

Abstract
Protein misfolding is now recognized as playing a crucial role in both normal and pathogenic folding reactions. An interesting example of misfolding at the earliest state of a natural folding reaction is provided by the alpha subunit of tryptophan synthase, a (β/α)8 TIM barrel protein. The molecular basis for the formation of this off-pathway misfolded intermediate, IBP, and a subsequent on-pathway intermediate, I1, was probed by mutational analysis of 20 branched aliphatic side chains distributed throughout the sequence. The elimination of IBP and the substantial destabilization of I1 by replacement of a selective set of the isoleucine, leucine or valine residues (ILV) with alanine in a large ILV cluster external-to-the-barrel and spanning the N- and C-termini (Cluster 2) implies tight-packing at most sites in both intermediates. Differential effects on IBP and I1 for replacements in α3, β4 and α8 at the boundaries of Cluster 2 suggest that their incorporation into I1 but not IBP reflects non-native folds at the edges of the crucial (β/α)1–2β3 core in IBP. The retention of IBP and the smaller and consistent destabilization of both IBP and I1 by similar replacements in an internal-to-the-barrel ILV cluster (Cluster 1) and a second external-to-the-barrel ILV cluster (Cluster 3) imply molten globule-like packing. The tight packing inferred in part for IBP or for all of I1 in Cluster 2, but not in Clusters 1 and 3, may reflect the larger size of Cluster 2 and/or the enhanced number of isoleucine, leucine and valine self-contacts in and between contiguous elements of secondary structure. Tightly-packed ILV- dominated hydrophobic clusters could serve as an important driving force for the earliest events in the folding and misfolding of the TIM barrel and other members of the (β/α)n class of proteins.
Keywords: Hydrophobic cluster, off-pathway intermediate, sub-millisecond reaction, TIM barrel, misfolding
Introduction
The folding reactions of small proteins and protein domains (< ∼ 100 amino acids) are often well-described by a simple exponential response thought to reflect the conformational diffusion of the unfolded polypeptide across an energy barrier separating the native and unfolded manifolds of structures.1,2 The folding of these proteins is only limited by the intrinsic features of the energy surface defined primarily by the chain topology3 and modulated by the amino acid sequence.4,5
By contrast, the folding reactions of larger proteins often display complex kinetics indicative of a significant role for alternative conformational states.1,2,6–9 These “frustrated” folding reactions10 have been ascribed to a variety of sources, including slowly-interconverting cis/trans prolyl peptide bond isomers in the unfolded state,11-14 mis-ligated side chain-heme complexes in early folding intermediates for cytochrome c,15 on-pathway, stable intermediates,9,16–18 alternative partially-folded states in parallel pathways,19,20 maturation of multimeric intermediates that are pre-cursors for native multimers,21–23 mandatory non-native conformers,24–28 misfolded off-pathway intermediates,29–33 and the sub-millisecond formation of marginally-stable conformers that are rich in secondary structure34
The case of misfolded off-pathway intermediates is particularly interesting because, contrary to the expectations of an optimized energy landscape, the protein has to unfold these partially-structured states to some extent before proceeding to the native conformation. In the case of chemotaxis protein Y (CheY), a β/α sheet protein, the off-pathway intermediate is thought to reflect the rapid formation of the complete complement of α helical segments. G -model simulations35 and phi analysis36 suggest that the C-terminal helices must unfold to access the rate-limiting transition state defined by the N-terminal strands and helices. Apo-flavodoxin, another member of this class, also misfolds to an off-pathway intermediate before accessing the native conformation; the structural basis for this misfolding reaction is not known.30,31
The α-subunit of tryptophan synthase (αTS) (Figure 1a), a member of the TIM barrel, (β/α)8, sub-class of the (β/α)n fold family, is known to fold to an off-pathway intermediate, IBP, in the sub-millisecond time range.18 A simplified folding mechanism, ignoring a set of four parallel pathways that correspond to several cis/trans prolyl peptide bond isomerizations,13,38,39 is shown in Scheme I:
Figure 1.
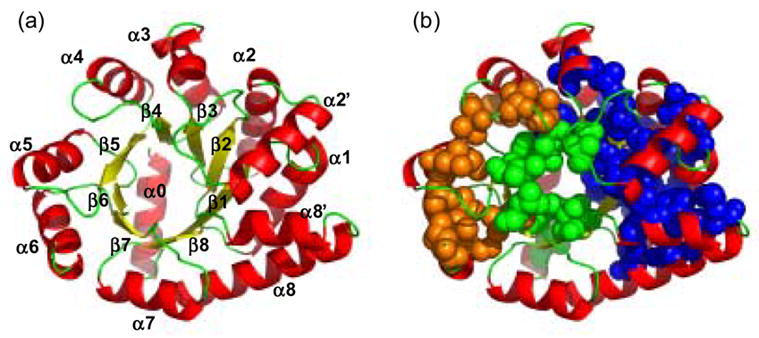
Ribbon diagrams of αTS (a) showing the elements of secondary structure and (b) highlighting the three hydrophobic clusters formed by the I, L and V residues: Cluster 1 (green), Cluster 2 (blue) and Cluster 3 (orange), respectively, obtained using a 4.2 Å cutoff distance. L7 and L11, V214 and V220, which form two small isolated clusters, are not shown. Coordinates of the α subunit of tryptophan synthase from S. typhimurium, which shares 85% sequence identity with E. coli αTS, were used to generate the figure from a refined version of PDB file 1BKS63 using Pymol.37 57 out of 63 ILV residues are conserved in E.coli; the six differences primarily involve I/V switches located at V52I (β2), I148V (loop before β5), V166I (α5), I197V (α6), V224I (α7) and L245I (loop between α8' and α8).
Scheme 1.
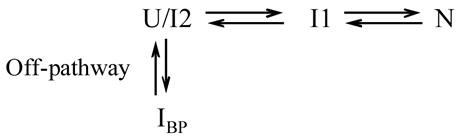
U/I2 represents a pair of rapidly inter-converting, thermodynamically-distinct unfolded, U, or unfolded-like, I2, states that are highly populated under denaturing conditions. IBP represents a transient misfolded intermediate with substantial secondary structure and appreciable stability.18,32 I1 is a stable intermediate whose conversion to the native state, N, is the rate- limiting step in folding. The mandatory unfolding of IBP under strongly refolding conditions is an early rate-limiting and distinguishing feature of an off-pathway kinetic intermediate.
In a previous study on αTS,32 it was shown that alanine replacements at three sites where amide hydrogens are extremely slow to exchange with solvent, I37 (α1), L50 (β2) and L99 (β3), eliminate the misfolded IBP intermediate while replacements at more rapidly-exchanging nonpolar sites, L127 (β4) and L176 (β6), do not. A potential explanation for these differential effects can be found in the crystal structure of αTS, which shows three distinct and substantive hydrophobic clusters comprised of the large branched aliphatic side chains isoleucine, leucine and valine (Figure 1b). I37, L50 and L99 are a part of the largest cluster, Cluster 2, which is outside the β barrel and inside the helical shell, spans the N- and C-termini, and contains 31 ILV side chains from β1, α1, β2, α2, β3, α3, β4, α8’ and α8. L127 belongs to Cluster 1, an internal-to-the-barrel cluster, which is defined by 8 ILV side chains from β1, β3, β4, β5, β6, β7, β8 and capping helix α0. L176 belongs to cluster 3, a C-terminal external-to-the-barrel cluster, which is formed by 12 ILV side chains from β5, α5, β6 and α6. These clusters account for 51 of 63 (81%) of the total number of ILV residues, which together comprise 42% of the nonpolar side chains in αTS and are distributed throughout the sequence.
The dramatically different effects on IBP of alanine replacements for leucines or isoleucines in Cluster 2 vs. Clusters 1 and 3 prompted a comprehensive mutational analysis of the relationship of these clusters to the formation of IBP and to the stability of the equilibrium intermediate, I1.
Results
Cluster assignment
The ILV clusters were visualized with PyMOL37 by generating the 3D map of all van der Waals contacts within 4.2 Å between heavy atoms of only the ILV side chains. Clusters were defined as those sets of contacts involving a minimum of 6 ILV side chains arranged in a compact, self-contained structure. The choice of the minimum size was based upon MD simulations in which 30 methanes or 6 isobutylenes were required to form a stable hydrophobic cluster in water.40 Cluster boundaries were identified by the absence of intervening ILV-ILV contacts. Based on these criteria three discernable clusters can be identified (Figure 1 and Table 1S). Cluster 1, containing 8 residues, is internal-to-the barrel. Cluster 2, containing 31 ILV residues, and Cluster 3, containing 12 residues, are outside the β barrel and inside the helical shell. All these contacts are defined as ILV self-contacts, to distinguish them with the contacts of nonpolar side chains from alanine, cysteine, methionine, phenylalanine and tyrosine and nonpolar components of otherwise polar side chains with the I, L and V residues.
Secondary and tertiary structures of the variants are largely conserved
Far-UV and near-UV circular dichroism (CD) spectra were used to monitor the perturbation in the secondary and tertiary structure introduced by the individual replacement of 20 ILV residues with alanine. The far-UV CD spectra of most of the αTS variants closely resemble the wild-type spectrum with a minimum near 222 nm, a shoulder near 208 nm, and a maximum near 195 nm (Figure 2). The mean residue ellipticities at 222 nm for these variants were reduced by less than 20%, suggesting a relatively small perturbation in the secondary structure. Although the far-UV CD spectra of L25A, L48A and I232A indicate a greater loss of secondary structure, all three variants retain the same apparent 3-state equilibrium folding model displayed by wild-type αTS and the rate-limiting unfolding reactions characteristic of the native conformation (see below). Inspection of the near-UV CD spectra (data not shown) also shows that the packing of the aromatic residues is conserved for the majority of the variants. Those for which the aromatic packing is disrupted to some extent also retain the three-state folding model and rate-limiting unfolding reactions. Thus, the evident perturbations in structure for a subset of the αTS variants do not preclude the interpretation of their thermodynamic properties in terms of its native and stable intermediates states.
Figure 2.
