

Summary
Two immunoresistant (IR) glioma cell variants, 13-06-IR29 and 13-06-IR30, were cloned from 13-06-MG glioma cell populations after receiving continuous immunoselective pressure from multiple alloreactive cytotoxic T lymphocyte (aCTL) preparations. Reapplication of aCTL immunoselective pressure to the IR clones, displaying a partial regain in sensitivity to aCTL after removal of the selective pressure, restored the resistance. The IR variants exhibited cross-resistance to non-human leukocyte antigen (HLA)-restricted effector cells and γ-irradiation, but not to carmustine. The IR clones were characterized for factors that might contribute to the immunoresistance. The aCTL adhesion to extracellular matrix extracts derived from either the IR clones or the parental cells was similar and not impaired. Furthermore, aCTL binding to parental cells and IR clones was equal. Down-regulation of the cell recognition molecules, class I HLA or intercellular adhesion molecule-1 (ICAM-1), that would inhibit their recognition by aCTL was not observerd on the IR clones. The down-regulation of Fas by the IR clones correlated with their resistance to FasL-induced apoptosis. HLA-G or FasL that might provide an immunotolerant environment or provide a means of counterattack to aCTL, respectively, were not associated with the IR phenotype. The aCTL, coincubated with the IR clones and parental cells, displayed up-regulation of multiple secreted cytokines. A significant up-regulation of bioactive transforming growth factor (TGF)-β was observed in the IR clones compared with the parental cells. These data suggest that increased secretion of bioactive TGF-β may inhibit aCTL lysis of the IR clones. Disruption of the TGF-β signaling pathway may circumvent the resistance.
Keywords: IL-6, IL-8, apoptosis, astrocytomas, CTL
Malignant glioma patients demonstrate a limited response to conventional therapies that include surgery, radiation, and/or chemotherapy.1,2 The location of gliomas within a semiprivileged immune site,3 T helper immune cell defects of the patients,4,5 immunosuppressive glucocorticoid therapy to alleviate vasogenic edema, and glioma-derived immunosuppressive factors prevent the host from mounting effective antitumor responses.6,7 Consequently, the prognosis for these patients still remains poor, with survival times of 12 to 14 months, even with aggressive treatment regimens.2,8 To overcome these obstacles, researchers have devised strategies to stimulate and/or enhance antitumoral immune responses in glioma patients and to circumvent glioma-induced immunosuppression.9
The same mechanisms in place to prevent host-mediated tumor rejection may also impede adjuvant immunotherapies from suppressing tumor growth.10,11 Notably, tumors used multiple tactics to evade attack by cytotoxic effector cells that include production of glycosaminoglycan coats, down-regulation of class I human leukocyte antigens (HLA) or intercellular adhesion molecules (ICAM) to prevent their adhesion to and recognition by T cells.12-14 Furthermore, tumors may either down-regulate their Fas (CD95) expression to prevent their destruction, or up-regulate their Fas ligand (FasL) molecules to counterattack the T lymphocytes.15
A cellular therapy approach tested by our research group in a phase 1 clinical trial involved local adoptive transfer of recombinant human interleukin-2 (IL-2) and ex vivo activated alloreactive cytotoxic T lymphocytes (aCTL). Repeated administrations of aCTL, generated in hollow fiber bioreactors and sensitized to the patient HLA antigens, were placed into the resected tumor beds of recurrent malignant glioma patients.16-18 Three of 6 patients treated may have responded.18 Although the results of the phase 1 trial were promising, we decided to explore plausible explanations to account for disease progression in the nonresponders. One explanation, among others, was the existence of intrinsically immunoresistant (IR) glioma cells. We cloned aCTL resistant glioma cells for study after applying aCTL immunoselection in vitro.19
Two IR glioma cell clones, 13-06-IR29 and 13-06-IR30, were isolated from continuously immunoselected 13-06-MG glioma cell populations and cytogenetically characterized.19 The IR variants were present at a low frequency within the immunoselected cell populations and resisted lysis by multiple aCTL populations in the absence of immunoselective pressure.19 We have determined that the IR clones display a reduced induction to aCTL-induced apoptosis, and microarray analyses have implicated multiple down-regulated proapoptotic factors in both clones.20
Here we examined the susceptibility of the IR clones to other apoptotic/necrotic stimuli, such as non-HLA–restricted effector cells, carmustine and γ-irradiation and to FasL-mediated apoptosis. Our experimental approach to clarify the mechanism(s) of immunoresistance involved a survey for the possible employment of strategies by the IR clones to create an immunotolerant microenvironment, by examining factors that prevent apoptotic induction in the clones, by inducing effector aCTL apoptosis, or by disrupting aCTL adhesion/recognition to the IR clones. In addition, we examined the cytokines secreted by the IR clones, parental cells, and aCTL when cultured alone and upon coincubation of the aCTL with parental cells and IR clones.
MATERIALS AND METHODS
Cell Culture
The Jurkat T cell leukemia line, erythroleukemic K562 cells, 13-06-MG glioma cells and in vitro aCTL-immunoselected clones, 13-06-IR29 and 13-06-IR30 derived from continuously immunoselected 13-06-MG cultures,19 were maintained in RPMI 1640 (Mediatech, Herdon, VA) containing 10% fetal bovine serum (FBS, Gemini-Bioproducts, Woodland, CA) in a 37°C humidified chamber with 5% CO2. The DBTRG-05 MG glioma cells and the TALL-104 leukemic T cell line were cultured as described.21,22 The HLA class I antigens displayed on 13-06-MG cells were A1, A2, B44, B57, and Bw4, and the class I antigens displayed on DBTRG-05 MG cells were A2, A68, B35, B38, Bw4, and Bw6, as determined by molecular typing using polymerase chain reaction sequence-specific-primers and sequence-based typing (ClinImmune Labs, Aurora, CO). TALL-104 cells were positive for the following HLA antigens: class I HLA-A11 and A29, HLA-B8 and B58, HLA-DR3 and 8, and HLA-DQ 2 and 4.23 G10 cells, which are K652 cells stably transfected with the human FasL gene,24 were maintained in Dulbecco’s modified Eagle’s medium containing 10% FBS and 1.0 mg/mL G418 (Invitrogen, Carlsbad, CA). Cells were negative for mycoplasma by Hoechst 33342 (Sigma, St Louis, MO) staining.19
Activation and Culture of aCTL, Allogeneic Lymphokine-activated Killer (LAK), and TALL-104 Cells
Under institutional regulatory body approval, precursor aCTL were obtained from either leucopaks derived from aphaeresis procedures, or from heparinized whole blood drawn from healthy volunteers. Sensitization of the precursor aCTL to glioblastoma patient 13-06 HLA occurred as previously described.19,25 aCTL cultures were restimulated with OKT-3 (10 ng/mL; Ortho Pharmaceutical, Raritan, NJ) every 12 to 14 days. The cytokines secreted by aCTL upon coincubation with target cells and their lytic activity was tested between 14 and 28 days after the initial sensitization.
For activation and generation of LAK cells, cryopreserved peripheral blood mononuclear cells isolated from the same blood donors used to generate aCTL cells, were thawed and incubated with 600 IU/mL of IL-2 in RPMI 1640 containing 10% FBS. After 4 to 5 days, the ability of LAK cells to lyse glioma parental cells and IR clones was determined. The cultured TALL-104 cells were cryopreserved after lethal irradiation (400 rads) with a 137Cs source.22,26 The cryopreserved cells were quick-thawed and washed twice in phosphate-buffered saline (PBS) before placing into cytotoxicity assays as previously described.22,26
Cytotoxicity and Survival Assays to Measure Sensitivity of Gliomas to Non-HLA–restricted Effectors, Carmustine, and Radiation
The 51Cr release assay was used to determine the lytic activity when effector aCTL were coincubated with glioma cells for 4 hours at various effector to target (E:T) ratios.26 In some experiments, blockade of the granule-exocytosis pathway was achieved by adding 10 mM ethylene glycolbis(2-aminoethyl)N,N,N′,N′-tetra-acetic acid (EGTA, Calbiochem, LaJolla, CA), a calcium chelator, to the assay medium.27 The percentage of specific release was calculated by the formula: [(cpmexperimental − cpmspontaneous)/ (cpmmaximal − cpmspontaneous)] × 100%. Spontaneous release was the counts per minute (cpm) of the targets in assay medium alone and maximal release was produced by incubation of the targets with 2% Triton X-100 (Sigma). The data collected were analyzed by paired t test or the nonparametric Kruskal-Wallis test.
Glioma cells (2.5 × 103) were plated into each of 6 wells in round bottom 96-well plates and cultured for 2 days. Cells were treated in serum free RPMI 1640 medium for 1 hour at 37°C in 5% CO2 with the nitrosourea, carmustine (Sigma) at different concentrations (0, 0.31, 0.63, 10, and 40 μg/mL) to obtain dose-response curves. Surviving cells were allowed to proliferate after carmustine treatment for 3 days. MTT dye solution [5 mg/mL of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide, Sigma) was diluted 10-fold in RPMI 1640 containing 10% FBS and added to wells of the plates for 4 hours at 37°C before reading absorbance at 562 nm. To obtain a relative measure of survival, the mean absorbances of the carmustine-treated cells were expressed as percentages of the mean absorbance of untreated cells for each glioma cell type at the different drug concentrations. The data collected for each glioma were statistically compared by the analysis of variance.
