

Summary
Reactions using fluorous reagents and scavengers are compared side-by-side with their solid-supported counterparts. Fluorous triphenylphosphine is used in the bromination reaction of alcohols, fluorous thiol is used as an electrophile scavenger for α-bromoketones, fluorous isatoic anhydride is used as a nucleophile scavenger for primary and secondary amines. Reactions involving fluorous reagents and scavengers occur in homogeneous media with solution-phase reaction kinetics. Reactions with solid-supported reagents and scavengers occur in a heterogeneous media, and the reaction kinetics is greatly affected by the nature of the solid-support and reaction environment. Significantly larger amounts of reagents and longer time are usually needed to complete the solid-supported reaction.
Keywords: fluorous reagents, fluorous scavengers, solid-supported reagents, solid-supported scavengers, triphenylphosphine, isatoic anhydride, thiol, reaction rate, reaction kinetics
Introduction
With the increasing practice of combinatorial and parallel synthesis to generate large numbers of compounds for biological screening, there are continuous needs to improve the efficiency of both the reaction and separation aspects of organic synthesis. An efficient way to improve the low conversion of reactions is to use reagent in excess. But this excess has an adverse effect on the efficiency of the product purification. To facilitate product purification, immobilized reagents and scavengers have been introduced in so called solid-supported solution-phase synthesis [1]. Excess reagents and scavenged byproducts on the solid support are readily separated from the reaction mixture by filtration. Since solid-supported solution-phase reactions are performed in a heterogeneous media, the advantage of easy separation can be counterbalanced by slow reaction, limitation on solvent selection, and the need to use large excess of the solid-bound reagent because not all the active sites on the solid-support are equally accessible.
The recent success of fluorous technologies has provided synthetic chemists opportunities to combine the advantages of solution-phase reactions and solid-phase separations [2]. Perfluoroalkyl chains instead of solid-supports are employed as “phase tags” to attach to substrates, reagents, or scavengers. The electronic effect of the perfluoroalkyl chain is minimized by insertion of an ethylene or propylene spacer between the tag and the functional group. Fluorous tags play a role similar to solid-supports in controlling the purification. Unique fluorous liquid-liquid extractions or fluorous silica gel-based solid-phase extractions efficiently separate fluorous molecules from non-fluorous molecules [3]. In addition to high solubility in organic solvents, fluorous tags are chemically inert and have good thermal stability for reactions conducted under microwave irradiation [4, 5a].
The development of fluorous scavengers for electrophiles, nucleophiles, and dienophiles has been accomplished by the Curran group at the University of Pittsburgh [5], the Lindsley group at Merck [6], and our group at Fluorous Technologies, Inc [7]. We have recently reported the synthesis of fluorous thiol [7b], fluorous isocyanate, and fluorous isatoic anhydride [7c] and their applications to scavenge activated halides and amines, respectively. We found that only a slight excess amount of scavengers was needed in fluorous scavenging. The quenched materials were efficiently separated by fluorous solid-phase extraction to give products in 90% or even higher purities. Our preliminary results also indicated that fluorous scavenging is faster than the solid-supported scavenging [7b]. We report herein quantitative comparisons of two fluorous scavengers with their solid-bound counterparts in terms of reactivity and conversion rate. The experiments described in this paper focus on the quenching step of the scavenging reactions. Results on post-reaction workup and separation, reaction yields, and product purities associated with the scavenging reactions have been reported in the previous papers [7b–c].
Results and Discussion
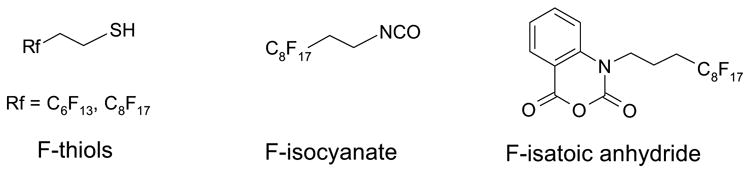
In the thiol quenching study, the structural difference between the immobilized thiol and fluorous thiol was minimized by choosing fluorous 2-(perfluoroctyl)ethanethiol (F-thiol) 1 and silica gel-bound mercaptopropane (Si-thiol) 2. Another commercially available polymer-bound thiol (PS-CH2NHCH2CH2SH) was not selected because of significant structural difference (an amino group in the linker). The thiol quenching was performed by the reaction of 1.0 equiv of 2-bromo-4’-methoxyacetophenone 3a with an excess amount of thiols 1 or 2 using diisopropylethylamine (DIPEA) as a base to promote the reaction and THF as the solvent. The reaction was carried out at room temperature. The reaction progress was monitored by taking aliquots from the reaction mixture at regular time intervals and was analyzed by GC with dodecane as an internal standard to determine the conversion of the bromide.
