

Abstract
β-adrenergic receptor (β-AR) stimulation induces apoptosis in adult rat ventricular myocytes (ARVM). β1 integrin signaling plays a protective role in β-AR-stimulated apoptosis. Glycogen synthase kinase-3β (GSK-3β), a multifunctional serine/threonine kinase, negatively regulates cardiac hypertrophy. Here we show that β-AR stimulation (isoproterenol; 15 min) increases tyr216 phosphorylation and GSK-3β activity. Inclusion of LiCl, inhibitor of GSK-3β, in the reaction mix or expression of catalytically inactive GSK-3β (KM-GSK) inhibited β-AR-stimulated GSK-3β activity. Inhibition of tyrosine kinase using genistein or chelation of intracellular Ca2+ using BAPTA-AM inhibited β-AR-stimulated increases in tyr216 phosphorylation and GSK-3β activity. Inhibition of GSK-3β using pharmacological inhibitors or infection with KM-GSK decreased β-AR-stimulated cytosolic cytochrome C release and apoptosis. Expression of β1 integrins increased ser9 phosphorylation and inhibited β-AR-stimulated increase in GSK-3β activity. Wortmannin, inhibitor of PI3-kinase, reversed the effects of β1 integrins on GSK-3β activity and apoptosis. Purified active matrix metalloproteinase-2 (MMP-2), shown to interfere with β1 integrin signaling, increased GSK-3β activity, while inhibition of MMP-2 inhibited β-AR-stimulated increases in GSK-3β activity. β-AR stimulation induced nuclear accumulation of GSK-3β. β-AR stimulation (3 h) increased the expression of transcription factor Gadd153 (growth arrest- and DNA damage-inducible gene 153). These data suggest that β-AR stimulation increases GSK-3β activity. Activation of GSK-3β plays a pro-apoptotic role in β-AR stimulated apoptosis via the involvement of mitochondrial death pathway. β1 integrins inactivate GSK-3β and play an anti-apoptotic role via the involvement of PI3-kinase pathway. The apoptotic effects of GSK-3β may be mediated, at least in part, via its nuclear localization and induction of pro-apoptotic genes, such as Gadd153.
Keywords: GSK-3β, integrins, adult myocytes, apoptosis, Gadd153
Introduction
Apoptosis occurs in the myocardium of patients with end-stage heart failure and myocardial infarction (MI), and in animal models of myocardial hypertrophy and failure [1–3]. Stimulation of β-adrenergic receptor (β-AR) induces apoptosis in cardiac myocytes in vitro and in vivo [4]. Recently, we have shown that stimulation of β1 integrin signaling protects ARVM against β-AR-stimulated apoptosis [5,6].
GSK-3β was originally identified based on its kinase activity towards an in vitro substrate, glycogen synthase [7]. Since then, it has been shown to regulate a wide range of cellular functions, including metabolism, gene expression and cytoskeletal integrity [8]. Activity of GSK-3β is regulated by phosphorylation and interaction with inhibitory proteins. The phosphorylation at a tyrosine residue (tyr216) located in the kinase domain increases its activity, while phosphorylation of an N-terminal serine residue (ser9) inactivates GSK-3β [9]. Protein kinases, including PI3-kinase, Akt, protein kinase A, protein kinase C and integrin-linked kinase are implicated in ser9 phosphorylation and inactivation of GSK-3β [10]. In mammalian cells, MEK1 and Ca2+-dependent tyrosine kinase PYK2 are implicated in tyr216 phosphorylation of GSK-3β [11–13].
GSK-3β is shown to play a protective as well as pro-apoptotic role in various cells [10]. GSK-3β may promote cell death induced by the mitochondrial intrinsic apoptotic pathway, but inhibit the death receptor-mediated extrinsic apoptotic signaling pathway [14]. In cardiac myocytes, inhibition of GSK-3β contributes to the development of cardiac hypertrophy [15]. Transgenic mice expressing wild-type GSK-3β in the heart exhibit dramatic impairment of normal post-natal cardiomyocyte growth as well as abnormal cardiac contractile function [16]. The protective effects of Kallikrein/kinin in ischemia/reperfusion-induced apoptosis are suggested to involve Akt-mediated inactivation of GSK-3β [17]. Inhibition of GSK-3β is also shown to inhibit hypoxia/reperfusion-induced cardiac myocyte apoptosis [18].
This study was undertaken to study the activation of GSK-3β in ARVM and its role in βAR-stimulated apoptosis. We hypothesized that GSK-3β plays a pro-apoptotic role in β-AR-stimulated apoptosis, while β1 integrins inactivate GSK-3β via the activation of PI3-kinase pathway and confer protection against β-AR-stimulated apoptosis. Our results show that β-AR stimulation increases tyr216 phosphorylation and activity of GSK-3β. Using several different strategies, we show that activation of GSK-3β plays a pro-apoptotic role in β-AR stimulated apoptosis via the involvement of mitochondrial death pathway. β1 integrins may play a protective role in β-AR stimulated apoptosis via the activation of PI3-kinase and inactivation of GSK-3β. Localization to the nucleus and the increased expression of a stress-induced transcription factor, Gadd153, may be one of the mechanisms by which GSK-3β induces apoptosis.
Materials and Methods
Cell isolation and culture
Calcium-tolerant ARVM were isolated from the hearts of adult male Sprague-Dawley rats (240–280 g) as described [6]. ARVM were plated in Dulbecco’s modified Eagle’s medium (DMEM; Mediatech) supplemented with HEPES (25 mM), BSA (0.2%), creatine (5 mM), L-carnitine (2 mM), taurine (5 mM) and 0.1% penicillin-streptomycin at a density of 30–50 cells/mm2 on 100-mm tissue culture dishes (Fisher Scientific) or coverslips precoated with laminin (1μg/cm2). The chemicals, if not mentioned otherwise, were purchased from Sigma-Aldrich, USA. The animal experimentation was conducted in conformity with the recommendation from the Declaration of Helsinki and the Guiding Principles in the Care and Use of Animals as endorsed by the International Society for Heart Research.
Cell treatment
ARVM, cultured for 24 h, were treated with isoproterenol (ISO; 10 μM) in the presence of ascorbic acid (100 μM) for different time periods (15 min – 24 h). ARVM were treated with the GSK-3β inhibitors [LiCl (1 mM), SB216763 (20 μM) or SB415286 (20 μM)], gelatinase inhibitor SB3CT (1 nM), tyrosine kinase inhibitor genistein (100 μM) or the Ca2+ chelator BAPTA-AM (100 μM) for 30 min before treatment with ISO. To inhibit PI3-kinase, cells were pretreated with wortmannin (0.5 μM) for 30 min before treatment with ISO and/or infection with adenoviruses. The inhibitors (purchased from Calbiochem) were maintained in the medium during the treatment period. Cells were also treated with purified active MMP-2 (1 nM) to measure GSK-3β activity.
Adenovirus infection of ARVM
ARVM were infected with adenoviruses expressing catalytically inactive form of GSK-3β (KM-GSK); [courtesy of Dr. Morris J Birnbaum, Howard Hughes Medical Institute, University of Pennsylvania-School of Medicine, Philadelphia], green fluorescence protein (GFP) and wild type human β(1A) integrin (Adβ1A) at a multiplicity of infection of 50–100 for a total period of 48 h.