The radiosensitivity of the IR clones was determined by exposure of the cells to several doses of γ-irradiation (range 5 to 20 Gy) as has been described before.28 Cells were plated 3 hours after irradiation into 10 wells of 96-well plates and allowed to recover for 5 days. A relative measure of cell survival was determined with the MTT assay as described above. Because single unfractionated radiation doses at 15 Gy or less induced minimal IR clone cell death, cells were treated with a single dose at 20 Gy. The data collected for each glioma were statistically compared by the Kruskal-Wallis test.
Adhesion of aCTL to the Extracellular Matrix (ECM) Proteins Derived From Parental Glioma Cells and IR Clonal Variants
Wells coated with glioma-derived ECM proteins were obtained with parental cells or IR clones (2.5 × 103) plated to confluency in flat-bottom, 96-well plates.29 The cell monolayers were washed once with PBS, treated with 0.5% Triton X-100 for 30 minutes, exposed to 0.1 M NH4OH for 2 minutes, and rinsed 4 times with PBS. aCTL (5 × 104) were added to wells (n = 6) for each of the individual glioma cell types, and incubated on ice for 30 minutes. Next, the plates were incubated for 30 minutes at 37°C, placed on a horizontal shaker at 210 rpm for 6 minutes, then rinsed with PBS. Adherent aCTL were fixed in 1% glutaraldehyde for 10 minutes and stained with a 0.1% crystal violet solution. Adhesion of the aCTL to the glioma-derived ECM was quantified by spectrophotometric absorbance of stained nuclei measured at 562 nm.30 Data derived from the experiments were analyzed by the Kruskal-Wallis test.
Glioma:aCTL Conjugate Formation Assays
To determine the ability of the aCTL to form conjugates with parental glioma cells or IR clones, glioma cells (5 × 104) were plated onto Lab-Tek 4-well glass chamber slides (Nalge Nunc International, Naperville, IL) for 48 hours before the addition of aCTL (5 × 105). To distinguish glioma cells from aCTL, glioma cells were labeled with 0.25 μM carboxyfluorescein diacetate succinimidyl ester (CFSE) dye and aCTL were labeled with 0.2 μM CellTracker Orange dye in PBS following manufacturer’s instructions (Molecular Probes, Eugene, OR). The cell mixtures were incubated in a humidified 37°C, 5% CO2 chamber for 30 minutes. The medium was removed and the chambers gently rinsed with PBS before the cells were fixed in 4% paraformaldehyde for 15 minutes. Confocal micrographs were acquired with a 60 × /0.17 numerical aperture oil objective lens on a BX61 microscope (Olympus America, Inc, Melville, NY). The percentages of glioma cells that were bound by aCTL were determined by examination of 5 random 20 × fields for each glioma cell type incubated with aCTL. The percentages of glioma cells bound by aCTL, irrespective of the total number of aCTL cells binding a single glioma cell ± SEM were determined. Data derived from the experiments were analyzed by the Kruskal-Wallis test.
Flow Cytometry
Adherent cell cultures at ~80% confluency were disaggregated with PBS containing 10 mM ethylene diamine tetra-acetic acid. Cells (5 × 105) were pelleted at 200 × g for 5 minutes, supernatants decanted and the cells were resuspended in 50 μL of PBS containing 1% FBS. After washing, cells were then incubated on ice for 20 minutes with monoclonal antibodies specific for HLA class I, HLA-G, ICAM-1, leukocyte function antigen-1 (LFA-1), and Fas and FasL antigens. Purified primary antibodies were mouse antihuman ICAM-1 (RR1/1.1.1, Dr Robert Rothlein), LFA-1 (R3.1.E2.G8.C8, Dr Rothlein), Fas (DX2, BD Pharmingen, San Diego, CA), and FasL (NOK-1, BD Pharmingen). Unbound primary antibody was washed away before a 20-minute incubation of the cells with R-phycoerythrin (RPE)-conjugated secondary antimouse IgG (H+L) (eBioscience, San Diego, CA). Cells were also incubated with RPE-conjugated mouse antihuman class I HLA-ABC (W6/32, Dako, Carpinteria, CA) or isotype-matched RPE-conjugated mouse IgG2a (Coulter, Hialeah, FL) antibodies. After staining cells with fluorochrome-conjugated antibodies, cells were washed with PBS containing 1% FBS and pelleted at 200 × g for 5 minutes. The stained cells were then fixed with 1% paraformaldehyde (Sigma) in PBS and kept refrigerated until analysis on EPICS XL-MCL (Coulter) or FacScan (BD Pharmingen) flow cytometers. For detection of HLA-G, tumor cells were fixed and permeabilized (Cytofix Cytoperm, BD Pharmingen) before incubating with purified mouse antihuman HLA-G antibody (4H84, BD Pharmingen) and a secondary RPE-conjugated antibody. From the flow cytometric analysis, we determined the percentage of positive cells and the mean fluorescence intensity (MFI).
Detection of Tumor Cell Injury Using 7-Amino Actinomycin D (7AAD)
Tumor cell injury was detected by 7AAD incorporation as described with minor modification.26 Glioma cells were seeded into 6-well plates and cultured for 48 hours before labeling for 1 hour with 0.25 μM CFSE in PBS. Adherent glioma cells were then coincubated with FasL-expressing G10 cells at a 1:5 ratio for 18 hours at 5% CO2 in a 37°C humidified chamber. For a positive control, nonadherent Fas-expressing Jurkat T cells were labeled with CFSE before their coincubation with effector G10 cells in polystyrene tubes (12 × 75 mm, BD Biosciences, Franklin Lakes, NJ). After coincubation, the cells in suspension were collected and combined with adherent cells that were harvested by treatment with 0.1% trypsin. After centrifugation at 200 × g for 5 minute, cells were incubated in 100 μL of 7AAD (20 μg/mL, Sigma) in PBS for 20 minutes at 4°C. The percentages of CFSE-labeled glioma cells within the segregated live, apoptotic, and late apoptotic/necrotic populations on scattergrams were determined as previously described.26
Detection of Secreted Cytokines
To determine the rates of transforming growth factor (TGF)-β1 and TGF-β2 secretion by the glioma cells, single cell suspensions (105 cells/mL) were added to 60 mm dishes and cultured for 48 to 72 hours. Cell culture medium was collected, clarified by centrifugation at 400 × g for 10 minutes, and stored in microcentrifuge tubes at − 80°C until analysis. The total cell number in each dish at the end of the incubation period was determined by hemocytometer counts after collecting the adherent cells with 0.25% trypsin, pelleting by centrifugation at 200 × g for 10 minutes, and resuspending in RPMI 1640 culture medium. The amount of activated TGF-β1 and TGF-β2 secreted into the medium was determined using sandwich enzyme-linked immunosorbent assay (ELISA) kits according to manufacturer’s instructions (R&D Systems, Inc, Minneapolis, MN). To determine the total amount of TGF-β1 and TGF-β2 (activated and latent forms) secreted, samples were treated with 1 N HCL for 10 minutes at room temperature. The pH was neutralized by the addition of an equal volume of 1.2 N NaOH containing 0.5 M N-2-hydroxyethylpiperazine-N-2-ethanesulfonic acid. Rates of secretion from 3 independent experiments were normalized and expressed as pg TGF-β/106 cells/24 h ± SEM. Data obtained were statistically analyzed by the nonparametric Kruskal-Wallis test.
Detection of a panel of TH1 and TH2 cytokines that included IL-1α, -1β, -2, -4, -5, -6, -8, -10, -13, IFN-γ, TNF-α, and TNF-β was achieved using the Q-plex Human Cytokine Array Screen (Biolegend, Inc, San Diego, CA). The concentrations of cytokines in supernates obtained from aCTL, glioma cells, and coincubates of aCTL with glioma cells were determined against a 5 point standard curve using the Q-view software program according to manufacturer’s instructions. To determine the cell type contributing to the release of the cytokines, either the aCTL or the glioma cells were pretreated with Brefeldin A (BrfA, BD Pharmingen) before their coincubation. BrfA was added at a concentration of 10 μL BrfA/mL of culture medium.31 After 2 hours, BrfA treated cells were washed once before coincubation and collection of the supernatants as described earlier. The results obtained for IL-6 and IL-8 with the Q-plex array were individually verified in 2 independent experiments using sandwich ELISA kits (Biolegend).
RESULTS
IR Phenotype of the IR Clones is Restored Upon Application of Selective Pressure
The 13-06-IR29 and 13-06-IR30 IR clones maintained their resistance to aCTL lysis in the absence of immunoselective pressure for 40 and 13 cell doublings, respectively, before displaying a partial regain in sensitivity to aCTL lysis as described.19 aCTL selective pressure was reapplied intermittently to IR cell cultures displaying the restored sensitivity. The immunoresistance was reassessed by cytotoxicity assays after the clones had undergone about 13 to 15 cell doublings (35 d after starting selective pressure). The susceptibility of the IR clones to aCTL lysis was significantly reduced (P ≤ 0.05) once again compared with lysis of the 13-06-MG parental cells at multiple E:T cell ratios (Fig. 1). The lysis of the IR clones was fairly equivalent to that of partially relevant DBTRG-05 MG glioma cell targets that displayed only 2 class I HLA alleles in common with 13-06-MG cells. Lysis of the IR clones and DBTRG-05 MG targets was minimal and statistically distinct from the parental 13-06-MG target cells (P ≤ 0.05). The data demonstrate that the IR phenotype is restored upon application of intermittent selective pressure.
FIGURE 1.