Results shown in Figure 1 demonstrate that when 1.5 equiv of F-thiol 1 was used (square-dot line), greater than 95% conversion was achieved in less than 40 min. The reaction with Si-thiol 2 (round-dot line) had much lower conversion; 50% of the halide was still unreacted even after 80 min. By doubling the amount of Si-thiol to 3.0 equiv, the reaction was significantly improved to gave 95% conversion after 60 min (diamond-dot line).
Figure 1.
Reactions of α-bromoketone 3a with thiols 1 or 2
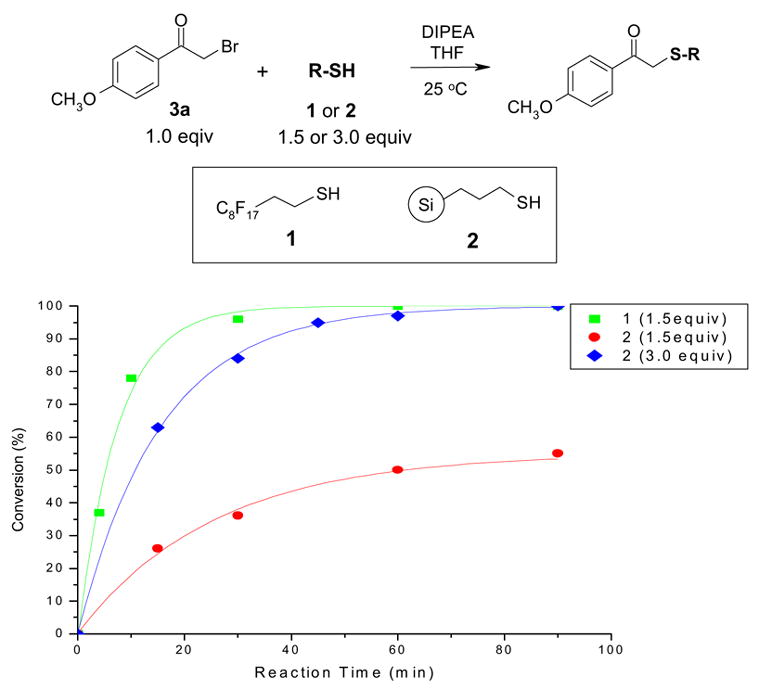
A similar study was conducted to compare reactions of F-isatoic anhydride 4 and PS-isatoic anhydride 5 [8] (cross-linked polystyrene with 1% DVB) with N-phenylpiperazine 7a. Compound 6 bearing a C8H17 group was synthesized by N-alkylation of isatoic anhydride and also included in the comparison experiment. Of course, compound 6 has no phase tag so it has no utility in actual scavenging applications; it only serves as a control to illustrate normal solution-phase reactivity. Reactions were performed in CH2Cl2 at room temperature. The data compiled in Figure 2 shows that reactions using 1.5 equiv of fluorous compound 4 (square-dot line) or non-fluorous compound 6 (triangle-dot line) were very similar. The fluorous reaction was slightly faster than the non-fluorous reaction. We attribute the fast reaction rate to the electronic-withdrawing effect of the fluorous tag which enhances the electrophilicity of compound 4 despite the propylene spacer [9]. A similar reaction with polymer-supported compound 5 proceeded much slower (round-dot line). Doubling the amount of 5 to 3.0 equiv increased the reaction rate (diamond-dot line), but it was still not as fast as either the reaction with 1.5 equiv of F-isatoic anhydride 4 or octylated isatoic anhydride 6.
Figure 2.
Reactions of N-phenylpiperazine 7a with isatoic anhydrides 4–6
We have compared the reaction rate of different scavengers as the function of time (Figures 1 and 2). We also evaluated the performance of different scavengers with different substrates. Three α-bromoketones 3a–c with different substituents on the benzene ring were selected to react with thiols and three amines 7a–c including one primary and two secondary amines were selected to react with isatoic anhydrides.