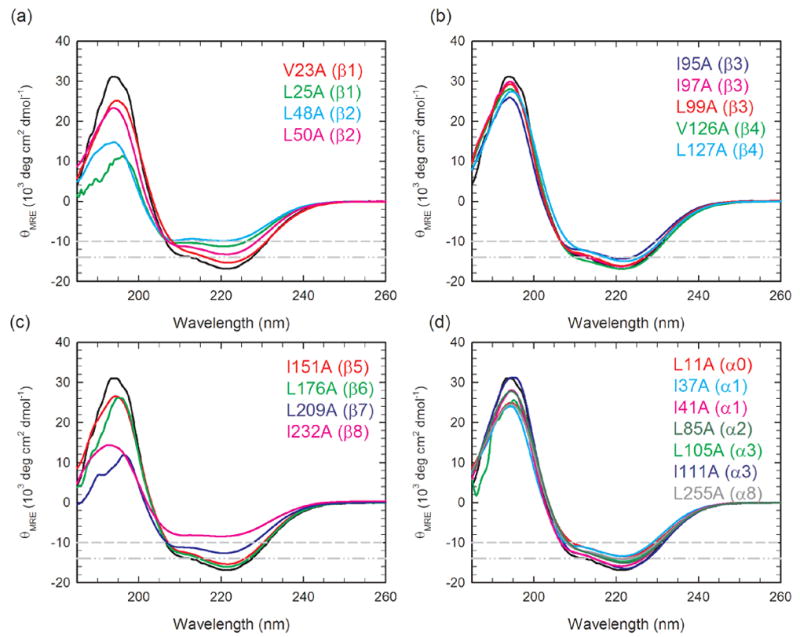
Far-UV circular dichroism spectra of αTS variants grouped by replacements in elements of secondary structure (a) β1 and β2, (b) β3 and β4 (c) β5 to β8 and (d) α0, α1, α3 and α8. The far-UV CD spectrum for the wild-type protein is also shown in each panel with a black line, for reference. The gray dash-dot-dot and dash guidelines indicate a 15% and 40% decrease in signal at 222 nm, relative to wild-type αTS, respectively. The protein concentration ranged from 5 to 7 μM. Buffer conditions: 10 mM potassium phosphate, pH 7.8, 0.2 mM K2EDTA, and 1 mM β-mercaptoethanol. Data were collected at 25 °C.
Differential effects on the stability of αTS
To examine the effects of these replacements on the thermodynamic properties of αTS, the urea-induced unfolding was monitored by CD spectroscopy in the range from 215 nm to 265 nm. Equilibrium unfolding transition curves obtained by monitoring the urea-dependence of the mean residue ellipticity at 222 nm for wild-type and variants are shown in Figure 3. Similar to wild-type αTS, the equilibrium unfolding reactions of all of these variants are best described by a three-state model, N⇌I1⇌I2/U. Because the ellipticity of I2 is indistinguishable from the U state, I2/U can be treated as a single apparent thermodynamic state for titrations monitored by CD spectroscopy. Displacements of the midpoints for both the N⇌I1 and I1⇌I2/U transitions to lower urea concentrations for almost all of the variants (Figure 3) showed that the replacement of these buried isoleucine, leucine and valine side chains with alanine destabilizes both the N and the I1 states. The free energy differences between N and I1 and between I1 and I2/U in the absence of denaturant are displayed in bar graph form in Figure 4a and Figure 4b, respectively, for all of the variants. These values and the urea dependence of the free energy differences, the m values, as well as the corresponding midpoints, Cm, are also provided in Table 1.
Figure 3.
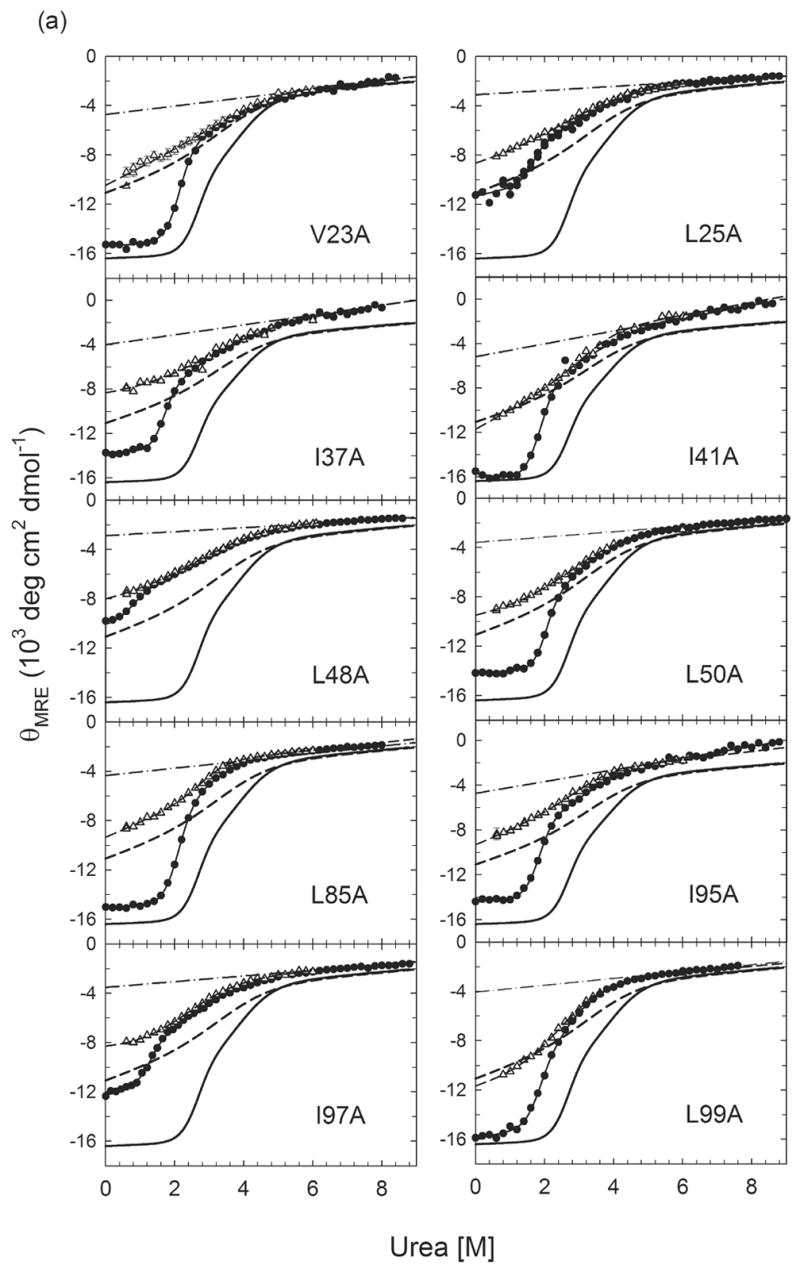
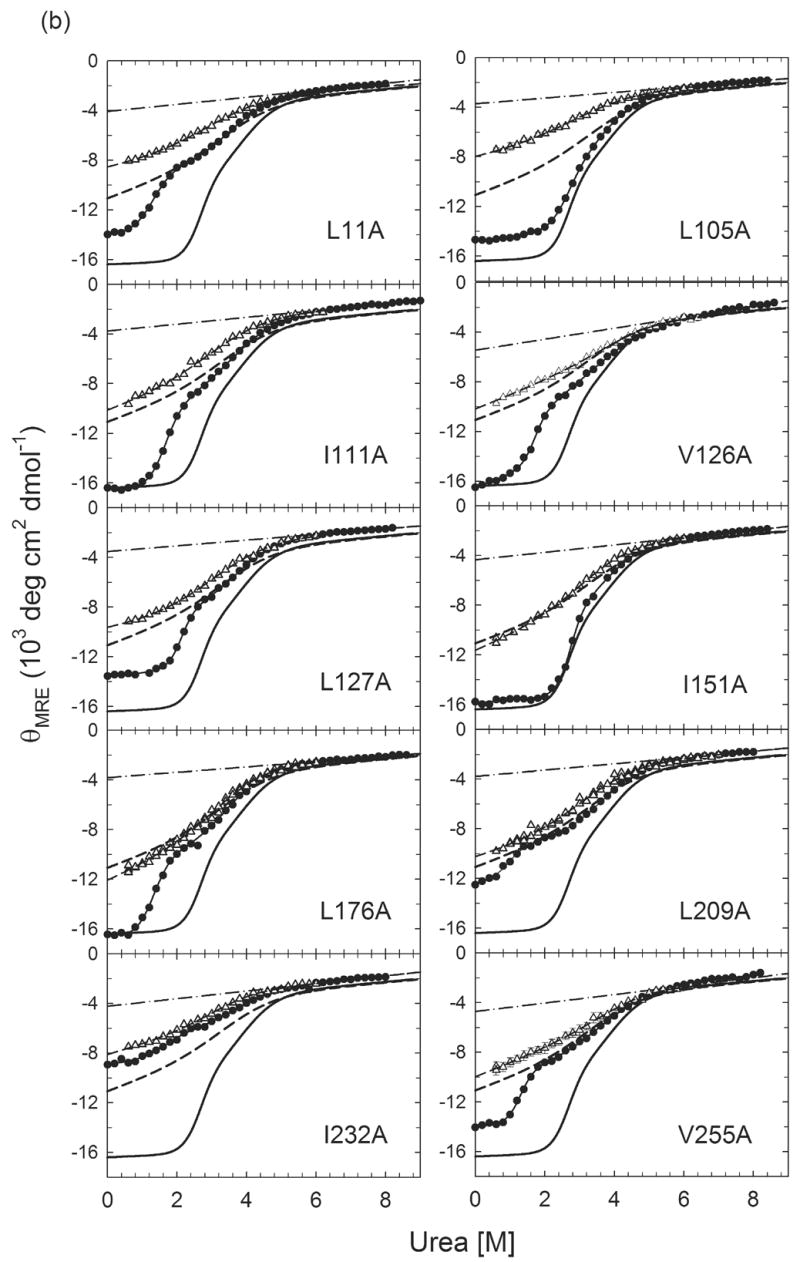
Urea-induced equilibrium unfolding reactions (●)refolding burst-phase amplitudes (△) from stopped-flow CD measurements initiated from the unfolded state at 6 M urea for (a) Group I αTS variants and (b) Group II αTS variants. For I37A, L50A, L99A, L127A and L176A, data are from Wu et al.32 The unfolded baselines are shown as dashed-dotted lines. The solid lines represent the fits of the data to a three-state equilibrium model. The burst-phase amplitudes for all proteins were fit to a two-state model and are represented by the dashed lines. The equilibrium unfolding profile of wild-type αTS and its burst-phase intermediate are shown as thick black full and dashed lines, respectively, for reference. Buffer conditions are described in the caption for Figure 2.