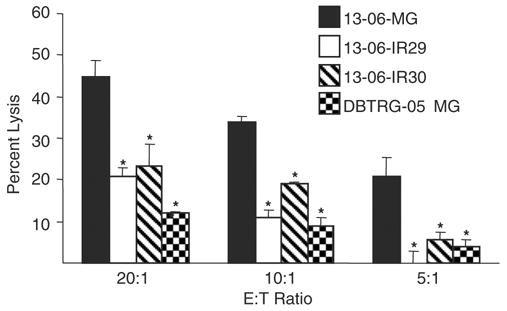
Reapplication of immunoselective pressure restores the IR phenotype to IR cultures displaying a partial reversion to aCTL resistance. The 13-06-IR29 and 13-06-IR30 clones previously cultured in the absence of immunoselective pressure for 40 and 13 cell doublings, respectively, were thawed and given intermittent aCTL selective pressure for 35 days. After removal of the selective pressure, the susceptibility of the IR clones and parental cells to aCTL lysis was determined at 3 E:T ratios in cytotoxicity assays. Lysis of a partially relevant glioma target, DBTRG-05 MG that displayed only 2 class I HLA alleles in common with 13-06-MG cells was also tested. Asterisks indicate a statistically significant (P ≤ 0.05) decrease in aCTL lysis compared with the parental glioma cells as determined by Student’s paired t test.
IR Clones Display Cross-resistance to Non-HLA–restricted Allogeneic LAK and TALL-104 Effector Cells and to γ-irradiation, but not to Carmustine
After cancer cells receive selective pressure in vitro or in vivo, the phenomenon of cross-resistance of the cells to other cytotoxic therapies/agents has been observed.32 We hypothesized that the IR clones that had received in vitro selective pressure, might exhibit cross-resistance to other applied cytotoxic selective pressures, compared with the parental cells that had not received in vitro selective pressure; the parental cells also had come from a patient at initial diagnosis such that no in vivo selective pressure had been applied to them. We tested the parental cells and the IR clones for cross-resistance to other non-HLA–restricted allogeneic effector cells, to γ-irradiation, and to the chemotherapeutic agent carmustine (Fig. 2).
FIGURE 2.
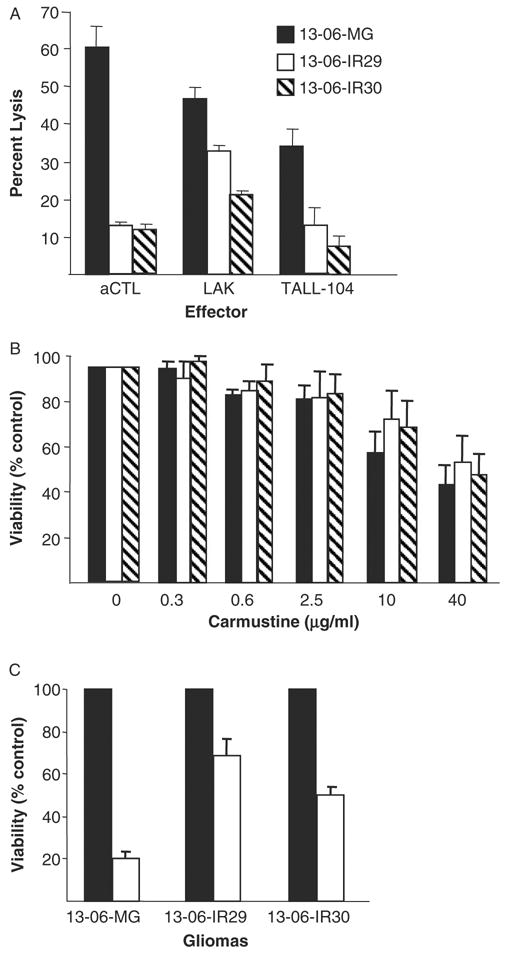
Assessments for cross-resistance of the IR clones to non-HLA–restricted, allogeneic LAK and TALL-104 cells, to carmustine, and to γ-irradiation treatments. A, The percentage lysis ± SEM obtained for parental 13-06-MG glioma cells and IR clones to aCTL, to LAK cells (generated from the peripheral blood mononuclear cells of the aCTL blood donor), and to lethally irradiated TALL-104 cell effectors, all at a 12.5:1 E:T ratio. B, Sensitivity of the parental and variant glioma cells to carmustine at 5 drug concentrations, shown as percentages of viability ± SEM relative to untreated control cells at 100%. C, Sensitivity of the parental and variant glioma cells treated with an unfractionated dose of γ-irradiation (20 Gy). The percentages of viability ± SEM of the radiation treated cells (□) for each glioma and for nontreated control cells (■) ± SEM are given.
The lysis of the parental glioma cells and IR clones caused by non-HLA–restricted LAK and TALL-104 cell preparations were compared with that of the 13-06 HLA–restricted aCTL preparation. The aCTL were generated from the same blood donor as the LAK cells and were at 14 culture days from the initial sensitization in a 1-way mixed lymphocyte reaction. The TALL-104 cells were used immediately after washing, taken from a cryopreserved state. Lytic data from a representative 4 hour cytotoxicity assay are shown for each of the effector cell types at a 12.5:1 E:T ratio (Fig. 2A). The IR clones demonstrated a greater degree of resistance to LAK and TALL-104 cell lysis compared with the parental cells. Analysis of data from multiple experiments revealed that LAK cell lysis of both IR clones was significantly reduced (P ≤ 0.04) compared with lysis of the parental cells. The data shown for lethally irradiated TALL-104 cells are from one of 2 independent experiments. In both experiments, the IR clones similarly demonstrated significantly reduced lysis caused by the TALL-104 cells. Cumulatively, the data show that the aCTL-resistant variants display cross-resistance to non-HLA–restricted allogeneic cell effectors.
The susceptibility of the IR clones to carmustine, a chemotherapeutic agent commonly used to treat gliomas,33 and to γ-irradiation was also determined. At the multiple carmustine doses tested, significant differences in cell survival 3 days after carmustine treatment were not observed in the IR clones relative to the parental cells (Fig. 2B). The data additionally show that the glioma cells and clonal variants need exposure at fairly high concentrations (≥ 10 μg/mL) to be susceptible to the cytotoxic action by the chemotherapeutic agent. In contrast, substantially higher fractions of the IR clones survived γ-irradiation treatment and therefore are more resistant to the single 20 Gy dose of γ-irradiation (P ≤ 0.02) (Fig. 2C). The data indicate that the IR clones are less sensitive to γ-irradiation but do not display cross-resistance to carmustine.
aCTL Adhesion to IR Clone-derived ECM is not Impaired Relative to the Parental Glioma-derived ECM
ECM proteins are produced by glioma cells, albeit the specific types and ratios may vary from one to another.29,30 It was possible that ECM components and secretion by the IR clones was altered to inhibit the effector aCTL from adhering to them.12,34,35 To test this idea, we performed assays to detect differences in aCTL adherence to the ECM proteins derived from the parental cells and IR clones. No impairment of aCTL adhesion to the IR clones ECM was detected; aCTL sensitized to the HLA antigens of glioma patient 13-06 bound equally well to ECM protein extracts from the IR clones and parental cells (Fig. 3A).
FIGURE 3.
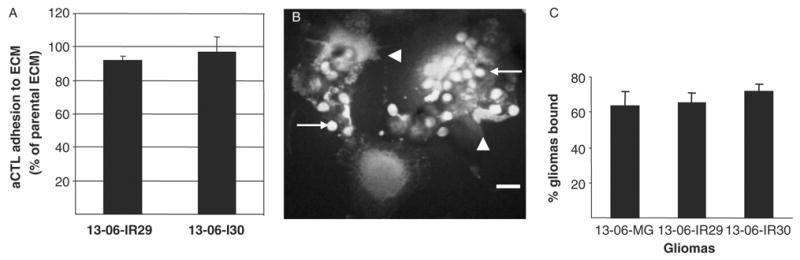
aCTL adhesion to glioma-derived ECM proteins and conjugate formation between aCTL and glioma cells. A, The adhesion of aCTL to IR clone-derived ECM proteins are expressed as a percentage ± SEM of the aCTL adhered to parental ECM proteins, as derived from absorbance measurements. B, Confocal micrograph demonstrates conjugates of adherent CFSE-labeled 13-06-IR29 glioma cells with CellTracker Orange-labeled aCTL. Multiple smaller sized aCTL (arrows) are associated with the larger individual 13-06-IR29 cells. Damage to the glioma cells is readily visualized by the membrane blebbing (arrowheads). Bar = 20 μm. C, Percentages of parental glioma cells and IR clones bound to aCTL ± SEM, as determined by confocal microscopic examination of 5 random 20 × fields for each glioma cell type incubated with aCTL for 30 minutes.
Conjugate Formation Between aCTL and the Parental Glioma Cells and IR Clones is Similar
Tumors can avoid immune recognition by modulating their expression of adhesion molecules required for establishing cell to cell contacts with effector cells.13 Because adhesion and conjugate formation must precede lysis, we determined the ability of aCTL to establish whole cell to cell contacts with the IR clonal variants. Confocal microscopic imaging was used to assess cell conjugate formation upon coincubation of CFSE-labeled glioma cells with CellTracker Orange-labeled aCTL. It was readily apparent shortly after adding the aCTL to the glioma cultures, that multiple aCTL formed conjugates with a single glioma cell (Fig. 3B). Examination of multiple microscopic fields showed that high percentages (≥ 60%) of the plated parental glioma cells and IR clones formed conjugates with aCTL. Quantitative differences in the adhesion of aCTL to the parental glioma cells versus the IR clones were not observed (Fig. 3C), even though morphologic signs of glioma cell injury (Fig. 3B) were apparent more so in coincubates of aCTL with parental cells compared with those with the IR variants.
