

Abstract
The parathyroid hormone 2 (PTH2) receptor is a Family B G-protein coupled receptor most highly expressed within the brain. Current evidence suggests that tuberoinfundibular peptide of 39 residues (TIP39) is the PTH2 receptor’s endogenous ligand. To facilitate investigation of the physiological function of the PTH2 receptor/TIP39 system we have developed a novel PTH2 receptor antagonist, by changing several residues within the amino terminal domain of TIP39. Histidine4, tyrosine5, tryptophan6, histidine7-TIP39 binds the PTH2 receptor with high affinity, has over 30-fold selectivity for the rat PTH2 receptor over the rat PTH1 receptor and displays no detectable agonist activity. This ligand should be useful for in vivo investigation of PTH2 receptor function.
Keywords: Peptide antagonist, site-directed mutagenesis, tuberoinfundibular peptide of 39 residues, parathyroid hormone 2 receptor
1. Introduction
The parathyroid hormone 2 (PTH2) receptor is a member of the Family B group of G-protein coupled receptors (GPCRs) [6]. Its distribution has been mapped in detail, both in the central nervous system and in a number of peripheral organs [16, 18]. Based on its distribution in the brain it may play a role in regulation of neuroendocrine, emotion, auditory, and pain related processes. Its peripheral distribution suggests involvement in endocrine, cardiovascular and reproductive function [20]. However there is relatively little experimental data relevant to its physiological role. The few published studies have used administration of pharmacological doses of its putative agonist, tuberoinfundibular peptide of 39 residues (TIP39) [1, 9, 10, 13, 14, 21], or sequestration of TIP39 with an antibody [1]. A potent and specific antagonist would be a particularly useful reagent for investigation of PTH2 receptor function.
The PTH2 receptor has about 50% sequence identity with the PTH1 receptor [17]. The PTH1 receptor is also referred to as the PTH/PTHrP receptor. It is the receptor responsible for parathyroid hormone (PTH) regulation of calcium homeostasis, and for many effects of parathyroid hormone-related protein (PTHrP) on skeletal development and tissue remodeling [11]. PTH1 receptors are potently activated by both PTH and PTHrP. The human PTH2 receptor is potently activated by both PTH and TIP39 [19], whereas PTH is a weak partial agonist at the rat PTH2 receptor and TIP39 a potent agonist [15]. PTHrP does not cause detectable activation of PTH2 receptors. Peptides containing the first 34 or 36 residues of PTH or PTHrP have full potency and affinity at the PTH1 receptor, and have been used in most studies of receptor function. The carboxyl terminal half of these peptides is largely responsible for high affinity binding to the receptor and the amino terminal half for activation of the receptor [2]. Ligand residues that provide PTH2 receptor discrimination between PTH and PTHrP have been identified. Either a hybrid peptide composed of the first 21 residues of PTHrP followed by 13 residues of PTH or an analog of PTHrP in which the phenylalanine normally present at residue 23 of PTHrP is replaced by tryptophan, which is present at that position in PTH, bind to but do not activate the human PTH2 receptor [4]. These human PTH2 receptor antagonists are full and potent agonists at the human PTH1 receptor. Their properties at the rat PTH2 receptor have not been described. They may not be ideal reagents for investigating PTH2 receptor function in rodents because of the relatively low potency of PTH for the rat PTH2 receptor, and because of the confound created by their activation of the PTH1 receptor.
Deletion of residues from the amino terminus of PTH or PTHrP reduces their ability to activate the PTH1 receptor while having relatively small effects on their binding affinity. PTH(7–34), PTHrP(7–36), and several analogs of these peptides are PTH1 receptor antagonists ([3]and references therein). We reasoned that similar modifications of TIP39 might generate a PTH2 receptor antagonist. Removing amino terminal residues from TIP39 did reduce its ability to activate the PTH2 receptor, but it also reduced the peptide’s affinity for the PTH2 receptor such that a peptide with no detectable agonist activity, TIP(7–39), had too low an affinity to be useful as a PTH2 receptor antagonist [7]. Surprisingly, removing amino terminus residues from TIP39 increased its affinity for the PTH1 receptor, and TIP(7–39) is a potent PTH1 receptor antagonist [8].