Terminal deoxynucleotidyl transferase-mediated nick end labeling (TUNEL) assay
TUNEL-staining was performed on ARVM plated on thermanox coverslips using in situ death detection kit according to the manufacturer’s instructions (Roche Molecular Biochemicals). The percentage of TUNEL-positive cells (relative to total ARVM) was determined by counting ~200 cells in 10 randomly chosen fields per coverslip for each experiment.
Analysis of cytosolic cytochrome C
Cytosolic fractions are prepared as described [6]. The cytosolic fractions were analyzed by western blot using anti-cytochrome C antibody (Santa Cruz).
Western blot analyses
ARVM were lysed in RIPA buffer (10 mM Tris HCl [pH 7.2], 158 mM NaCl, 1 mM EGTA, 1 mM sodium orthovanadate, 0.1% SDS, 1.0% Triton X-100, 1% Sodium deoxycholate and 1 mM phenylmethylsulfonyl fluoride). Equal amount of proteins (50–100 μg) were analyzed by western blot as described [6]. The primary antibodies used were P-ser9 GSK-3β, (Cell Signaling), P-tyr279/216 GSK-3α/β (Upstate), Gadd153, or cytochrome C (Santa Cruz). The membranes were stripped and probed with anti-actin (Chemicon) or anti-GSK-3β antibodies (Santa Cruz) to optimize protein loading.
Measurement of GSK-3β activity
GSK-3β activity was measured by immune-complex kinase assay using phosphoglycogen synthase peptide-2 (Upstate) as described [19].
Immunocytochemical analysis
Immuncytochemical analysis was carried out as described [20]. Briefly, ARVM plated on coverslips were fixed in 3.7% formaldehyde and permeabilized using 1% triton X-100. The coverslips were then incubated with 10% goat serum for 1 h. After washing with phosphate-buffered saline, the coverslips were incubated overnight with monoclonal anti-GSK-3β antibodies (1:100, BD Biosciences). After incubation with FITC-conjugated secondary antibody, the coverslips were counterstained with propidium iodide (PI), mounted and visualized using confocal microscope (Nikon, Tokyo).
Statistical analyses
All data are expressed as mean ± SE. Statistical analysis was performed using the student’s t test or a one-way analysis of variance (ANOVA) and a post hoc Tukey’s test. Probability (p) values of <0.05 were considered to be significant. The number (n) indicates the number of biological replicates.
Results
β-AR stimulation increases the activity of GSK-3β
A time course analysis of GSK-3β activity using immune-complex kinase assay indicated that β-AR stimulation using isoproterenol (10 μM) for 15 min increases GSK-3β activity by 4.6±1.2-fold (p<0.01 vs. control, n=10; Fig 1A). The activity of GSK-3β declined thereafter, reaching to basal levels within 1 h of β-AR stimulation. To confirm that the kinase activity is indeed due to GSK-3β, we added LiCl (a known inhibitor of GSK-3β) to the reaction mix. LiCl almost completely inhibited β-AR stimulated increases in GSK-3β activity (fold increase vs. CTL, ISO, 2.2±0.11*; LiCl+ISO, 1.08±0.46#; *p<0.05 vs. CTL; #p<0.05 vs. ISO, n=3; Fig 1B). To further confirm GSK-3β activity, ARVM were infected with adenoviruses expressing GFP or catalytically inactive GSK-3β (KM-GSK). GFP had no effect on β-AR-stimulated increases in GSK-3β activity. However, KM-GSK inhibited β-AR-stimulated increases in GSK-3β activity (fold increase vs. GFP, GFP+ISO, 2.6±0.3*; KM-GSK+ISO, 1.1±0.3#; *p<0.05 vs. GFP; n=7; #p<0.05 vs. GFP+ISO, n=3; Fig 1C). Phosphorylation of ser9 inactivates GSK-3β [9]. Ser9 phosphorylation was also increased within 15 min of β-AR stimulation. However, maximum increase in ser9 phosphorylation was observed 3 h after β-AR stimulation (Fig 1D). Since phosphorylation of tyr216 increases GSK-3β activity [9], we next measured the levels of tyr216 phosphorylation using phospho-specific antibodies by western blot analysis. This analysis demonstrated that β-AR stimulation (15 min) increases tyr216 phosphorylation of GSK-3β (Fig 2A). β-AR-stimulated apoptosis in ARVM requires calcium entry via voltage-dependent calcium channels [4]. Calcium-dependent kinase, pyk2, is suggested to increase tyr216 phosphorylation and activity of GSK-3β in neuronal cells [12]. Pretreatment with genistein (tyrosine kinase inhibitor) and BAPTA-AM (Bapta; intracellular Ca2+ chelator) inhibited β-AR-stimulated increases in GSK-3β tyr216 phosphorylation (fold change vs. CTL, ISO, 1.5±0.3*; Gen+ISO, 0.5±0.1#; Bapta+ISO, 0.8±0.2#; *p<0.05 vs. CTL; #p<0.05 vs. ISO, n=4; Fig 2A) and activity (fold change vs. CTL, ISO, 2.3±0.35*; Bapta+ISO, 0.99±0.2#; Gen+ISO, 0.88±0.18#; *p<0.05 vs. CTL; #p<0.05 vs. ISO, n=4; Fig 2B).
Fig 1. β-AR stimulation increases GSK-3β activity.

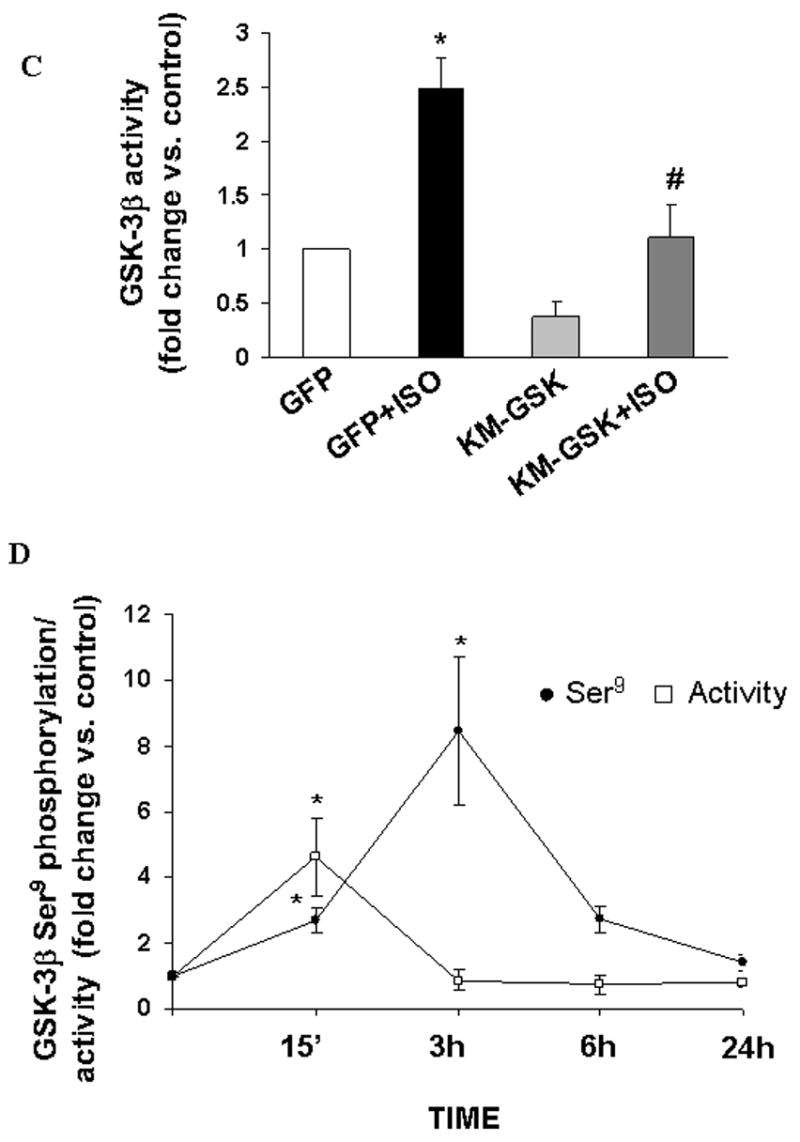
A. ARVM were treated with isoproterenol (ISO; 10 μM) for 15 min (15’), 30 min (30’), 1 h, 3 h, 6 h and 24 h. Activity of GSK-3β was measured using immune-complex kinase assay as described in the methods. The results are expressed as fold change in activity vs. control (CTL). *p<0.05 vs. CTL; n=4–10. Addition of LiCl (1 mM) in the reaction mix (B) or infection with KM-GSK (C) inhibits β-AR stimulated increases in GSK-3β activity. *p<0.05 vs. control (CTL/GFP); n=3–7; #p<0.05 vs. ISO/GFP+ISO; n=3. D. ARVM were treated with ISO for 15 min (15’), 3 h, 6 h and 24 h. Cell lysates were analyzed by western blot using phospho-specific (ser9) GSK-3β antibodies, while activity of GSK-3β was measured using immune-complex kinase assay. *p<0.05 vs. CTL; n=3–10.