Figure 4.
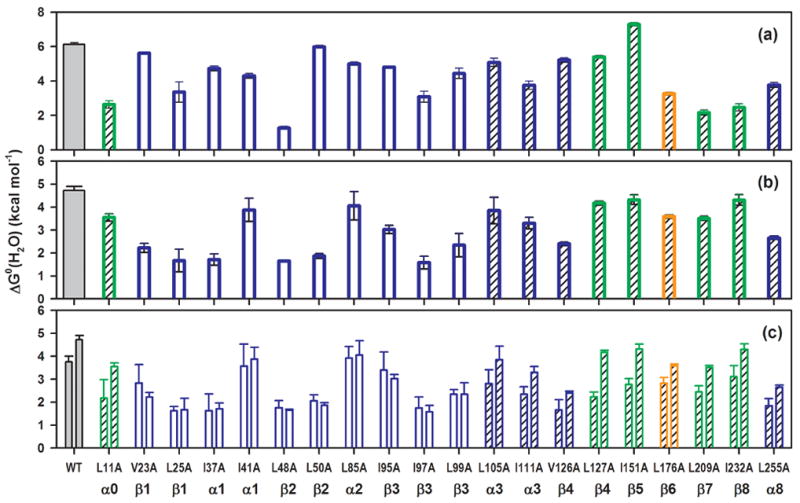
Bar graph of the free energy difference in the absence of urea for (a) N ⇌ I1, (b) I1 ⇌ I2/U and (c) IBP ⇌ I2/U vs. I1 ⇌ I2/U transitions for Group I (open bars) and Group II (hatched bars) αTS variants. For (c), the bars on the left refer to the stabilities of the burst phase intermediates (IBP) obtained from stopped−flow CD measurements; the bars on the right represent the stabilities of the I1 species measured from equilibrium unfolding experiments. The green, blue and orange bars represent Cluster 1, 2 and 3 αTS variants, respectively. The values for wild-type αTS are shown in gray for reference. The secondary structure assignment for each mutation site is labeled underneath the corresponding variant.
Table 1.
Thermodynamic parametersa for urea-induced unfolding of variants and wild-type αTS at pH 7.8 and 25 °Cb
| Secondary Structure | Variants | Kinetic Response | Equilibrium Unfolding
|
Refolding Burst Phase
|
|||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| N⇌I1 transition
|
I1⇌ I2/U transition
|
ΔGo (H2O) | − m | Cm | |||||||
| ΔGo (H2O) | −m | Cm | ΔGo (H2O) | − m | Cm | ||||||
| Cluster 2 | WT | 6.13 ± 0.09 | 2.29 ± 0.04 | 2.68 ± 0.06 | 4.72 ± 0.1 | 1.20 ± 0.04 | 3.93 ± 0.20 | 3.75 ± 0.25 | 1.08 ± 0.04 | 3.47 ± 0.26 | |
|
| |||||||||||
| β1 | V23A | Group I | 5.62 ± 0.06 | 2.58 ± 0.02 | 2.18 ± 0.03 | 2.22 ± 0.20 | 0.66 ± 0.04 | 3.36 ± 0.36 | 2.84 ± 0.80 | 0.64 ± 0.20 | 4.43 ± 1.86 |
| L25A | 3.36 ± 0.59 | 2.16 ± 0.31 | 1.56 ± | 1.67± 0.50 | 0.64 ± 0.11 | 2.61 ± 0.90 | 1.63 ± 0.18 | 0.55 ± 0.03 | 2.96 ± 0.36 | ||
| α1 | I37Ac | 4.72 ± 0.14 | 2.72 ± 0.08 | 1.74 ± 0.07 | 1.71 ± 0.25 | 0.64 ± 0.06 | 2.67 ± 0.46 | 1.63 ± 0.73 | 0.55 ± 0.16 | 2.96 ± 1.58 | |
| I41A | 4.304.30 | 2.28 ± 0.07 | 1.89 ± 0.08 | 3.88 ± 0.51 | 1.09 ± 0.12 | 3.56 ± 0.61 | 3.57 ± 0.96 | 1.03 ± 0.21 | 3.47 ± 1.17 | ||
| β2 | L48A | 1.27 ± 0.09 | 1.81 ± 0.07 | 0.70 ± 0.06 | 1.65 ± 0.04 | 0.64 ± 0.01 | 2.58 ± 0.07 | 1.76 ± 0.31 | 0.59 ± 0.04 | 2.98 ± 0.56 | |
| L50Ac | 5.99 ± 0.07 | 2.87 ± 0.04 | 2.09 ± 0.04 | 1.87 ± 0.11 | 0.70 ± 0.02 | 2.67 ± 0.17 | 2.06 ± 0.26 | 0.71 ± 0.05 | 2.90 ± 0.41 | ||
| α2 | L85A | 5.00 ± 0.11 | 2.33 ± 0.05 | 2.15 ± 0.07 | 4.05 ± 0.62 | 1.23 ± 0.14 | 3.29 ± 0.63 | 3.92 ± 0.50 | 1.34 ± 0.15 | 2.92 ± 0.49 | |
| β3 | I95A | 4.80 ± 0.07 | 2.59 ± 0.04 | 1.85 ± 0.04 | 3.03 ± 0.18 | 0.97 ± 0.04 | 3.12 ± 0.23 | 3.40 ± 0.78 | 1.05 ± 0.21 | 3.24 ± 0.98 | |
| I97A | 3.08 ± 0.32 | 2.38 ± 0.23 | 1.29 ± 0.18 | 1.58 ± 0.28 | 0.63 ± 0.07 | 2.51 ± 0.53 | 1.75 ± 0.48 | 0.77 ± 0.12 | 2.27 ± 0.71 | ||
| L99Ac | 4.44 ± 0.30 | 2.39 ± 0.18 | 1.86 ± 0.19 | 2.34 ± 0.51 | 0.94 ± 0.11 | 2.49 ± 0.62 | 2.34 ± 0.21 | 0.90 ± 0.04 | 2.60 ± 0.26 | ||
|
| |||||||||||
| α3 | L105A | Group II | 5.07 ± 0.24 | 1.91 ± 0.10 | 2.65 ± 0.19 | 3.85 ± 0.58 | 1.03 ± 0.11 | 3.74 ± 0.69 | 2.81 ± 0.60 | 0.79 ± 0.11 | 3.56 ± 0.91 |
| I111A | 3.76 ± 0.24 | 2.20 ± 0.14 | 1.71 ± 0.15 | 3.30 ± 0.25 | 0.94 ± 0.06 | 3.51 ± 0.35 | 2.35 ± 0.32 | 0.72 ± 0.05 | 3.26 ± 0.50 | ||
| β4 | V126A | 5.21 ± 0.13 | 3.03 ± 0.08 | 1.72 ± 0.06 | 2.40 ± 0.08 | 0.69 ± 0.02 | 3.48 ± 0.15 | 1.66 ± 0.44 | 0.60 ± 0.05 | 2.77 ± 0.77 | |
| α8 | L255A | 3.77 ± 0.14 | 2.91 ± 0.10 | 1.30 ± 0.07 | 2.66 ± 0.08 | 0.75 ± 0.02 | 3.55 ± 0.14 | 1.84 ± 0.31 | 0.53 ± 0.03 | 3.47 ± 0.62 | |
| Cluster 1 | |||||||||||
| α0 | L11A | 2.63 ± 0.21 | 2.02 ± 0.14 | 1.30 ± 0.14 | 3.55 ± 0.16 | 1.02 ± 0.04 | 3.48 ± 0.21 | 2.18 ± 0.80 | 0.71 ± 0.20 | 3.07 ± 1.42 | |
| β4 | L127Ac | 5.40 ± 0.08 | 2.55 ± 0.04 | 2.12 ± 0.05 | 4.17 ± 0.09 | 1.10 ± 0.02 | 3.79 ± 0.11 | 2.24 ± 0.20 | 0.71 ± 0.03 | 3.15 ± 0.31 | |
| β5 | I151A | 7.29 ± 0.10 | 2.66 ± 0.04 | 2.74 ± 0.06 | 4.32 ± 0.21 | 1.13 ± 0.04 | 3.82 ± 0.23 | 2.78 ± 0.24 | 0.82 ± 0.04 | 3.39 ± 0.34 | |
| β7 | L209A | 2.16 ± 0.15 | 2.36 ± 0.14 | 0.92 ± 0.08 | 3.51 ± 0.09 | 0.95 ± 0.02 | 3.69 ± 0.12 | 2.45 ± 0.26 | 0.73 ± 0.04 | 3.36 ± 0.40 | |
| β8 | I232A | 2.46 ± 0.21 | 1.49 ± 0.12 | 1.65 ± 0.19 | 4.31 ± 0.23 | 1.15 ± 0.05 | 3.75 ± 0.26 | 3.11 ± 0.48 | 0.94 ± 0.09 | 3.31 ± 0.60 | |
| Cluster 3 | |||||||||||
| β6 | L176Ac | 3.25 ± 0.09 | 2.16 ± 0.05 | 1.50 ± 0.05 | 3.59 ± 0.07 | 1.01 ± 0.02 | 3.55 ± 0.10 | 2.82 ± 0.25 | 0.80 ± 0.04 | 3.53 ± 0.40 | |
The equilibrium unfolding data were fit to a 3-state model, N⇌I1⇌I2/U, while the refolding burst phase data were fit to a 2-state model, N⇌U, assuming a linear dependence of the free energy of unfolding on the denaturant concentration, ΔGo=ΔGo (H2O)+m[urea].41 ΔGo , ΔGo (H2O), m and Cm represent the energy of unfolding in the presence of urea, the free energy of unfolding in the absence of urea, the urea dependence of the free energy of unfolding and the urea concentration at the midpoint of transition, respectively. Units are as follows: ΔGo(H2O), kcal mol−1 (M urea)−1; Cm, M urea. Errors for ΔGo(H2O) and m are standard errors from the fits. Errors in Cm are obtained by standard error propagation of the equation: Cm=(H2O)/m.