We have now taken a third approach to development of a PTH2 receptor antagonist. Instead of removing residues from the amino terminus, receptor activating, domain of TIP39 we examined the effect of changing some of the residues to ones with different charge or bulk. Using this approach we have developed a potent and selective PTH2 receptor antagonist.
2. Materials and Methods
2.1 Preparation of TIP39 mutations
A bovine TIP39 cDNA was cloned into the vector pet32 (Novagen, EMD Biosciences, San Diego CA) to produce a hexahistidine containing thioredoxin fusion protein. Site-directed mutagenesis was performed using the Stratagene Quick-Change mutagenesis strategy. A 50 μl PCR reaction mixture contained 50 ng wild type pet-TIP39 plasmid DNA, 125 ng of designed mutagenic upper and lower primers, 10 mM dNTP’s, 5 units of PfuTurbo DNA Polymerase (Stratagene, LaJolla CA), and reaction buffer supplied with enzyme. Following incubation at 95°C for 1 minute, PCR was performed for 18 cycles at 95°C for 50 seconds, 56.7°C for 50 seconds, and 68°C for 12 minutes, followed by 68°C for 7 minutes. PCR products were digested with DpnI restriction enzyme at 37°C for 2 hours and then BL21-gold (DE3) competent cells (Stratagene) were transformed. Individual clones from the transformations were grown in 3 ml of LB-Ampicillin at 37°C overnight, and plasmid DNA was prepared using a QIAprep Spin Miniprep Kit (Qiagen, Valencia CA). Mutations were identified by sequencing with either pet-TIP39 vector sense primer (GCAGCCGGATCTCAGTGC), or pet-TIP39 vector anti-sense primer (GATGCGTCCGGCGTAGAG). Histidine4, tyrosine5, tryptophan6, histidine7-TIP39 was custom synthesized by Biomolecules Midwest (Waterloo, Il).
2.2 Purification of mutant proteins
A single colony containing a selected TIP39 mutation was inoculated into 10 ml of LB-Ampicillin, and incubated with shaking at 37°C until the density reached an OD600 of 0.5 ~ 0.6. The bacteria were collected by centrifugation and resuspended into 500 ml of LB-Ampicillin. The bacteria were then grown until the OD600 reached 0.4 ~ 0.5. Protein expression was then induced with 1 mM IPTG for 3 hours at 37°C. The bacteria were harvested by centrifugation at 5,000 × g for 15 minutes at 4°C and stored at −80°C until use. Induced bacteria were lysed in 25 ml BugBuster™ reagent (Novagen) containing 25 μl of Benzonase (Novagen) by sonication. Following centrifugation, the histidine-tagged proteins were isolated by applying the supernatant to HisTrap FF columns (Amersham Biosciences, Piscataway NJ). The columns were initially equilibrated in 5 mM imidazol, 0.5 M NaCl, 20 mM Tris-Cl pH 7.9, and 10% DMSO. After loading the samples the colums were washed with five column volumes of equilibration buffer, and the histidine-tagged proteins were then eluted from column with 100 mM EDTA, 0.5 M NaCl, 20 mM Tris-Cl pH 7.9, 10% DMSO. Histidine-tagged proteins were digested with Factor-Xa enzyme (Novagen; 0.2 μg enzyme per 100 μg of histidine-tagged protein, overnight at room temperature), and TIP39 mutant proteins were purifed by reverse-phase HPLC on a Vydac C4 column (Grace Vydac, Columbia MD) with an acetonitrile gradient in 0.1% trifluroacetic acid. The molecular weights of TIP39 mutation proteins were then determined by MALDI-TOF mass spectroscopy on a Voyager-DE™ STR BioSpectrometry™ Workstation (PerSeptive Biosystems, Framingham MA). Protein concentrations were determined by the BCA method (Pierce, Rockford IL) using bovine serum albumin as the standard.
