

Abstract
Background
Filamentous fungi such as Aspergillus niger have a high capacity secretory system and are therefore widely exploited for the industrial production of native and heterologous proteins. However, in most cases the yields of non-fungal proteins are significantly lower than those obtained for fungal proteins. One well-studied bottleneck appears to be the result of mis-folding of heterologous proteins in the ER during early stages of secretion, with related stress responses in the host, including the unfolded protein response (UPR). This study aims at uncovering transcriptional and translational responses occurring in A. niger exposed to secretion stress.
Results
A genome-wide transcriptional analysis of protein secretion-related stress responses was determined using Affymetrix DNA GeneChips and independent verification for selected genes. Endoplasmic reticulum (ER)-associated stress was induced either by chemical treatment of the wild-type cells with dithiothreitol (DTT) or tunicamycin, or by expressing a human protein, tissue plasminogen activator (t-PA). All of these treatments triggered the UPR, as shown by the expression levels of several well-known UPR target genes. The predicted proteins encoded by most of the up-regulated genes function as part of the secretory system including chaperones, foldases, glycosylation enzymes, vesicle transport proteins, and ER-associated degradation proteins. Several genes were down-regulated under stress conditions and these included several genes that encode secreted enzymes. Moreover, translational regulation under ER stress was investigated by polysomal fractionation. This analysis confirmed the post-transcriptional control of hacA expression and highlighted that differential translation also occurs during ER stress, in particular for some genes encoding secreted proteins or proteins involved in ribosomal biogenesis and assembly.
Conclusion
This is first genome-wide analysis of both transcriptional and translational events following protein secretion stress. Insight has been gained into the molecular basis of protein secretion and secretion-related stress in an effective protein-secreting fungus, and provides an opportunity to identify target genes for manipulation in strain improvement strategies.
Background
Many species of filamentous fungi such as Aspergillus niger are effective secretors of, mainly, hydrolytic enzymes to facilitate their saprophytic lifestyles by providing substrates from polymeric organic materials. This high capacity secretory system has driven the exploitation of filamentous fungi as cell factories for provision of enzymes used in a wide variety of applications [1]. Since the availability of gene-transfer systems, several fungal species have become potentially excellent hosts for achieving commercial yields of heterologous proteins. However, the yields of recombinants enzymes are often lower than desired, and this is especially so when the donor organism is not a fungus [2]. Many approaches have been used to overcome the bottlenecks to achieving high-secreted yields of heterologous proteins from fungi [2,3] but the levels do not reach the same level as the best native protein.
Several data suggest that bottlenecks mainly exist at the post-transcriptional level, and most probably within the secretory pathway [4,5]. The ER orchestrates the folding and some post-translational modifications of proteins that reside in, or pass through, the endomembrane system of a eukaryotic cell. In expression systems, the large flux of proteins being translocated into the ER generates a need to enhance the efficiency of protein folding and transport as well as the quality control of the synthesized proteins. Increased flux of proteins through the ER, especially those which do not fold correctly, or at least with the required kinetics, lead to the induction of stress responses that are collectively called protein secretion stress or ER stress. Mechanisms that allow the cell to sense the state of the lumen and to respond to ER stress conditions have been characterized in detail in the yeast Saccharomyces cerevisiae and in mammalian cells [6]. The first of these mechanisms is the unfolded protein response (UPR), a complex intracellular signaling pathway that increases the transcriptional activity of a number of genes involved in protein folding, glycosylation and transport [6,7]. The UPR also appears to be intimately linked to the ER-associated (protein) degradation (ERAD) pathway [7,8], in which misfolded proteins in the ER lumen are retrotranslocated (dislocated) through the translocon to the cytoplasm, and are ubiquitinated and degraded by the proteasome [9]. Recently, a novel type of feed-back mechanism, termed repression under secretion stress (RESS), has been suggested in filamentous fungi [10,11]. It functions during secretion stress and down-regulates the transcript levels of some genes. A gene array study under ER stress conditions has recently been published with A. nidulans where ca. 30% of the predicted genes were represented on the array [12]. They described the first attempt to analyze in part the influence of the production and secretion of a heterologous protein on the cellular transcript profile of a filamentous fungus. A complete analysis of ER stress responses in the yeast S. cerevisiae, a fungus that does not secrete proteins as effectively as A. niger has also been described [7].
The ER surveillance system continuously coordinates the activity and participation of the processing and degradation pathways for unfolded proteins. Upon accumulation of unfolded proteins in the ER lumen, the UPR is activated, reducing the amount of new protein translocated into the ER lumen, increasing dislocation of proteins from the lumen of the ER and, then, their degradation, and bolstering the protein-folding capacity of the ER. Thus, understanding the process of folding and the stress responses in filamentous fungi may hold the major key to improving their use as cell factories for production of recombinant proteins. A genome-wide expression analysis of these secretion-related stress responses has never been reported in industrially-exploited species, since genome sequence data has not previously been available. Recently, the sequencing and annotation of the genome of A. niger have been completed [13]. DNA GeneChips have been made available and provide an unprecedented resource for exploring expression profiles in response to particular environmental cues. Here, we report the first gene expression analysis studies showing the response to various secretion stresses and assessing the breadth of the UPR in A. niger.
Results
Transcriptional analysis of the ER stress response
To gain further insight into the UPR of filamentous fungi, we identified transcriptional targets of this signaling pathway in A. niger by monitoring mRNA levels using oligonucleotide arrays on Affymetrix GeneChips. The UPR was induced by treating mycelium cultures for 2 h with two chemical agents which disrupt protein folding in the ER. Dithiothreitol (DTT) is a strong reducing agent that prevents disulfide bond formation. Tunicamycin is a drug that inhibits N-linked glycosylation by preventing core oligosaccharide addition to nascent polypeptides and thereby blocks protein folding and transit through the ER [14]. Finally, a strain [15] producing recombinant tissue plasminogen activator (t-PA), a serine protease, was chosen to assess the UPR under conditions of heterologous protein production. Previous studies indicated that the expression of t-PA in A. niger leads to the appearance of the spliced form of hacA mRNA (hacAi) (which is translated to yield the UPR-mediating transcription factor HacA) and the simultaneous up-regulation of bipA and pdiA [15,16]. Unlike with S. cerevisiae [7] it was not possible to include strains of A. niger that are devoid of genes encoding key mediators of the UPR such as ireA and hacA because such strains are not available despite the cloning and functional characterization of those genes [7,17]. Deletion of ireA in A. niger has not been achieved and the only ΔhacA strain described to date grows very differently from the wild-type strain of A. niger (H. Mulder, personal communication) and, anyway, is not generally available.
Replicate experiments were performed for each of the three stress responses. All (experimental and control) chips were normalized/scaled to a target intensity of 100 by global scaling (details regarding Scaling and Normalization are listed in the Affymetrix Microarray Suite User Guide Version 5.0, Appendix D) with scaling factors being comparable for all comparisons [see Additional file 1]. Next, intra-experiment and inter-experiment GeneChip comparisons were processed for each of the 3 stress conditions and all genes with a detected transcript in at least one condition were checked for even distribution of intensities. For all the probe sets on the Affymetrix GeneChip, we determined the fold change in expression due to each treatment by comparing its expression level in the treated sample to its level in the untreated control. Two comparisons were processed for each of the 3 stress conditions. Increased expression of bipA, pdiA (two genes expected to be up-regulated by ER stress) and an uncharacterised A. niger gene (An02g13410, putative homolog of an acetyl CoA transporter) shown on the GeneChips to be up-regulated, was confirmed in response to both chemical treatments and to t-PA production by Northern hybridisation (Fig. 1A). Increased levels of hacAi mRNA was also confirmed by RT-PCR across the hacA mRNA intron, and RT-PCR was also used to confirm both the GeneChip and Northern hybridisation data showing that the mRNA levels of bipA and pdiA were increased under stress (Fig. 1B). For tunicamycin treatment especially but also t-PA expression, overall variations in gene expression levels were generally small, whereas DTT treatment showed a relatively large reponse [see Additional files 2, 3, 4]. With standard criteria of a fold change of 2, less than 10 differentially-expressed genes would be detected in the tunicamycin treatment and therefore selection criteria were adjusted to a less stringent 1.5-fold change for all treatments. According to the analytical criteria adopted (at least 1.5-fold change), a list of induced genes was produced with 79 independent entries for the heterologous protein production condition, 38 entries for the tunicamycin treatment and 865 entries for the DTT treatment. The repressed gene sets identified with these restrictions constituted 110, 11 and 774 independent entries from the t-PA producing strain and for the tunicamycin and DTT treatments respectively. Tables 1 and 2 present the lists of genes that were up-regulated or down-regulated by at least 2 of the 3 treatments respectively. A more complete list of genes differentially expressed following each treatment (tunicamycin and t-PA production) is provided in the supplementary information [see Additional files 5 and 6]. Moreover, an additional GeneChip experiment was conducted after a 1 h exposure to tunicamycin and results are also included in those Tables.
Figure 1.
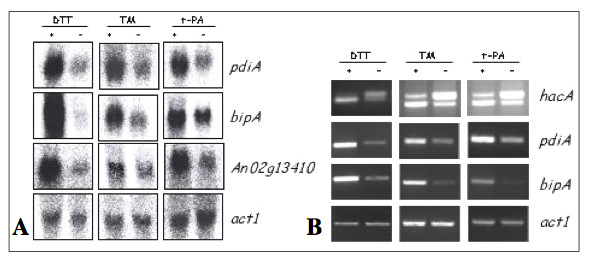
GeneChip results were confirmed for some genes using Northern blotting and RT-PCR. Examples are provided for both Northern blots (A) and RT-PCR (B). Note that the RT-PCR for the hacA mRNA was designed to indicate enhanced splicing of the hacA mRNA intron under stress conditions (DTT, tunicamycin and production of t-PA). This is shown as a relative increase in the amount of the processed (lower band) hacAi form of the mRNA compared to the unprocessed higher band (hacAu). Probing or PCR for an actin gene was used as a non-stress-responsive control transcript.