Fig 2. Tyr216 phosphorylation and activity of GSK-3β.
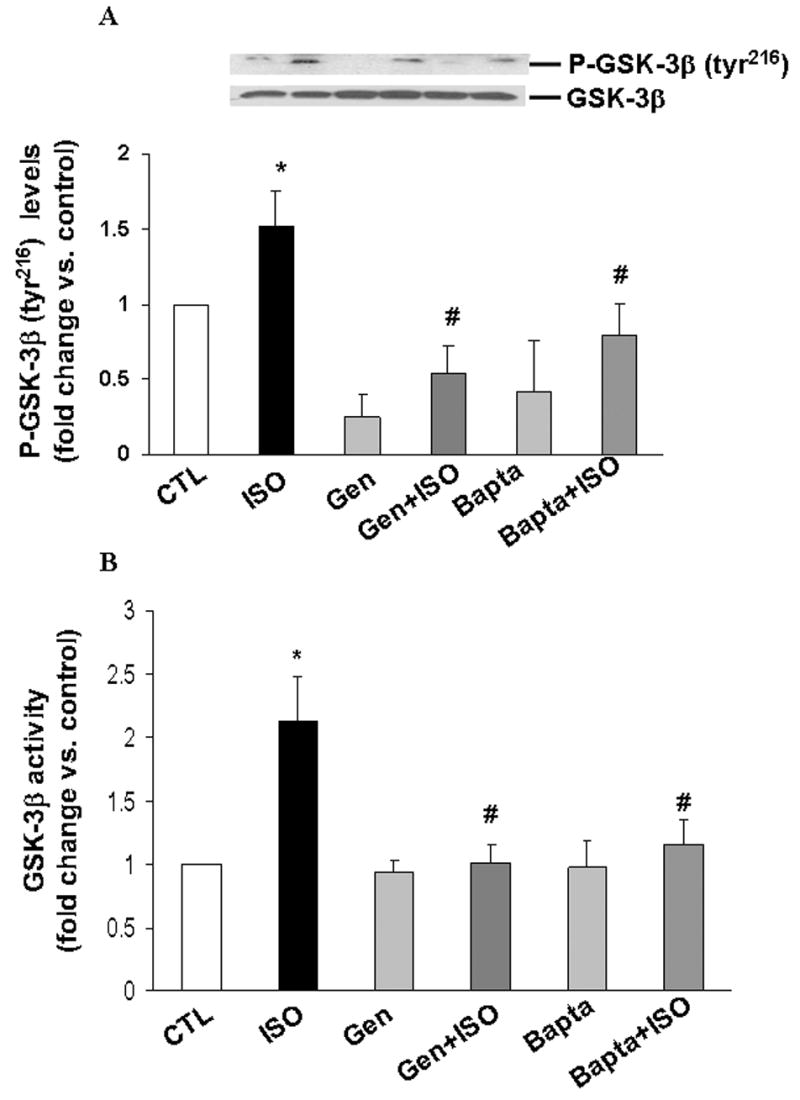
ARVM were pretreated with Genistein (Gen) or BAPTA-AM (Bapta) for 30 min followed by treatment with ISO for 15 min. Cell lysates were analyzed by western blot using phospho-specific (tyr216) GSK-3β antibodies (A) or immune-complex kinase assay (B). Equal protein loading (A) was verified using anti-GSK-3β antibodies. *p<0.05 vs. CTL; #p<0.05 vs. ISO; n=4.
Inhibition of GSK-3β inhibits β-AR-stimulated increases in cytosolic cytochrome C and apoptosis
To study the role of GSK-3β in mitochondrial death pathway of apoptosis, we measured levels of cytosolic cytochrome C using western blot analysis. β-AR simulation significantly increased levels of cytosolic cytochrome C by 2.5±0.3-fold (p<0.05 vs. CTL; n=6; Fig 3A). Inhibition of GSK-3β using SB216763 decreased β-AR stimulated cytosolic cytochrome C levels to 1.4±0.3-fold (p<0.05 vs. ISO, n=3; Fig 3A). Adenovirus-mediated expression of KM-GSK also decreased cytosolic cytochrome C levels (fold change vs. GFP; GFP+ISO, 2.5±0.33*; KM-GSK, 1.35±0.16; KM-GSK+ISO, 1.47±0.31#; *p<0.05 vs. GFP; #p<0.05 vs. GFP+ISO, n=3; Fig 3B). As shown previously [21], β-AR stimulation for 24 h increased the number of apoptotic ARVM (percent apoptosis; CTL, 7.5±0.8; ISO, 18.6±1.1*; *p<0.001; n=19). Pretreatment with LiCl or two potent and selective cell permeable inhibitors of GSK-3β, SB216763 and SB415286, decreased β-AR-stimulated increases in apoptotic ARVM (percent apoptosis; ISO+LiCl, 5.3±1.05#; ISO+SB216763, 10.3±3.2#; ISO+SB415286, 11.9±1.23#; #p<0.05 vs. ISO; n=3; Fig 4A). To confirm the role of GSK-3β in β-AR-stimulated apoptosis, cells were infected with adenoviruses expressing KM-GSK followed by β-AR stimulation. Measurement of apoptosis using TUNEL-staining assay indicated that expression of KM-GSK also inhibits β-AR-stimulated increases in apoptotic ARVM (percent apoptosis; ISO+KM-GSK, 10.4±0.8#; #p<0.05 vs. ISO; n=7; Fig 4B).
Fig 3. Inhibition of GSK-3β inhibits β-AR-stimulated increases in cytosolic cytochrome C levels.

ARVM were pretreated with SB216763 (20 μM; A) for 30 min or infected with adenoviruses expressing catalytically inactive GSK-3β (KM-GSK; B) for 42 h. The cells were then treated with ISO for 6 h. Cytosolic cytochrome C levels were measured using western blot analysis. Actin immunostaining indicates protein loading. *p<0.05 vs. control (CTL/GFP); n=3; #p<0.05 vs. ISO/GFP+ISO; n=3.