Conditions: 10 mM potassium phosphate, pH 7.8, 0.2 mM K2EDTA, and 1 mM β-mercaptoethanol.
Data from ref.32
The decreases in the free energy difference between the N and I1 states for the variants range from 0.14 kcal mol−1 for L50A to 4.86 kcal mol−1 for L48A; I151A was the only replacement that increased the free energy difference, by 1.16 kcal mol−1. The variations in the stability of N relative to I1 were similar across the entire sequence and for replacements in all three hydrophobic clusters (Figure 4a). By contrast, the majority of the replacements in Cluster 2 markedly decreased the stability of I1 relative to I2/U while all of those in Clusters 1 and 3 had a lesser effect (Figure 4b). For example, the striking contrast between the substantial destabilization by V126A and the minimal perturbation by L127A appears to be related to their participation in Cluster 2 and Cluster 1, respectively. Excluding the four outliers in Cluster 2, I41A, L85A, L105A and I111A (which are all located in surface α helices), the average decrease in stability of the I1 state with respect to the I2/U state for the Cluster 2 variants is 2.61 ± 0.27 kcal mol−1; the average decrease experienced by the Cluster 1 and 3 variants is 0.74 ± 0.25 kcal mol−1. The small decrease in the stability of I1 for the four Cluster 2 outliers, 0.95 ± 0.46 kcal mol−1, may reflect their participation in surface helices that might individually reorient to minimize the disruption in stability. It is also possible that the enhanced helical propensity of alanine vs. leucine and isoleucine42 mitigates the decrease in stability at all of these sites.
Differential effects on the kinetic folding properties of αTS
As previously reported, alanine replacements for leucine and isoleucine residues in α1, β2 and β3 have two distinct effects on the properties of the early folding events in αTS.32 The distinguishing features are the effects on the stability of the product of the burst-phase reaction compared to the on-pathway equilibrium intermediate and the presence (Figure 5a and Figure 5c) or absence (Figure 5b) of a subsequent unfolding reaction in the hundreds of milliseconds time range under refolding conditions. A lower stability for IBP relative to I1 and the presence of this unfolding reaction are indicative of an off-pathway burst-phase species similar to that observed for wild-type αTS. Similar stabilities for the product of the burst-phase reaction and I1 as well as the absence of this kinetic phase indicate that the folding reaction can proceed directly to the on-pathway equilibrium intermediate, I1, within the dead time of the stopped-flow measurement (< 5 ms) (Scheme I).
Figure 5.
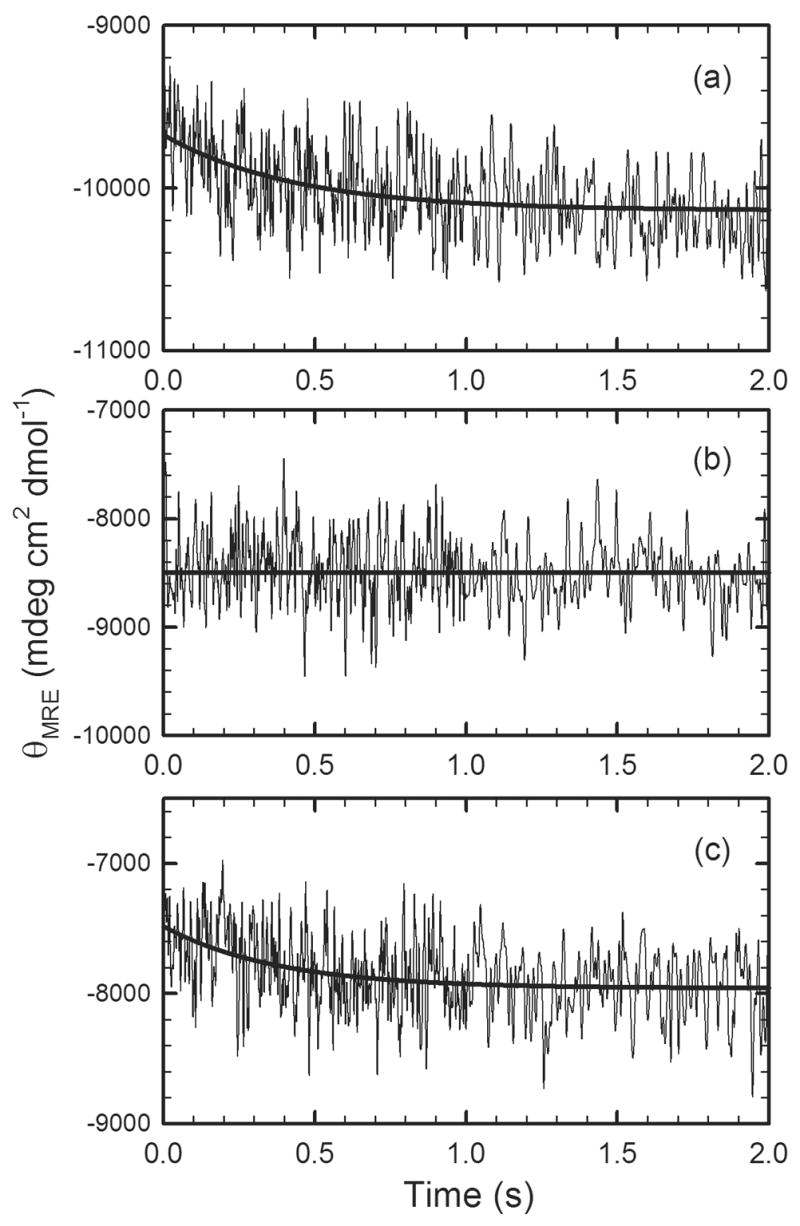
The refolding kinetic traces of (a) wild-type αTS, (b) L99A αTS and (c) L105A αTS monitored by stopped-flow CD for refolding jumps from 6.0 M to 1.0 M urea in standard buffer, described in the caption for Figure 2
Based upon the kinetic and thermodynamic responses, the 20 αTS variants in this study can be classified into two groups: Group I eliminates the off-pathway intermediate and Group II retains this misfolded species. Chevron plots show that 10 of the 20 variants, specifically, V23A (β1), L25A (β1), I37A (α1), I41A (α1), L48A (β2), L50A (β2), L85A (α2), I95A (β3), I97A (β3) and L99A (β3), lack the hundreds of milliseconds unfolding reaction under strongly refolding conditions (Figure 6a). Further, the denaturation profiles of their CD burst-phase amplitudes are coincident with those for the unfolding of the equilibrium intermediate (Figure 3a). A demonstration of the equivalent stabilities of I1 and the burst-phase intermediate, relative to the apparent I2/U state, for the entire set of Group I replacements is shown in the bar graph in Figure 4c and in Table 1. The retention of the unfolding kinetic phase under refolding conditions and the uniformly lower stability of the burst-phase species compared to the I1 species for L11A (α0), L105A (α3), I111A (α3), V126A (β4), L127A (β4), I151A (β5), L176A (β6), L209A (β7), I232A (β8), and L255A (α8) (Figure 4c and Table 1) define these 10 variants as members of the Group II category. The branched aliphatic side chains at these positions are not essential for the formation of the misfolded intermediate. Interestingly, the stability of the IBP species for the Group II variants is reduced by a small but consistent amount relative to the wild-type protein (Figure 4c), 1.16 ± 0.42 kcal mol−1.
Figure 6.