Table 1.
Genes induced by at least 2 treatments.
| DSM code | Predicted protein | ||||
| Protein Folding | Tun 1 h | Tun 2 h | DTT | tPA | |
| An01g13220 | similar to the chaperone LHS1 | 3.4 ± 0.5 | 1.5 ± 0.7 | 2.5 ± 0.3 | 1.7 ± 0.8 |
| An02g14800 | Protein disulfide isomerase PDIA | 1.7 ± 0.1 | 2.6 ± 0.1 | 2.2 ± 0.004 | 1.9 ± 0.01 |
| An01g04600 | Protein disulfide isomerase PRPA | 3.8 ± 0.2 | 2.6 ± 0.3 | 4.0 ± 0.3 | 2.2 ± 0.2 |
| An01g08420 | calnexin CNXA | 3.0 ± 0.3 | 2.3 ± 0.1 | 3.2 ± 0.5 | 2.2 ± 0.1 |
| An11g04180 | chaperone BIPA | 3.2 ± 0.7 | 2.6 ± 0.6 | 4.7 ± 0.6 | 2.4 ± 0.008 |
| An16g07620 | similar to ER oxidising protein ERO1 | 1.9 ± 0.7 | 2.6 ± 0.4 | 3.1 ± 0.9 | 3.1 ± 0.5 |
| An18g02020 | Protein disulfide isomerase TIGA | *1.3 ± 0.5 | 1.7 ± 0.4 | *1.7 ± 0.03 | 1.8 ± 0.02 |
| An11g11250 | similar to the chaperone P58IPK Homo sapiens | 2.5 ± 0.6 | 1.9 ± 0.07 | *1.3 ± 0.3 | 1.5 ± 0.05 |
| An05g00880 | similar to dnaJ protein homolog SCJ1 | 2.5 ± 0.3 | 1.5 ± 0.06 | 2.1 ± 0.3 | *1.6 ± 0.2 |
| Translocation/signal peptidase complex | |||||
| An01g13070 | similar to ER protein-translocation complex subunit SEC63 | *1.3 ± 0.5 | 1.6 ± 0.09 | *1.7 ± 0.2 | 2.5 ± 0.1 |
| An16g08830 | similar to component of subcomplex SEC71 | 1.8 ± 0.2 | *1.4 ± 0.08 | 1.9 ± 0.4 | *1.7 ± 0.04 |
| An17g00090 | similar to translocation complex subunit SEC72 | 1.7 ± 0.2 | 1.6 ± 0.2 | 2.1 ± 0.5 | *1.3 ± 0.1 |
| An01g11630 | similar to translocation complex component SSS1 | 2.5 ± 0.2 | 1.6 ± 0.3 | 1.6 ± 0.04 | *1.6 ± 0.1 |
| An09g05420 | similar to signal peptidase subunit SPC3 | 2.7 ± 0.3 | 1.8 ± 0.2 | 1.5 ± 0.2 | 1.8 ± 0.8 |
| An01g00560 | similar to signal peptidase subunit SEC11 | 2.2 ± 0.4 | 1.9 ± 0.06 | *1.1 ± 0.2 | 1.7 ± 0.009 |
| An15g06470 | similar to signal sequence receptor α-subunit | 1.8 ± 0.6 | 1.5 ± 0.07 | *1.5 ± 0.4 | 1.5 ± 0.08 |
| Glycosylation | |||||
| An14g05910 | similar to mannosyltransferase ALG2 | 2.3 ± 0.5 | *2.9 ± 1.1 | *1.7 ± 0.2 | 1.5 ± 0.1 |
| An18g02360 | similar to mannosyltransferase ALG3 | 2.4 ± 0.4 | 4.8 ± 2.1 | *-1.4 ± 0.3 | 2.0 ± 0.3 |
| An03g04410 | similar to glucosyltransferase ALG5 | 1.8 ± 0.3 | *1.7 ± 0.001 | 2.2 ± 0.4 | 1.7 ± 0.1 |
| An02g03240 | similar to N-acetylglucosaminephosphotransferase ALG7 | 1.8 ± 0.3 | 2.8 ± 0.5 | 2.1 ± 0.1 | *1.7 ± 0.2 |
| An07g04190 | similar to glycosyltransferase WBP1 | 1.9 ± 0.2 | 1.8 ± 0.09 | *1.4 ± 0.2 | 1.7 ± 0.09 |
| An02g14560 | oligosaccharyltransferase alpha subunit OSTA | 1.7 ± 0.09 | 1.6 ± 0.06 | *-1.4 ± 0.007 | 1.6 ± 0.4 |
| An18g03920 | similar to oligosaccharyltransferase subunit OST2 | 1.9 ± 0.4 | 1.5 ± 0.2 | 1.5 ± 0.1 | *1.2 ± 0.2 |
| An18g04260 | similar to UDP-galactose transporter HUT1 | 2.3 ± 0.2 | *-1.0 ± 0.5 | 2.1 ± 0.3 | 1.9 ± 0.2 |
| An18g06220 | similar to alpha-mannosidase MNS1 | *1.1 ± 0.5 | 2.4 ± 0.6 | *1.2 ± 0.004 | 1.8 ± 0.1 |
| An13g00620 | similar to beta subunit of an ER alpha-glucosidase | *1.4 ± 0.5 | 1.9 ± 0.2 | *-1.8 ± 0.1 | 1.7 ± 0.2 |
| An15g01420 | similar to glucosidase I CWH41 | *1.3 ± 0.5 | 1.7 ± 0.5 | *-1.8 ± 0.2 | 1.6 ± 0.01 |
| An04g05250 | similar to dolichol synthesis protein RER2 | 1.7 ± 0.1 | *1.3 ± 0.08 | 2.2 ± 0.1 | *1.2 ± 0.1 |
| An02g14940 | similar to flippase RFT1 | *1.5 ± 0.5 | *-1.0 ± 0.9 | 1.7 ± 0.1 | 1.7 ± 0.1 |
| Vesicle trafficking/transport | |||||
| An03g04940 | similar to COPII vesicle coat component protein ERV41 | 2.2 ± 0.07 | 1.9 ± 0.08 | 2.4 ± 0.8 | 2.3 ± 0.6 |
| An01g04320 | similar to COPII vesicle coat component protein ERV46 | 2.1 ± 0.2 | 2.4 ± 0.3 | *1.5 ± 0.1 | 2.1 ± 0.05 |
| An02g04250 | similar to ER protein P58 (lectin family) Rattus norvegicus | 1.7 ± 0.5 | 1.8 ± 0.2 | *1.2 ± 0.01 | 1.5 ± 0.3 |
| An02g08450 | secretory gene product NSFA | 1.5 ± 0.2 | *1.2 ± 0.006 | 2.5 ± 0.06 | *1.1 ± 0.06 |
| An08g06780 | similar to ER to Golgi transport protein USO1 | 2.0 ± 0.1 | *6.5 ± 3.2 | 5.3 ± 3.6 | *1.7 ± 0.6 |
| Proteolytic degradation | |||||
| An16g06750 | similar to D-stereospecific aminopeptidase | *2.0 ± 0.5 | 2.1 ± 0.1 | 14.0 ± 8.0 | *2.1 ± 0.2 |
| An08g09000 | similar to ubiquitin like protein DSK2 | 1.7 ± 0.4 | *1.0 ± 0.1 | 3.0 ± 0.2 | *-1.1 ± 0.2 |
| An09g00950 | similar to aminopeptidase DAP Ochrobactrum anthropi | 1.6 ± 0.2 | *-1.7 ± 0.3 | 6.2 ± 1.4 | *1.1 ± 0.2 |
| Lipid/Inositol metabolism | |||||
| An02g13410 | similar to acetyl-coenzyme A transporter AT-1 | 2.4 ± 0.04 | 1.8 ± 0.1 | 5.4 ± 0.3 | 2.3 ± 0.2 |
| An11g02990 | similar to cytochrome P450 DIT2 | *-1.2 ± 0.5 | *1.2 ± 0.002 | 2.0 ± 0.3 | 1.6 ± 0.03 |
| An14g07030 | similar to carboxylesterase Aspergillus fumigatus | *1.6 ± 0.5 | *-1.9 ± 1.2 | 43.0 ± 5.3 | 1.7 ± 0.2 |
| Cellular transport | |||||
| An12g03150 | similar to multidrug resistance protein ATRD | 2.0 ± 0.4 | *-1.1 ± 0.3 | 10.1 ± 2.5 | *-1.1 ± 0.1 |
| Translation | |||||
| An14g06610 | similar to elongation factor Eef1 alpha-A chain TEF2 | 1.6 ± 0.1 | *1.6 ± 0.3 | 8.6 ± 1.3 | *1.4 ± 0.3 |
| Stress related | |||||
| An12g03580 | similar to glutathione S-transferase 3 MGST3 H. sapiens | *1.2 ± 0.5 | *1.3 ± 0.1 | 3.2 ± 0.5 | 5.1 ± 1.4 |
| An01g14100 | weakly similar to stress protein HERP Mus musculus | 2.0 ± 0.1 | *1.5 ± 0.2 | 1.5 ± 0.3 | *1.8 ± 0.3 |
| Cell Cycle and DNA processing | |||||
| An01g08170 | similar to DNA repair endonuclease RAD1 S. pombe | 1.8 ± 0.5 | *1.8 ± 0.2 | 2.2 ± 1.1 | *1.6 ± 0.2 |
| C-compound and carbohydrate metabolism | |||||
| An09g06400 | similar to chitinase CHIA Aspergillus nidulans | -2.1 ± 0.5 | *1.0 ± 0.008 | 2.4 ± 0.3 | 2.3 ± 0.8 |
| An16g09070 | similar to glucosamine-6-phosphate deaminase A. fumigatus | *-1.2 ± 0.5 | *1.4 ± 0.2 | 2.1 ± 0.08 | 2.4 ± 0.7 |
| Unclassified | |||||
| An08g03960 | hypothetical endoplasmic reticulum associated protein | 2.1 ± 0.09 | 1.7 ± 0.07 | 3.3 ± 1.0 | 1.6 ± 0.06 |
| An02g00120 | hypothetical protein | *2.4 ± 0.5 | *1.2 ± 0.3 | 2.0 ± 0.5 | 1.8 ± 0.07 |
| An08g03970 | hypothetical protein | *1.7 ± 0.5 | 2.0 ± 0.4 | *1.2 ± 0.02 | 1.8 ± 0.1 |
| An09g00650 | hypothetical protein | 2.0 ± 0.3 | *-1.30.4 | 2.7 ± 0.2 | 4.0 ± 1.4 |
| An17g00660 | hypothetical protein | *-1.1 ± 0.2 | *-1.5 ± 0.6 | 5.0 ± 2.7 | 1.6 ± 0.1 |