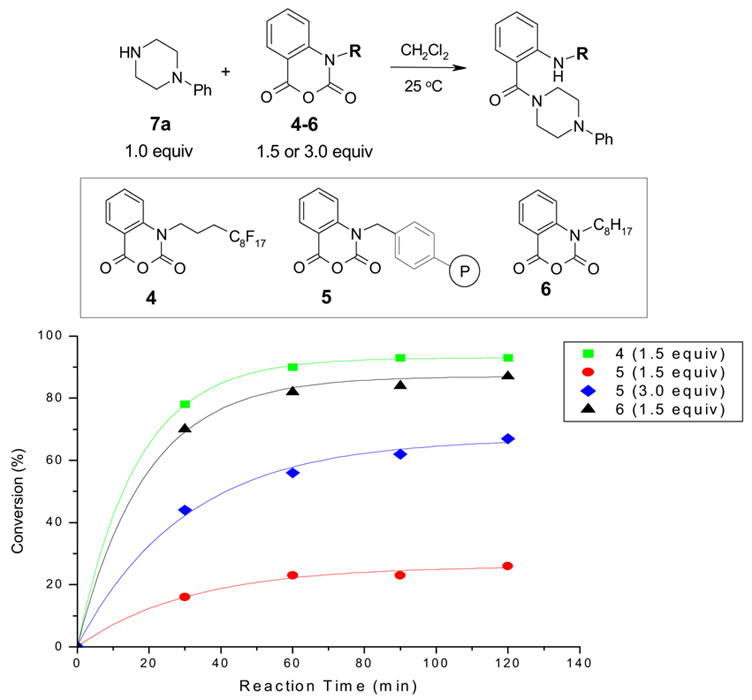
Reactions of α-bromoketones 3a–c with F-thiol 1 and Si-thiol 2 were performed under the same conditions: 1.0 equiv of 3 was reacted with 1.5 or 3.0 equiv of thiols 1 or 2 using DIPEA as a base and THF as a solvent. The reaction was carried out at room temperature for 1 h. The reaction mixture was analyzed by GC with dodecane as an internal standard to determine the conversion of α-bromoketones 3a–c. The data compiled in Figure 3 are consistent with the results observed in the previous experiment. F-thiol 1 had better performance than Si-thiol 2 with all three substrates. Doubling the amount of Si-thiol 2 was necessary to catch up to the performance of the F-thiol 1. The reaction of 3a with F-thiol 1 was completion, but the reaction with Si-thiol 2 was only 50% complete and with 3.0 equiv of Si-thiol was only 97% complete. The reaction of α-bromoketone 3b suggested that even 3.0 equiv of Si-thiol 2 was not enough to drive the conversion to completion in 1 h.
Figure 3.
Reactions of three α-bromoketones 3a–c with F-thiol 1 and Si-thiol 2. Bars in 3 different patterns represent 2 different thiols and different equiv. The first group represents the data obtained with 3a, the second group obtained with 3b, and the third, 3c
Reactions of three different amines 7a–c with F-isatoic anhydride 4 or PS-isatoic anhydride 5 were performed at 55 °C (Figure 4). The reaction of 7a with F-isatoic anhydride 4 went to completion within 30 min whereas that with the PS-isatoic anhydride 5 only went 32% completion. Doubling the amount of 5 to 3.0 equiv only pushed the reaction to 64% completion. In all these three cases, 3.0 equiv of PS-isatoic anhydride 5 was not enough to drive the reaction to completion whereas only 1.5 equiv of F-isatoic anhydride 4 was enough to complete the reaction.
Figure 4.
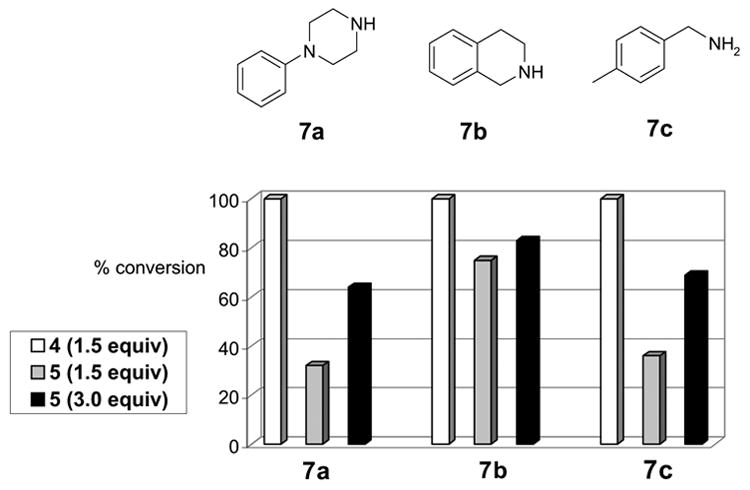
Reactions of amines 7a–c with F-isatoic anhydride 4 or PS-isatoic anhydride 5. Bars in 3 different patterns represent 2 different thiols at different equiv. The first group represents the data obtained with 7a, the second group obtained with 7b, and the third, 7c
In addition to evaluation of supported scavengers, we also used triphenylphosphine (TPP) as an example to evaluated supported reagents. TPP has been involved in many important organic transformations such as Wittig, Mitsunobu, Staudinger reactions, halogenation reactions, and Pd-catalyzed reactions [10]. Separation of triphenylphosphine oxide from the TPP reaction usually requires chromatography. Recently, PS-TPP [11] and F-TPP [12] have been used to simplify the byproduct separation process. The Lindsley group studied the Staudinger reaction using F-TPP 8 and two polymeric TPPs with cross-linked polystyrene 9a and cross-linked NovaGel™ 9b, respectively [13]. In THF solution, it took 1 h for F-TPP 8 to convert the azide to the phosphoazide, an intermediate for the amine product, compared to 36 h for polymer-bound TPPs 9a and 9b to complete the same process. The reaction with F-TPP was not only faster than its resin-bound counterparts, it also gave 100% conversion with product purity greater than 98%. Polystyrene-bound TPP gave 26% conversion with >86% product purity and NovaGel-bound TPP gave 60% conversion with the same purity.
