII region of the Ras protein for interactions with specific downstream targets. Oncogene. 1995;11:447–454. [PubMed] [Google Scholar]
- 56.Clark GJ, Drugan JK, Terrell RS, Bradham C, Der CJ, Bell RM, Campbell S. Peptides containing a consensus Ras binding sequence from Raf-1 and theGTPase activating protein NF1 inhibit Ras function. Proc Natl Acad Sci USA. 1996;93:1577–1581. doi: 10.1073/pnas.93.4.1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Vojtek AB, Hollenberg SM, Cooper JA. Mammalian Ras interacts directly with the serine/threonine kinase Raf. Cell. 1993;74:205–214. doi: 10.1016/0092-8674(93)90307-c. [DOI] [PubMed] [Google Scholar]
- 58.Ogier-Denis E, Bauvy C, Houri J, Codogno P. Evidence for a dual control to macroautophagic sequestration and intracellular trafficking of N-linked glycoproteins by thr trimeric Gi3 protein in HT-29 cells. Biochem Biophy Res Commun. 1997;235:166–170. doi: 10.1006/bbrc.1997.6727. [DOI] [PubMed] [Google Scholar]
- 59.Ogier-Denis E, Pattingre S, El Benna J, Codogno P. Erk1/2-dependent phosphorylation of Galpha-interacting protein stimulates its GTPase accelerating activity and autophagy in human colon cancer cells. J Biol Chem. 2000;275:39090–39095. doi: 10.1074/jbc.M006198200. [DOI] [PubMed] [Google Scholar]
- 60.Pattingre S, Bauvy C, Codogno P. Amino acids interfere with the ERK1/2-dependent control of macroautophagy by controlling the activation of Raf-1 in human colon cancer HT-29 cells. J Biol Chem. 2003;278:16667–16674. doi: 10.1074/jbc.M210998200. [DOI] [PubMed] [Google Scholar]
- 61.Corcelle E, Nebout M, Bekri S, Gauthier N, Hofman P, Poujeol P, Fenichel P, Mograbi B. Disruption of autophagy at the maturation step by the carcinogen lindane is associated with the sustained mitogen-activated protein kinase/extracellular signal-regulated kinase activity. Cancer Res. 2006;66:6861–6870. doi: 10.1158/0008-5472.CAN-05-3557. [DOI] [PubMed] [Google Scholar]
- 62.Rodriguez-Viciana P, Warne PH, Dhand R, Vanhaesebroeck B, Gout I, Fry MJ, Waterfield MD, Downward J. Phosphatidylinositol-3-OH kinase as a direct target of Ras. Nature. 1994;370:527–532. doi: 10.1038/370527a0. [DOI] [PubMed] [Google Scholar]
- 63.Petiot A, Ogier-Denis E, Blommaart EF, Meijer AJ, Codogno P. Distinct classes of phosphatidylinositol 3'-kinases are involved in signaling pathways that control macroautophagy in HT-29 cells. J Biol Chem. 2000;275:992–998. doi: 10.1074/jbc.275.2.992. [DOI] [PubMed] [Google Scholar]
- 64.Arico S, Petiot A, Bauvy C, Dubbelhuis PF, Meijer AJ, Codogno P, Ogier-Denis E. The tumor suppressor PTEN positively regulates macroautophagy by inhibiting the phosphatidylinositol 3-kinase/protein kinase B pathway. J Biol Chem. 2001;276:35243–35246. doi: 10.1074/jbc.C100319200. [DOI] [PubMed] [Google Scholar]
- 65.Mochizuki T, Asai A, Saito N, Tanaka S, Katagiri H, Asano T, Nakane M, Tamura A, Kuchino Y, Kitanaka C, Kirino T. Akt protein kinase inhibits non-apoptotic programmed cell death induced by ceramide. J Biol Chem. 2002;277:2790–2797. doi: 10.1074/jbc.M106361200. [DOI] [PubMed] [Google Scholar]