| An11g06970 | hypothetical protein | *3.1 ± 0.3 | *1.7 ± 0.7 | 17.3 ± 6.0 | 2.3 ± 0.6 |
| An16g00070 | hypothetical protein | *-1.4 ± 0.5 | *1.1 ± 0.1 | 5.1 ± 0.1 | 1.5 ± 0.2 |
| An16g08470 | hypothetical protein | *1.8 ± 0.3 | *1.1 ± 0.2 | 1.5 ± 0.1 | 1.5 ± 0.05 |
| An02g14500 | hypothetical protein | *-1.2 ± 0.4 | *1.0 ± 0.1 | 9.3 ± 2.7 | 1.7 ± 0.1 |
| An03g00720 | hypothetical protein | *1.1 ± 0.5 | *-1.2 ± 0.06 | 2.6 ± 1.1 | 1.9 ± 0.7 |
| An04g02250 | questionable ORF | *2.2 ± 0.3 | 2.0 ± 0.2 | 20.2 ± 10.3 | *1.3 ± 0.006 |
| An14g01990 | hypothetical protein | 3.0 ± 0.4 | *-1.4 ± 0.2 | 4.3 ± 0.9 | *-1.1 ± 0.03 |
| An08g04260 | hypothetical protein | 2.0 ± 0.6 | *1.7 ± 0.3 | 2.4 ± 0.01 | *1.3 ± 0.07 |
| An07g10280 | hypothetical protein | 2.0 ± 0.7 | *1.6 ± 0.08 | 1.8 ± 0.9 | *1.6 ± 0.2 |
| An09g06130 | hypothetical protein | 2.0 ± 0.3 | *2 ± 0.2 | 1.6 ± 0.4 | *1.4 ± 0.09 |
| An08g09860 | hypothetical protein | 2.3 ± 0.6 | *1.2 ± 0.4 | 7.4 ± 1.0 | *-1.4 ± 0.1 |
| An18g06120 | similar to secreted protein vc33_1 | 1.9 ± 0.2 | *-1.1 ± 0.1 | 2.2 ± 0.1 | *1.0 ± 0.05 |
| An14g06550 | hypothetical protein | 2.1 ± 0.3 | *1.2 ± 0.1 | 2.8 ± 0.6 | *1.2 ± 0.1 |
| An15g02650 | hypothetical protein | 2.8 ± 1.5 | *1.1 ± 0.2 | 2.8 ± 0.6 | *1.1 ± 0.1 |
| An11g10800 | similar to probable isochorismatase | 2.3 ± 0.3 | *1.0 ± 0.002 | 11.5 ± 2.2 | *1.2 ± 0.2 |
| An18g01690 | hypothetical protein | 2.3 ± 1.5 | *2.1 ± 0.5 | 2.4 ± 0.9 | *1.1 ± 0.1 |
| An16g08680 | hypothetical protein | 1.7 ± 0.4 | *-1.1 ± 0.2 | 6.7 ± 0.5 | *1.1 ± 0.07 |
The fold changes in expression are indicated for all the treatments (Tun = tunicamycin, DTT = dithiothreitol, tPA = production of t-PA) and are averaged over two experiments. Similarities are expressed in comparison with the Saccharomyces cerevisiae genome except when indicated. The symbol* indicates that the value does not meet one of the 3 restrictive criteria defined in the Method section. The genes induced by the three treatments are marked in bold.
Table 2.
Genes repressed by at least 2 treatments.
| DSM code | Predicted protein | ||||
| Protein Fate | Tun 1 h | Tun 2 h | DTT | tPA | |
| Proteolytic Degradation | |||||
| An12g05960 | similar to dipeptidyl peptidase II DPPII Rattus norvegicus | *1.3 ± 0.5 | *1.6 ± 0.2 | -1.9 ± 0.3 | -1.5 ± 0.6 |
| An03g01660 | similar to vacuolar aminopeptidase Y APE3 | -2.1 ± 0.3 | *1.0 ± 0.05 | -1.7 ± 0.7 | *1.1 ± 0.02 |
| An03g05200 | similar to carboxypeptidase S1 Penicillium janthinellum | -2.1 ± 0.4 | *1.1 ± 0.4 | -1.8 ± 0.6 | *-1.4 ± 0.08 |
| An04g01440 | similar to vacuolar aspartyl protease PEP4 | -2.1 ± 0.3 | *1.1 ± 0.2 | -1.6 ± 0.04 | *-1.3 ± 0.03 |
| An08g08750 | carboxypeptidase Y CPY | -1.9 ± 0.2 | *1.0 ± 0.06 | -2.2 ± 0.02 | *-1.3 ± 0.04 |
| An14g00620 | similar to aminopeptidase Aspergillus oryzae | -2.4 ± 0.5 | *-1.0 ± 0.01 | -2.3 ± 0.1 | *-1.1 ± 0.2 |
| Lipid metabolism | |||||
| An16g01880 | similar to lysophospholipase Aspaergillus foetidus | *-2.2 ± 0.5 | -1.7 ± 0.3 | -99.8 ± 5.6 | 1.4 ± 0.8 |
| An02g09540 | similar to choline permease HNM1 | *1.1 ± 0.5 | *1.0 ± 0.6 | -2.4 ± 0.9 | -1.9 ± 0.1 |
| An16g06090 | similar to choline permease HNM1 | *-1.3 ± 0.3 | *1.1 ± 0.01 | -1.9 ± 0.2 | -2.2 ± 0.5 |
| An15g06810 | similar to cytochrome-b5 reductase MCR1 | *1.1 ± 0.2 | *1.6 ± 0.1 | -1.7 ± 0.3 | -2.6 ± 011 |
| An01g03350 | similar to C-8 sterol isomerase ERG1 Neurospora crassa | -1.9 ± 0.3 | *1.2 ± 0.2 | -2.1 ± 0.6 | *-1.2 ± 0.3 |
| An02g03580 | similar to lipid metabolism protein ERG28 | -1.8 ± 0.2 | *1.2 ± 0.02 | -2.0 ± 0.2 | *-1.3 ± 0.06 |
| An01g07000 | similar to C-14 sterol reductase ERG24 | -1.7 ± 0.1 | *1.2 ± 0.2 | *3.9 ± 0.4 | -1.9 ± 0.08 |
| Cell wall | |||||
| An16g03370 | similar to protein involved in of cell wall biogenesis CWH43 | -2.0 ± 0.2 | *1.2 ± 0.1 | -2.4 ± 0.1 | *-1.2 ± 0.01 |
| An16g07040 | similar to cell wall glucanase SCW10 | -1.7 ± 0.1 | *-1.1 ± 0.07 | -4.0 ± 0.5 | *-1.0 ± 0.04 |
| Amino acid metabolism | |||||
| An04g00990 | NADP-dependent glutamate dehydrogenase GDHA | *1.1 ± 0.3 | *-3.5 ± 2.0 | -3.6 ± 0.7 | -1.5 ± 0.005 |
| An16g01610 | similar to hydroxylase BBH Rattus norvegicus | *-1.2 ± 0.5 | -2.5 ± 0.7 | -13.5 ± 0.1 | *-1.0 ± 0.5 |
| An01g14730 | similar to lactonizing enzyme I TcMLE Trichosporon cutaneum | -1.6 ± 0.1 | *-1.3 ± 0.1 | -1.5 ± 0.2 | *1.3 ± 0.2 |
| An02g00190 | similar to enantiomer-selective amidase AMDA Rhodococcus sp. | -2.9 ± 0.5 | *1.1 ± 0.2 | -2.3 ± 0.3 | *-1.3 ± 0.5 |
| An12g10000 | similar to GABA permease GABA Aspergillus nidulans | *-1.0 ± 0.08 | *1.0 ± 0.1 | -5.5 ± 1.9 | -1.8 ± 0.09 |
| Phosphate Metabolism | |||||
| An12g01910 | similar to phytase PHYA3 Aspergillus fumigatus | -1.5 ± 0.1 | -1.6 ± 0.3 | -21.1 ± 2.5 | *-1.0 ± 0.1 |
| C-compound and carbohydrate metabolism | |||||
| An09g00270 | alpha-galactosidase AGLC | *-1.2 ± 0.1 | -2.0 ± 0.5 | -5.9 ± 2.0 | *1.1 ± 0.3 |
| An09g00260 | alpha-galactosidase AGLC | *-2.6 ± 0.5 | -2.1 ± 0.3 | -6.0 ± 4.3 | *1.1 ± 0.2 |
| An01g01540 | similar to alpha.alpha-trehalase TREA A. nidulans | -3.0 ± 0.6 | *-1.1 ± 0.1 | -2.7 ± 0.6 | *1.2 ± 0.02 |
| An01g00780 | endo-1.4-xylanase XYNB | -3.4 ± 0.6 | *1.5 ± 0.4 | -4.3 ± 0.9 | *1.0 ± 0.1 |
| An05g00930 | similar to mitochondrial malic enzyme MAE1 | -2.1 ± 0.2 | *1.2 ± 0.1 | -4.0 ± 1.0 | *-1.0 ± 0.04 |
| An19g00090 | similar to exo-beta-1.3-glucanase CMG1 Coniothyrium minitans | -2.6 ± 0.3 | *1.6 ± 0.1 | -4.6 ± 1.0 | *3.7 ± 0.06 |
| An11g04780 | similar to protein involved in active glycerol uptake GUP1 | -1.7 ± 0.2 | *1.2 ± 0.2 | -2.4 ± 0.3 | *1.0 ± 0.1 |
| metabolism of vitamins. cofactors. and prosthetic groups | |||||
| An01g04250 | similar to uroporphyrinogen decarboxylase HEM12 | *-1.2 ± 0.06 | *1.1 ± 0.04 | -1.6 ± 0.1 | -1.6 ± 0.1 |
| Cellular transport | |||||
| An12g10320 | similar to high-affinity zinc transport protein ZRT1 | *1.1 ± 0.05 | *1.4 ± 0.04 | -2.2 ± 0.04 | -3.2 ± 1.1 |
| An16g06740 | similar to cadmium resistance protein YCF1 | *-1.4 ± 0.2 | *-1.7 ± 0.5 | -1.7 ± 0.2 | -1.7 ± 0.3 |
| Cell Rescue. Defense and Virulence | |||||