We conducted a comparison experiment by reaction of 1.5 equiv of different TPPs (F-TPP 8, PS-TPP 9, and normal TPP 10) with 1.0 equiv of 2,4-dimethoxybenzyl alcohol 11 and 1.5 equiv of CBr4 in toluene by following a literature procedure reported by the Sinou group [12h]. The reaction mixture was shaken at room temperature and monitored by GC using dodecane as an internal standard to determine the conversion of 11. Results presented in Figure 4 shows reactions with F-TPP (square-dot line) were slightly slower than the normal TPP (triangle-dot line), but they have very similar reaction pattern; a 90% conversion was achieved after 180 min for both reactions. We were interested to find that during the first 10–15 min, the reaction with 1.5 equiv of PS-TPP 10 (round-dot line) proceeded a little faster than reactions with F-TPP or normal TPP. However, the reaction with PS-TPP slowed down and the conversion was less than 80% after 180 min. The reaction with 3.0 equiv of PS-TPP gave a significantly better result (diamond-dot line); the conversion was over 95% in less than 100 min.
The experimental results suggest that reaction kinetics with PS-TPP is different from that with fluorous and normal TPPs. The polymer-supported reagent does not always react slower than the fluorous counterpart at a given reaction time, but the conversion rate is low because not all functional groups on the polymer bead are easily accessible. Polymer-supported reagents are greatly affected by the nature of polymer support and reaction solvent system which made polymer-supported reactions less predictable. The influence of polymer supports on reaction rate and kinetics have been extensively studied [14].
Conclusions
These comparison experiments have demonstrated that when equal molar amounts of material are used, reactions with fluorous reagents and scavengers usually proceed faster and give higher conversion than reactions with solid-supported analogs. Fluorous reagents and scavengers are real molecules, they can be used in a stoichiometric manner and analyzed by standard methods such as LCMS and NMR. Solid-supported reagents and scavengers are functionalized materials, their chemical and physical properties such as loading, swelling, particle size, and percentage of cross-linking usually have a range of variation and are difficult to be assessed by the users. It is not uncommon that because of these uncertainties and unfavorable heterogeneous reaction kinetics, chemists use a large excess (3.0 equiv or even more) of solid-supported reagents and scavengers to “over kill” the reactions. This kind of practice results in high cost on reagents as well as high volume of solvent used for reaction and resin washes after the reaction. These are found not to be the issues in fluorous-supported reactions.
Experimental section
Procedure for reactions of halides 3 with thiols 1 or 2
Fluorous thiol 1 (Fluorous Technologies, Inc. 72.3 mg, 0.15 mmol) was dissolved in 1.0 mL THF in a vial. The internal standard dodecane (23.0 μL, 0.14 mmol), diisopropylethylamine (26.0 μL, 0.15 mmol), and 2-bromo-4’-methoxyacetophenone 3a (23.0 mg, 0.1 mmol) were added. The mixture was shaken on a MS 1 Minishaker at 600 rpm. Prior to the addition of the base, an aliquot of 10.0 μL was taken, diluted in 300 iL of THF and injected into GC to obtain the ratio of α-bromoketone to the standard from the peak areas. This ratio was taken as the starting point (t0 = H0/S0 = X0 ; where H0 = peak area of the α-bromoketone and S0 = peak area of the standard, and X0 = ratio). Aliquots of 10.0 μL were taken at given time intervals, diluted in 300 μL of THF and injected into the GC for ratio analysis. The ratio of the α-bromoketone to the standard at subsequent time (tn = Hn/Sn = Yn) was used to calculate the remaining amount of α-bromoketone as a percentage (Yn/X0)100 = Zn% which gives the reaction completion (100 – Zn)%. GC conditions: 100% dimethylpolysiloxane, 30m x 0.32 mm, 25 micron column; initial time 1 min at 150 °C, 100 to 250 ºC at 20 °C/min, remain at 250 ºC for 10 min. Similar reactions and data collection processes were applied to silica-supported thiol 2 (Aldrich cat # 53808-6, 200–400 mesh, 0.3–0.9 mmol S/g, average loading 0.6 mmol/g was used for calculations). The percent completion was plotted against time (min) to give the graphs in Figure 1. Comparison reactions of thiols 1 or 2 with different bromides 3a–c were conducted at room temp under stirring. Reaction mixtures were analyzed at 60 min. The conversion data were collected and presented in Figure 4.