Fig 4. Inhibition of GSK-3β inhibits β-AR stimulated apoptosis.

A. ARVM were pretreated with LiCl (1 mM), SB216763 (20 μM) or SB415286 (20 μM) for 30 min followed by treatment with isoproterenol (ISO) for 24 h. B. ARVM were infected adenoviruses expressing catalytically inactive GSK-3β (KM-GSK) for 24 h followed by treatment with ISO for 24 h. The numbers of apoptotic cells were measured using TUNEL-staining assay. *p<0.05 vs. control (CTL); n=19; #p<0.05 vs. ISO; n=3–7.
β1 integrins inhibits β-AR-stimulated increases in GSK-3β activity
Overexpression of β1 integrins inhibits β-AR-stimulated apoptosis [6]. Therefore, we next examined the role of β1 integrins in β-AR-stimulated increases in GSK-3β activity. For this, ARVM were infected with adenoviruses expressing wild type human β(1A) integrin (Adβ1A). Adβ1A significantly increased ser9 phosphorylation of GSK-3β alone or in combination with ISO (fold increase vs. GFP; GFP+ISO, 2.42±0.3*; Adβ1A, 3.9±0.6*#; Adβ1A+ISO, 4.9±0.5*#; *p<0.01 vs. GFP; #p<0.01 vs. GFP+ISO; n=6; Fig 5A). Measurement of GSK-3β activity using immune-complex kinase assay confirmed β1 integrin-mediated inactivation of GSK-3β (fold increase vs. GFP, GFP+ISO, 2.6±0.3*; Adβ1A+ISO, 1.3± 0.2#; *p<0.01 vs. GFP; n=7; #p<0.05 vs. GFP+ISO; n=4; Fig 5B). Pretreatment with wortmannin (Wort), a specific PI3-kinase inhibitor decreased ser9 phosphorylation of GSK-3β (fold increase vs. GFP, Adβ1A+Wort+ISO, 1.9±0.19; p<0.05 vs. Adβ1A+ISO; n=4; Fig 5A) while increasing its activity (fold increase vs. GFP, Adβ1A+Wort+ISO, 9.8±4.7; p<0.05 vs. Adβ1A+ISO; n=4; Fig 5B). Wortmannin in the presence of ISO exhibited no significant change in GSK-3β activity when compared to ISO. Previously, we have provided evidence that MMP-2 interferes with the β1 integrin-mediated survival signals [6]. Analysis of GSK-3β activity indicated that inhibition of MMP-2 using SB3CT [6] inhibits βAR-stimulated increases in GSK-3β activity (fold increase vs. CTL, ISO, 2.3±0.35*; SB3CT+ISO, 1.3±0.35#; *p<0.05 vs. CTL; #p<0.05 vs. ISO, n=4; Fig 5C). On the other hand, treatment of ARVM with purified active MMP-2 protein increased GSK-3β activity (fold increase vs. CTL, MMP-2, 1.89±0.3*; *p<0.05 vs. CTL; n=4; Fig 5D).
Fig 5. Overexpression of β integrins or inhibition of MMP-2 inhibits β-AR-stimulated increases in GSK-3β activity.
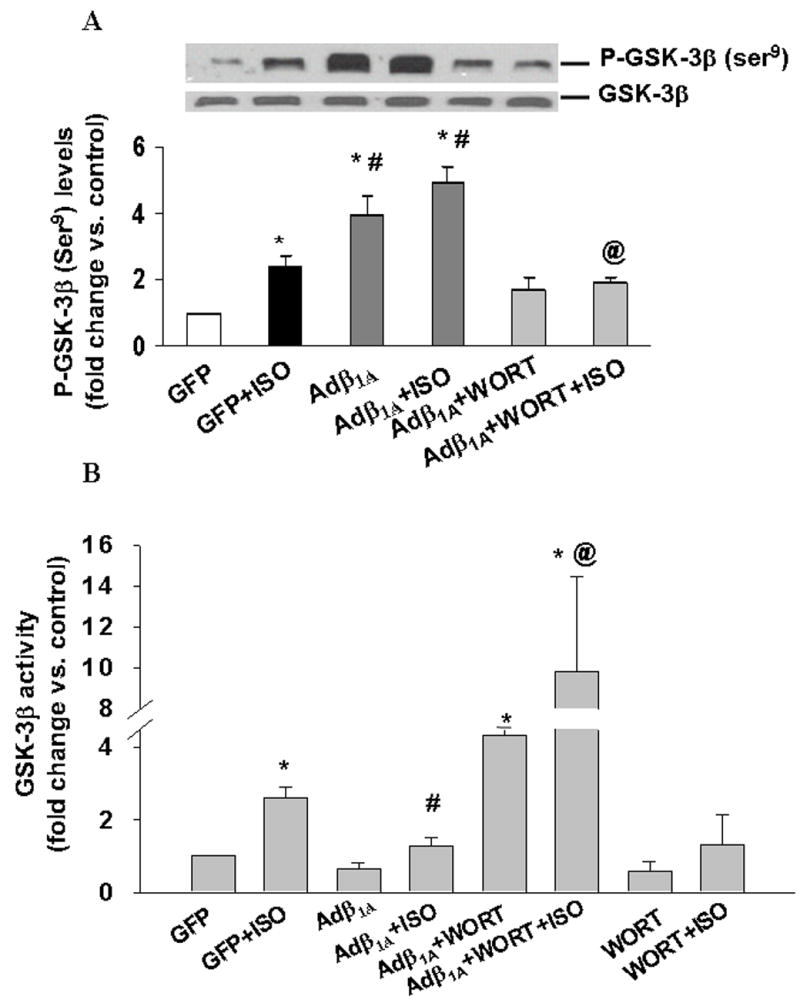

A and B. ARVM pretreated with wortmannin (Wort; 0.5 μM) were infected with Adβ1A or GFP for 48 h followed by treatment with ISO for 15 min. Cell lysates were analyzed by (A) western blot using phospho-specific (ser9) GSK-3β antibodies or (B) immune-complex kinase assay to measure GSK-3β activity. Equal protein loading (A) was verified using anti-GSK-3β antibodies. Phosphorylation and activity of GSK-3β are expressed as fold vs. GFP. *p<0.05 vs. GFP; n=6–7; #p<0.05 vs. GFP+ISO; n=4; @p<0.05 vs. Adβ1A+ISO; n=3–4. C and D. ARVM were pretreated with SB3CT for 30 min followed by treatment with ISO for 15 min (C) or treated with MMP-2 for 15 min (D). Cell lysates were analyzed by immune-complex kinase assay. *p<0.05 vs. CTL; #p<0.05 vs. ISO; n=4.
Inhibition of PI3-kinase inhibits the protective effects of β1 integrin
Pretreatment with wortmannin alone had no effect on β-AR stimulated apoptosis. However, wortmannin almost completely reversed the protective effect of β1 integrins on β-AR stimulated apoptosis (percent apoptosis; CTL, 5±1.7; ISO, 16.0±2.5*; ISO+Wort, 14.4±0.7*; Adβ1A+ISO, 6.2±2#; Adβ1A+Wort+ISO, 20.5± 2.6*@; *p<0.05 vs. CTL; #p<0.05 vs. ISO; @p<0.05 vs. Adβ1A+ISO; n=3–6; Fig 6).
Fig 6. Inhibition of PI3-kinase pathway reverses the anti-apoptotic effect of β1 integrins.