| An07g00570 | similar to membrane protein PTH11 Magnaporthe grisea | *-1.1 ± 0.1 | *-1.0 ± 0.04 | -2.7 ± 0.06 | -2.0 ± 0.1 |
| An18g00980 | similar to membrane protein PTH11 M. grisea | *-3.0 ± 0.5 | *1.4 ± 0.2 | -4.6 ± 0.2 | -2.0 ± 0.1 |
| An16g05920 | similar to membrane protein PTH11 M. grisea | *1.1 ± 0.2 | -3.9 ± 1.1 | -5.5 ± 1.1 | *1.0 ± 0.08 |
| Nucleotide/nitrogen and sulfur metabolism | |||||
| An10g00800 | similar to purine nucleoside permease NUP Candida albicans | -5.4 ± 0.8 | *-1.5 ± 0.07 | -31.9 ± 2.7 | *1.2 ± 0.1 |
| An18g01220 | similar to allantoate permease DAL5 | -1.6 ± 0.1 | *1.6 ± 0.2 | -4.2 ± 0.06 | *-1.2 ± 0.05 |
| Other | |||||
| An07g06530 | similar to multicopy suppressor SUR7 | *2.0 ± 0.4 | *1.7 ± 0.2 | -5.0 ± 1.9 | -1.6 ± 0.1 |
| An16g05910 | similar to putative cytochrome P450 A. fumigatus | *-1.2 ± 0.08 | -2.3 ± 0.5 | -5.8 ± 0.2 | *2.2 ± 0.05 |
| An11g09870 | similar to palmitoleyltransferase ERF2 | -1.6 ± 0.1 | *1.4 ± 0.2 | -2.1 ± 0.3 | *1.0 ± 0.01 |
| Unclassified | |||||
| An02g03570 | similar to membrane protein YBR159w | -2.1 ± 0.2 | *-1.3 ± 0.3 | *-1.9 ± 1.0 | -1.6 ± 0.2 |
| An11g07340 | similar to hypothetical O-methyl transferase ENCK | *1.3 ± 0.1 | *1.3 ± 0.5 | -4.2 ± 0.6 | -4.2 ± 1.7 |
| An12g09640 | similar to hypothetical GTP cyclohydrolase | *2.6 ± 0.5 | *1.3 ± 0.4 | -3.3 ± 0.2 | -1.9 ± 0.5 |
| An01g09220 | weakly similar to tyrosinase MELC2 | *-2.1 ± 0.5 | *2.1 ± 0.2 | -5.2 ± 1.7 | -1.8 ± 0.4 |
| An03g00770 | similar to allergic allergen rAsp f 4 A. fumigatus | *-2.3 ± 0.6 | *-1.1 ± 0.3 | -2.6 ± 0.1 | -2.8 ± 0.6 |
| An08g03760 | hypothetical protein | *-1.1 ± 0.1 | *-1.3 ± 0.07 | -4.0 ± 1.8 | -2.4 ± 0.3 |
| An02g08330 | hypothetical protein | *1.2 ± 0.2 | *-17 ± 11.3 | 13.5 ± 5.2 | -1.8 ± 0.1 |
| An06g01000 | hypothetical protein | *-1.1 ± 0.1 | *-1.0 ± 0.05 | -1.83 ± 0.5 | -2.3 ± 0.1 |
| An11g07020 | hypothetical protein | *1.3 ± 0.3 | *1.2 ± 0.02 | -2.1 ± 0.3 | -1.8 ± 0.2 |
| An02g08300 | hypothetical protein | *2.2 ± 0.5 | *-1.7 ± 0.3 | -3.2 ± 0.8 | -8.6 ± 0.9 |
| An03g00840 | hypothetical protein | 1.8 ± 0.2 | *-1.2 ± 0.2 | *1.3 ± 0.1 | -2.4 ± 0.9 |
| An18g01000 | hypothetical protein | *-2.4 ± 0.5 | *1.1 ± 0.1 | -4.6 ± 0.5 | -1.8 ± 0.2 |
| An13g01520 | hypothetical protein | -1.5 ± 0.1 | *-2.9 ± 0.2 | *-1.0 ± 0.4 | -1.9 ± 0.2 |
| An02g00120 | hypothetical protein | -2.4 ± 0.5 | *1.2 ± 0.3 | -2.0 ± 0.5 | *-1.8 ± 0.07 |
| An04g01690 | hypothetical protein | -2.1 ± 0.4 | *-1.0 ± 0.3 | -3.1 ± 1.1 | *-1.1 ± 0.03 |
| An05g01770 | hypothetical protein | -1.5 ± 0.1 | *-1.5 ± 0.3 | -5.8 ± 1.0 | *1.0 ± 0.5 |
| An07g05160 | hypothetical protein | -1.7 ± 0.1 | *-1.2 ± 0.2 | -2.1 ± 0.05 | *1.0 ± 0.08 |
| An07g08400 | strong similarity to allergen rAsp f 4 A. fumigatus | -1.8 ± 0.2 | *1.3 ± 0.2 | -4.2 ± 1.2 | *1.1 ± 0.07 |
| An08g08600 | similar to RTN2 | -1.9 ± 0.2 | *-1.0 ± 0.01 | -3.3 ± 0.6 | *-1.0 ± 0.1 |
| An12g10590 | hypothetical protein | -2.6 ± 0.4 | *1.1 ± 0.03 | -12.0 ± 2.1 | *1.0 ± 0.02 |
| An15g01740 | similar to ER protein of unknown function | -1.6 ± 0.1 | *1.4 ± 0.1 | -2.1 ± 0.3 | *1.0 ± 0.1 |
The fold changes in expression are indicated for all the treatments (Tun = tunicamycin, DTT = dithiothreitol, tPA = production of t-PA) and are averaged over two experiments. Similarities are expressed in comparison with the Saccharomyces cerevisiae genome except when indicated. The symbol* indicates that the value does not meet one of the 3 restrictive criteria defined in the Method section.
Comparisons of ER stress induced under different conditions
Comparison of the signal intensities on the GeneChips showed that, while the replicates for each stress condition clustered closely together, the DTT stress was more distant from the t-PA and tunicamycin stresses which were more closely clustered together (Fig. 2). The numbers of genes that were induced or repressed under each of the conditions, together with a summary of those regulated in more than one condition, are shown in Fig. 3. Of the up-regulated genes, only ten were found in all three conditions and these are indicated in Table 1. This list is dominated by genes that are expected to be directly influenced by the UPR, e.g. pdiA, prpA, bipA, clxA, and lhsA. It is striking that the majority of the DTT-induced genes were not anticipated ER stress genes and we conclude that, although DTT can induce the expression of a large number of genes it is not the most appropriate stress agent for studies of ER stress, a conclusion reached independently elsewhere [12,18]. In contrast, most of the tunicamycin-induced genes are also induced by t-PA and/or DTT and about half of the t-PA-induced genes are also induced by either DTT or tunicamycin. As previously reported in yeast [7], our results show that ER stress responses affect multiple ER and secretory pathway functions. As expected, we observe induction of ER-resident chaperones and other proteins involved in protein folding. However, these represent only a fraction of this set of target genes and we also found several categories of induced genes with other functions throughout the secretory pathway including translocation, protein glycosylation, vesicular transport, ER-associated degradation and lipid metabolism.
Figure 2.
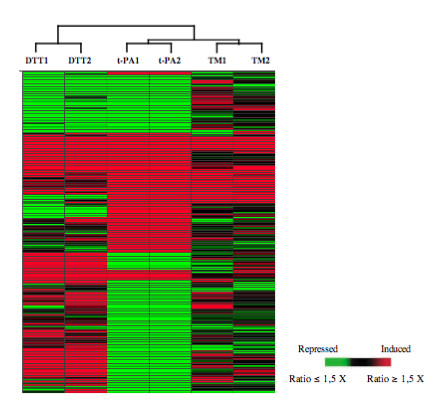
Hierarchical clustering of records in a dendrogram (tree graph) based on the similarity of the signal log ratios obtained in each of the duplicate stress studies. Records (188) were selected based on differential expression in the tPA comparisons. This tree is representative for multiple clusterings performed using signals or signal log ratios. The genes have been rearranged into their cluster order and are represented on the vertical axis. The experiments are represented on the horizontal axis. The significance of the colour scale is indicated.
Figure 3.
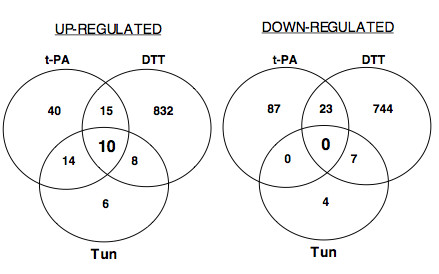
Venn diagrams of the numbers of overlapping and non-overlapping induced (≥ 1.5 fold) or repressed (≤ 1.5 fold) genes on the A. niger array after exposure to DTT or tunicamycin (Tun) and in the t-PA producing strain (t-PA).