General procedure for the reaction of amine 7 with isatoic anhydrides 4–6
F-isatoic anhydride 4 (Fluorous Technologies, Inc., 144.0 mg, 0.23 mmol) was dissolved in 1.0 mL of CH2Cl2 in a vial. The internal standard dodecane (24.0 iL, 0.14 mmol) and phenyl piperazine 7a (24.0 μL, 0.15 mmol) were added and the reaction mixture was shaken on a MS 1 Minishaker at 600 rpm at room temperature. Prior to the addition of the amine, an aliquot of 30.0 μL was taken out, diluted with 1.0 mL of CH2Cl2 and injected into the GC to obtain X0. At intervals of 30 minutes an equal amount of aliquot is taken out from the reaction vial and subjected to GC analysis to obtain Yn. Same GC analysis and data collection methods as described above were used in this experiment. Similar reactions with PS-isatoic anhydride 5 (Aldrich, part # 51,437–3, 1% DVB, 200–400 mesh, 2.0–2.5 mmol N/g, average loading 0.23 mmol was used for calculations) and N-octyl isatoic anhydride 6 were conducted. The data of the percent completion were collected and plotted against time (min) to give the graphs in Figure 2.
Reactions of isatoic anhydrides 4 or 5 with different amines 7a–c were conducted at 55°C under stirring. Reactions with 7a and 7c were analyzed at 30 min while the reaction with 7b was analyzed at 60 min because of low reactivity of this substrate. The conversion data is presented in Figure 4.
General procedure for reactions of 2,4-dimethoxybenzyl alcohol 11 with triphenylphosphines 8, 9a, or10
Fluorous triphenylphosphine 8 (Fluorous Technologies, Inc., 318.0 mg, 0.45 mmol) was dissolved in 2 mL of toluene in a vial. The internal standard dodecane (25.0 iL, 0.15 mmol), 2,4-dimethoxybenzyl alcohol (50.0 mg, 0.30 mmol), and CBr4 (149.0 mg, 0.45 mmol) were added at room temperature. The reaction mixtures were shaken on a MS 1 Minishaker at 600 rpm. Prior to the addition of CBr4, an aliquot of 30 μL was taken out, diluted with 1.0 mL of toluene and injected into the GC to obtain X0. At different reaction time, an equal amount of aliquot is taken out from the reaction vial and subjected to GC analysis to obtain Yn. Same GC analysis and data collection methods described above were used in this experiment. Similar reactions with PS-triphenylphosphine 9a (Argonaut, part # 800378, 2.1 mmol/g) and normal triphenylphosphine 10 were conducted. The data was collected and plotted against time (min) to give the graphs in Figure 5.
Figure 5.
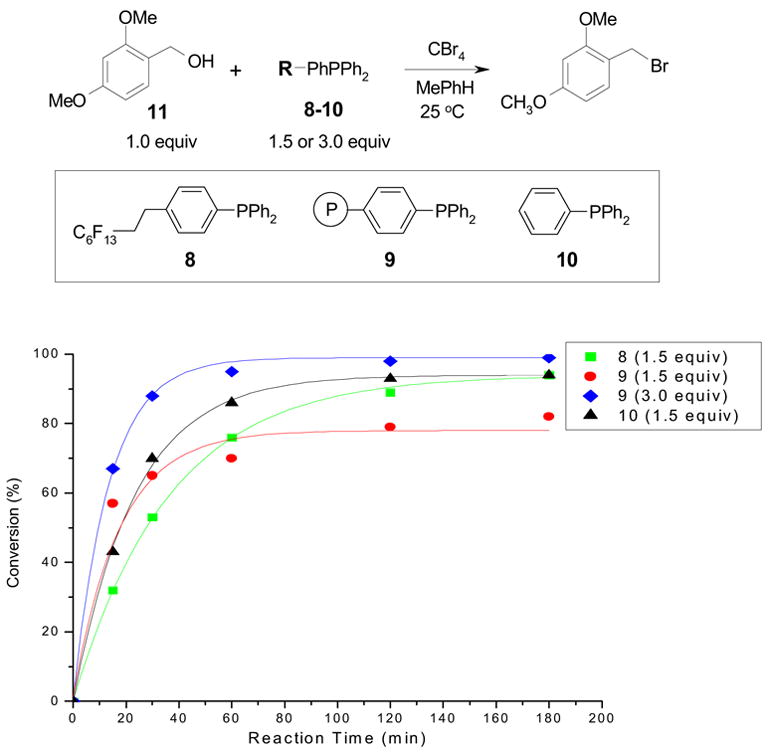
Reactions of 2,4-dimethoxybenzyl alcohol 11 with triphenylphosphines 8–10 and CBr4
Scheme 1.