HLA Class I, ICAM-1, Fas and Fas-L Expression by the Parental Cells and IR Glioma Clones
The parental and IR cells were characterized for their expression of cell surface proteins, HLA-ABC, ICAM-1, and Fas, known to be required for efficient CTL recognition, adhesion, and destruction of tumor cells.25,36 In addition, the gliomas were characterized for Fas-L and HLA-G expression. The Fas-L and HLA-G molecules induce T cell apoptosis and inhibit allospecific T cell killing of gliomas, respectively.15,37,38
After confirming that the majority of cells within the anti-13-06 aCTL preparations (80% to 85%) used in these experiments expressed T cell receptor (TCR) α/β and LFA-1 (data not shown), we analyzed the cell surface expressions of HLA-ABC and ICAM-1 on the parental cells and IR clones by flow cytometry (Fig. 4) to determine whether down-regulation of either class I HLA or ICAM-1 molecules on the IR clone surfaces had occurred. Down-regulation of one or both of these molecules might be expected if the IR clones used this tactic to evade recognition by the aCTL. Nearly all of the parental cells and clonal variants were highly positive for HLA-ABC class I antigens in 3 independent experiments (92% to 99% positive, all at high relative antigen densities with MFIs of 21 to 34). Although ICAM-1 was expressed on higher percentages of the IR clones (45% and 62% for 13-06-IR29 and 13-06-IR30, respectively) compared with parental cells (34% positive), the antigen density levels were low to moderate (MFIs at 4 to 12) on all when examined on 2 separate occasions. The down-regulated expression of these 2 molecules known to participate in E:T cell recognition was not observed on the IR clones. We conclude that the IR clones did not use down-regulations of HLA-ABC or ICAM-1 as mechanisms of resistance.
FIGURE 4.
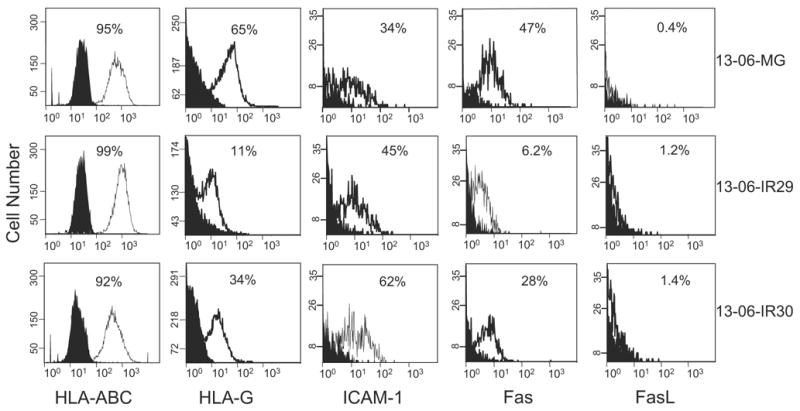
Expression of classic HLA-ABC class I antigen, nonclassic HLA-G, ICAM-1, and Fas and FasL by parental 13-06-MG glioma cells and 13-06-IR29 and 13-06-IR30 IR clones by flow cytometric analyses. The histograms show the nonspecific binding of isotype and secondary antibodies (filled peaks) and the degree of specific staining (unfilled peaks). The percentages of positive cells are indicated and the relative antigen densities can be ascertained by placement and height of the peak on the abscissa.
Because we also showed that apoptosis induction was inhibited in the IR clones when incubated with aCTL compared with the parental cells,20 we explored the possibility that the Fas pathway was disrupted in the IR clones. In 2 independent experiments, we found that the percentages of positive cells and levels of Fas molecule cell surface expression were lower on the IR clones (13-06-IR29 6.2%, MFI = 1.6 and 13-06-IR30 28%, MFI = 3) compared with the parental cells (13-06-MG 47%, MFI = 4.5) (Fig. 4), suggesting down-regulation of Fas as a probable mechanism of aCTL evasion.
Although multiple aCTL cell populations maintained in culture for 14 to 21 days variably expressed Fas (22% to 82% Fas positive cells), FasL was not expressed by the IR clones or the parental cells (Fig. 4). Thus, it is unlikely that the IR clones escape lysis by inducing apoptosis of the aCTL effectors. In support of this conclusion, incorporation of the cell viability dye, 7AAD, was not observed in the CFSE-negative gated aCTL population coincubated for 4 hours with the CFSE-labeled IR clones (data not shown).
For the detection of HLA-G, an antibody was used that would detect all HLA-G isoforms. We found that only low to moderate levels of HLA-G were expressed by the parental glioma and IR variants, albeit in the experiment we present (Fig. 4), we observed it on a higher percentage of the parental cells (65% positive, MFI = 12) compared with the clones (13-06-IR29 11%, MFI = 4 and 13-06-IR30 34%, MFI = 12). Data obtained from 3 independent experiments analyzed by the Kruskal-Wallis test, however, demonstrated that HLA-G expression by the parental cells and IR clones was not statistically significant (P ≤ 0.84). Thus, the prediction that up-regulated HLA-G expression would be associated with the IR phenotype did not hold.
IR Clones Exhibit Decreased Susceptibility to FasL-induced Apoptosis
To determine if the reduced expression of Fas on the IR clones also correlated with a reduced susceptibility to FasL-induced apoptosis, we performed 2 independent 7AAD flow cytometric cell injury assays. Small percentages of aCTL (1% to 4%) expressed Fas-L 14 to 28 days after activation (data not shown), as a consequence, G-10 cells (K562 cells transfected with human Fas-L gene)24 served as the effector cells in the experiments. The G10 cells were coincubated with Fas-expressing Jurkat T cells for 18 hours in each experiment. G10 cells were also coincubated with parental 13-06-MG glioma cells, and IR 13-06-IR29 and 13-06-IR30 clones. Representative data from one experiment are shown (Fig. 5). Jurkat T cells were highly viable (96%) when incubated alone (Fig. 5A), but underwent extensive injury (86% cell injury) when coincubated with G10 cells (Fig. 5B). The parental 13-06-MG glioma cell injury increased 20% upon their coculture with G10 cells (Figs. 5C, D). In contrast, IR clone 13-06-IR29 demonstrated a minimal increase in cell death (Figs. 5E, F) and clone 13-06-IR30 was completely insensitive to Fas/FasL-induced cell death (Figs. 5G, H). Thus, only a fifth of the cells within the parental glioma population are susceptible to apoptosis by the Fas/FasL pathway even though half of the population displays Fas, whereas, few to none of the IR clones undergo apoptosis by this pathway.
FIGURE 5.
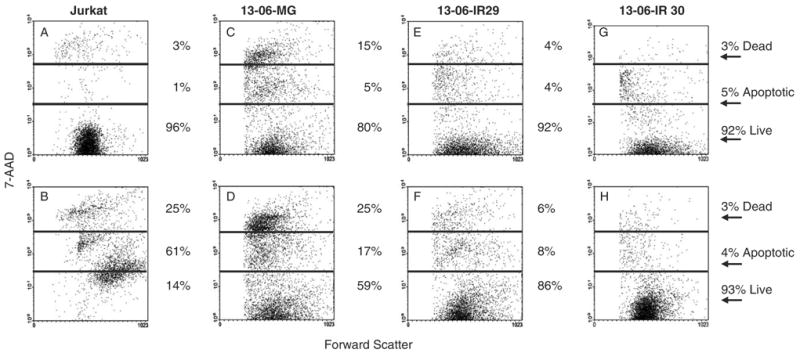
Flow cytometric scattergrams are shown for CFSE-labeled tumor cells that were (bottom row) or were not (top row) coincubated with FasL positive G10 cells for 18 hours at a 5:1 E:T ratio. The percentages of cells that were live, apoptotic, or dead (late apoptotic/necrotic) are given. A, Fas positive Jurkat cells, (B) Jurkat+G10 cells, (C) 13-06-MG cells, (D) 13-06-MG+G10 cells, (E) 13-06-IR29 cells, (F) 13-06-IR29+G10 cells, (G) 13-06-IR30 cells, and (H) 13-06-IR30+G10 cells.
Lysis of Parental Cells and IR Clones is Largely Calcium Dependent
Interactions of glioma cell HLA-ABC with the TCR on T cells trigger T cell exocytosis of granzyme and perforin molecules. The exocytosis of perforin and granzyme molecules is calcium dependent, in contrast to the calcium-independent Fas/FasL pathway.27 We explored whether the addition of the calcium chelator, EGTA, to the cytotoxicity assay medium would inhibit aCTL-mediated lysis of the parental cells and IR clones. Experiments with EGTA added to the cytotoxicity assay medium established that the majority (80% to 90%) of lysis to the parental cells or to the IR clones caused by the aCTL occurred by a calcium-dependent mechanism(s) (Fig. 6).
FIGURE 6.
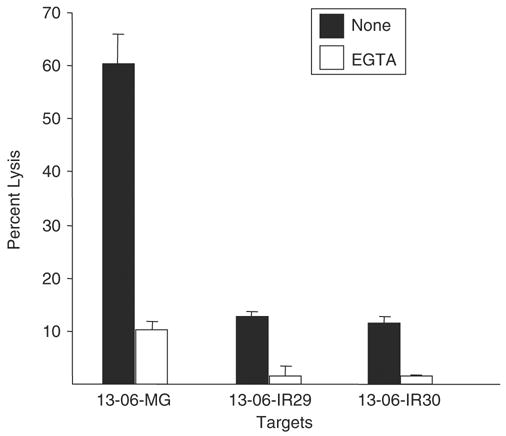
Parental glioma cells and IR clones are sensitive to calcium-dependent lysis by aCTL. Parental 13-06-MG cells and IR clones, 13-06-IR29 and 13-06-IR30, were coincubated with anti-13-06 aCTL at a 12.5:1 E:T ratio for 4 hours. In some wells the calcium-chelator, EGTA, was added to inhibit perforin and granzyme-mediated cell lysis. The percentages of glioma cells lysed ± SEM, obtained in the presence or absence of EGTA, are shown.