Fewer genes were down-regulated overall than were up-regulated and there were no genes down-regulated in all conditions. Exposure to tunicamycin for 2 h caused down-regulation of only eleven genes in total compared to 38 up-regulated. Over one hundred genes (t-PA) and nearly 800 genes (DTT treatment) were down-regulated (Table 2) and 23 of these were common to both the t-PA and DTT conditions. Among the 61 genes that we found to be down-regulated by more than one stress condition, we identified 30 sequences containing a signal peptide, 6 genes encoding extracellular proteins and 4 genes related to the cell wall. At least 5 other genes involved in cell wall biosynthesis or function were additionally repressed by t-PA production or tunicamycin treatment.
Translational regulations during the ER stress response
ER stress is known to lead to differential up- and down-regulation of transcription, but differential translation has yet to be explored in filamentous fungi although a few reports are available with S. cerevisiae [19,20]. A direct analysis of translational control can be achieved by fractionation of cytoplasmic extracts in sucrose gradients, based on the methods described for polysome analysis [21], which involves size separation of large cellular components and monitoring the A254 across the gradient. This method enables the separation of free mRNPs (ribonucleoprotein particles) from mRNAs fully loaded with ribosomes (i.e. polysomes). As only polysomes represent actively translated transcripts, this fraction should be directly correlated with the set of de novo synthesized proteins in a particular cellular state and enables the determination of the translation efficiencies, which are characteristic for each transcript in a cell [22]. In addition, changes in the distribution of a given mRNA indicate how this translational efficiency can vary under different conditions. Because it is generally accepted that translational control predominantly occurs at the initiation step [23], the number of mRNA molecules engaged in polysomes should be a robust indicator of the synthesis rate of the corresponding protein.
Cytoplasmic extracts from DTT treated or untreated cells were loaded onto sucrose gradients and twenty fractions were collected from each one. RNA was extracted from each fraction and an aliquot was subjected to electrophoresis through a formaldehyde gel (Fig. 4B). As expected, 25S and 18S ribosomal RNAs were the prominent species. As described [21,24], the assignment of OD254 peaks corresponding to the 40S and 60S subunits and to intact ribosomes was confirmed with 18S and 25S RNAs distribution (Fig. 4A). There was no marked net change in the absorbance profile of DTT-treated samples compared to the control, indicating that the ER stress did not cause a global change of translational activity.
Figure 4.
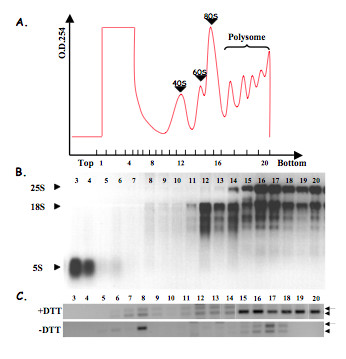
(A) Representative absorbance profile for RNA separated by velocity sedimentation through a 15–60% sucrose gradient. Fractions are numbered from the top to the bottom of the gradient. (B) RNA was extracted from each fraction and subjected to electrophoresis through a formaldehyde gel. The ribosomal RNA distribution profile (25S, 18S and 5S rRNA; indicated by arrowheads) enables the the assignment of OD254 peaks, corresponding to the 40S and 60S ribosomal subunits and to intact ribosomes (80S). (C) RT-PCR analysis with hacA primers were performed from each fraction of collected gradients from treated and non-treated cells. The full length hacAu mRNA (arrow) as well as low-molecular-weight version of hacAi (arrowhead) can be visualized.
Furthermore, the distribution of spliced (hacAi) and unspliced hacA mRNA (hacAu) over the gradients was analysed by RT-PCR with hacA primers amplifying a fragment across the 20 nt intron region (Figure 4C). In A. niger control samples, a low level of hacAu transcripts was detected, which sedimented with both polysomal and non-polysomal fractions. Low level amplifications from the spliced form were also obtained in the polysomal fractions, suggesting that splicing occurs to a low level even in non-stressed samples. In DTT-treated samples, spliced and unspliced transcripts were detected but the spliced form was clearly predominant and was mainly recovered from the bottom of the gradient and therefore was ribosome-associated.
RNA fractions were pooled into non-polysomal and polysomal samples according to OD254 profiles and were used as a matrix for GeneChip hybridisation. Translational regulation of each mRNA was assessed by measuring the relative proportions of each mRNA in the polysomal and non-polysomal RNA fractions and then determining changes to this ratio. The ratio was determined by calculation of "DTT-induced shift from non-polysomal to polysomal samples": for each transcript microarray expression values were analysed by calculating (DTT-treated polysomal/DTT-treated non-polysomal)/(control polysomal/control non-polysomal). Several of these translational regulated genes are listed in the Table 3. Twenty six genes showed enhanced translation during DTT-treatment (a > 2 fold shift from non-polysomal to polysomal fractions). Several of the predicted proteins function as part of the secretory system including a signal peptidase and a proteasome protein. Two hundred and fifty three genes showed reduced translation (a < 2 fold shift from polysomal to non-polysomal fractions) including 108 hypothetical protein encoding genes. DTT treatment resulted in translational repression of a large number of genes with functions in ribosomal biogenesis and assembly. Included were several mRNAs encoding both large and small ribosomal subunit proteins (RPL and RPS families). This result may support a hypothesis of a translational repression as a mechanism of reducing ER throughput during ER stress. Three RPS and 2 RPL encoding genes are included in Table 3, but 9 other RPS and 23 other RPL were affected by this regulation, suggesting that RP mRNAs were coordinately regulated at the translational level. We also found that several mRNAs encoding secreted proteins were also redistributed from polysomes to monosomes and untranslated mRNPs. These results suggest that, in addition to the transcriptional repression mechanism called RESS, another feed-back mechanism could occur upon ER stress at the translational level and lead to a reduced amount of new protein translocated into the ER lumen. Our results also indicate that DTT repressed the translational activity of a number of genes belonging to other functional categories including the ERAD pathway, lipid metabolism or cell wall biogenesis (Table 3).
Table 3.
Examples of genes for which DTT treatment resulted in > 2 fold shift of transcripts from the non-polysomal to polysomal samples, or vice versa, relative to untreated controls.
| DSM code | Predicted protein | Polysomal/non-polysomal Control | Polysomal/non-polysomal DTT treated | DTT induced shift |
| From non-polysomal to polysomal fractions | ||||
| An09g05420 | similar to signal peptidase subunit SPC3 | 4.61 | 0.64 | 7.20 |
| An15g01130 | similar to brefeldin A resistance protein BFR1 (S. pombe) | 0.79 | 0,26 | 3.04 |
| An01g14260 | similar to delta subunit of the coatomer delta-coat protein COPD (Bos. taurus) | 1.79 | 0.68 | 2.63 |
| An17g00270 | similar to 26S ATP/ubiquitin-dependent proteinase chain S4 (S. pombe) | 3.13 | 1.28 | 2.45 |
| From polysomal to non-polysomal fractions | ||||
| Translation | ||||
| An02g13840 | similar to cytoplasmic ribosomal protein of the small subunit S9 (H. sapiens) | 0.47 | 27.28 | 57.91 |
| An18g05810 | similarity to cytoplasmic ribosomal protein of the small subunit S26 (H. sapiens) | 0.51 | 11.22 | 22,03 |
| An08g00970 | ribosomal protein of the small subunit rps28 | 0.71 | 3.48 | 4.91 |
| An11g09570 | similar to cytoplasmic ribosomal protein of the large subunit L37.b | 0.57 | 8.33 | 14.62 |
| An12g04860 | similar to cytoplasmic ribosomal protein of the large subunit L30 | 0.37 | 4.18 | 11.42 |
| An01g02880 | similar to cytoplasmic ubiquitin/ribosomal fusion protein CEP52 | 0.67 | 4.61 | 6.92 |
| An18g04570 | similar to cleavage/polyadenylation factor IA subunit CLP1 | 0.71 | 1.56 | 2.20 |
| An02g09260 | similar to nucleolar protein NOP5 | 2.12 | 9.22 | 4.34 |
| An18g04840 | similar to elongation factor 1 alpha (Podospora anserina) | 1.88 | 6.02 | 3.21 |
| An15g00750 | Similar to elongation factor 1-gamma 1 TEF3 | 2.16 | 5.35 | 2.47 |
| An07g02650 | similar to translation elongation factor 3 YE3 | 2.96 | 23.88 | 8.07 |
| Proteolytic degradation | ||||
| An11g01760 | similar to proteasome 20S subunit PRE2 | 3.79 | 20.52 | 5.42 |
| An02g07010 | similar to ubiquitin UBI1 (A. nidulans) | 1.1 | 7.5 | 7.12 |
| Lipid metabolism | ||||
| An08g10110 | similar to lipid transfer protein POX18 (C. tropicalis) | 0.96 | 4.04 | 4.23 |
| An03g06410 | similar to methyl sterol oxidase ERG25 | 1.23 | 2.84 | 2.32 |
| An12g01890 | similar to squalene synthase ERG9 (C. utilis) | 1.01 | 2.21 | 2.18 |
| An02g10350 | similar to farnesyl-pyrophosphate synthase FPPS | 1.90 | 4.13 | 2.17 |
| An07g09570 | similar to phosphatidate cytidylyltransferase CDS1 | 0.84 | 1.73 | 2.06 |
| Cell Wall | ||||
| An03g02400 | similar to hydrophobin DEWA (A. nidulans) | 0.51 | 1.59 | 3.10 |
| An06g01550 | similar to glucan synthase FKS (Paracoccidioides brasiliensis) | 0.46 | 1.33 | 2.89 |
| An11g00270 | similar to lanosterol 14 alpha-demethylase CYP51 (H. sapiens) | 0.64 | 1.44 | 2.25 |
| An02g02360 | similar to chitin synthase (A. nidulans) | 0.43 | 0.96 | 2.24 |
| Secreted proteins | ||||
| An12g01910 | similar to phytase PHYA3 (A. fumigatus) | 0.36 | 4.03 | 11.1 |
| An03g06550 | Glucoamylase GLAA | 0.57 | 17.69 | 30.98 |
| An02g13220 | similar to lysophospholipase phospholipase B (P. notatum) | 0.55 | 2.90 | 5.23 |
| An09g00270 | alpha-galactosidase AGLC | 0.38 | 1.65 | 4.3 |
| An02g01550 | similar to serine protease (Coccidioides immitis) | 1.78 | 6.69 | 3.76 |
| An02g04690 | similar to serine-type carboxypeptidase I CDPS (A. saitoi) | 1.71 | 3.56 | 2.08 |
The DTT induced shift was determined by calculating (DTT treated polysomal/DTT treated non-polysomal)/(control polysomal/control non-polysomal).