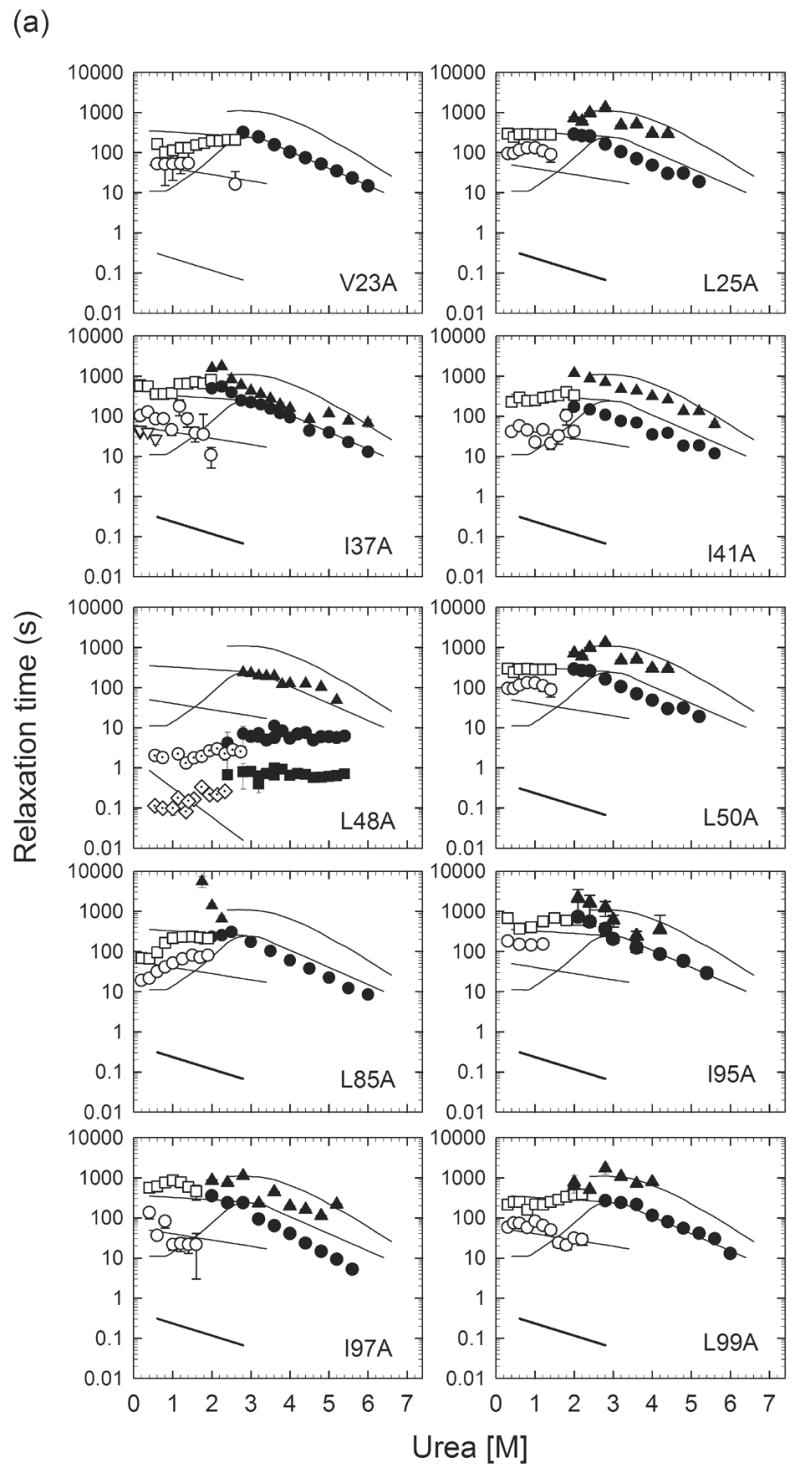
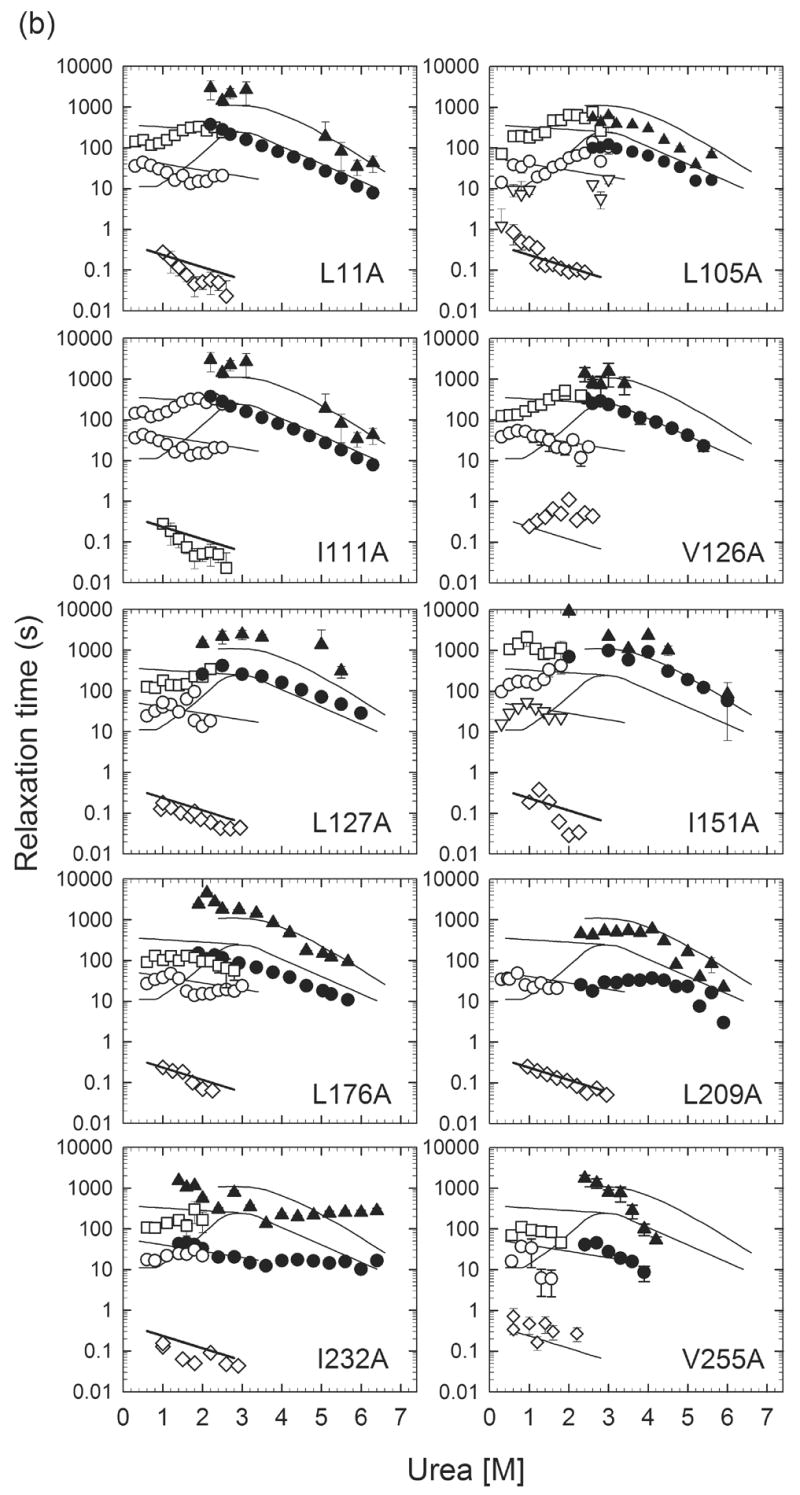
Urea-dependence of the observed refolding phases monitored by manual-mixing CD (○▽□) and stopped-flow fluorescence (◇), as well as unfolding phases monitored by manual-mixing CD (●▲) for (a) Group I αTS variants and (b) Group II αTS variants. The solid lines represent observed relaxation times for wild-type αTS. The diamonds (◇) indicate the behavior of the several hundred millisecond refolding phase corresponding to the unfolding of the off-pathway, burst-phase intermediate. The open triangles (▽), open circles (○) and open squares (□) reflect the fast, intermediate and slow refolding phases attributed to prolyl isomerization. The filled circles (●) and triangles (▲) represent the major and minor unfolding phases. In the case of L48A, the observation of a sub-millisecond phase (◈) reflects the refolding of the I2/U to I1 reaction, based on its continuous linkage with the unfolding reaction of the I1 species (■).
DISCUSSION
The role of hydrophobic clusters in the early misfolding reaction of αTS
The replacement of a series of isoleucine, leucine and valine residues in αTS with alanine has revealed distinctively different roles for their resident hydrophobic clusters in the formation of an early off-pathway misfolded intermediate and in the stability of the subsequent on-pathway equilibrium intermediate. Replacements in Cluster 2, in β1, α1, β2, α2 and β3, eliminate the off-pathway burst-phase species and enable direct access to the on-pathway intermediate in the sub-millisecond time frame; replacements in α3, β4 and α8 do not eliminate the burst-phase species. Replacements in both Clusters 1 and 3, including residues in α0, β4, β5, β6, β7, β8 and α8, retain the off-pathway intermediate but decrease its stability by an average value of 1.16 ± 0.42 kcal mol−1. Thus, the N-terminal region of Cluster 2, formed by branched aliphatic side chains protruding from 5 consecutive elements of secondary structure defining the β barrel and the surrounding helical shell, i.e., the (β/α)1–2β3 segment, is crucial for the misfolding reaction of αTS.
The alanine replacements for the Group I variants could exert their effects by significantly destabilizing the misfolded species or by increasing the free energy of the barrier between the U or I2 precursor and IBP.32 Either explanation requires a destabilization exceeding several kcal mol−1 to eliminate IBP, implying tight packing around all of these side chains. It has been previously shown that the replacement of branched aliphatic side chains with alanine in the interior of staphylococcal nuclease can destabilize the native conformation by up to 5.8 kcal mol−1.43 Similar results were obtained in a more limited study on barnase;44 the decrease in stability was equivalent to 1.0 – 1.6 kcal mol−1 per methylene removed.45
The small but non-negligible perturbations in the stability of IBP for the Group II variants show that the C-terminal region of Cluster 2 and both Clusters 1 and 3 have also formed in the millisecond time frame. The close agreement between the average decrease in stability, 1.16 ± 0.42 kcal mol−1, and the differences in the free energy of transfer between nonpolar and aqueous solvents for isoleucine, leucine and valine vs. alanine, 1.3, 1.3 and 1.0 kcal mol−1, respectively,43,46 suggest that these clusters resemble molten globule-like states.47
The role of hydrophobic clusters in stabilizing the on-pathway equilibrium intermediate in αTS
Inspection of the perturbations on the stability of the I1 intermediate for the Cluster 1, 2 and 3 variants (Figure 4b and Table 1) shows a trend that is strikingly similar to that for the misfolded IBP state. The external-to-the-barrel side chains in β1, α1 (I37), β2, β3, β4 (V126) and α8 are tightly-packed in I1 while the internal-to-the-barrel side chains in α0, β4 (L127), β5, β7 and β8 and the external-to-the-barrel side chains in β6 occupy molten globule-like environments. The average decrease in stability of the I1 state for these latter variants is 0.74 ± 0.25 kcal mol−1. Differentiating I1 from IBP is the implied tight-packing for V126 (β4) and L255 (α8) in I1, both of which participate in Cluster 2 but are not members of the Group I variants.
The role of hydrophobic clusters in stabilizing the rate-limiting transition state in the folding of αTS
In striking contrast to the differential effects of the ILV replacements on the free energies of IBP and I1, 17 of the 20 variants (with the exception of I151A in β5, I232A in β8 and L255A in α8) display a major slow unfolding reaction that is indistinguishable from that for wild-type αTS (Figure 6). Given the observed destabilization of the native state in the variants (Table 1), these results demonstrate that very native-like packing is maintained in all three hydrophobic clusters in the transition state for the rate-limiting N→I1 unfolding step48 in Scheme I. By inference from the chevron shape49 for the urea dependence of the relaxation times for the reversible N⇌I1 reaction (Figure 3), native-like tight packing is achieved at all of these positions in the transition state for the I1→ N reaction. Thus, the ILV side chains play significant roles in stabilizing this transition state and, thereby, potentially accelerating the final step in the folding reaction. The results also show that the transition state is formed by side chains that are distributed throughout the sequence, a finding that may be related to its rate-limiting role in folding.
Why is misfolding favored over productive folding for wild- type αTS?