Cytokines are Secreted by aCTL When Cocultured With Glioma Cells
We previously demonstrated that among a panel of cytokines analyzed, IL-6 and IL-8 were secreted by aCTL, parental glioma cells, and the immunoselected polyclonal glioma cell pools and that the 2 cytokines were up-regulated upon coincubation of the glioma cells with aCTL.19 Therefore, we analyzed a more extensive panel of cytokines (IL-1α, -1β, -2, -4, -5, -10, -13, IFN-γ, TNF-α, and TNF-β) and also included IL-6 and IL-8, in supernatants obtained individually from the aCTL, the parental cells, the IR clones, and their coincubates with the effector aCTL.
The glioma cells alone secreted minimal amounts of IL-1α, -1β, -2, -4, -5, -13, IFN-γ, TNF-α, and TNF-β (Table 1 and data not shown). Similarly, the aCTL produced minimal levels of all cytokines with the exception of IL-8. The IL-6, IL-8, and IL-10 cytokines were produced by the parental and IR variant cells. The secretions of IL-6 and IL-8 in cocultures of aCTL with the parental cells and IR clones were up-regulated and consistent with our previous findings,19 as was IL-10 secretion (Table 1).
TABLE 1.
aCTL Up-regulate Secretion of Cytokines When Coincubated With Glioma Cells
| Cells | Cells Treated w/BrfA* | IFN-γ (pg/mL) | IL-6 (pg/mL) | IL-8 (pg/mL) | IL-10 (pg/mL) | IL-13 (pg/mL) |
|---|---|---|---|---|---|---|
| aCTL | None | 10 | 7 | 678 | 29 | 5 |
| 13-06-MG | None | 25 | 86 | 11474 | 415 | 36 |
| 13-06-MG+aCTL | None | 48 | 1187 | 17166 | 853 | 86 |
| 13-06-MG+aCTL | aCTL | bld† | 6 | 462 | 15 | 1.6 |
| 13-06-MG+aCTL | Glioma | 124 | 4924 | 17169 | 812 | 257 |
| 13-06-IR29 | None | 9 | 416 | 2464 | 123 | 20 |
| 13-06-IR29+aCTL | None | 38 | 1195 | 6911 | 277 | 29 |
| 13-06-IR29+aCTL | aCTL | bld | 33 | 299 | 6 | bld |
| 13-06-IR29+aCTL | Glioma | 100 | 4711 | 17169 | 820 | 268 |
| 13-06-IR30 | None | 4 | 247 | 1211 | 56 | 6 |
| 13-06-IR30+aCTL | None | 22 | 1795 | 6841 | 293 | 36 |
| 13-06-IR30+aCTL | aCTL | 1.7 | 16 | 195 | 6 | bld |
| 13-06-IR30+aCTL | Glioma | 131 | 4665 | 17171 | 849 | 209 |
To determine the contribution of each cell type to the release of IFN-γ, IL-6, IL-8, IL-10, and IL-13 either aCTL or glioma cells were treated with BrfA for 2 h before their coincubation at a 10:1 E:T ratio for 18 h. Clarified supernatants obtained from the cocultures were subjected to Q-plex/Q-View analysis to determine the quantities of cytokines secreted.
bld, below limit of detection by Q-plex/Q-View analysis.
To determine which cell type, the effector or target, contributed to the up-regulated release of cytokines upon coincubation, either the aCTL or the glioma cells were pretreated with BrfA, a secretion inhibitor, for 2 hours before the coincubation. When BrfA pretreatment of aCTL was used to indicate cytokine release by the gliomas during coincubation, surprisingly significant decreases in the cytokines released were detected (Table 1). When BrfA pretreatment of the gliomas was used to indicate cytokine release by the aCTL during coincubation, the secretions of IL-6, -8, -10, -13 and IFN-γ were usually enhanced, suggesting that the gliomas produce a factor to inhibit aCTL secretion of these cytokines. Thus, the aCTL were largely responsible for the production of the cytokines during coincubation with the relevant target cells.
IR Clones Display an Increased Rate of Biologically Active TGF-β Secretion
Our cytokine analyses suggested that a factor produced by the gliomas inhibited aCTL secretion of proinflammatory cytokines. Given the known ability of TGF-β to inhibit expression of proinflammatory cytokines upon priming and restimulation of T lymphocytes with antigen,39 we analyzed cell culture supernatants obtained from parental cells and IR clones by ELISA to determine the rates of secretion (pg/106 cells/24 h) of the TGF-β1 and TGF-β2 isoforms by the cells. The antibodies used in the ELISA assays bind to the activated forms of TGF-β1 and TGF-β2 but not to their latent forms. To determine the total amounts of TGF-β secreted, latent TGF-β present in the conditioned medium was activated by treatment with 1 M HCL and then analyzed by ELISA.
Both clones exhibited a statistically significant (P ≤ 0.02) increase in the rate of active TGF-β1 and total TGF-β1 secretion compared with the parental cells (Fig. 7A). Although not statistically significant, the data showed greater levels of activated and total TGF-β2 secretion by the IR clones (Fig. 7B). The cumulative amount of active TGF-β (TGF-β1+TGF-β2) secreted by the IR clones was statistically greater than the secretion observed by the parental cells (P ≤ 0.02). The data demonstrate that the IR phenotype is associated with an increased rate of bioactive TGF-β production.
FIGURE 7.
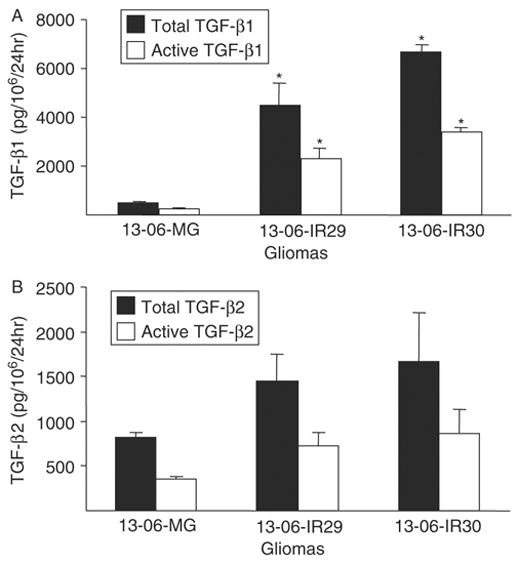
Detection of TGF-β isoforms secreted by monolayers of the parental glioma cells or IR clones. The rates of total (latent plus active forms) and active forms of (A) TGF-β1 and (B) TGF-β2 secretions were determined by ELISA. Rates of TGF-β secretion are expressed as pg TGF-β/106 cells/24 h ± SEM. Asterisks indicate statistically significant production (P ≤ 0.02) relative to the parental glioma cells by the nonparametric Kruskal-Wallis test.
DISCUSSION
In this study, we demonstrated that intermittent in vitro immunoselective pressure applied to 2 aCTL resistant glioma cell clones, after they displayed a partial loss of the resistance, restored the IR phenotype, indicating that the resistance is inducible. We observed that the IR variants also displayed cross-resistance to non-HLA–restricted allogeneic LAK and TALL-104 cells and to γ-irradiation. Observations of cross-resistant cancer cells suggest an impingement on common cellular and molecular apoptotic targets by various cytotoxic agents.32,40 This phenomenon likely occurs through the activation of a compensatory apoptotic pathway(s).41
One interaction required for efficient aCTL lysis of glioma cells is aCTL adhesion to glioma cell surfaces. Glioma-derived ECM protein production has been shown to interfere with this process.12,34 Glioma cells producing thick glycosaminoglycan coats, composed predominately of hyaluronic acid, are protected from allogeneic CTL responses.42 Furthermore, ECM produced by the tumor stroma may contribute to tumor-mediated immunosuppression.43 Therefore, our findings where binding of aCTL to the ECM of parental glioma cells and the IR clones were similar, and that conjugate formation between aCTL with parental cells and IR clones were not different indicated that protective ECM coats did not mechanistically provide immunoresistance for the clones.