ARVM were pretreated with wortmannin (Wort; 0.5 μM) were infected with Adβ1A for 24 h followed treatment with ISO for 24 h. The number of apoptotic cells was measured using TUNEL-staining assay. *p<0.05 vs. control (CTL); n=3–6; #p<0.05 vs. ISO; n=4; @p<0.05 vs. Adβ1A+ISO; n=4.
β-AR stimulation induces nuclear translocation of GSK -3β
In Wnt signaling pathway, GSK-3β is suggested to negatively regulate the levels of transcriptional activator, β-catenin [22]. Inactivation of GSK-3β by hypertrophic stimuli increases β-catenin levels and transcriptional activity of β-catenin [22]. To study β-catenin levels in ARVM, cells were treated with ISO for different time periods (15 min, 3 h, 6 h or 24 h). Analysis of total cell lysates using western blot indicated no significant change in β-catenin protein levels following β-AR stimulation [fold change vs. CTL; ISO (15 min), 1.15±0. 06; ISO (3 h), 1.14±0.06; ISO (6 h), 0.99±0.07; ISO (24 h), 1.08± 0.09; p=NS vs. CTL; n=3–4; Fig 7A]. Pro-apoptotic stimuli are suggested to induce nuclear accumulation of GSK-3β in human neuroblastoma cells [23]. To study nuclear accumulation of GSK-3β, ARVM were treated with ISO for 3h, immunostained with anti-GSK-3β antibody and visualized by confocal microscopy. Control cells exhibited mainly cytoplasmic and sarcomeric staining for GSK-3β. β-AR stimulation induced nuclear localization of GSK-3β (Fig 7B).
Fig 7. β-AR stimulation does not alter β-catenin levels but induces nuclear accumulation of GSK-3β.
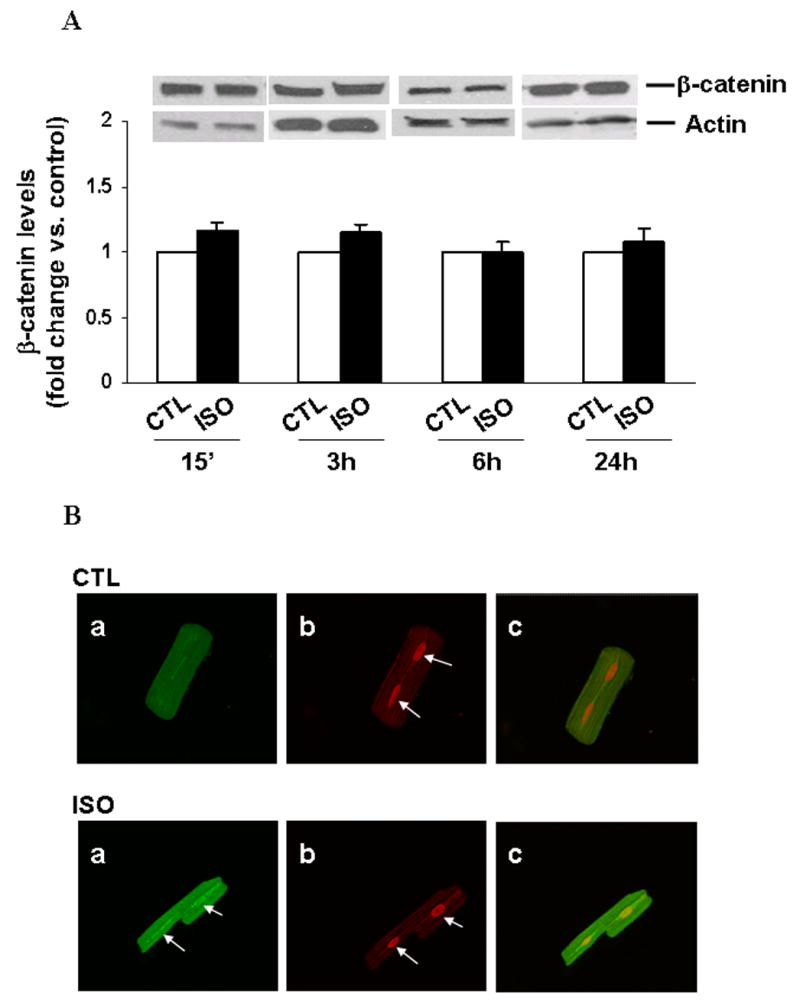
A. ARVM were treated with ISO (15 min, 3 h, 6 h and 24 h). Cell lysates were analyzed by western blot using anti β-catenin antibodies. Equal protein loading in each lane was verified using actin antibodies. B. ARVM were treated with isoproterenol (ISO; 10 μM) for 3 h, immunostained with anti-GSK-3β antibody and subjected to confocal microscopic analyses. Green fluorescence indicates GSK-3β immunostaining (Panel a). Red staining indicates PI-stained nuclei (Panel b). Panel c demonstrates overlay images of panels a and b. Arrows indicate nuclear staining. Negative control performed with the omission of primary antibodies exhibited no staining. The experiments were repeated three times with similar results.
Inhibition of GSK-3β or overexpression of β1 integrins inhibits β-AR-stimulated increases in Gadd153 levels
GSK-3β is shown to play a role in cardiac myocyte hypertrophy via transcription factors such as NF-AT and GATA4 [24,25]. Transcription factor Gadd153 (Growth arrest and DNA damage-inducible gene 153; a member of C/EBP transcription factor family) is suggested to play an important role in apoptosis [26]. Therefore, we next analyzed expression of Gadd153. A time course analysis (3, 6, 24 h) indicated maximal increase in Gadd153 protein levels 3 h after β-AR stimulation. Protein levels of Gadd153 decreased thereafter (data not shown). β-AR stimulation (3 h) increased Gadd153 protein levels by 1.5±0.07-fold (p<0.05 vs. CTL, n=3; Fig 8A). Inhibition of GSK-3β using SB216763 or LiCl completely inhibited β-AR-stimulated increases in Gadd153 protein levels (fold change vs. CTL; SB+ISO, 0.7±0.11#; Li+ISO, 0.7±0.08#; #p<0.05 vs. ISO, n=3; Fig 8A). Likewise, adenovirus mediated overexpression of β1 integrins significantly decreased the levels of Gadd153 (fold increase vs. GFP; GFP+ISO, 1.46±0.17*; Adβ1A, 0.93±0.17; Adβ1A+ISO, 0.88±0.16#; *p<0.01 vs. GFP; #p<0.01 vs. GFP+ISO; n=3; Fig 8B).
Fig 8. Inhibition of GSK-3β and overexpression of β1 integrins decreases β-AR stimulated increases in Gadd153.