2.3 Assay of receptor blockade
Cloned rat PTH2 and cloned human PTH1 receptors were expressed from CMV promoter based vectors in HEK293 cells, as previously described [5]. The ROS 17/2.8 cell line (Gift of G. Rodan) [12] was used as a rat PTH1 receptor source. The day prior to assay cells were plated in 96-well tissue culture plates. For the assay, the cells were rinsed with PBS and then incubated in stimulation buffer (0.03 mM RO-1724, 0.2 mM AEBSF, 2 μg/ml Bacitracin, and 0.2% BSA in DMEM), containing varying concentrations of TIP39 or PTH plus a mutant protein, for 30 minutes at room temperature. The reactions were terminated by addition of HCl to 0.2N and the amount of cAMP in the cell lysate determined by ELISA. Dose-response curves were fitted to a four parameter logistic equation (Y=min + (max-min)/(1+10^((LogEC50-X)*n)) where X is the logarithm of the ligand concentration, y is the amount of cAMP produced at a given peptide concentration, min is the cAMP level in the absence of ligand and max is the maximum level produced) using GraphPad Prism (GraphPad Software, San Diego CA) as previously described [8]. pA2 values (negative logarithm of the concentration of antagonist required to produce a 2-fold shift in the agonist concentration response curve) were determined in Prism using the equation (log(DR - 1) = n * log[antagonist] + pA2 where DR is the dose ratio (EC50 in the presence of antagonist divided by EC50 in the absence of antagonist), and n is the gradient.
3. RESULTS
We performed site directed mutagenesis on a bovine TIP39 cDNA cloned as a histidine tagged fusion protein in a bacterial expression vector. After identification of mutants by sequencing the inserts in selected clones, protein expression was induced in bacterial cultures, the expressed proteins were purified on a nickel column, and then digested with Factor Xa. The desired peptides were then purified by reverse-phase HPLC. Their purity was assessed and sequence confirmed by high resolution MALDI-TOF mass spectrometry. Peptide analogs were initially screened by examining their stimulation of cAMP accumulation in tissue culture cells expressing the rat PTH2 receptor. Candidates of interest were tested for inhibition of cAMP accumulation stimulated by TIP39 and for their effect at the PTH1 receptor.
Previous work has shown that the first two residues of TIP39 can be removed with little effect on PTH2 receptor activation or affinity [7]. We initially replaced each of the non-alanine residues in positions 3 through 11 with alanine (Table 1). All of these peptides were full agonists, suggesting that the side chain of no individual amino acid in TIP39’s amino terminal region is absolutely essential for receptor activation. Because of the potential importance of the amino terminal structure we next prepared several analogs in which an additional residue was added before the normal amino terminal serine residue. All of these analogs were also full agonists.
Table 1.
TIP39 analogs screeneda
| −1 | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | % Maximal PTH2-R activationb | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| S | L | A | L | A | D | D | A | A | F | R | E | 1E-06 | 1E-07 | 1E-08 | |
| A | 100 | 100 | 79 | ||||||||||||
| A | 100 | 100 | 100 | ||||||||||||
| A | 100 | 85 | 0 | ||||||||||||
| A | 100 | 100 | 100 | ||||||||||||
| A | 100 | 100 | 100 | ||||||||||||
| E | 100 | 100 | 100 | ||||||||||||
| F | 100 | 100 | 100 | ||||||||||||
| H | 100 | 100 | 100 | ||||||||||||
| I | 100 | 100 | 100 | ||||||||||||
| V | 100 | 100 | 100 | ||||||||||||
| W | 100 | 100 | 100 | ||||||||||||
| Y | 100 | 100 | 100 | ||||||||||||
| V | 87 | 77 | 62 | ||||||||||||
| W | 69 | 63 | 45 | ||||||||||||
| P | 72 | 67 | 18 | ||||||||||||
| L | 41 | 52 | 51 | ||||||||||||
| H | 56 | 59 | 52 | ||||||||||||
| F | 60 | 46 | 41 | ||||||||||||
| S | 50 | 61 | 55 | ||||||||||||