We found that ICAM-1 and HLA class I expressions were equivalently high in the IR clones as they were in the parental cells. Thus, down-modulation of ICAM-1 and/or HLA-ABC by the IR clones that would interfere with LFA-1/ICAM-1 and TCR/HLA effector cell to target cell recognition25,36 were also not suspect in providing immune resistance for the clones, although HLA class I down-regulation or loss does not always result in immunoresistance.44-46 Of course, even providing demonstration of allelic dropout47,48 in the IR clones would be insufficient evidence alone that this is a mechanism of resistance, as exceptions to CTL susceptibility were observed when this occurred. For instance, chondrosarcoma cells with allelic loss were susceptible to autologous CTL lysis, indicating that the tumor cells were sufficiently able to present antigenic peptides with the remaining HLA allele.49
Nonclassic HLA-G molecules displayed by glioma cells have been implicated in immunotolerant functions50 and inhibition of allospecific CTL killing of glioma cells.37,38 However, HLA-G up-regulation in the IR clones was not exhibited, as one might predict if the glioma cells used it to avoid immune recognition and destruction.38,51
Interestingly, tumor cells adapted to cell death induced by the Fas/FasL pathway by their expression of FasL, which rendered them capable of inducing apoptosis of Fas-expressing tumor infiltrating T lymphocytes.15 However, the IR variants did not express membrane bound FasL, indicating they do not employ the ‘‘counterattack’’ strategy for immune cell resistance. We did observe a decreased expression of Fas by the IR clones, implicating the down-regulation of Fas as a probable mechanism of aCTL-resistance. When incubated with FasL positive G10 effector cells minimal apoptosis was detected in the IR clones relative to the parental cells. However, few aCTL expressed membrane-bound FasL, suggesting that aCTL lysis of the glioma variants did not involve the Fas apoptosis pathway. Inclusion of EGTA in our cytotoxicity assays to inhibit the calcium-dependent lysis established that the majority of the parental cells and IR clones were lysed by the aCTL through calcium-dependent exocytosis of perforin and granzyme molecules. The cells that remained within the cell cultures (10% to 20%) likely were the ones susceptible to killing by the Fas/FasL pathway. It is also plausible that secretion of IFN upon prolonged aCTL/glioma cell exposure may lead to up-regulated Fas and FasL on glioma cells and aCTL, respectively.52
Gliomas and immune cells are capable of secreting IL-6 and IL-8 cytokines.19,53-58 Gliomas might have secreted these cytokines in response to coincubation with aCTL, because they are known to stimulate glioma cell proliferation and survival.59 However, experiments with the secretion inhibitor, BrfA, indicated that aCTL produced the up-regulated cytokines during the coincubations. The aCTL may have produced these 2 pleiotropic cytokines to counter-act the immunosuppressive activity of soluble glioma-derived factors such as TGF-β. In coincubates containing aCTL and BrfA-treated glioma cells, significant increases in IL-6 and IL-8 were noted, suggesting that the gliomas produced a soluble factor that inhibited the ability of aCTL to secrete proinflammatory cytokines. In vivo IL-6 and IL-8 are known to recruit inflammatory cells that may participate in suppressing tumor growth.60 Supporting this idea, T9 glioma cells engineered to secrete IL-6 were rejected by a neutrophil-dependent mechanism when implanted into syngeneic Fischer rats.61 We also observed up-regulations of IFN-γ, IL-10, and IL-13 by the aCTL upon coincubation with the glioma cells. Nonrobust levels of IFN-γ were secreted by the aCTL when cocultured with glioma target cells. Flow cytometric analysis of a small CD3+/ CD8+/CD69+ subset within aCTL preparations were found to express IFN-γ at high levels that were greatly increased upon coincubation with parental 13-06-MG cells.62 The IL-10 and IL-13 cytokines may mediate proinflammatory or anti-inflammatory responses. For example, melanoma vaccines employing gene-modified gliomas producing IL-13 combined with tumor-lysate stimulated dendritic cells resulted in clinical responses,63 whereas IL-10 inhibited brain tumor growth in vivo.64
There are 3 closely related mammalian TGF-β isoforms (TGF-β1, 2, and 3), all of which seem to have the similar effects on immune cells in vitro.65 TGF-β2 was reported as the major isoform expressed by glioblastomas,66 although more recent studies indicate that TGF-β1 expression is predominately restricted to glioblastomas.67,68 Unlike gliomas, normal glial cells secrete TGF-β1 and TGF-β2 in a latent form that must be proteolytically cleaved to have biologic activity.67,68 TGF-β inhibits T cell activation, proliferation, perforin mRNA synthesis, and the maturation and function of professional antigen presenting cells.69-72 As well, TGF-β inhibits activated T cell effector functions.73,74 In vitro silencing of TGF-β expression promoted glioma cell recognition and lysis by CD8+ T and natural killer cells.75 Data from a number of studies suggest that immune therapy for gliomas might be potentiated by inhibition of TGF-β.6,76,77 ELISA analyses of supernatants from parental gliomas and IR cultures demonstrated that the gliomas secreted abundant amounts of activated TGF-β1 and TGF-β2. This finding was not surprising because chromosomes 19 and 1 that encode for TGF-β178 and TGF-β2,79 respectively, were amplified to a similar degree in parental and IR cell metaphase spreads.19 The production of activated TGF-β was significantly higher in the IR clones, linking high TGF-β secretion by glioma cells to their resistance to CTL lysis. To our knowledge, this is the first report linking increased synthesis of biologically active TGF-β with glioma cell resistance to aCTL lysis. The introduction of antisense TGF-β vectors6 or the TGF-β antagonist decorin gene80 into the IR variants may facilitate a determination of the role of TGF-β in the IR phenotype. Future studies can address whether disrupting glioma secretion of TGF-β or TGF-β-mediated T cell signaling events can overcome the resistance or improve upon the current aCTL therapy.
Acknowledgments
German G. Gomez performed the experimental work in partial fulfillment of his doctoral dissertation work for the Experimental Pathology Program at the University of Colorado. Chiron, Inc, supplied the IL-2 for these studies. The G10 cells, which are K562 erythroleukemic cells transfected with the human FasL gene, were generously provided by Dr Donald Bellgrau (Integrated Department of Immunology, University of Colorado and National Jewish Medical Research Center, Denver, CO). Purified mouse antihuman RR1/1.1.1 ICAM-1 and R3.1.E2.G8.C8 LFA-1 were obtained from Dr Robert Rothlein (TransTech Pharma, Highpoint, NC). The authors thank Dr Tom Nicholas for his suggestions regarding statistical evaluation of the data and Dr L. E. Gerschenson for critically reading the manuscript.
Footnotes
Financial disclosure statement: The authors declare there are no financial conflicts of interest related to the above work.
Supported by National Institutes of Health grants F31 CA94834 to GGG, NS-046463 and NS-056300 to CAK, The R. Herbert and Alma S. Manweiler Memorial Research Fund at the University of Colorado Foundation, and The La Jolla Foundation for Molecular Medicine Research.
References
- 1.CBTRUS. Statistical Report: Primary Brain Tumors in the United States, 1998–2002. Central Brain Tumor Registry of the United States; 2005. [Google Scholar]
- 2.Prados MD, Berger MS, Wilson CB. Primary central nervous system tumors: advances in knowledge and treatment. CA Cancer J Clin. 1998;48:331–360. 321. doi: 10.3322/canjclin.48.6.331. [DOI] [PubMed] [Google Scholar]
- 3.Medewar P. Immunity to homologous grafted skin. III. The fate of skin homografts transplanted to the brain, to subcutaneous tissue and to the anterior chamber of the eye. Br J Exp Pathol. 1948;29:58–69. [PMC free article] [PubMed] [Google Scholar]
- 4.Dix AR, Brooks WH, Roszman TL, et al. Immune defects observed in patients with primary malignant brain tumors. J Neuroimmunol. 1999;100:216–232. doi: 10.1016/s0165-5728(99)00203-9. [DOI] [PubMed] [Google Scholar]
- 5.Kruse CA, Mitchell DH, Lillehei KO, et al. Interleukin-2-activated lymphocytes from brain tumor patients. A comparison of two preparations generated in vitro. Cancer. 1989;64:1629–1637. doi: 10.1002/1097-0142(19891015)64:8<1629::aid-cncr2820640813>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]
- 6.Fakhrai H, Dorigo O, Shawler DL, et al. Eradication of established intracranial rat gliomas by transforming growth factor beta antisense gene therapy. Proc Natl Acad Sci U S A. 1996;93:909–2914. doi: 10.1073/pnas.93.7.2909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Baxevanis CN, Reclos GJ, Gritzapis AD, et al. Elevated prostaglandin E2 production by monocytes is responsible for the depressed levels of natural killer and lymphokine-activated killer cell function in patients with breast cancer. Cancer. 1993;72:491–501. doi: 10.1002/1097-0142(19930715)72:2<491::aid-cncr2820720227>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- 8.Stupp R, Mason WP, van den Bent MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352:987–996. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 9.Virasch N, Kruse CA. Strategies using the immune system for therapy of brain tumors. Hematol Oncol Clin N Am. 2001;15:1053–1071. doi: 10.1016/s0889-8588(05)70267-7. [DOI] [PubMed] [Google Scholar]