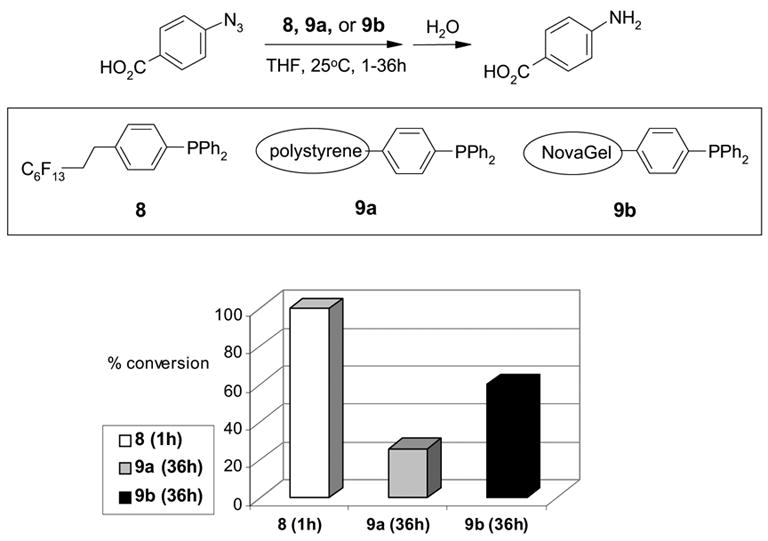
Staudinger reactions with triphenyl phosphines 8, 9a and 9b
Acknowledgments
We thank the National Institutes of General Medical Sciences for the SBIR funding (2R44GM067326-02A1); Mr. Yimin Lu for analytical data support; Dr. Phil Yeske, Tadamichi Nagashima, and Prof. Dennis Curran for their comments on the manuscript.
References
- 1.Selected reviews on polymer-assisted organic synthesis:Hodge P. Organic synthesis using polymer-supported reagents, catalysts and scavengers in simple laboratory flow systems. Curr Opin Chem Biol. 2003;7:362–373. doi: 10.1016/s1367-5931(03)00052-8.Kirschning A, Monenschein H, Wittenberg R. Functionalized polymers – emerging versatile tools for solution-phase chemistry and automated parallel synthesis. Angew Chem Int Ed. 2001;40:650–679.Eames J, Watkinson M. Polymeric Scavenger Reagents in Organic Synthesis. Eur J Org Chem. 2001:1213–21224.Thompson LA. Recent applications of polymer-supported reagents and scavengers in combinatorial, parallel, or multistep synthesis. Curr Opin Chem Biol. 2000;4:324–337. doi: 10.1016/s1367-5931(00)00096-x.Ley SV, Baxendale IR, Bream RN, Jackson PS, Leach AG, Longbottom DA, Nesi M, Scott JS, Storer RI, Taylor SJ. Multi-step organic synthesis using solid-supported reagents and scavengers: a new paradigm in chemical library generation. J Chem Soc, Perkin Trans 1. 2000;23:3815–4195.Booth RJ, Hodges JC. Solid-supported reagent strategies for rapid purification of combinatorial synthesis products. Acc Chem Res. 1999;32:18–26.Parlow JJ, Devraj RV, South MS. Solution-phase chemical library synthesis using polymer-assisted purification techniques. Curr Opin Chem Biol. 1999;3:320–336. doi: 10.1016/S1367-5931(99)80049-0.Flynn DL, Devraj RV, Naing W, Parlow JJ, Weidner JJ, Yang S. Polymer-assisted solution phase (PASP) chemical library synthesis. Med Chem Res. 1998;8:219–243.