| L | 3 | 0 | 0 | ||||||||||||
| K | 50 | 0 | 0 | ||||||||||||
| S | 54 | 1 | 0 | ||||||||||||
| V | 5 | 0 | 0 | ||||||||||||
| E | 82 | 65 | 71 | ||||||||||||
| Q | 65 | 26 | 0 | ||||||||||||
| R | 0 | 0 | 0 | ||||||||||||
| A | 100 | 0 | 0 | ||||||||||||
| W | 0 | 0 | 0 | ||||||||||||
| M | 73 | 0 | 0 | ||||||||||||
| T | 23 | 0 | 0 | ||||||||||||
| H | 23 | 6 | 1 | ||||||||||||
| W | H | 0 | 1 | 0 | |||||||||||
| Y | W | H | 0 | 1 | 0 | ||||||||||
| P | Y | W | H | 1 | 1 | 1 | |||||||||
| H | Y | W | H | 0 | 0 | 0 | |||||||||
| H | Y | W | G | 5 | 4 | 3 | |||||||||
| H | Y | W | D | 10 | 6 | 9 | |||||||||
| H | Y | W | A | 0 | 3 | 5 | |||||||||
| H | Y | W | V | 9 | 7 | 5 | |||||||||
| H | Y | W | I | 4 | 3 | 5 | |||||||||
| H | Y | W | T | 9 | 7 | 9 | |||||||||
| H | Y | W | N | 6 | 7 | 9 | |||||||||
The first 12 amino acid residues of TIP39 are indicated at the top. The analogs screened are indicated on subsequent rows. Either a single addition or substitution, or several substitutions, were incorporated as indicated. Standard single letter amino acid code.
The percentage of the amount of cAMP accumulation produced by 1nM unmodified TIP39, in the presence of 10−6, 10−7 or 10−8 mutant peptide, in single screening experiments.
Next we selected positions 3 and 6 for substitution by different residues using degenerate mutagenic primers to create a collection of modified peptides. The rationale for mutation at position 3 was based on the observations that the amino terminus is important for receptor activation and that removing the residues at positions 1 and 2 has little effect on receptor activation, but removing the residue at position 3 decreases the peptides potency [7]. Thus, introducing a residue with a side chain in this position to replace the alanine normally present might interfere with receptor activation. Position 6 was selected because alanine6-TIP39 appeared to be somewhat less potent than wild-type TIP39 and because it was the first charged residue in the peptide. The position 3 mutants that we screened appeared to have decreased potency, but all displayed significant agonist activity. Several of the position 6 mutants had very little intrinsic agonist activity. Arg6-TIP39 and Trp6-TIP39 caused no measurable stimulation of cAMP accumulation at the rat PTH2 receptor in the initial screen. When these peptides were screened on the PTH1 receptor Arg6-TIP39 was a potent agonist but Trp6-TIP39 did not measurably increase cAMP. However, Trp6-TIP39 appeared to be a relatively weak inhibitor of TIP39 activation of the PTH2 receptor. It decreased cAMP accumulation stimulated by 1nM TIP39 by about 60% at 1micromolar.
One of the most significant differences between the amino terminal halves of PTH and PTHrP is at the position corresponding to the aspartate residue at position 7 of TIP39. Previous work had shown that introducing histidine, which is normally present at this position in PTHrP, into PTH decreased its ability to activate the human PTH2 receptor [4]. We prepared His7-TIP39 and in an initial screen observed that it was a very weak agonist. We then made a cDNA encoding both the tryptophan and histidine substitutions. This peptide exhibited some weak agonism so we began preparing a series of further mutations using oligonucleotide primers that contained degenerate bases that would create substitutions at positions 4, 5, and 7 using the tryp6, his7-TIP39 cDNA as the template, based on the idea that this cluster of residues might interact with a particular receptor domain. We screened the Trp6 his7-TIP39 analogs for inhibition of TIP39 activation of the rat PTH2 receptor, and inhibition of PTH activation of the rat PTH1 receptor. One, His4, Tyr5, Trp6, His7-TIP39 (HYWH-TIP39) was particularly promising based on its potency and selectivity, and was selected for more complete characterization following its chemical synthesis.