- 10.Restifo NP, Marincola FM, Kawakami Y, et al. Loss of functional beta 2-microglobulin in metastatic melanomas from five patients receiving immunotherapy. J Natl Cancer Inst. 1996;88:100–108. doi: 10.1093/jnci/88.2.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rosenberg SA, Yang JC, Robbins PF, et al. Cell transfer therapy for cancer: lessons from sequential treatments of a patient with metastatic melanoma. J Immunother. 2003;26:385–393. doi: 10.1097/00002371-200309000-00001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gately CL, Muul LM, Greenwood MA, et al. In vitro studies on the cell-mediated immune response to human brain tumors. II. Leukocyte-induced coats of glycosaminoglycan increase the resistance of glioma cells to cellular immune attack. J Immunol. 1984;133:3387–3395. [PubMed] [Google Scholar]
- 13.Fiore E, Fusco C, Romero P, et al. Matrix metalloproteinase 9 (MMP-9/gelatinase B) proteolytically cleaves ICAM-1 and participates in tumor cell resistance to natural killer cell-mediated cytotoxicity. Oncogene. 2002;21:5213–5223. doi: 10.1038/sj.onc.1205684. [DOI] [PubMed] [Google Scholar]
- 14.Ruiz-Cabello F, Cabrera T, Lopez-Nevot MA, et al. Impaired surface antigen presentation in tumors: implications for T cell-based immunotherapy. Sem Cancer Biol. 2002;12:15–24. doi: 10.1006/scbi.2001.0406. [DOI] [PubMed] [Google Scholar]
- 15.Walker PR, Saas P, Dietrich PY. Role of Fas ligand (CD95L) in immune escape: the tumor cell strikes back. J Immunol. 1997;158:4521–4524. [PubMed] [Google Scholar]
- 16.Kruse CA, Beck LT. Artificial-capillary-system development of human alloreactive cytotoxic T-lymphocytes that lyse brain tumours. Biotechnol Appl Biochem. 1997;25:197–205. [PubMed] [Google Scholar]
- 17.Kruse CA, Cepeda L, Owens B, et al. Treatment of recurrent glioma with intracavitary alloreactive cytotoxic T lymphocytes and interleukin-2. Cancer Immunol Immunother. 1997;45:77–87. doi: 10.1007/s002620050405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kruse CA, Rubinstein D. Cytotoxic T-lymphocytes reactive to patient major histocompatibility complex proteins for therapy of brain tumors. In: Liau LM, Becker DP, Cloughesy TF, et al., editors. Brain Tumor Immunotherapy. Totowa, NJ: Humana Press; 2001. pp. 149–170. [Google Scholar]
- 19.Gomez GG, Varella-Garcia M, Kruse CA. Isolation of immunoresistant human glioma cell clones after selection with alloreactive cytotoxic T lymphocytes: cytogenetic and molecular cytogenetic characterization. Cancer Genet Cytogenet. 2006;165:121–134. doi: 10.1016/j.cancergencyto.2005.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gomez GG, Kruse CA. Immunoresistant human glioma cell clones selected with alloreactive cytotoxic T lymphocytes: downregulated Apaf-1 is implicated with immune resistance. Submitted 2006. [Google Scholar]
- 21.Kruse CA, Mitchell DH, Kleinschmidt-DeMasters BK, et al. Characterization of a continuous human glioma cell line DBTRG-05 MG: growth kinetics, karyotype, receptor expression, and tumor suppressor gene analyses. In vitro Cell Dev Biol. 1992;28A:609–614. doi: 10.1007/BF02631035. [DOI] [PubMed] [Google Scholar]
- 22.Kruse CA, Visonneau S, Kleinschmidt-DeMasters BK, et al. The human leukemic T-cell line, TALL-104, is cytotoxic to human malignant brain tumors and traffics through brain tissue: implications for local adoptive immunotherapy. Cancer Res. 2000;60:5731–5739. [PubMed] [Google Scholar]
- 23.O’Connor R, Cesano A, Lange B, et al. Growth factor requirements of childhood acute T-lymphoblastic leukemia: correlation between presence of chromosomal abnormalities and ability to grow permanently in vitro. Blood. 1991;77:1534–1545. [PubMed] [Google Scholar]
- 24.Modiano JF, Sun J, Lang J, et al. Fas ligand-dependent suppression of autoimmunity via recruitment and subsequent termination of activated T cells. Clin Immunol. 2004;112:54–65. doi: 10.1016/j.clim.2004.03.011. [DOI] [PubMed] [Google Scholar]
- 25.Read SB, Kulprathipanja NV, Gomez GG, et al. Human alloreactive CTL interactions with gliomas and with those having upregulated HLA expression from exogenous IFN-gamma or IFN-gamma gene modification. J Int Cyt Res. 2003;23:379–393. doi: 10.1089/107999003322226032. [DOI] [PubMed] [Google Scholar]
- 26.Gomez GG, Read SB, Gerschenson LE, et al. Interactions of the allogeneic effector leukemic T cell line, TALL-104, with human malignant brain tumors. Neuro-Oncol. 2004;6:83–95. doi: 10.1215/S1152851703000140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Frost P, Caliliw R, Belldegrun A, et al. Immunosensitization of resistant human tumor cells to cytotoxicity by tumor infiltrating lymphocytes. Int J Oncol. 2003;22:431–437. [PubMed] [Google Scholar]
- 28.Ohnishi T, Taki T, Hiraga S, et al. In vitro and in vivo potentiation of radiosensitivity of malignant gliomas by antisense inhibition of the RAD51 gene. Biochem Biophys Res Commun. 1998;245:319–324. doi: 10.1006/bbrc.1998.8440. [DOI] [PubMed] [Google Scholar]
- 29.Giese A, Loo MA, Tran N, et al. Dichotomy of astrocytoma migration and proliferation. Int J Cancer. 1996;67:275–282. doi: 10.1002/(SICI)1097-0215(19960717)67:2<275::AID-IJC20>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 30.Mattern RH, Read SB, Pierschbacher MD, et al. Glioma cell integrin expression and their interactions with integrin antagonists: research article. Cancer Ther. 2005;3A:325–340. [PMC free article] [PubMed] [Google Scholar]
- 31.Simon MM, Waring P, Lobigs M, et al. Cytotoxic T cells specifically induce Fas on target cells, thereby facilitating exocytosis-independent induction of apoptosis. J Immunol. 2000;165:3663–3672. doi: 10.4049/jimmunol.165.7.3663. [DOI] [PubMed] [Google Scholar]
- 32.Kamarajan P, Sun NK, Chao CC. Up-regulation of FLIP in cisplatin-selected HeLa cells causes cross-resistance to CD95/Fas death signalling. Biochem J. 2003;376:253–260. doi: 10.1042/BJ20030659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Norman SA, Rhodes SN, Treasurywala S, et al. Identification of transforming growth factor-beta1-binding protein overexpression in carmustine-resistant glioma cells by MRNA differential display. Cancer. 2000;89:850–862. [PubMed] [Google Scholar]
- 34.Gately MK, Glaser M, McCarron RM, et al. Mechanisms by which human gliomas may escape cellular immune attack. Acta Neurochir (Wien) 1982;64:175–197. doi: 10.1007/BF01406052. [DOI] [PubMed] [Google Scholar]
- 35.Oberc-Greenwood MA, Muul LM, Gately MK, et al. Ultrastructural features of the lymphocyte-stimulated halos produced by human glioma-derived cells in vitro. J Neuro-oncol. 1986;3:387–396. doi: 10.1007/BF00165589. [DOI] [PubMed] [Google Scholar]
- 36.Schiltz PM, Gomez GG, Read SB, et al. Effects of IFN-gamma and interleukin-1beta on major histocompatibility complex antigen and intercellular adhesion molecule-1 expression by 9L gliosarcoma: relevance to its cytolysis by alloreactive cytotoxic T lymphocytes. J Int Cyt Res. 2002;22:1209–1216. doi: 10.1089/10799900260475731. [DOI] [PubMed] [Google Scholar]
- 37.Riteau B, Menier C, Khalil-Daher I, et al. HLA-G inhibits the allogeneic proliferative response. J Reprod Immunol. 1999;43:203–211. doi: 10.1016/s0165-0378(99)00034-0. [DOI] [PubMed] [Google Scholar]
- 38.Wiendl H, Mitsdoerffer M, Hofmeister V, et al. A functional role of HLA-G expression in human gliomas: an alternative strategy of immune escape. J Immunol. 2002;168:4772–4780. doi: 10.4049/jimmunol.168.9.4772. [DOI] [PubMed] [Google Scholar]
- 39.Lin JT, Martin SL, Xia L, et al. TGF-beta 1 uses distinct mechanisms to inhibit IFN-gamma expression in CD4+ T cells at priming and at recall: differential involvement of Stat4 and T-bet. J Immunol. 2005;174:5950–5958. doi: 10.4049/jimmunol.174.10.5950. [DOI] [PubMed] [Google Scholar]
- 40.Medema JP, de Jong J, van Hall T, et al. Immune escape of tumors in vivo by expression of cellular FLICE-inhibitory protein. J Exp Med. 1999;190:1033–1038. doi: 10.1084/jem.190.7.1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hiraki A, Ono T, Tanaka M, et al. Generation of cytotoxic T lymphocytes against autologous lung cancer cells resistant to apoptosis. Anticancer Res. 2001;21:2561–2567. [PubMed] [Google Scholar]
- 42.Dick SJ, Macchi B, Papazoglou S, et al. Lymphoid cell-glioma cell interaction enhances cell coat production by human gliomas: novel suppressor mechanism. Science. 1983;220:739–742. doi: 10.1126/science.6220469. [DOI] [PubMed] [Google Scholar]
- 43.Hemesath TJ, Marton LS, Stefansson K. Inhibition of T cell activation by the extracellular matrix protein tenascin. J Immunol. 1994;152:5199–5207. [PubMed] [Google Scholar]