Discussion
This is the first complete analysis of ER stress in A. niger. It has been made possible by the availability of the complete annotated genome sequence [13] and genome-wide GeneChips, which include built-in control sequences. The global ER stress response has already been reported in S. cerevisiae [7] but equivalent studies in the filamentous fungi have been hampered by the lack of complete genome sequences and the non-availability of gene arrays. Such bottlenecks are being removed and we have already seen the description of ER stress responses in Trichoderma reesei [25] and A. nidulans using arrays that cover approximately one third of the predicted open reading frames [12]. A. nidulans is scarcely exploited for its capacity to secrete enzymes because other filamentous fungi, including A. niger, have proved to hold advantages in terms of the range and yield of secreted enzymes. Previous analyses of secretion stress in A. niger have relied either on analysis of specified target genes [2,26] or have used cDNA subtraction libraries [18,27] which, in contrast to GeneChips, reveal only a small fraction of differentially expressed genes.
A low variation (a technical standard deviation of 0.16 and a biological standard deviation of 0.25 on average for the signal log ratio of 'present' genes) was shown for GeneChip replicates (data not shown). Generally, dependent on growth conditions, 5000–6000 genes were found to be expressed, with 10–20 genes detected as false positives in a single experiment. In any global transcriptomic study it is essential to have biological replicates that provide assurance of the validity of the conclusions reached. The biological replicates used in this study showed a small variation compared to the experimentally induced variation except for the tunicamycin study where the experimentally induced variation was small. Both for the chemostat-cultivated tPA strain and the shake-flask-cultivated tunicamycin-exposed cells, less than 4% and 1% of the expressed genes, respectively, were determined as changed by at least 1.5-fold.
We confirmed that each of the stress conditions led to induction of the UPR as judged by the transcriptional induction of A. niger genes known to be affected by ER stress, bipA [28] and pdiA [29]. In addition, we confirmed splicing of the 20bp intron in the A. niger hacA mRNA [10,16]. It has recently been established that the UPR does not just involve the simple switch based around the synthesis of the mediating transcription factor but that there is further complexity under some conditions [30-32]. In A. niger [10], Trichoderma reesei [11] and S. cerevisiae [33] it is known that ER stress can lead to the transcriptional down-regulation of some genes encoding secreted proteins and this effect, termed repression under ER stress (RESS) [11], may be independent of the UPR [10]. ER stress due to over-expression of membrane proteins can elicit the transcriptional up-regulation of bipA without apparent splicing of the hacA intron in A. niger [34]. This effect, together with RESS, indicates that there is complexity in ER stress responses in filamentous fungi. Our results confirm the existence of the RESS mechanism in A. niger since the transcription of several major secreted proteins encoding genes was clearly repressed by at least 2 treatments. Moreover, in common with the data generated in Arabidopsis thaliana [35], many repressed genes in A. niger encode membrane proteins and transporter proteins that may be essential for the maintenance of cellular ion homeostasis. Thus, the list presented in the additional file 6 contains several putative zinc, iron, calcium and manganese transporters and the zinc-regulated transcription factor Zap1, which were mainly repressed by t-PA production. Previous studies showed that both calcium [36] and zinc [37] are required for ER function in yeast and that their deficiency induces the UPR.
Analysis of the impact of secretion stress on the genes encoding components of the secretory system in A. niger is summarized in Fig. 5. Few of the translocon genes (encoding components of both the co- and post-translational translocation system), which were largely not represented in the A. nidulans arrays [12], were induced under two or more stress conditions. The signal sequence recognition system was transcriptionally unaffected except in one component but there was more response from components of the signal peptidase complex. Several of the translocon genes and one component of signal peptidase were induced under UPR in S. cerevisiae [7]. A. niger homologues of the S. cerevisiae SEC11, SPC2 and SPC3 were all up-regulated under ER stress. Of the predicted ER-resident chaperones, bipA was induced under all conditions as expected [28]. The calnexin-encoding clxA gene [38], where the production of prochymosin was previously shown to induce its transcription, was shown here to be additionally induced by both DTT and tunicamycin. A homolog of the S. cerevisiae LHS1 gene, lhsA, has not been previously described in A. niger but was annotated in the genome of A. niger and represented on the GeneChips. As with bipA, lhsA was transcriptionally up-regulated under all 3 conditions compared to controls. In S. cerevisiae, the chaperone cycle involves products encoded by KAR2 (encodes a Bip-like chaperone) and LHS1 (where the ATPase activities of these two Hsp70p chaperones are coordinately regulated) as well as nucleotide exchange activity provided by Sil1p and DNAJ proteins such as Scj1p and Jem1p [39]. The ER-resident chaperone and foldase system was generally induced under UPR conditions in S. cerevisiae even if KAR2 and PDI1 failed to meet the stringent criteria applied [7]. Although lhsA has been identified here as a stress-responsive gene, as was at least one DNAJ protein, no candidate genes have been found for the nucleotide exchange factor homolog of yeast SIL1. On the presumption that this functionality exists, it may reside in a protein with low sequence identity to Sil1p or in another component, e.g. LhsA. A putative ortholog of a mammalian p58-encoding gene (similarity to human p58 with e value of 4e-59, showing 32% amino acid identity over 450 residues) was induced by both t-PA and tunicamycin. P58 is involved in translational regulation in mammalian cells, its induction is mediated by ATF6 and it plays a role in regulating the PERK/eIF2alpha/ATF4 pathway [40]. Homologs of PERK and ATF4/6 appear to be absent from A. niger so the role of the putative p58 in A. niger is intriguing.
Figure 5.
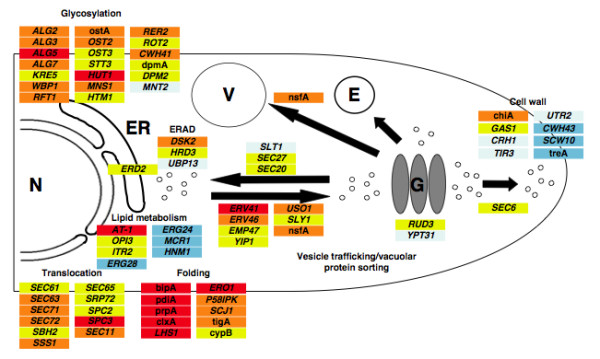
Model of the secretory pathway under different ER stress conditions (t-PA secretion, tunicamycin and DTT) together with examples of genes that are transcriptionally induced or repressed. The A. niger gene designation is provided where previously known or, otherwise, the S. cerevisiae gene name is provided. Red, genes up-regulated by 3 conditions; orange, genes up-regulated by 2 conditions; yellow, genes up-regulated by 1 condition; light blue, genes down-regulated by 1 condition; blue, genes down-regulated by 2 conditions. N, nucleus; ER, endoplasmic reticulum; E, endosome; V, vacuole; G, Golgi. ERAD is ER-associated degradation.
Manipulations of the ER lumenal environment have been previously examined in Aspergillus with the aim of improving the secreted yield of heterologous proteins. This has been attempted with individual genes such as bipA [41] and pdiA [42] as well as through manipulation of the UPR [43]. Detailed knowledge of the responses of individual genes to different stresses should permit refinements to these approaches that would be more consistently beneficial. The complexity of the chaperone cycle has already been mentioned but the formation of disulfide bonds is another key example. Although the pdiA gene encodes the principal foldase in A. niger [29], other genes encoding members of this foldase/isomerase family have also been described [44,45]. A fuller description of their responses to different stresses is provided by the GeneChip studies described here and shows that all three foldase genes are up-regulated by all stresses except for tigA by DTT. The contributions to foldase activity provided by the three identified lumenal foldases (PdiA, TigA, PrpA) is not known although, in S. cerevisiae, Pdi1p is known to contribute more activity than the other yeast foldases [46]. In S. cerevisiae, Ero1p is an essential lumenal protein involved in electron transfer during the formation of disulfide bonds and its homolog in A. niger, eroA, was induced under all stress conditions. Interestingly, Ero1p is retained in the lumen by attachment to the ER membrane in a manner that is not wholly understood [47] whereas the A. niger EroA is predicted to contain a conventional C-terminal ER retention sequence. The receptor for retention of C-terminal (H/K)DEL-containing proteins (Erd2p in S. cerevisiae) was induced under ER stress in both A. niger and S. cerevisiae [7]. In other areas, several genes involved in glycosylation of secretory proteins were induced under ER stress in both S. cerevisiae [7] and in A. niger (Fig. 5) and included a homolog of the yeast RFT1 gene that is responsible for translocation of lipid-linked glycan intermediates into the ER [48].