HYWH-TIP39 was able to completely block activation of the rat PTH2 receptor by TIP39. In the presence of 3.2 nM TIP39, which causes approximately 80% maximal stimulation of cAMP under the conditions of the experiment, 64nM HYWH-TIP39 decreased cAMP accumulation by 50% (Figure 1). In contrast, approximately 1microM HYWH-TIP39 was required to decrease cAMP accumulation by 50% via the rat PTH1 receptor in the presence of 3.2nM PTH, which also cause an approximately 80% maximal increase. In the same experiments TIP(7–39) had an IC50 of approximately 40 nM at the rat PTH1 receptor and 200 nM at the rat PTH2 receptor, demonstrating the opposite selectivity, as expected from previous data [8]. HYWH-TIP39 was even less potent at the human PTH1 receptor, causing less than 50% inhibition at 1 micromolar. No increase over basal levels of cAMP were detected in cells expressing either receptor when incubated with up to 1 micromolar HYWH-TIP39, under conditions in which increases stimulated by 10 picomolar TIP39 and PTH were detected.
Figure 1.
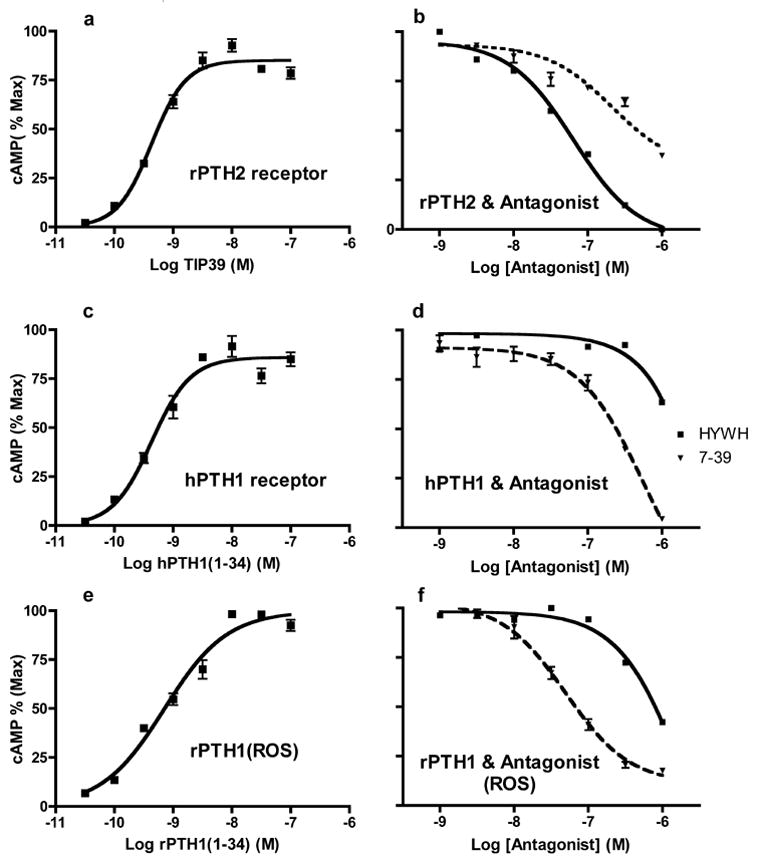
Inhibition of receptor activation by HYWH-TIP39. Cloned rat PTH2 (a, b) and human PTH1 (c, d) receptors stably expressed in HEK293 cells and the rat PTH1 (e, f) receptor natively expressed in ROS cells were used to establish agonist concentration-response relationships, and then inhibition of activation by 3.2 nM agonist by varying concentrations of HYWH-TIP39 (solid lines) and TIP(7–39) (dashed lines). cAMP accumulation was expressed as the percentage of the maximal accumulation stimulated by agonist alone in a particular experiment. The plotted data are the mean +/− standard error of individual data points from two separate experiments, each performed in triplicate.
We analyzed the shift in the TIP39 agonist dose-concentration curve at the rat PTH2 receptor produced by several concentrations of HYWH-TIP39 (Figure 2). HYWH-TIP39 produced rightward shifted agonist dose-response curves and a decrease in the maximal amount of cAMP accumulation. The calculated pA2 value was 7.61 (25 nM).