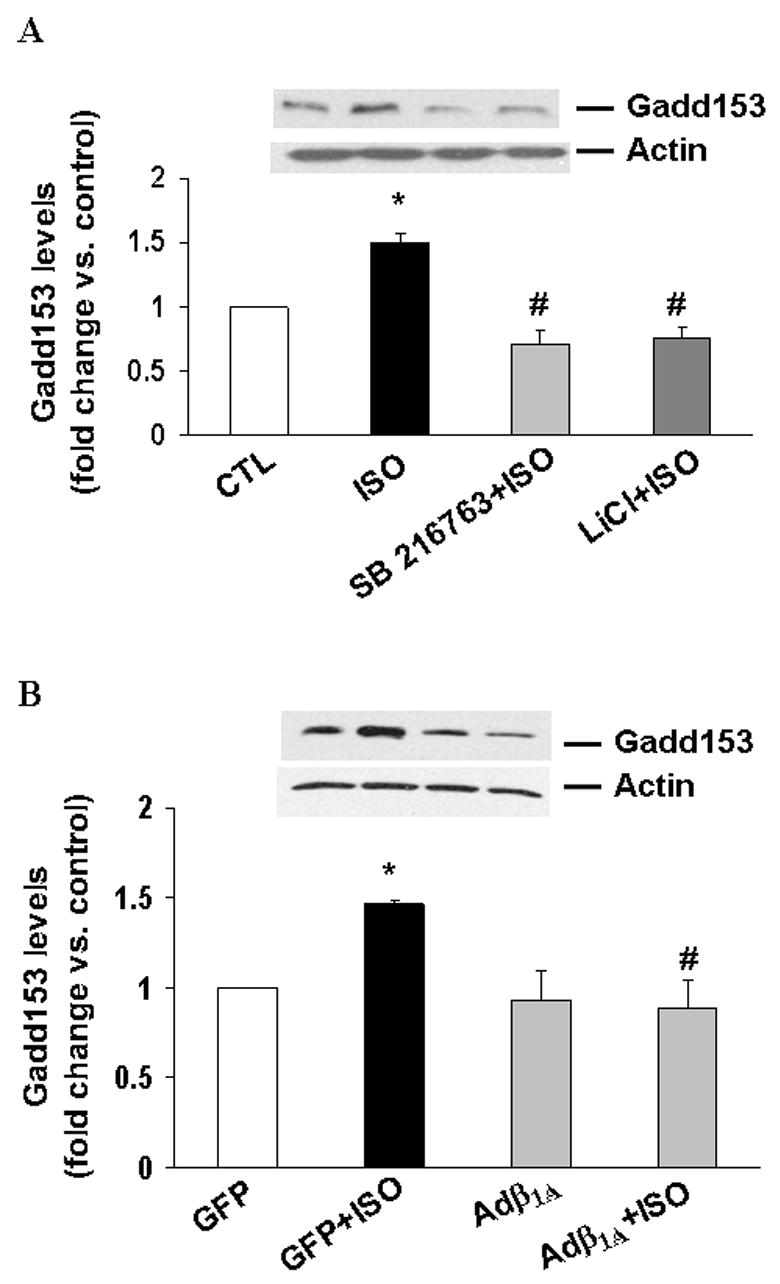
ARVM were pretreated with SB216763 (20 μM; SB) or LiCl (1 mM) for 30 min (A) or infected with Adβ1A or GFP for 45 h (B) followed by treatment with ISO (3 h). Cell lysates were analyzed by western blot using anti Gadd153 antibodies. Actin immunostaining indicates protein loading. *p<0.05 vs. control (CTL/GFP); #p<0.05 vs. ISO/GFP+ISO; n=3.
Discussion
β-AR stimulation induces cardiac myocyte apoptosis in vitro and in vivo [4]. β1 integrins protect ARVM against β-AR stimulated apoptosis [5,6]. Here, we provide evidence that β-AR stimulation increases GSK-3β activity. Inhibition of GSK-3β inhibits β-AR-stimulated increases in GSK-3β activity, levels of cytosolic cytochrome C and apoptosis. Expression of β1 integrins or inhibition of MMP-2 inhibits β-AR-stimulated activation of GSK-3β. Inhibition of PI3-kinase reversed the effects of β1 integrins on GSK-3β activity, and inhibited the protective effect of β1 integrins. Furthermore, we provide evidence for nuclear translocation of GSK-3β and show that GSK-3β increases the expression of Gadd153, a pro-apoptotic protein.
GSK-3β is shown to be a negative regulator of cardiac myocyte hypertrophy [15]. Transgenic mice expressing wild-type GSK-3β in the heart show a dramatic impairment of normal cardiac myocyte growth and contractile function [16]. In the present study, we show that β-AR stimulation increases GSK-3β activity in ARVM. This increase in the activity was inhibited by addition of LiCl in the reaction mix or infection with adenoviruses expressing dominant negative GSK-3β. Western blot analysis indicated increased tyr216 phosphorylation of GSK-3β. In neonatal cardiac myocytes, β-AR stimulation (10 min) is shown to inhibit GSK-3β activity [15]. These differential effects of β-AR stimulation on GSK-3β activity may reflect fundamental differences in the cell biology of neonatal versus adult cardiac myocytes and/or adrenergic physiology [21]. In ARVM, β-AR stimulation also increased phosphorylation of inhibitory ser9 on GSK-3β. However, maximum increase in GSK-3β activity was observed 15 min after β-AR-stimulation, while maximal increase in ser9 phosphorylation was observed 3 h after β-AR-stimulation. The phenomenon of increased tyr216 and ser9 phosphorylation with a net increase in GSK-3β activity has also been observed in neuroblastoma cells in response to other G protein-coupled receptor agonist, lysophosphatidic acid (LPA) [12]. LPA is shown to increase tyrosine phosphorylation of GSK-3β via the Ca2+-sensitive tyrosine kinase, Pyk2. The data presented here demonstrate that BAPTA-AM inhibits β-AR stimulated increases in tyrosine phosphorylation and activity of GSK-3β. β-AR stimulation is known regulate Ca2+ entry via L-type Ca2+ channels in adult heart [4]. Inhibition of L-type Ca2+ channels is shown to inhibit βAR-stimulated apoptosis in ARVM [4]. Collectively, these data suggest the involvement of a Ca2+-sensitive tyrosine kinase, possibly Pyk2, in β-AR stimulated increases in GSK-3β activity. However, further investigations are needed to prove this thesis.
Using pharmacological inhibitors and adenoviral strategies, we provide evidence that inhibition of GSK-3β inhibits β-AR-stimulated apoptosis. GSK-3β is shown to promote apoptosis in other cell types [10]. Activated GSK-3β also facilitates death by anoikis, which results from disruption of cell-matrix interactions [10]. Conversely, GSK-3β is shown to be essential for survival [10]. GSK-3β inhibition reduced hypoxia/reperfusion-induced mitochondrial permeability transition pore and apoptosis in cardiac myocytes [18]. Furthermore, direct inhibition of GSK-3β reduces infarct size and improves post-ischemic heart function [27]. Collectively these data suggest activation of GSK-3β may play an important role in cardiac myocyte apoptosis and heart function.
The PI3-kinase/Akt pathway is activated by a wide range of extracellular stimuli, including the integrins [28]. Previously, we have shown that β1 integrins play a protective role in β-AR-stimulated apoptosis [6]. Transgenic mice overexpressing melusin, a muscle specific β1 integrin interacting protein, increased GSK-3β (ser9) phosphorylation and reduced cardiac myocyte apoptosis in response to chronic pressure overload [29]. Here, we show that expression of β1 integrins inhibits β-AR-stimulated increases in GSK-3β activity. Inhibition of PI3-kinase using wortmannin increased GSK-3β activity, while preventing the protective effects of β1 integrins. These data suggest involvement of PI3-kinase in β1 integrin signaling pathway leading to inactivation of GSK-3β and inhibition of β-AR-stimulated apoptosis. The involvement of β1 integrins in the inhibition of GSK-3β activity is further supported by our data on MMP-2. Previously, we have shown that MMP-2 plays a pro-apoptotic role in β-AR-stimulated apoptosis [20]. MMP-2 interacts with β1 integrins and interferes with β1 integrin-mediated survival signals, leading to the activation of JNK and mitochondrial death pathway [6]. Here we show that purified active MMP-2 protein increases GSK-3β activity, while inhibition of MMP-2 inhibits β-AR-stimulated increases in GSK-3β activity. In neuronal cultures, stimulation with MMP-1 is shown to be associated with a rapid reduction in the ser9 phosphorylation of GSK-3β, eventually leading to cell death [30].