- 2.Recent reviews on fluorous synthesis:Gladysz JA, Hovath I, Curran DP, editors. The handbook of fluorous chemistry. Wiley-VCH; New York: 2004. Zhang W. Fluorous synthesis of heterocyclic systems. Chem Rev. 2004;104:2531–2556. doi: 10.1021/cr030600r.Zhang W. Fluorous technologies for solution-phase high-throughput organic synthesis. Tetrahedron. 2003;59:4475–4489.Gladysz JA, Curran DP. Fluorous chemistry: from biphasic catalysis to a parallel chemical universe and beyond. Tetrahedron. 2002;58:3823–3825.Curran DP. In: Stimulating Concepts in Chemistry. Stoddard F, Reinhoudt D, Shibasaki M, editors. Wiley-VCH; New York: 2000. pp. 25–37.Curran DP. Strategy-Level separations in organic synthesis: from planning to practice. Angew Chem, Int Ed. 1998;37:1175–1196. doi: 10.1002/(SICI)1521-3773(19980518)37:9<1174::AID-ANIE1174>3.0.CO;2-P.
- 3.Curran DP. Fluorous reverse phase silica gel. a new tool for preparative separations in synthetic organic and organofluorine chemistry. Synlett. 2001:1488–1496. [Google Scholar]
- 4.a) Zhang W, Tempest P. Highly efficient microwave-assisted fluorous Ugi and post-condensation reactions for benzimidazoles and quinoxalinones. Tetrahedron Lett. 2004;45:6757–6760. [Google Scholar]; b) Zhang W, Nagashima T, Lu Y, Chen CHT. A traceless perfluorooctylsulfonyl tag for deoxygenation of phenols under microwave irradiation. Tetrahedron Lett. 2004;45:4611–4613. [Google Scholar]; c) Zhang W, Chen CHT, Lu Y, Nagashima T. A Highly efficient microwave-assisted Suzuki coupling reaction of aryl perfluorooctylsulfonates with boronic acids. Org Lett. 2004;6:1473–1476. doi: 10.1021/ol0496428. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Zhang W, Lu Y, Chen CHT. Combination of microwave reactions with fluorous separations in the palladium-catalyzed synthesis of aryl sulfides. Mol Diversity. 2003;7:199–202. doi: 10.1023/b:modi.0000006825.12186.5f. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Larhed M, Hoshino M, Hadida S, Curran DP, Hallberg A. Rapid fluorous Stille coupling reactions conducted under microwave irradiation. J Org Chem. 1997;62:5583–5587. [Google Scholar]; f) Olofsson K, Kim SY, Larhed M, Curran DP, Hallberg A. High-speed, highly fluorous organic reactions. J Org Chem. 1999;64:4539–4541. [Google Scholar]
- 5.a) Linclau B, Singh AK, Curran DP. Organic-fluorous phase switches: a fluorous amine scavenger for purification in solution phase parallel synthesis. J Org Chem. 1999;64:2835–2842. doi: 10.1021/jo9823442. [DOI] [PubMed] [Google Scholar]; b) Curran DP, Hadida S, Kim SY, Luo ZY. Fluorous tin hydrides: a new family of reagents for use and reuse in radical reactions. J Am Chem Soc. 1999;121:6607–6615. [Google Scholar]; c) Werner S, Curran DP. Fluorous dienophiles are powerful diene scavengers in Diels-Alder reactions. Org Lett. 2003;5:3293–3296. doi: 10.1021/ol035214a. [DOI] [PubMed] [Google Scholar]
- 6.a) Lindsley CW, Zhao Z, Leister W. In: Fluorous scavengers in The handbook of fluorous chemistry. Gladysz JA, Hovath I, Curran DP, editors. Wiley-VCH; New York: 2004. pp. 236–246. [Google Scholar]; b) Lindsley CW, Zhao Z, Leister W. Fluorous-tethered quenching reagents for solution phase parallel synthesis. Tetrahedron Lett. 2002;43:4225–4228. [Google Scholar]; c) Lindsley CW, Zhou Z, Leister WH, Strauss KA. Fluorous-tethered amine bases for organic and parallel synthesis: scope and limitations. Tetrahedron Lett. 2003;44:3619–3623. [Google Scholar]
- 7.a) Zhang W. The Worldwide PharmaChem Directory. 2003. Fluorous scavengers for solution-phase synthesis; pp. 18–20. [Google Scholar]; b) Zhang W, Curran DP, Chen CHT. Use of fluorous silica gel to separate fluorous thiol quenching derivatives in solution-phase parallel synthesis. Tetrahedron. 2002;58:3871–3875. [Google Scholar]; c) Zhang W, Chen CHT, Nagashima T. Fluorous electrophilic scavengers for solution-phase parallel synthesis. Tetrahedron Lett. 2003;44:2065–2068. [Google Scholar]
- 8.For polymer-supported isatoic anhydride, seeCoppola GM. A new scavenger resin for amines. Tetrahedron Lett. 1998;39:8233–8236.