Figure 2.
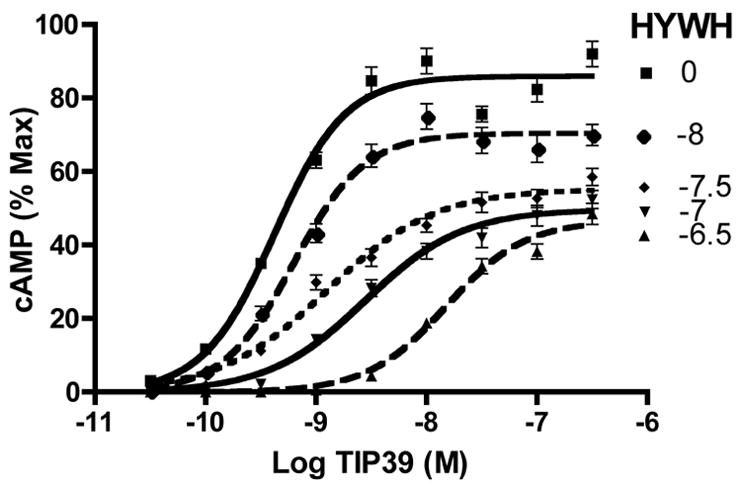
Agonist dose-response shifts produced by HYWH-TIP39. Concentration-response relationships were determined for the cloned rat PTH2 receptor using the indicated concentrations of TIP39 as the agonist. Responses were determined in absence, or presence of one of five concentrations of the TIP39 analog HYWH-TIP39 (0, none;−8, 1 × 10−8;−7.5, 3.16 × 10−8;−7, 1 × 10−7;−6.5, 3.16 × 10−7). Data are the mean +/− standard error from six separate experiments each containing three replicates.
4. Discussion
We have identified a peptide analog of TIP39 that is a potent and selective antagonist of the rat PTH2 receptor. It has over 30-fold selectivity for the rat PTH2 receptor over the rat PTH1 receptor and causes no detectable activation of either receptor in our very sensitive assays. The antagonist has significantly greater affinity for the PTH2 receptor than previously described antagonists. Previously described PTH analogs are also either not selective for the PTH2 receptor or act as agonists at the PTH1 receptor.
The antagonist derived from a “semi-rational” approach. Previous work established that the amino terminal domains of PTH and PTHrP are critical for receptor activation but make relatively small contributions to their receptor affinity [2]. The amino terminal domain of TIP39 makes a relatively greater contribution to its affinity for the PTH2 receptor but is also critical for receptor activation [7]. Our initial alanine scan of TIP39’s amino terminal end showed that no individual amino acid side chain residue was essential for receptor activation, and that extending TIP39 with an additional amino terminal residue did not block receptor activation. We hypothesized that replacing one or more residues in the amino terminal region by residues with very different side chains might result in an analog without a large loss in PTH2 receptor affinity that did lose its ability to activate the receptor. By retaining sequences in the amino terminal domain that are quite different from those of PTH and PTHrP we hoped that this analog would also fail to bind the PTH1 receptor. We somewhat arbitrarily picked residues for site-directed mutagensis and when we identified mutants with favorable properties we made several additional analogs based on these mutants. While our screen was far from complete the affinity and selectivity of the antagonist we obtained suggest that we are unlikely to produce a much better antagonist through this approach. Since we used site-directed mutagenesis and bacterial expression the approach is limited to natural amino acids.
We characterized the antagonists using inhibition of agonist activity. This is ultimately what the antagonists will be used for and provided a simple and robust assay. Surprisingly, when we attempted to perform Schild analysis to measure the antagonist’s receptor affinity we observed an apparent non-competitive interaction. Because HYWH-TIP39 is a relatively simple modification of TIP39 and contains the sequences thought to contribute most of the receptor binding affinity we think it is unlikely to bind to a site distinct from TIP39. Using radioligand binding we have previously described a relatively slow dissociation rate for TIP(7–39) from the PTH1 receptor (t1/2 14 min; [8]) and observed a comparably slow dissociation of TIP39 from the PTH2 receptor (S. Hoare, unpublished). Thus, we hypothesize that the decrease in maximal cAMP accumulation results from relatively slow dissociation of HYWH-TIP39 from the PTH2 receptor. While this precludes an accurate determination of an affinity constant from functional data, the data clearly demonstrate that the ligand is a potent rat PTH2 receptor antagonist.