The similar but not identical responses of IBP and I1 to the alanine replacements beg the question as to why the unfolded state of wild-type αTS initially misfolds. Why does U/I2 not proceed directly to the on-pathway equilibrium intermediate? The answer appears to reside in the peculiar responses of V126A and L255A. Both replacements substantially decrease the stability of the I1 state, similar to most of the Group I variants, but retain the off-pathway burst- phase intermediate, a defining feature of the Group II variants. The differential effects on the IBP and I1 intermediates imply that both side chains are excluded from the hydrophobic cluster crucial to the formation of the off-pathway species but are intimately involved in stabilizing the on-pathway intermediate. One interpretation of these results is that the folding of the region from β1 to β3 is driven by the sub-millisecond formation of a localized and self-contained structure whose boundaries, unlike those in the native TIM barrel, allow favorable interactions with the solvent. The putative non-native structure at the boundaries does not provide suitable docking sites for β4 and α8, requiring the misfolded IBP species to at least partially unfold before recruiting both of these elements of secondary structure into the I1 intermediate. The failure to incorporate α8 into IBP may also be related to its distal position in sequence, where chain diffusion might limit access to a receptive (β/α)1–2β3 platform.
Differential packing in hydrophobic clusters in the off- and on-pathway intermediates for αTS
The heterogeneous response of the hydrophobic clusters in αTS to alanine replacements implies tight packing for side chains in Cluster 2 (with the exception of α3, β4 and α8 in IBP) and looser, molten globule-like packing for Clusters 1 and 3 in both IBP and I1 (Table 1). The striking segregation of the ILV side chains into three distinct clusters (Figure 1b) motivated a closer inspection of these clusters and their interactions with other nearby side chains (Table 2). The 31 ILV side chains in Cluster 2 make 90 self-contacts, assuming a 4.2 Å cut-off distance between carbon atoms, the 8 ILV side chains in Cluster 1 make 14 self-contacts and the 12 ILV side chains in Cluster 3 make 20 self-contacts. Thus, the average number of self-contacts per residue is about twice as large for Cluster 2 than for Clusters 1 and 3. This distinction disappears if other nonpolar side chains from alanine, cysteine, methionine, phenylalanine and tyrosine and nonpolar components of otherwise polar side chains in contact with the ILV residues in the clusters are included in this analysis (Table 2). Thus, the differential properties of the three clusters do not appear to be related to the density of van der Waals contacts when all buried side chains are considered.
Table 2.
Summary of the side chain contacts within 4.2 Å for nonpolar side chains and the aliphatic component of polar side chains in the three αTS hydrophobic clustersa
| ILV | ACFILMPVY | ALLb | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| # contacts | # residues | # contacts/ residue |
# contacts | # residues | # contacts/ residue |
# contacts | # residues | # contacts/ residue |
||
| Cluster 1 | 14 | 8 | 1.8 | 42 | 12 | 3.5 | 48 | 13 | 3.7 | |
| Cluster 2 | 90 | 31 | 2.9 | 182 | 57 | 3.2 | 287 | 100 | 2.9 | |
| Cluster 3 | 20 | 12 | 1.7 | 85 | 35 | 2.4 | 144 | 52 | 2.8 | |
|
| ||||||||||
| Total | 124 | 51 | 2.4 | 309 | 104 | 3.0 | 479 | 165 | 2.9 | |
As noted in the legend to Figure 1, the side chain contacts are calculated based on the coordinates of the α subunit of tryptophan synthase from S. typhimurium (1BKS63), which shares 85% sequence identity with E. coli αTS. ILV residues are largely conserved in E. coli, except the six I/V switches: V52I (β2), I148V (loop before β5), V166I (α5), I197V (α6), V224I (α7) and L245I (loop between α8' and α8).
All residues in the sequence except glycine.
Two previous observations on αTS suggest that the differential properties might be related to a special role for leucines and isoleucines in stabilizing partially-folded states. An HX- NMR study revealed that 7 of the 11 amide hydrogens most resistant to exchange with solvent deuterium are leucines or isoleucines located in α1, β2 and β3.32 A network of L - L and L - I interactions between these 7 side chains appear to stabilize the core of the N-terminal hydrophobic cluster, (β/α)1-2β3, that is central to both the IBP and I1 intermediates. Further support for a unique role for leucine and isoleucine residues can be found in a Heteronuclear Single Quantum Coherence NMR spectrum of selectively 15N-Leu or 15N-Ile labeled αTS at 5 M urea where the possible I2 precursor to IBP is highly populated (Vadrevu, R., Wu, Y. and Matthews, C. R., manuscript in preparation). In the absence of well-defined secondary structure, ∼ 3 leucines, including L99 of β3, and ∼ 4 isoleucines are exchange broadened by the formation of a previously-identified hydrophobic cluster;50 phenylalanines and valines do not participate in the dynamic broadening process. The persistent interactions between these leucine and isoleucine residues under strongly-denaturing conditions imply that mutual interactions between large branched aliphatic side chains in the absence of denaturant may be the primary source of the differential behavior of the three clusters in αTS.
Branden and Tooze51 have previously noted a strong bias towards ILV residues in TIM barrel proteins. This bias may reflect a distinct preference observed for ILV side chains with ILV neighbors in adjacent parallel strands in TIM barrel and parallel (β/α)n sheet proteins.52 Simulations designed to optimize side chain placements in predicted protein structures53 found that aliphatic pairs adopt restricted orientations in actual structures that differentiate them from other pairs of nonpolar side chains. Along with pairing and conformational preferences, the ILV side chains are 3 of the 4 side chains (cysteine is the fourth) that are most effective at screening the underlying peptide hydrogen-bond from solvent.54 As a result, the hydrogen-bond is increased in strength and the peptide linkage is decreased in volume. If the increase in packing density is propagated to the attached side chains, the ILV side chains might pack more tightly than other nonpolar side chains. The increase in hydrogen bond strength and decreased accessibility to solvent would both be expected to contribute to enhanced protection against exchange of amide hydrogens with solvent.55 This conjecture is consistent with HX-MS results that show that the strongest protection against exchange was observed for the (β/α)1–3β4 segment under conditions favoring the I1 species in αTS.56
Given the unique roles of the (β/α)1–2β3 segment in the misfolded IBP intermediate and the (β/α1–3β4 and α8 segments in the on-pathway I1 intermediate, it appears that a critical number of ILV residues with a significant fraction of self-contacts is required for the formation of a tightly- packed hydrophobic cluster in these intermediates. Smaller ILV clusters or clusters with a reduced fraction of self-contacts among the ILV subset form molten globule-like structures that play a lesser role in defining the free energy surface on which the αTS folds.
Branched Aliphatic Side Chain Hypothesis and partially-folded states in TIM barrel and (β/α)n proteins
Taken together, these observations led to the proposal of the Branched Aliphatic Side Chain (BASiC) hypothesis. This hypothesis posits a unique role for isoleucines, leucines and valines in forming tightly-packed hydrophobic clusters that guide the formation of structure in TIM barrel and (β/α)n parallel sheet proteins. Based on the results for αTS, these clusters contain >12 ILV residues and correspond to segments of >80 residues. From the perspective of secondary structure, this requirement translates to a minimum βαβαβ module whose hydrogen-bonding network would enable, and be strengthened by, the tight packing of the branched aliphatic side chains between the strands and the helices. Less than a critical mass of ILV residues or dilution with other residues, so as to diminish the ILV self interactions below a critical level, can result in loosely-packed molten globule-like structures (Table 2). Although the molten globule-like clusters may not guide the folding reaction beyond reducing conformational space, they could serve to decrease the propensity for self-association and aggregation during folding.
Preliminary application of this hypothesis to two other TIM barrel proteins, phosphoribosyl anthranilate isomerase,57 and yeast triose phosphate isomerase,58 revealed good agreement between the location of tightly-packed ILV-dominated hydrophobic clusters and stable folding cores. Similar to αTS, the minimal core for both proteins encompasses a βαβαβ module. Regarding the off-pathway misfolded intermediate in CheY, examination of the structure shows two tightly-packed ILV clusters, one on either side of the parallel β-sheet, which span the N- and C-termini. If one or both of the clusters stabilizes the misfolded species, the C- terminus would have to at least partially unfold to access a transition state known to be defined by the N-terminal β strands and α helices.36
The relatively high content of isoleucine, leucine and valine residues in all proteins, ∼20%,46,59 suggests that hydrophobic clusters formed by mutual interactions between these residues might play important roles in guiding the early folding events of other motifs. These same branched aliphatic side chains might also be crucial to the development of extremely potent intermolecular interactions that drive the formation of a subset of the aggregates and amyloids thought to be involved in a host of human pathologies.60
Materials and Methods
Chemicals and reagents
Ultra-pure urea was purchased from ICN Biomedicals (Costa Mesa, California). For all experiments, urea was freshly purified from ionic degradation products using a column containing AG® 501-XB (D) resin purchased from Bio-Rad laboratories (Hercules, California). All other chemicals were reagent grade.
Oligonucleotide-directed mutagenesis, protein expression and purification
Oligonucleotides used for mutagenesis were obtained from Operon (Alameda, CA). E. coli strain CB149 served as the host for expressing wild-type and variant αTS proteins. Mutations were introduced by the Stratagene (La Jolla, CA) Quikchange™ site-directed mutagenesis method. Mutations were verified by DNA sequence analysis. Over-expression and purification of the wild- type and the variant αTS proteins are described elsewhere.32,48 Purified protein, which was confirmed by the presence of a single band on a 15% sodium dodecyl sulfate polyacrylamide gel, was stored in 80% ammonium sulfate solutions at 4 °C and dialyzed just before use against the standard buffer used for all folding experiments (10 mM potassium phosphate, pH 7.8, 0.2 mM K2EDTA and 1 mM β-mercaptoethanol). Protein concentration was determined from the UV absorbance at 278 nm, using an extinction coefficient of 12,600 M−1 cm−1 61 for both wild-type and variant αTS proteins. The possible effects of amino acid replacements near tyrosine residues on the extinction coefficients were accounted for by direct measurements of the extinction coefficients by the method of Gill et al.62
Equilibrum and kinetic experiments
CD measurements for equilibrium and manual-mixing kinetic experiments were carried out on a Jasco Model J-810 spectropolarimeter equipped with a thermoelectric cell holder as described previously.13 The dead-time for manual mixing was 3 s. Unfolding jumps were from 0 M urea to various final concentrations of urea (2.0 M – 7.0 M, and refolding jumps were performed from 8 M or 6 M to various final urea concentrations (0.3 M – 3.4 M). The final protein concentration was 3 to 5 μM. The stopped-flow CD refolding experiments were performed with an Aviv model 202SF stopped-flow CD spectrometer, with a 5 ms dead-time for mixing. All of the tyrosine fluorescence experiments were performed with an Applied Photophysics SX-18MV stopped-flow fluorometer. The excitation pathlength was 10 mm, and the emission pathlength was 2 mm. The excitation wavelength was set at 280 nm with a 2.32 nm bandwidth. The emission above 320 nm was detected with an appropriate cut-off filter.