- 44.Slingluff CL, Jr, Colella TA, Thompson L, et al. Melanomas with concordant loss of multiple melanocytic differentiation proteins: immune escape that may be overcome by targeting unique or undefined antigens. Cancer Immunol Immunother. 2000;48:661–672. doi: 10.1007/s002620050015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rivoltini L, Gambacorti-Passerini C, Supino R, et al. Generation and partial characterization of melanoma sublines resistant to lymphokine activated killer (LAK) cells. Relevance to doxorubicin resistance. Int J Cancer. 1989;43:880–885. doi: 10.1002/ijc.2910430524. [DOI] [PubMed] [Google Scholar]
- 46.Topalian SL, Kasid A, Rosenberg SA. Immunoselection of a human melanoma resistant to specific lysis by autologous tumor-infiltrating lymphocytes. Possible mechanisms for immunotherapeutic failures. J Immunol. 1990;144:4487–4495. [PubMed] [Google Scholar]
- 47.Chang CC, Campoli M, Restifo NP, et al. Immune selection of hot-spot beta 2-microglobulin gene mutations, HLA-A2 allospecificity loss, and antigen-processing machinery component down-regulation in melanoma cells derived from recurrent metastases following immunotherapy. J Immunol. 2005;174:1462–1471. doi: 10.4049/jimmunol.174.3.1462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Marincola FM, Shamamian P, Alexander RB, et al. Loss of HLA haplotype and B locus down-regulation in melanoma cell lines. J Immunol. 1994;153:1225–1237. [PubMed] [Google Scholar]
- 49.Hiraki A, Ikeda K, Yoshino T, et al. Tumor-specific cytotoxic T lymphocyte responses against chondrosarcoma with HLA haplotype loss restricted by the remaining HLA class I allele. Biochem Biophys Res Commun. 2001;286:786–791. doi: 10.1006/bbrc.2001.5411. [DOI] [PubMed] [Google Scholar]
- 50.Onno M, Guillaudeux T, Amiot L, et al. The HLA-G gene is expressed at a low mRNA level in different human cells and tissues. Hum Immunol. 1994;41:79–86. doi: 10.1016/0198-8859(94)90089-2. [DOI] [PubMed] [Google Scholar]
- 51.Riteau B, Faure F, Menier C, et al. Exosomes bearing HLA-G are released by melanoma cells. Hum Immunol. 2003;64:1064–1072. doi: 10.1016/j.humimm.2003.08.344. [DOI] [PubMed] [Google Scholar]
- 52.Mullbacher A, Lobigs M, Hla RT, et al. Antigen-dependent release of IFN-gamma by cytotoxic T cells up-regulates Fas on target cells and facilitates exocytosis-independent specific target cell lysis. J Immunol. 2002;169:145–150. doi: 10.4049/jimmunol.169.1.145. [DOI] [PubMed] [Google Scholar]
- 53.Bass HZ, Yamashita N, Clement LT. Heterogeneous mechanisms of human cytotoxic T lymphocyte generation. II. Differential effects of IL-6 on the helper cell-independent generation of CTL from CD8+ precursor subpopulations. J Immunol. 1993;151:2895–2903. [PubMed] [Google Scholar]
- 54.Van Meir E, Ceska M, Effenberger F, et al. Interleukin-8 is produced in neoplastic and infectious diseases of the human central nervous system. Cancer Res. 1992;52:4297–4305. [PubMed] [Google Scholar]
- 55.Van Meir E, Sawamura Y, Diserens AC, et al. Human glioblastoma cells release interleukin 6 in vivo and in vitro. Cancer Res. 1990;50:6683–6688. [PubMed] [Google Scholar]
- 56.Lichtor T, Libermann TA. Coexpression of interleukin-1 beta and interleukin-6 in human brain tumors. Neurosurg. 1994;34:669–672. doi: 10.1227/00006123-199404000-00015. [DOI] [PubMed] [Google Scholar]
- 57.Choi C, Gillespie GY, Van Wagoner NJ, et al. Fas engagement increases expression of interleukin-6 in human glioma cells. J Neuro-oncol. 2002;56:13–19. doi: 10.1023/a:1014467626314. [DOI] [PubMed] [Google Scholar]
- 58.Choi C, Xu X, Oh JW, et al. Fas-induced expression of chemokines in human glioma cells: involvement of extracellular signal-regulated kinase 1/2 and p38 mitogen-activated protein kinase. Cancer Res. 2001;61:3084–3091. [PubMed] [Google Scholar]
- 59.Brat DJ, Bellail AC, Van Meir EG. The role of interleukin-8 and its receptors in gliomagenesis and tumoral angiogenesis. Neuro-Oncol. 2005;7:122–133. doi: 10.1215/S1152851704001061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ward SG, Westwick J. Chemokines: understanding their role in T-lymphocyte biology. Biochem J. 1998;333(Pt 3):457–470. doi: 10.1042/bj3330457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Graf MR, Prins RM, Merchant RE. IL-6 secretion by a rat T9 glioma clone induces a neutrophil-dependent antitumor response with resultant cellular, antiglioma immunity. J Immunol. 2001;166:121–129. doi: 10.4049/jimmunol.166.1.121. [DOI] [PubMed] [Google Scholar]
- 62.Gomez GG, Kruse CA. Subsets within alloreactive CTL exhibit upregulated proinflammatory cytokine secretion: aCTL response to coincubation with irrelevant and relevant parental and immunoresistant gliomas. 2006 Experimental Biology meeting abstracts [on CD-ROM] FASEB J. 2006;20 Abstract 697.4. [Google Scholar]
- 63.Salcedo M, Bercovici N, Taylor R, et al. Vaccination of melanoma patients using dendritic cells loaded with an allogeneic tumor cell lysate. Cancer Immunol Immunother. 2005;55:1–11. doi: 10.1007/s00262-005-0078-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Book AA, Fielding KE, Kundu N, et al. IL-10 gene transfer to intracranial 9L glioma: tumor inhibition and cooperation with IL-2. J Neuroimmunol. 1998;92:50–59. doi: 10.1016/s0165-5728(98)00172-6. [DOI] [PubMed] [Google Scholar]
- 65.Gorelik L, Fields PE, Flavell RA. Cutting edge: TGF-beta inhibits Th type 2 development through inhibition of GATA-3 expression. J Immunol. 2000;165:4773–4777. doi: 10.4049/jimmunol.165.9.4773. [DOI] [PubMed] [Google Scholar]
- 66.Bodmer S, Strommer K, Frei K, et al. Immunosuppression and transforming growth factor-beta in glioblastoma. Preferential production of transforming growth factor-beta 2. J Immunol. 1989;143:3222–3229. [PubMed] [Google Scholar]
- 67.Constam DB, Philipp J, Malipiero UV, et al. Differential expression of transforming growth factor-beta 1, -beta 2, and -beta 3 by glioblastoma cells, astrocytes, and microglia. J Immunol. 1992;148:1404–1410. [PubMed] [Google Scholar]
- 68.Kjellman C, Olofsson SP, Hansson O, et al. Expression of TGF-beta isoforms, TGF-beta receptors, and SMAD molecules at different stages of human glioma. Int J Cancer. 2000;89:251–258. doi: 10.1002/1097-0215(20000520)89:3<251::aid-ijc7>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- 69.Gorelik L, Flavell RA. Abrogation of TGFbeta signaling in T cells leads to spontaneous T cell differentiation and autoimmune disease. Immunity. 2000;12:171–181. doi: 10.1016/s1074-7613(00)80170-3. [DOI] [PubMed] [Google Scholar]
- 70.Geissmann F, Revy P, Regnault A, et al. TGF-beta 1 prevents the noncognate maturation of human dendritic Langerhans cells. J Immunol. 1999;162:4567–4575. [PubMed] [Google Scholar]
- 71.Ranges GE, Figari IS, Espevik T, et al. Inhibition of cytotoxic T cell development by transforming growth factor beta and reversal by recombinant tumor necrosis factor alpha. J Exp Med. 1987;166:991–998. doi: 10.1084/jem.166.4.991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Smyth MJ, Strobl SL, Young HA, et al. Regulation of lymphokine-activated killer activity and pore-forming protein gene expression in human peripheral blood CD8+ T lymphocytes. Inhibition by transforming growth factor-beta. J Immunol. 1991;146:3289–3297. [PubMed] [Google Scholar]
- 73.Lin CM, Wang FH, Lee PK. Activated human CD4+ T cells induced by dendritic cell stimulation are most sensitive to transforming growth factor-beta: implications for dendritic cell immunization against cancer. Clin Immunol. 2002;102:96–105. doi: 10.1006/clim.2001.5151. [DOI] [PubMed] [Google Scholar]
- 74.Thomas DA, Massague J. TGF-beta directly targets cytotoxic T cell functions during tumor evasion of immune surveillance. Cancer Cell. 2005;8:369–380. doi: 10.1016/j.ccr.2005.10.012. [DOI] [PubMed] [Google Scholar]
- 75.Friese MA, Wischhusen J, Wick W, et al. RNA interference targeting transforming growth factor-beta enhances NKG2D-mediated antiglioma immune response, inhibits glioma cell migration and invasiveness, and abrogates tumorigenicity in vivo. Cancer Res. 2004;64:7596–7603. doi: 10.1158/0008-5472.CAN-04-1627. [DOI] [PubMed] [Google Scholar]
- 76.Jachimczak P, Bogdahn U, Schneider J, et al. The effect of transforming growth factor-beta 2-specific phosphorothioate-antisense oligodeoxynucleotides in reversing cellular immunosuppression in malignant glioma. J Neurosurg. 1993;78:944–951. doi: 10.3171/jns.1993.78.6.0944. [DOI] [PubMed] [Google Scholar]
- 77.Witham TF, Villa L, Yang T, et al. Expression of a soluble transforming growth factor-beta (TGFbeta) receptor reduces tumorigenicity by regulating natural killer (NK) cell activity against 9L gliosarcoma in vivo. J Neurooncol. 2003;64:63–69. doi: 10.1007/BF02700021. [DOI] [PubMed] [Google Scholar]
- 78.Weinshenker BG, Hebrink D, Kantarci OH, et al. Genetic variation in the transforming growth factor beta1 gene in multiple sclerosis. J Neuroimmunol. 2001;120:138–145. doi: 10.1016/s0165-5728(01)00424-6. [DOI] [PubMed] [Google Scholar]
- 79.Nishimura DY, Purchio AF, Murray JC. Linkage localization of TGFB2 and the human homeobox gene HLX1 to chromosome 1q. Genomics. 1993;15:357–364. doi: 10.1006/geno.1993.1068. [DOI] [PubMed] [Google Scholar]
- 80.Munz C, Naumann U, Grimmel C, et al. TGF-beta-independent induction of immunogenicity by decorin gene transfer in human malignant glioma cells. Eur J Immunol. 1999;29:1032–1040. doi: 10.1002/(SICI)1521-4141(199903)29:03<1032::AID-IMMU1032>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]