The major potential use for HYWH-TIP39 is to facilitate the investigation of TIP39’s physiological role by using it within in vivo experiments. Our group is particularly interested in the function of TIP39 in the brain. We have only characterized HYWH-TIP39 in vitro. We do not know its refractoriness to peptidase inactivation and it may not penetrate the blood brain barrier well. However we anticipate that it will be a useful reagent for investigation of the TIP39-PTH2 receptor system when delivered through intracerebral cannulas either acutely or using extended delivery via osmotic minipumps.
Acknowledgments
This research was supported by the intramural program of the NIH, National Institute of Mental Health. DNA sequencing was provided by the National Institute of Neurological Diseases Intramural Sequencing Core Facility. Mass Spectrometry was performed in the National Institute of Neurological Diseases Intramural Protein/Peptide Sequencing Facility. We thank Samuel Hoare for helpful discussion.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Dobolyi A, Ueda H, Uchida H, Palkovits M, Usdin TB. Anatomical and physiological evidence for involvement of tuberoinfundibular peptide of 39 residues in nociception. Proceedings of the National Academy of Sciences, U S A. 2002;99:1651–6. doi: 10.1073/pnas.042416199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gardella TJ, Juppner H. Interaction of PTH and PTHrP with their receptors. Rev Endocr Metab Disord. 2000;1:317–29. doi: 10.1023/a:1026522619828. [DOI] [PubMed] [Google Scholar]
- 3.Gardella TJ, Luck MD, Jensen GS, Schipani E, Potts JT, Jr, Juppner H. Inverse agonism of amino-terminally truncated parathyroid hormone (PTH) and PTH-related peptide (PTHrP) analogs revealed with constitutively active mutant PTH/PTHrP receptors. Endocrinology. 1996;137:3936–41. doi: 10.1210/endo.137.9.8756569. [DOI] [PubMed] [Google Scholar]
- 4.Gardella TJ, Luck MD, Jensen GS, Usdin TB, Juppner H. Converting parathyroid hormone-related peptide (PTHrP) into a potent PTH-2 receptor agonist. Journal of Biological Chemistry. 1996;271:19888–93. doi: 10.1074/jbc.271.33.19888. [DOI] [PubMed] [Google Scholar]
- 5.Goold CP, Usdin TB, Hoare SR. Regions in rat and human parathyroid hormone (PTH) 2 receptors controlling receptor interaction with PTH and with antagonist ligands. Journal of Pharmacology and Experimental Therpeutics. 2001;299:678–90. [PubMed] [Google Scholar]
- 6.Harmar AJ. Family-B G-protein-coupled receptors. Genome Biol. 2001;2:REVIEWS3013. doi: 10.1186/gb-2001-2-12-reviews3013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hoare SR, Clark JA, Usdin TB. Molecular determinants of tuberoinfundibular peptide of 39 Residues (TIP39) selectivity for the parathyroid hormone-2 (PTH2) receptor: N-terminal truncation of TIP39 reverses PTH2 receptor/PTH1 receptor binding selectivity. Journal of Biological Chemistry. 2000;275:27274–83. doi: 10.1074/jbc.M003910200. [DOI] [PubMed] [Google Scholar]
- 8.Hoare SR, Usdin TB. Tuberoinfundibular peptide (7–39) [TIP(7–39)], a novel, selective, high-affinity antagonist for the parathyroid hormone-1 receptor with no detectable agonist activity. J Pharmacol Exp Ther. 2000;295:761–70. [PubMed] [Google Scholar]