In fungi, the only reported translational control during the UPR concerns the expression of the yeast transcription factor Hac1p [20]. This control is mediated by a base-pairing interaction between an intron at the 3'end and the 5' untranslated region, which represses mRNA translation. Splicing of this unconventional intron is sufficient to relieve this translational block. It has been reported that the HAC1u mRNA in yeast was stable, located in the cytosol and associated with ribosomes, yet did not produce protein, indicating that the ribosomes engaged on the mRNA were stalled and that translation was attenuated at the elongation step [20]. Our results showed that hacAu mRNA could also sediment in non-polysomal fractions, suggesting that translation of this mRNA species may be blocked at the translational initiation step in addition to the previously reported elongation step, as also shown in yeast [49]. DTT treatment resulted in translational repression of a large number of genes with functions in ribosomal biogenesis and assembly (Table 3). This result may support the hypothesis of a translational repression as a mechanism of reducing ER throughput. It is known that the abundance of RP mRNAs rapidly decreases when yeast cells encounter stress situations [50,51]. We also found that several mRNAs encoding secreted proteins were also redistributed from polysomes to monosomes after DTT exposure and the gene glaA encoding glucoamylase, which is a major secreted protein in A. niger, was one of them. Therefore, in addition to the transcriptional down-regulation of glaA due to DTT stress, our results suggest that a post-transcriptional regulatory mechanism negatively affects the translation of glaA mRNA in A. niger. For the first time in filamentous fungi, these results suggest the existence, in addition to the RESS, of another feed-back mechanism that occurs upon ER stress at the translational level and leads to a reduction in the amount of new protein translocated into the ER lumen.
Conclusion
This is the first complete analysis of ER stress in A. niger, a filamentous fungus used commercially for the secreted production of a range of native and heterologous proteins. It has been made possible by the availability of the complete annotated genome sequence and genome-wide GeneChips. We induced ER stress either by chemical treatments of the wild-type cells or by expressing a heterologous protein. Following the induction of ER stress, A. niger cells display a diverse array of adaptative changes in gene expression at both the transcriptional and translational levels. The transcriptional responses to each stress were compared and the overlaps common to these conditions led to the identification of robust sets of induced or repressed genes Thus, a range of transcriptional targets has been identified as putative candidates for secretion stress- related genes, including a large number of genes encoding components of the secretory system. Moreover, by combining polysomal fractionation with DNA GeneChip analyses, we have focused on the translational activity of several individuals mRNAs following DTT exposure. Although the observed polysome profiles were similar under control and ER stress conditions, we confirmed the post-transcriptional control of hacA expression and highlighted that differential translation could also occur during ER stress at least for some genes encoding secreted or ribosomal proteins. Many of the genes identified in this study provide targets for improving A. niger as a cell factory for protein production.
Methods
Strains, culture conditions and treatments
The AB4.1 strain, a pyrG- strain that is auxotrophic for uridine [52], was used for the chemical treatment experiments. A protease-deficient strain (D15) of A. niger, derived from strain AB4.1, was used as a host for the production of human tissue plasminogen activator (t-PA). The t-PA was produced in the recombinant strain D15#25 as a fusion protein with the catalytic domain of the native glucoamylase protein. The gene encoding this fusion protein was expressed under control of the constitutive promoter for glyceraldehyde-3-phosphate dehydrogenase (gpdA).
The A. niger strains were maintained on potato dextrose agar slopes (Oxoid). All AB4.1 cultures were supplemented by 10 mM uridine (Sigma). Slopes were incubated at 30°C until the cultures had conidiated and spores were resuspended in 0.1% (v/v) Tween 20 (Sigma). Liquid batch cultures were inoculated with 3 × 105 spores/ml, grown for 44 h in 100 ml of starch-containing ACMN/N/P medium [53] in 250 ml conical flasks at 25°C and shaken at 150 r.p.m. Batch AB4.1 cultures were treated with ER stress reagent (DTT or tunicamycin) for 2 h. DTT stock was prepared in water and tunicamycin stock in DMSO. DTT or tunicamycin were added to the liquid medium at a final concentration of 20 mM or 10 μg/ml, respectively. Control cultures had an equivalent volume of sterile water or DMSO added. For tunicamycin treatment with germlings of A. niger, fresh spore cultures were prepared as previously described and incubated for 5 h. A sample was taken for microscopic examination of germination, which was in excess of 90%. Cultures were treated by tunicamycin (10 μg/ml) or DMSO for 1 h, after which the germlings were harvested by filtration and frozen in liquid nitrogen. Continuous cultures were carried out in an Applikon (FT Applikon Ltd., Tewkesbury, UK) bioreactor (2.1 l full working volume) according to published methods [54]. The medium (one tenth strength SCM contained 5 gl-1 maltose as the carbon source. Cultures were inoculated with 5 × 40 ml shake flask pre-cultures, which had been growing in medium containing 10 gl-1 of maltose for approximately 24 hours. Cultures were maintained at 30°C ± 1°C and pH 5.5 ± 0.1, agitated at 1200 rpm with a 3 six-bladed (48 mm diameter) Rushton turbine impeller and aerated with 0.7 vvm (l air [l culture]-1min-1). Foaming was controlled by continuous addition of a mixture of polypropylene glycol (PPG) of different molecular weights PPG 1025 (BDH): PPG 2025 (BDH): FoamMaster PPG (mixed molecular weight; Henkel Performance Chemicals, Leeds, UK) in the ratio 2:2:1 [15] to give a final concentration of approximately 0.01% (v/v) PPG. The dilution rate was kept constant at 0.080 ± 0.006 h-1. In order to reduce the degree of attachment of biomass to the surfaces inside the bioreactor, the impeller speed was increased to 1500 rpm once a day for about 15 minutes after sampling. The maximum specific growth rate (μmax) during the batch growth phase in bioreactors was estimated from the CO2 output using an ADC 7000 infrared Gas Analyzer (The Analytical Development Co. Ltd.; U.K) and CO2 evolution rate was also measured online throughout the whole experiment to monitor steady state.
Total RNA extraction
Total RNA was prepared from each condition according to the TRIzol reagent protocol (Invitrogen). Each sample was treated with RQ1 RNase-Free DNase (Promega) according to the manufacturer's instructions, followed by a phenol:chloroform extraction and ethanol precipitation. An additional cleanup was performed using the RNEasy Mini Kit (Qiagen), following the manufacturer's RNA Clean-up protocol.
Polysome analysis
Ribosomal fractions were prepared according to the method described for polysome analysis [21], modified as follows. At the time of harvest, cycloheximide was added to a final concentration of 0.1 mg/ml to trap elongating ribosomes. The cultures were swirled rapidly and chilled on ice for 10 minutes. Fungal material was pelleted by centrifugation at 11000 g for 10 min at 4°C. The pellet was then resuspended in 5 ml of polysome extraction buffer (20 mM Tris-HCl pH 8.0, 140 mM KCl, 1.5 mM MgCl2, 1% Triton X-100, 0.1 mg/ml cycloheximide, 1.0 mg/ml heparin, 0.5 mM DTT) and sedimented. This washing step was repeated and cells were frozen in liquid nitrogen and stored at -80°C.
Approximately 0.25 g of cells was ground in liquid nitrogen with a mortar and a pestle, and the powder was resuspended in 750 μl of ice-cold polysome extraction buffer. Excess cell debris were removed by sedimentation at 4000 g for 5 min at 4°C. The supernatant was clarified by further centrifugation (12000 g, 10 min, 4°C). Each sample was loaded on an 8 ml 15 to 60% (w/v) sucrose gradient and sedimented at 150000 g (55000 rpm) and 4°C in a Beckman MLA-80 rotor for 135 min. Sucrose solutions were prepared in 50 mM Tris acetate pH 7.0, 50 mM NH4Cl, 12 mM MgCl2, 1 mM DTT. The gradient was fractionated with a density gradient fractionator (Foxy Jr. Fraction Collector, ISCO), whilst monitoring absorbance at 254 nm using a UA-6 UV detector (ISCO). 0.5 ml fractions were collected from the top of the gradient directly into 1 ml volume of 6 M guanidine hydrochloride. RNA was precipitated by adding an equal volume of 100% ethanol and resuspended in Tris-EDTA (TE, pH 8.0). The RNA was again precipitated by addition of 50 μl of 3 M sodium acetate (pH 5.2) and 1 ml of 100% ethanol, and resuspended in TE (pH 8.0).
For RNAs destined for microarray analyses, the polysomal fractions and non-polysomal fractions were pooled respectively. Each fraction was treated with RQ1 RNase-Free DNase (Promega) and was again cleaned up by applying the samples to an RNeasy mini column (Qiagen).
Northern analysis and RT-PCR amplification analyses
Northern blot hybridization was performed using the formaldehyde-based system NorthernMax as described by manufacturer and transfer was achieved to BrightStar-Plus membranes (Ambion Inc., Austin, Tex.). [α-32P]dUTP-labeled antisense RNA probes were prepared with the Lig'nScribe and the Strip-EZ RNA kits according to the manufacturer's instructions (Ambion Inc., Austin, Tex.). Coding sequences for differentially expressed genes were downloaded from the DSM A. niger database [13] and PCR primers for each selected gene were designed with Primers 3.0 (Whitehead Institute, Massachusetts Institute of Technology, Boston, MA). The PCR primers used to generate probes for Northern blotting and for RT-PCR (including for RT-PCR across the hacA mRNA intron) are listed in Table 4. Probes were generated from genomic DNA of A. niger strains by PCR amplification. The PCR conditions were as follows: an initial denaturation of 94°C for 3 min was followed by 30 cycles of 94°C for 30 s, 57°C for 1 min and 72°C for 1 min. For densitometric analysis, signals were quantified with a Fuji film BAS2000 phosphorimaging system.
Table 4.