Active GSK-3β induces degradation of β-catenin [31], while inactivation of GSK-3β is suggested to stabilize β-catenin levels. Stabilized β-catenin can then translocate to the nucleus and activate specific target genes. The data presented here demonstrate that β-AR stimulation does not affect β-catenin protein levels, suggesting involvement of β-catenin-independent mechanisms in β-AR-stimulated apoptosis. GSK-3β is predominantly located in cytosol. It is also shown to be present in nuclei and mitochondria. Apoptotic signals are demonstrated to selectively activate GSK-3β in the nucleus and mitochondria, but not the cytosol [32]. GSK-3β is reported to phosphorylate and activate over a dozen of transcription factors including AP-1, NFAT, NF-κB, C/EBP, GATA-4, etc [10]. C/EBP homologous protein, also known as growth arrest- and DNA damage-inducible gene 153 (Gadd153), encodes a nuclear, pro-apoptotic, bZIP transcription factor of the CCAAT/enhancer-binding protein (C/EBP) transcription factor family. Expression of Gadd153 is often induced by cellular stress [33]. Due to the presence of ER-stress responsive elements (ERSE) in the promoter, Gadd153 expression is generally associated with endoplasmic reticulum (ER) stress [26]. However, Gadd153 promoter also has AP-1 site and CEBP/ATF composite site [33]. The AP-1 site is described to have a significant role in transcriptional activation of Gadd153 [33]. We observed increased nuclear accumulation of GSK-3β and Gadd153 protein levels following β-AR stimulation. Inhibition of GSK-3β as well as β1 integrin overexpression almost completely inhibited β-AR-stimulated increases in Gadd153 protein levels. These data suggest that GSK-3β may play an important role in the regulation of Gadd153 expression. However, further studies are needed to investigate the role of Gadd153 in β-AR-stimulated apoptosis.
Here we provide evidence that GSK-3β induces apoptosis via the activation of mitochondrial death pathway. GSK-3β is suggested to target several key proteins in the mitochondria that regulate signals leading to the disruption of mitochondria [reviewed in reference 14]. It can directly phosphorylate Bax (a pro-apoptotic Bcl-2 family member) resulting in the activation of Bax. GSK-3β is shown to be required for the stress-induced expression of Bim (a Bcl-2-interacting mediator of cell death) [14]. A few studies also support a link between Gadd153 and activation of mitochondrial pathway. Overexpression of Gadd153 resulted in the downregulation of Bcl-2 expression, depletion of glutathione levels and exaggerated production of reactive oxygen species, leading to sensitization of cells to endoplasmic reticulum (ER)-stress induced apoptosis [34]. In mouse embryonic fibroblasts, ER-stress-induced apoptosis and activation of caspase-12 is shown to occur downstream of mitochondrial apoptosis involving Apaf-1 [35].
The data presented here demonstrate that activation of GSK-3β plays a pro-apoptotic role in β-AR-stimulated apoptosis in ARVM. Further studies aimed at identifying the molecular targets involved in GSK-3β activation following β-AR-stimulation and how activation of GSK-3β activates mitochondrial death pathway in adult cardiac myocytes may provide potential new targets for prevention of heart failure.
Acknowledgments
This work is supported in part by National Institutes of Health; Grant numbers HL-071519 (KS), HL-57872 (RSR) and Department of Veterans Affairs, Merit Review Grant (KS).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Anversa P, Kajstura J, Olivetti G. Myocyte death in heart failure. Curr Opin Cardiol. 1996;11:245–51. doi: 10.1097/00001573-199605000-00004. [DOI] [PubMed] [Google Scholar]
- 2.Saraste A, Pulkki K, Kallajoki M, Henriksen K, Parvinen M, Voipio-Pulkki LM. Apoptosis in human acute myocardial infarction. Circulation. 1997;95:320–3. doi: 10.1161/01.cir.95.2.320. [DOI] [PubMed] [Google Scholar]
- 3.Teiger E, Than VD, Richard L, Wisnewsky C, Tea BS, Gaboury L, Tremblay J, Schwartz K, Hamet P. Apoptosis in pressure overload-induced heart hypertrophy in the rat. J Clin Invest. 1996;97:2891–7. doi: 10.1172/JCI118747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Singh K, Xiao L, Remondino A, Sawyer DB, Colucci WS. Adrenergic regulation of cardiac myocyte apoptosis. J Cell Physiol. 2001;189:257–65. doi: 10.1002/jcp.10024. [DOI] [PubMed] [Google Scholar]
- 5.Communal C, Singh M, Menon B, Xie Z, Colucci WS, Singh K. beta1 integrins expression in adult rat ventricular myocytes and its role in the regulation of beta-adrenergic receptor-stimulated apoptosis. J Cell Biochem. 2003;89:381–8. doi: 10.1002/jcb.10520. [DOI] [PubMed] [Google Scholar]
- 6.Menon B, Singh M, Ross RS, Johnson JN, Singh K. beta-Adrenergic receptor-stimulated apoptosis in adult cardiac myocytes involves MMP-2-mediated disruption of beta1 integrin signaling and mitochondrial pathway. Am J Physiol Cell Physiol. 2006;290:C254–C261. doi: 10.1152/ajpcell.00235.2005. [DOI] [PubMed] [Google Scholar]
- 7.Embi N, Rylatt DB, Cohen P. Glycogen synthase kinase-3 from rabbit skeletal muscle. Separation from cyclic-AMP-dependent protein kinase and phosphorylase kinase. Eur J Biochem. 1980;107:519–27. [PubMed] [Google Scholar]
- 8.Grimes CA, Jope RS. The multifaceted roles of glycogen synthase kinase 3beta in cellular signaling. Prog Neurobiol. 2001;65:391–426. doi: 10.1016/s0301-0082(01)00011-9. [DOI] [PubMed] [Google Scholar]
- 9.Wang QM, Fiol CJ, DePaoli-Roach AA, Roach PJ. Glycogen synthase kinase-3 beta is a dual specificity kinase differentially regulated by tyrosine and serine/threonine phosphorylation. J Biol Chem. 1994;269:14566–74. [PubMed] [Google Scholar]
- 10.Jope RS, Johnson GV. The glamour and gloom of glycogen synthase kinase-3. Trends Biochem Sci. 2004;29:95–102. doi: 10.1016/j.tibs.2003.12.004. [DOI] [PubMed] [Google Scholar]
- 11.Takahashi-Yanaga F, Shiraishi F, Hirata M, Miwa Y, Morimoto S, Sasaguri T. Glycogen synthase kinase-3beta is tyrosine-phosphorylated by MEK1 in human skin fibroblasts. Biochem Biophys Res Commun. 2004;316:411–5. doi: 10.1016/j.bbrc.2004.02.061. [DOI] [PubMed] [Google Scholar]