- 9.Gladysz JA. Ponytails: structural and electronic considerations. In: Gladysz JA, Hovath I, Curran DP, editors. The handbook of fluorous chemistry. Wiley-VCH; New York: 2004. pp. 41–55. [Google Scholar]
- 10.Appel R, Halstenberg M. In: Organophosphorous reagents in organic synthesis. Cadogan JIG, editor. Academic; New York: 1979. pp. 387–431. [Google Scholar]
- 11.For applications of PS-TPP, see:Harrison CR, Hodge P, Hunt BJ, Khoshdel E, Richardson G. Preparation of alkyl chlorides, acid chlorides, and amides using polymer-supported phosphines and carbon tetrachloride: mechanism of these reactions. J Org Chem. 1983;48:3721–3728.Caldarelli M, Habermann J, Ley SV. Clean five-step synthesis of an array of 1,2,3,4-tetra-substituted pyrroles using polymer-supported reagents. J Chem Soc Perkin Trans. 1999;1:107–110.Porwanski S, Kryczka B, Marsura A. A polymer-supported ‘one-pot’ phosphine imide reaction on cyclodextrins. Tetrahedron Lett. 2002;43:8441–8443.
- 12.For applications of F-TPPs bearing one-, two-, or three-fluorous tags, see:Grigg R, York M. Bimetallic catalytic cascade ring closing metathesis - intramolecular Heck reactions using a fluorous biphasic solvent system or a polymer-supported palladium catalyst. Tetrahedron Lett. 2000;41:7255–7258.Schneider S, Bannwarth W. Repetitive application of perfluoro-tagged Pd complexes for Stille couplings in a fluorous biphasic system. Angew Chem Int Ed. 2000;39:4142–4145. doi: 10.1002/1521-3773(20001117)39:22<4142::aid-anie4142>3.0.co;2-0.Richter B, de Wolf E, van Koten G, Deelman BJ. Synthesis and properties of a novel family of fluorous triphenylphosphine derivatives. J Org Chem. 2000;65:3885–3893. doi: 10.1021/jo991548v.Schneider S, Bannwarth W. Application of the fluorous biphase concept to palladium-catalyzed Suzuki couplings. Helv Chim Acta. 2001;84:735–742.Soós T, Bennett BL, Rutherford D, Barthel-Rosa LP, Gladysz JA. Synthesis, Reactivity, and Metal Complexes of Fluorous Triarylphosphines of the Formula P(p-C6H4(CH2)3(CF2)n–1CF3)3 (n = 6, 8, 10) Organometallics. 2001;20:3079–3086.Dandapani S, Curran DP. Fluorous Mitsunobu reagents and reactions. Tetrahedron. 2002;58:3855–3864.Parallel fluorous biphasic synthesis of 3H-quinazolin-4-ones by an Aza-Wittig reaction employing perfluoroalkyl-tagged triphenylphosphine. Barthélémy S, Schneider S, Bannwarth W. Tetrahedron Lett. 2002;43:807–810.Desmaris L, Percina N, Cottier L, Sinou D. Conversion of alcohols to bromides using a fluorous phosphine. Tetrahedron Lett. 2003;44:7589–7591.
- 13.Lindsley CW, Zhao Z, Newton RC, Leister W, Strauss KA. A general protocol for solution-phase parallel synthesis. Tetrahedron Lett. 2002;43:4467–4470. [Google Scholar]
- 14.Selected reviews:Walsh D, Wu D, Chang YT. Understanding the effects of the polymer support on reaction rates and kinetics: knowledge toward efficient synthetic design. Curr Opin Chem Biol. 2003;7:353–361. doi: 10.1016/s1367-5931(03)00054-1.Yan B. Single-bead analysis in combinatorial chemistry. Curr Opin Chem Biol. 2002;6:328–332. doi: 10.1016/s1367-5931(02)00325-3.Rana S, White P, Bradley M. Influence of resin cross-linking on solid-phase chemistry. J Comb Chem. 2001;3:9–15. doi: 10.1021/cc0000592.Groth T, Grotli M, Meldal M. Diffusion of reagents in macrobeads. J Comb Chem. 2001;3:461–468. doi: 10.1021/cc000106q.Vaino AR, Janda KD. Solid-phase organic synthesis: a critical understanding of the resin. J Comb Chem. 2000;2:579–596. doi: 10.1021/cc000046o.Bing Y. Monitoring the progress and the yield of solid phase organic reactions directly on resin supports. Acc Chem Res. 1998;31:621–630.