Data analysis
The refolding burst phase amplitudes as a function of urea and the equilibrium data were fit to a two-state and a three-state model, respectively, as described previously.32 The free energy changes were assumed to depend linearly on the denaturant concentration.41 The kinetic data obtained from unfolding or refolding experiments were fit to the sum of a minimum number of statistically significant exponentials and a constant.
Cluster assignment
The distance between side chain carbon atoms of the selected residue types was computed with an in-house program using a cutoff distance of 4.2 Å. The number of contacts between any two residues were counted and exported to Pymol for visualizing contacts in three dimensional format. Crystal structure of the α subunit of tryptophan synthase from S. typhimurium (PDB code 1BKS, a refined version of 1WSY63), which shares 85% sequence identity with E. coli αTS, was chosen for the cluster calculation, instead of the E. coli structure (PDB code 1V7Y and 1WQ564), because the crystal contacts in the E. coli αTS structure result in ambiguous results for the structure of the 52–77 and 178–192 regions in solution.
Supplementary Material
Acknowledgments
The authors wish to thank Drs. Jill Ann Zitzewitz and Osman Bilsel for critical reviews of the manuscript and many helpful comments. This work was supported by the National Institutes of Health through grant GM23303 to CRM.
Abbreviations
- ACFILMPVY
alanine, cysteine, phenylalanine, isoleucine, leucine, methionine, proline, valine and tyrosine residues
- αTS
α subunit of tryptophan synthase from E. coli
- HX
hydrogen exchange
- I1
equilibrium intermediate of αTS populated at ∼3 M urea
- I2
equilibrium intermediate of αTS populated at ∼5 M urea
- IBP
kinetic intermediate of αTS populated within the stopped-flow burst-phase (< 5 ms)
- ILV
isoleucine, leucine and valine residues
- NMR
Nuclear Magnetic Resonance
- N
native state
- U
unfolded state
- WT
wild type
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bilsel O, Matthews CR. Barriers in protein folding reactions. Adv Protein Chem. 2000;53:153–207. doi: 10.1016/s0065-3233(00)53004-6. [DOI] [PubMed] [Google Scholar]
- 2.Jackson SE. How do small single-domain proteins fold? Fold Des. 1998;3:R81–R91. doi: 10.1016/S1359-0278(98)00033-9. [DOI] [PubMed] [Google Scholar]
- 3.Alm E, Baker D. Matching theory and experiment in protein folding. Curr Opin Struct Biol. 1999;9:189–196. doi: 10.1016/S0959-440X(99)80027-X. [DOI] [PubMed] [Google Scholar]
- 4.Ferguson N, Capaldi AP, James R, Kleanthous C, Radford SE. Rapid folding with and without populated intermediates in the homologous four-helix proteins Im7 and Im9. J Mol Biol. 1999;286:1597–1608. doi: 10.1006/jmbi.1998.2548. [DOI] [PubMed] [Google Scholar]
- 5.Friel CT, Beddard GS, Radford SE. Switching two-state to three-state kinetics in the helical protein Im9 via the optimisation of stabilising non-native interactions by design. J Mol Biol. 2004;342:261–273. doi: 10.1016/j.jmb.2004.06.076. [DOI] [PubMed] [Google Scholar]
- 6.Matthews CR. Pathways of protein folding. Ann Rev Biochem. 1993;62:653–683. doi: 10.1146/annurev.bi.62.070193.003253. [DOI] [PubMed] [Google Scholar]
- 7.Baldwin RL. On-pathway versus off-pathway folding intermediates. Fold Des. 1996;1:R1–8. doi: 10.1016/S1359-0278(96)00003-X. [DOI] [PubMed] [Google Scholar]
- 8.Kamagata K, Arai M, Kuwajima K. Unification of the folding mechanisms of non-two-state and two-state proteins. J Mol Biol. 2004;339:951–965. doi: 10.1016/j.jmb.2004.04.015. [DOI] [PubMed] [Google Scholar]
- 9.Cecconi C, Shank EA, Bustamante C, Marqusee S. Direct observation of the three-state folding of a single protein molecule. Science. 2005;309:2057–2060. doi: 10.1126/science.1116702. [DOI] [PubMed] [Google Scholar]
- 10.Wolynes PG, Onuchic JN, Thirumalai D. Navigating the folding routes. Science. 1995;267:1619–1620. doi: 10.1126/science.7886447. [DOI] [PubMed] [Google Scholar]
- 11.Brandts JF, Halvorson HR, Brennan M. Consideration of the Possibility that the slow step in protein denaturation reactions is due to cis-trans isomerism of proline residues. Biochemistry. 1975;14:4953–4963. doi: 10.1021/bi00693a026. [DOI] [PubMed] [Google Scholar]
- 12.Maki K, Ikura T, Hayano T, Takahashi N, Kuwajima K. Effects of proline mutations on the folding of staphylococcal nuclease. Biochemistry. 1999;38:2213–2223. doi: 10.1021/bi981962+. [DOI] [PubMed] [Google Scholar]
- 13.Wu Y, Matthews CR. Parallel channels and rate-limiting steps in complex protein folding reactions: prolyl isomerization and the alpha subunit of Trp synthase, a TIM barrel protein. J Mol Biol. 2002;323:309–325. doi: 10.1016/s0022-2836(02)00922-1. [DOI] [PubMed] [Google Scholar]
- 14.Mello CC, Bradley CM, Tripp KW, Barrick D. Experimental characterization of the folding kinetics of the notch ankyrin domain. J Mol Biol. 2005;352:266–281. doi: 10.1016/j.jmb.2005.07.026. [DOI] [PubMed] [Google Scholar]
- 15.Sosnick TR, Mayne L, Hiller R, Englander SW. The barriers in protein folding. Nat Struct Biol. 1994;1:149–156. doi: 10.1038/nsb0394-149. [DOI] [PubMed] [Google Scholar]
- 16.Roder H, Elove GA, Englander SW. Structural characterization of folding intermediates in cytochrome c by H-exchange labelling and proton NMR. Nature. 1988;335:700–704. doi: 10.1038/335700a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Raschke TM, Kho J, Marqusee S. Confirmation of the hierarchical folding of RNase H: a protein engineering study. Nat Struct Biol. 1999;6:825–831. doi: 10.1038/12277. [DOI] [PubMed] [Google Scholar]
- 18.Bilsel O, Zitzewitz JA, Bowers KE, Matthews CR. Folding mechanism of the alpha-subunit of tryptophan synthase, an alpha/beta barrel protein: global analysis highlights the interconversion of multiple native, intermediate, and unfolded forms through parallel channels. Biochemistry. 1999;38:1018–1029. doi: 10.1021/bi982365q. [DOI] [PubMed] [Google Scholar]
- 19.Wallace LA, Matthews CR. Sequential vs. parallel protein-folding mechanisms: experimental tests for complex folding reactions. Biophys Chem. 2002;101–102:113–131. doi: 10.1016/s0301-4622(02)00155-2. [DOI] [PubMed] [Google Scholar]
- 20.Gianni S, Travaglini-Allocatelli C, Cutruzzola F, Brunori M, Shastry MC, Roder H. Parallel pathways in cytochrome c(551) folding. J Mol Biol. 2003;330:1145–1152. doi: 10.1016/s0022-2836(03)00689-2. [DOI] [PubMed] [Google Scholar]
- 21.Gloss LM, Matthews CR. Mechanism of folding of the dimeric core domain of Escherichia coli trp repressor: a nearly diffusion-limited reaction leads to the formation of an on-pathway dimeric intermediate. Biochemistry. 1998;37:15990–15999. doi: 10.1021/bi981511p. [DOI] [PubMed] [Google Scholar]
- 22.Topping TB, Hoch DA, Gloss LM. Folding mechanism of FIS, the intertwined, dimeric factor for inversion stimulation. J Mol Biol. 2004;335:1065–1081. doi: 10.1016/j.jmb.2003.11.013. [DOI] [PubMed] [Google Scholar]
- 23.de Prat-Gay G, Nadra AD, Corrales-Izquierdo FJ, Alonso LG, Ferreiro DU, Mok YK. The folding mechanism of a dimeric beta-barrel domain. J Mol Biol. 2005;351:672–682. doi: 10.1016/j.jmb.2005.05.070. [DOI] [PubMed] [Google Scholar]
- 24.Rothwarf DM, Scheraga HA. Role of non-native aromatic and hydrophobic interactions in the folding of hen egg white lysozyme. Biochemistry. 1996;35:13797–13807. doi: 10.1021/bi9608119. [DOI] [PubMed] [Google Scholar]
- 25.Colon W, Wakem LP, Sherman F, Roder H. Identification of the predominant non-native histidine ligand in unfolded cytochrome c. Biochemistry. 1997;36:12535–12541. doi: 10.1021/bi971697c. [DOI] [PubMed] [Google Scholar]
- 26.Arai M, Ikura T, Semisotnov GV, Kihara H, Amemiya Y, Kuwajima K. Kinetic refolding of beta-lactoglobulin. Studies by synchrotron X-ray scattering, and circular dichroism, absorption and fluorescence spectroscopy. J Mol Biol. 1998;275:149–162. doi: 10.1006/jmbi.1997.1456. [DOI] [PubMed] [Google Scholar]
- 27.Capaldi AP, Shastry MC, Kleanthous C, Roder H, Radford SE. Ultrarapid mixing experiments reveal that Im7 folds via an on-pathway intermediate. Nat Struct Biol. 2001;8:68–72. doi: 10.1038/83074. [DOI] [PubMed] [Google Scholar]
- 28.Cho JH, Sato S, Raleigh DP. Thermodynamics and kinetics of non-native interactions in protein folding: a single point mutant significantly stabilizes the N-terminal domain of L9 by modulating non-native interactions in the denatured state. J Mol Biol. 2004;338:827–837. doi: 10.1016/j.jmb.2004.02.073. [DOI] [PubMed] [Google Scholar]