- 9.LaBuda CJ, Dobolyi A, Usdin TB. Tuberoinfundibular peptide of 39 residues produces anxiolytic and antidepressant actions. Neuroreport. 2004;15:881–5. doi: 10.1097/00001756-200404090-00030. [DOI] [PubMed] [Google Scholar]
- 10.LaBuda CJ, Usdin TB. Tuberoinfundibular peptide of 39 residues decreases pain-related affective behavior. Neuroreport. 2004;15:1779–82. doi: 10.1097/01.wnr.0000134849.63755.15. [DOI] [PubMed] [Google Scholar]
- 11.Lanske B, Kronenberg HM. Parathyroid hormone-related peptide (PTHrP) and parathyroid hormone (PTH)/PTHrP receptor. Crit Rev Eukaryot Gene Expr. 1998;8:297–320. doi: 10.1615/critreveukargeneexpr.v8.i3-4.40. [DOI] [PubMed] [Google Scholar]
- 12.Majeska RJ, Rodan SB, Rodan GA. Parathyroid hormone-responsive clonal cell lines from rat osteosarcoma. Endocrinology. 1980;107:1494–503. doi: 10.1210/endo-107-5-1494. [DOI] [PubMed] [Google Scholar]
- 13.Ross G, Engel P, Abdallah Y, Kummer W, Schluter KD. Tuberoinfundibular peptide of 39 residues: a new mediator of cardiac function via nitric oxide production in the rat heart. Endocrinology. 2005;146:2221–8. doi: 10.1210/en.2004-1180. [DOI] [PubMed] [Google Scholar]
- 14.Sugimura Y, Murase T, Ishizaki S, Tachikawa K, Arima H, Miura Y, et al. Centrally administered tuberoinfundibular peptide of 39 residues inhibits arginine vasopressin release in conscious rats. Endocrinology. 2003;144:2791–6. doi: 10.1210/en.2002-0017. [DOI] [PubMed] [Google Scholar]
- 15.Usdin TB. The PTH2 receptor and TIP39: a new peptide-receptor system. Trends Pharmacol Sci. 2000;21:128–30. doi: 10.1016/s0165-6147(00)01455-3. [DOI] [PubMed] [Google Scholar]
- 16.Usdin TB, Bonner TI, Harta G, Mezey E. Distribution of parathyroid hormone-2 receptor messenger ribonucleic acid in rat. Endocrinology. 1996;137:4285–97. doi: 10.1210/endo.137.10.8828488. [DOI] [PubMed] [Google Scholar]
- 17.Usdin TB, Gruber C, Bonner TI. Identification and functional expression of a receptor selectively recognizing parathyroid hormone, the PTH2 receptor. Journal of Biological Chemistry. 1995;270:15455–8. doi: 10.1074/jbc.270.26.15455. [DOI] [PubMed] [Google Scholar]
- 18.Usdin TB, Hilton J, Vertesi T, Harta G, Segre G, Mezey E. Distribution of the parathyroid hormone 2 receptor in rat: immunolocalization reveals expression by several endocrine cells. Endocrinology. 1999;140:3363–71. doi: 10.1210/endo.140.7.6855. [DOI] [PubMed] [Google Scholar]
- 19.Usdin TB, Hoare SRJ, Wang T, Mezey É, Kowalak JA. TIP39: a new neuropeptide and PTH2 receptor agonist from hypothalamus. Nature Neuroscience. 1999;2:941–3. doi: 10.1038/14724. [DOI] [PubMed] [Google Scholar]
- 20.Usdin TB, Wang T, Hoare SR, Mezey E, Palkovits M. New members of the parathyroid hormone/parathyroid hormone receptor family: the parathyroid hormone 2 receptor and tuberoinfundibular peptide of 39 residues. Frontiers in Neuroendocrinology. 2000;21:349–83. doi: 10.1006/frne.2000.0203. [DOI] [PubMed] [Google Scholar]
- 21.Ward HL, Small CJ, Murphy KG, Kennedy AR, Ghatei MA, Bloom SR. The actions of tuberoinfundibular peptide on the hypothalamo-pituitary axes. Endocrinology. 2001;142:3451–6. doi: 10.1210/endo.142.8.8308. [DOI] [PubMed] [Google Scholar]