- 66.Rodriguez-Viciana P, Sabatier C, McCormick F. Signaling specificity by Ras family GTPases is determined by the full spectrum of effectors they regulate. Mol Cell Biol. 2004;24:4943–4954. doi: 10.1128/MCB.24.11.4943-4954.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tall GG, Barbieri MA, Stahl PD, Horazdovsky BF. Ras-activated endocytosis is mediated by the Rab5 guanine nucleotide exchange activity of RIN1. Dev Cell. 2001;1:73–82. doi: 10.1016/s1534-5807(01)00008-9. [DOI] [PubMed] [Google Scholar]
- 68.Lambert JM, Lambert QT, Reuther GW, Malliri A, Siderovski DP, Sondek J, Collard JG, Der CJ. Tiam1 mediates Ras activation of Rac by a PI(3)K-independent mechanism. Nat Cell Biol. 2002;4:621–625. doi: 10.1038/ncb833. [DOI] [PubMed] [Google Scholar]
- 69.Mitin NY, Ramocki MB, Zullo AJ, Der CJ, Konieczny SF, Taparowsky EJ. Identification and characterization of Rain, a novel Ras-interacting protein with a unique subcellular localization. J Biol Chem. 2004;279:22353–22361. doi: 10.1074/jbc.M312867200. [DOI] [PubMed] [Google Scholar]
- 70.Kuriyama M, Harada N, Kuroda S, Yamamoto T, Nakafuku M, Iwamatsu A, Yamamoto D, Prasad R, Croce C, Canaani E, Kaibuchi K. Identification of AF-6 and Canoe as putative targets for Ras. J Biol Chem. 1996;271:607–610. doi: 10.1074/jbc.271.2.607. [DOI] [PubMed] [Google Scholar]
- 71.Zhang Z, Rehmann H, Price LS, Riedl J, Bos JL. AF6 Negatively Regulates Rap1-induced cell adhesion. J Biol Chem. 2005;280:33200–33205. doi: 10.1074/jbc.M505057200. [DOI] [PubMed] [Google Scholar]
- 72.Han L, Wong D, Dhaka A, Afar D, White M, Xie W, Herschman H, Witte O, Colicelli J. Protein binding and signaling properties of RIN1 suggest a unique effector function. Proc Natl Acad Sci USA. 1997;94:4954–4959. doi: 10.1073/pnas.94.10.4954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Khokhlatchev A, Rabizadeh S, Xavier R, Nedwidek M, Chen T, Zhang XF, Seed B, Avruch J. Identification of a novel Ras-regulated proapoptotic pathway. Curr Biol. 2002;12:253–265. doi: 10.1016/s0960-9822(02)00683-8. [DOI] [PubMed] [Google Scholar]
- 74.Yan Z, Chen M, Perucho M, Friedman E. Oncogenic Ki-ras but not oncogenic Ha-ras blocks integrin beta1-chain maturation in colon epithelial cells. J Biol Chem. 1997;272:30928–30936. doi: 10.1074/jbc.272.49.30928. [DOI] [PubMed] [Google Scholar]
- 75.Holmen SL, Williams BO. Essential role for Ras signaling in glioblastoma maintenance. Cancer Res. 2005;65:8250–8255. doi: 10.1158/0008-5472.CAN-05-1173. [DOI] [PubMed] [Google Scholar]
- 76.Uhrbom L, Kastemar M, Johansson FK, Westermark B, Holland EC. Cell type-specific tumor suppression by Ink4a and Arf in Kras-induced mouse gliomagenesis. Cancer Res. 2005;65:2065–2069. doi: 10.1158/0008-5472.CAN-04-3588. [DOI] [PubMed] [Google Scholar]
- 77.Lefranc F, Brotchi J, Kiss R. Possible future issues in the treatment of glioblastomas: special emphasis on cell migration and the resistance of migrating glioblastoma cells to apoptosis. J Clin Oncol. 2005;23:2411–2422. doi: 10.1200/JCO.2005.03.089. [DOI] [PubMed] [Google Scholar]