- 29.Munoz V, Lopez EM, Jager M, Serrano L. Kinetic characterization of the chemotactic protein from Escherichia coli, CheY. Kinetic analysis of the inverse hydrophobic effect. Biochemistry. 1994;33:5858–5866. doi: 10.1021/bi00185a025. [DOI] [PubMed] [Google Scholar]
- 30.Fernandez-Recio J, Genzor CG, Sancho J. Apoflavodoxin folding mechanism: an alpha/beta protein with an essentially off-pathway intermediate. Biochemistry. 2001;40:15234–15245. doi: 10.1021/bi010216t. [DOI] [PubMed] [Google Scholar]
- 31.Bollen YJ, Sanchez IE, Van Mierlo CP. Formation of On- and Off-Pathway Intermediates in the Folding Kinetics of Azotobacter vinelandii Apoflavodoxin. Biochemistry. 2004;43:10475–10489. doi: 10.1021/bi049545m. [DOI] [PubMed] [Google Scholar]
- 32.Wu Y, Vadrevu R, Yang X, Matthews CR. Specific structure appears at the N terminus in the sub-millisecond folding intermediate of the alpha subunit of tryptophan synthase, a TIM barrel protein. J Mol Biol. 2005;351:445–452. doi: 10.1016/j.jmb.2005.06.006. [DOI] [PubMed] [Google Scholar]
- 33.Butler JS, Loh SN. Kinetic partitioning during folding of the p53 DNA binding domain. J Mol Biol. 2005;350:906–918. doi: 10.1016/j.jmb.2005.05.060. [DOI] [PubMed] [Google Scholar]
- 34.Roder H, Maki K, Cheng H. Early events in protein folding explored by rapid mixing methods. Chem Rev. 2006;106:1836–1861. doi: 10.1021/cr040430y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Clementi C, Nymeyer H, Onuchic JN. Topological and energetic factors: what determines the structural details of the transition state ensemble and "en-route" intermediates for protein folding? An investigation for small globular proteins. J Mol Biol. 2000;298:937–953. doi: 10.1006/jmbi.2000.3693. [DOI] [PubMed] [Google Scholar]
- 36.Lopez-Hernandez E, Serrano L. Structure of the transition state for folding of the 129 aa protein CheY resembles that of a smaller protein, CI-2. Fold Des. 1996;1:43–55. [PubMed] [Google Scholar]
- 37.DeLano WL. The PyMOL Molecular Graphics System. DeLano Scientific; 2002. [Google Scholar]
- 38.Wu Y, Matthews CR. A cis-prolyl peptide bond isomerization dominates the folding of the alpha subunit of Trp synthase, a TIM barrel protein. J Mol Biol. 2002;322:7–13. doi: 10.1016/s0022-2836(02)00737-4. [DOI] [PubMed] [Google Scholar]
- 39.Wu Y, Matthews CR. Proline replacements and the simplification of the complex, parallel channel folding mechanism for the alpha subunit of Trp synthase, a TIM barrel protein. J Mol Biol. 2003;330:1131–1144. doi: 10.1016/s0022-2836(03)00723-x. [DOI] [PubMed] [Google Scholar]
- 40.Tsai J, Gerstein M, Levitt M. Simulating the minimum core for hydrophobic collapse in globular proteins. Protein Sci. 1997;6:2606–2616. doi: 10.1002/pro.5560061212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schellman JA. Solvent denaturation. Biopolymers. 1978;17:1305–1322. doi: 10.1002/bip.360260408. [DOI] [PubMed] [Google Scholar]
- 42.Pace CN, Scholtz JM. A helix propensity scale based on experimental studies of peptides and proteins. Biophys J. 1998;75:422–427. doi: 10.1016/s0006-3495(98)77529-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shortle D, Stites WE, Meeker AK. Contributions of the large hydrophobic amino acids to the stability of staphylococcal nuclease. Biochemistry. 1990;29:8033–8041. doi: 10.1021/bi00487a007. [DOI] [PubMed] [Google Scholar]
- 44.Kellis JT, Jr, Nyberg K, Fersht AR. Energetics of complementary side-chain packing in a protein hydrophobic core. Biochemistry. 1989;28:4914–4922. doi: 10.1021/bi00437a058. [DOI] [PubMed] [Google Scholar]
- 45.Shortle D. Mutational studies of protein structures and their stabilities. Q sRev Biophys. 1992;25:205–250. doi: 10.1017/s0033583500004674. [DOI] [PubMed] [Google Scholar]
- 46.Creighton TE. Proteins: Structures and Molecular Properties. W. H. Freeman and Company; New York: 1993. [Google Scholar]
- 47.Arai M, Kuwajima K. Role of the molten globule state in protein folding. Adv Protein Chem. 2000;53:209–282. doi: 10.1016/s0065-3233(00)53005-8. [DOI] [PubMed] [Google Scholar]
- 48.Beasty AM, Hurle MR, Manz JT, Stackhouse T, Onuffer JJ, Matthews CR. Effects of the phenylalanine-22-leucine, glutamic acid-49-methionine, glycine-234-aspartic acid, and glycine-234-lysine mutations on the folding and stability of the alpha subunit of tryptophan synthase from Escherichia coli. Biochemistry. 1986;25:2965–2974. doi: 10.1021/bi00358a035. [DOI] [PubMed] [Google Scholar]
- 49.Matthews CR. Effect of point mutations on the folding of globular proteins. Methods Enzymol. 1987;154:498–511. doi: 10.1016/0076-6879(87)54092-7. [DOI] [PubMed] [Google Scholar]
- 50.Saab-Rincon G, Gualfetti PJ, Matthews CR. Mutagenic and thermodynamic analyses of residual structure in the alpha subunit of tryptophan synthase. Biochemistry. 1996;35:1988–1994. doi: 10.1021/bi951726o. [DOI] [PubMed] [Google Scholar]
- 51.Branden C, Tooze J. Introduction to Protein Structure. 2. Garland Science Publishing; 1999. [Google Scholar]
- 52.Fooks HM, Martin AC, Woolfson DN, Sessions RB, Hutchinson EG. Amino acid pairing preferences in parallel beta-sheets in proteins. J Mol Biol. 2006;356:32–44. doi: 10.1016/j.jmb.2005.11.008. [DOI] [PubMed] [Google Scholar]
- 53.Misura KM, Morozov AV, Baker D. Analysis of anisotropic side-chain packing in proteins and application to high-resolution structure prediction. J Mol Biol. 2004;342:651–664. doi: 10.1016/j.jmb.2004.07.038. [DOI] [PubMed] [Google Scholar]
- 54.Schell D, Tsai J, Scholtz JM, Pace CN. Hydrogen bonding increases packing density in the protein interior. Proteins. 2006;63:278–282. doi: 10.1002/prot.20826. [DOI] [PubMed] [Google Scholar]
- 55.Englander SW, Kallenbach NR. Hydrogen exchange and structural dynamics of proteins and nucleic acids. Q Rev Biophys. 1983;16:521–655. doi: 10.1017/s0033583500005217. [DOI] [PubMed] [Google Scholar]
- 56.Rojsajjakul T, Wintrode P, Vadrevu R, Robert Matthews C, Smith DL. Multi-state unfolding of the alpha subunit of tryptophan synthase, a TIM barrel protein: insights into the secondary structure of the stable equilibrium intermediates by hydrogen exchange mass spectrometry. J Mol Biol. 2004;341:241–253. doi: 10.1016/j.jmb.2004.05.062. [DOI] [PubMed] [Google Scholar]
- 57.Akanuma S, Yamagishi A. Identification and characterization of key substructures involved in the early folding events of a (beta/alpha)8-barrel protein as studied by experimental and computational methods. J Mol Biol. 2005;353:1161–1170. doi: 10.1016/j.jmb.2005.08.070. [DOI] [PubMed] [Google Scholar]
- 58.Silverman JA, Harbury PB. The equilibrium unfolding pathway of a (beta/alpha)8 barrel. J Mol Biol. 2002;324:1031–1040. doi: 10.1016/s0022-2836(02)01100-2. [DOI] [PubMed] [Google Scholar]
- 59.McCaldon P, Argos P. Oligopeptide biases in protein sequences and their use in predicting protein coding regions in nucleotide sequences. Proteins. 1988;4:99–122. doi: 10.1002/prot.340040204. [DOI] [PubMed] [Google Scholar]
- 60.Chiti F, Dobson CM. Protein misfolding, functional amyloid, and human disease. Annu Rev Biochem. 2006;75:333–366. doi: 10.1146/annurev.biochem.75.101304.123901. [DOI] [PubMed] [Google Scholar]
- 61.Matthews CR, Crisanti MM, Manz JT, Gepner GL. Effect of a single amino acid substitution on the folding of the alpha subunit of tryptophan synthase. Biochemistry. 1983;22:1445–1452. doi: 10.1021/bi00275a019. [DOI] [PubMed] [Google Scholar]
- 62.Gill SC, von Hippel PH. Calculation of protein extinction coefficients from amino acid sequence data. Anal Biochem. 1989;182:319–326. doi: 10.1016/0003-2697(89)90602-7. [DOI] [PubMed] [Google Scholar]
- 63.Hyde CC, Ahmed SA, Padlan EA, Miles EW, Davies DR. Three-dimensional structure of the tryptophan synthase alpha 2 beta 2 multienzyme complex from Salmonella typhimurium. J Biol Chem. 1988;263:17857–17871. [PubMed] [Google Scholar]
- 64.Nishio K, Morimoto Y, Ishizuka M, Ogasahara K, Tsukihara T, Yutani K. Conformational changes in the alpha-subunit coupled to binding of the beta 2-subunit of tryptophan synthase from Escherichia coli: crystal structure of the tryptophan synthase alpha-subunit alone. Biochemistry. 2005;44:1184–1192. doi: 10.1021/bi047927m. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.