- 12.Sayas CL, Ariaens A, Ponsioen B, Moolenaar WH. GSK-3 is activated by the tyrosine kinase Pyk2 during LPA1-mediated neurite retraction. Mol Biol Cell. 2006;17:1834–44. doi: 10.1091/mbc.E05-07-0688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hartigan JA, Xiong WC, Johnson GV. Glycogen synthase kinase 3beta is tyrosine phosphorylated by PYK2. Biochem Biophys Res Commun. 2001;284:485–9. doi: 10.1006/bbrc.2001.4986. [DOI] [PubMed] [Google Scholar]
- 14.Beurel E, Jope RS. The paradoxical pro- and anti-apoptotic actions of GSK3 in the intrinsic and extrinsic apoptosis signaling pathways. Prog Neurobiol. 2006;79:173–89. doi: 10.1016/j.pneurobio.2006.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hardt SE, Tomita H, Katus HA, Sadoshima J. Phosphorylation of eukaryotic translation initiation factor 2Bepsilon by glycogen synthase kinase-3beta regulates beta-adrenergic cardiac myocyte hypertrophy. Circ Res. 2004;94:926–35. doi: 10.1161/01.RES.0000124977.59827.80. [DOI] [PubMed] [Google Scholar]
- 16.Michael A, Haq S, Chen X, Hsich E, Cui L, Walters B, Shao Z, Bhattacharya K, Kilter H, Huggins G, Andreucci M, Periasamy M, Solomon RN, Liao R, Patten R, Molkentin JD, Force T. Glycogen synthase kinase-3beta regulates growth, calcium homeostasis, and diastolic function in the heart. J Biol Chem. 2004;279:21383–93. doi: 10.1074/jbc.M401413200. [DOI] [PubMed] [Google Scholar]
- 17.Yin H, Chao L, Chao J. Kallikrein/kinin protects against myocardial apoptosis after ischemia/reperfusion via Akt-glycogen synthase kinase-3 and Akt-Bad. 14-3-3 signaling pathways. J Biol Chem. 2005;280:8022–30. doi: 10.1074/jbc.M407179200. [DOI] [PubMed] [Google Scholar]
- 18.Juhaszova M, Zorov DB, Kim SH, Pepe S, Fu Q, Fishbein KW, Ziman BD, Wang S, Ytrehus K, Antos CL, Olson EN, Sollott SJ. Glycogen synthase kinase-3beta mediates convergence of protection signaling to inhibit the mitochondrial permeability transition pore. J Clin Invest. 2004;113:1535–49. doi: 10.1172/JCI19906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li M, Wang X, Meintzer MK, Laessig T, Birnbaum MJ, Heidenreich KA. Cyclic AMP promotes neuronal survival by phosphorylation of glycogen synthase kinase 3beta. Mol Cell Biol. 2000;20:9356–63. doi: 10.1128/mcb.20.24.9356-9363.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Menon B, Singh M, Singh K. Matrix metalloproteinases mediate beta-adrenergic receptor-stimulated apoptosis in adult rat ventricular myocytes. Am J Physiol Cell Physiol. 2005;289:C168–C176. doi: 10.1152/ajpcell.00606.2004. [DOI] [PubMed] [Google Scholar]
- 21.Communal C, Singh K, Pimentel DR, Colucci WS. Norepinephrine stimulates apoptosis in adult rat ventricular myocytes by activation of the beta-adrenergic pathway. Circulation. 1998;98:1329–34. doi: 10.1161/01.cir.98.13.1329. [DOI] [PubMed] [Google Scholar]
- 22.Haq S, Michael A, Andreucci M, Bhattacharya K, Dotto P, Walters B, Woodgett J, Kilter H, Force T. Stabilization of beta-catenin by a Wnt-independent mechanism regulates cardiomyocyte growth. Proc Natl Acad Sci U S A. 2003;100:4610–5. doi: 10.1073/pnas.0835895100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bijur GN, Jope RS. Proapoptotic stimuli induce nuclear accumulation of glycogen synthase kinase-3 beta. J Biol Chem. 2001;276:37436–42. doi: 10.1074/jbc.M105725200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Haq S, Choukroun G, Kang ZB, Ranu H, Matsui T, Rosenzweig A, Molkentin JD, Alessandrini A, Woodgett J, Hajjar R, Michael A, Force T. Glycogen synthase kinase-3beta is a negative regulator of cardiomyocyte hypertrophy. J Cell Biol. 2000;151:117–30. doi: 10.1083/jcb.151.1.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Morisco C, Seta K, Hardt SE, Lee Y, Vatner SF, Sadoshima J. Glycogen synthase kinase 3beta regulates GATA4 in cardiac myocytes. J Biol Chem. 2001;276:28586–97. doi: 10.1074/jbc.M103166200. [DOI] [PubMed] [Google Scholar]
- 26.Oyadomari S, Mori M. Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ. 2004;11:381–9. doi: 10.1038/sj.cdd.4401373. [DOI] [PubMed] [Google Scholar]
- 27.Tong H, Imahashi K, Steenbergen C, Murphy E. Phosphorylation of glycogen synthase kinase-3beta during preconditioning through a phosphatidylinositol-3-kinase--dependent pathway is cardioprotective. Circ Res. 2002;90:377–9. doi: 10.1161/01.res.0000012567.95445.55. [DOI] [PubMed] [Google Scholar]
- 28.Coppolino MG, Dedhar S. Bi-directional signal transduction by integrin receptors. Int J Biochem Cell Biol. 2000;32:171–88. doi: 10.1016/s1357-2725(99)00043-6. [DOI] [PubMed] [Google Scholar]
- 29.De Acetis M, Notte A, Accornero F, Selvetella G, Brancaccio M, Vecchione C, Sbroggio M, Collino F, Pacchioni B, Lanfranchi G, Aretini A, Ferretti R, Maffei A, Altruda F, Silengo L, Tarone G, Lembo G. Cardiac overexpression of melusin protects from dilated cardiomyopathy due to long-standing pressure overload. Circ Res. 2005;96:1087–94. doi: 10.1161/01.RES.0000168028.36081.e0. [DOI] [PubMed] [Google Scholar]
- 30.Conant K, St Hillaire C, Nagase H, Visse R, Gary D, Haughey N, Anderson C, Turchan J, Nath A. Matrix metalloproteinase 1 interacts with neuronal integrins and stimulates dephosphorylation of Akt. J Biol Chem. 2004;279:8056–62. doi: 10.1074/jbc.M307051200. [DOI] [PubMed] [Google Scholar]
- 31.Aberle H, Bauer A, Stappert J, Kispert A, Kemler R. beta-catenin is a target for the ubiquitin-proteasome pathway. EMBO J. 1997;16:3797–804. doi: 10.1093/emboj/16.13.3797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bijur GN, Jope RS. Glycogen synthase kinase-3 beta is highly activated in nuclei and mitochondria. Neuroreport. 2003;14:2415–9. doi: 10.1097/00001756-200312190-00025. [DOI] [PubMed] [Google Scholar]
- 33.Guyton KZ, Xu Q, Holbrook NJ. Induction of the mammalian stress response gene GADD153 by oxidative stress: role of AP-1 element. Biochem J. 1996;314 (Pt 2):547–54. doi: 10.1042/bj3140547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McCullough KD, Martindale JL, Klotz LO, Aw TY, Holbrook NJ. Gadd153 sensitizes cells to endoplasmic reticulum stress by down-regulating Bcl2 and perturbing the cellular redox state. Mol Cell Biol. 2001;21:1249–59. doi: 10.1128/MCB.21.4.1249-1259.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shiraishi H, Okamoto H, Yoshimura A, Yoshida H. ER stress-induced apoptosis and caspase-12 activation occurs downstream of mitochondrial apoptosis involving Apaf-1. J Cell Sci. 2006 doi: 10.1242/jcs.03160. [DOI] [PubMed] [Google Scholar]