Primers for probe generation by PCR and for RT-PCR.
| Gene | Primer pairs | Amplicon size (bp) |
| bipA1,2 | 5'-CAGAGACAGGCCACCAAGGACGCT-3' 5'-CGAACTTGGCGCGGGTGAGGGTCT-3' | 445 |
| pdiA1 | 5'-TATCTTTGCCGAGACCAAGG-3' 5'-AACAGGTTCCGACTTGATGC-3' | 298 |
| pdiA2 | 5'-TGATACCGAGTCTGAGTTA-3' 5'-CTCGATTCTGTTTGCCTAGC-3' | 303 |
| An02g1341011 | 5'-CGCATCCTACCCATACTCGT-3' 5'-GGAACCAGCGATTAGCAAAG-3' | 280 |
| hacA2 | 5'-CTTCTCCTACCCTAACTCCT-3' 5'-TCAAAGAGAGAGAGGGCAC-3' | 397 |
| act11 | 5'-GCCTTCTACGTCTCCATCCA-3' 5'-GAACAGAGCCTCAGGAGCAC-3' | 396 |
| act12 | 5'-TCATGATCGGTATGGGTCAG-3' 5'-ACGATGTTGCCGTACAGATC-3' | 765 |
1: primers designed for blot probes
2: primers designed for RT-PCR
Reverse transcriptions were carried out using the SuperScript II Reverse Transcriptase system (Invitrogen, Life Technologies) according to the manufacturer's instructions.
Genome sequence and microarray analysis
Processing of mRNA to cDNA, labelling, hybridization to A. niger Affymetrix GeneChips (Affymetrix, Inc., Santa Clara, CA) and fluorescence scanning were performed at the Nottingham Arabidopsis Stock Center (NASC, Loughborough, UK) Affymetrix facility. The Affymetrix Microarray Suite version 5.0 program was used to normalize microarray data and to calculate signal intensity, detection p-value, signal log ratio and change p-value using the statistical algorithms of version 5.0. The detection p-value was used to assign a call whether a measured transcript was detected at significant level as P (present), below detection level as A (absent) or in-between as M (marginal). The change p-value based on Wilcoxon's Signed Rank test of 12 probe pair comparisons, was used to provide a measure of the likelihood of change between two GeneChips and direction, assigning a change call as I (induced), D (decreased) or NC (not changed). The signal and signal log ratio were computed by taking probe pair intensities (each transcript is represented by 12 probe pairs) of one GeneChip or across two GeneChips, respectively. For each treatment condition, for all 14555 probe sets, the calculated signal log ratios (in base 2) were converted into fold change of mRNA level relative to untreated or control cultures, to make interpretation more intuitive.
In total, 12 Affymetrix Genechips measurements were obtained with total RNA samples from which 6 duplicated comparisons between the treated samples and the respective control were analysed: 2 for the DTT treatment, 2 for the tunicamycin treatment and 2 for the t-PA production. Genes were considered to be differentially expressed specifically by each treatment if (i) expression levels changed by at least 1.5 fold in both independent replicates, (ii) the mRNAs were assigned at least 2× p-values over the 4 detection calls, and (iii) the change call in gene expression was in the same direction (increased or decreased) in duplicated experiments. Additionally, a total of 8 Affymetrix Genechips were hybridized with polysomal and non-polysomal samples treated or not by DTT, each conditions being analysed twice. The Spotfire program was used to determine the list of genes meeting these restriction criteria and to determine the overlap in the gene sets. Finally, manual inspection was performed for each selected entry group to remove repeated or control entries. The Spotfire Decision Site for Functional Genomics was used to perform hierarchical clustering (Euclidean distance, UPGMA) and to group records according to their similarity in a dendrogram.
The A. niger genome and gene sequence can be accessed through [55].
Authors' contributions
This study was carried out predominantly by TG in the laboratory of DA. TG was funded by DSM Food Specialities and NvP was directly involved in the analysis of Affymetrix GeneChip data. Detailed discussions on this analysis were held at DSM and involved HS, NvP, TG and DA. KL and GR provided samples of A. niger biomass producing tPA. TGoosen and CvdH collaborated by undertaking some stress studies. All authors contributed in discussions of the approach and interpretation of results.
Supplementary Material
Scaling (normalization) factors for the GeneChips used in the stress studies. The figure provided show that overall signal intensities were comparable throughout all GeneChips.
Overall variations in gene expression levels induced by tunicamycin treatment. In panel A, signals of tunicamycin treatment (y-axis) vs DMSO (x-axis). In panel B, distribution of signal log ratio's (2 log).
Overall variations in gene expression levels induced by t-PA expression. In panel A, signals of t-PA treatment (y-axis) vs control strain (x-axis). In panel B, distribution of signal log ratio's (2log).
Overall variations in gene expression levels induced by DTT treatment. In panel A, signals of DTT (y-axis) vs control strain (x-axis). In panel B, distribution of signal log ratio's (2 log).
Genes induced by tPA and/or tunicamycin treatments. The fold changes in expression are indicated for all the treatments (Tun = tunicamycin, DTT = dithiothreitol, tPA = production of t-PA). Similarities are expressed in comparison with the Saccharomyces cerevisiae genome except when indicated. The symbol* indicates that the value do not meet the defined restrictive criteria. The genes induced by the three treatments are marked in bold.
Genes repressed by tPA and/or tunicamycin treatments. The fold changes in expression are indicated for all the treatments (Tun = tunicamycin, DTT = dithiothreitol, tPA = production of t-PA). Similarities are expressed in comparison with the Saccharomyces cerevisiae genome except when indicated. The symbol* indicates that the value do not meet the defined restrictive criteria.
Acknowledgments
Acknowledgements
We thank the Biotechnology and Biological Sciences Research Council for contributing financial support to DBA, GDR and KL.
Contributor Information
Thomas Guillemette, Email: thomas.guillemette@univ-angers.fr.
Noël NME van Peij, Email: Noel.Peij-van@DSM.COM.
Theo Goosen, Email: t.goosen@chello.nl.
Karin Lanthaler, Email: karin.lanthaler@man.ac.uk.
Geoffrey D Robson, Email: geoff.robson@man.ac.uk.
Cees AMJJ van den Hondel, Email: Hondel@rulbim.leidenuniv.nl.
Hein Stam, Email: Hein.Stam@DSM.COM.
David B Archer, Email: david.archer@nottingham.ac.uk.
References
- Archer DB. Filamentous fungi as microbial cell factories for food use. Curr Opin Biotechnol. 2000;11:478–483. doi: 10.1016/S0958-1669(00)00129-4. [DOI] [PubMed] [Google Scholar]
- MacKenzie DA, Jeenes DJ, Archer DB. Filamentous fungi as expression systems for heterologous proteins. In: Kück U, editor. The Mycota II. New York , Springer; 2004. pp. 289–315. [Google Scholar]
- Conesa A, Punt PJ, van Luijk N, van den Hondel CA. The secretion pathway in filamentous fungi: a biotechnological view. Fungal Genet Biol. 2001;33:155–171. doi: 10.1006/fgbi.2001.1276. [DOI] [PubMed] [Google Scholar]
- Archer DB, Peberdy JF. The molecular biology of secreted enzyme production by fungi. Crit Rev Biotechnol. 1997;17:273–306. doi: 10.3109/07388559709146616. [DOI] [PubMed] [Google Scholar]
- Gouka RJ, Punt PJ, van den Hondel CA. Efficient production of secreted proteins by Aspergillus: progress, limitations and prospects. Appl Microbiol Biotechnol. 1997;47:1–11. doi: 10.1007/s002530050880. [DOI] [PubMed] [Google Scholar]
- Mori K. Tripartite management of unfolded proteins in the endoplasmic reticulum. Cell. 2000;101:451–454. doi: 10.1016/S0092-8674(00)80855-7. [DOI] [PubMed] [Google Scholar]
- Travers KJ, Patil CK, Wodicka L, Lockhart DJ, Weissman JS, Walter P. Functional and genomic analyses reveal an essential coordination between the unfolded protein response and ER-associated degradation. Cell. 2000;101:249–258. doi: 10.1016/S0092-8674(00)80835-1. [DOI] [PubMed] [Google Scholar]
- Ng DT, Spear ED, Walter P. The unfolded protein response regulates multiple aspects of secretory and membrane protein biogenesis and endoplasmic reticulum quality control. J Cell Biol. 2000;150:77–88. doi: 10.1083/jcb.150.1.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonifacino JS, Weissman AM. Ubiquitin and the control of protein fate in the secretory and endocytic pathways. Annu Rev Cell Dev Biol. 1998;14:19–57. doi: 10.1146/annurev.cellbio.14.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Sheikh H, Watson AJ, Lacey GA, Punt PJ, MacKenzie DA, Jeenes DJ, Pakula T, Penttila M, Alcocer MJ, Archer DB. Endoplasmic reticulum stress leads to the selective transcriptional downregulation of the glucoamylase gene in Aspergillus niger. Mol Microbiol. 2004;53:1731–1742. doi: 10.1111/j.1365-2958.2004.04236.x. [DOI] [PubMed] [Google Scholar]
- Pakula TM, Laxell M, Huuskonen A, Uusitalo J, Saloheimo M, Penttila M. The effects of drugs inhibiting protein secretion in the filamentous fungus Trichoderma reesei. Evidence for down-regulation of genes that encode secreted proteins in the stressed cells. J Biol Chem. 2003;278:45011–45020. doi: 10.1074/jbc.M302372200. [DOI] [PubMed] [Google Scholar]
- Sims AH, Gent ME, Lanthaler K, Dunn-Coleman NS, Oliver SG, Robson GD. Transcriptome analysis of recombinant protein secretion by Aspergillus nidulans and the unfolded-protein response in vivo. Appl Environ Microbiol. 2005;71:2737–2747. doi: 10.1128/AEM.71.5.2737-2747.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pel HJ, de Winde JH, Archer DB, Dyer PS, Hofmann G, Schaap PJ, Turner G, de Vries RP, Albang R, Albermann K, Andersen MR, Bendtsen JD, Benen JA, van den Berg M, Breestraat S, Caddick MX, Contreras R, Cornell M, Coutinho PM, Danchin EG, Debets AJ, Dekker P, van Dijck PW, van Dijk A, Dijkhuizen L, Driessen AJ, d'Enfert C, Geysens S, Goosen C, Groot GS, de Groot PW, Guillemette T, Henrissat B, Herweijer M, van den Hombergh JP, van den Hondel CA, van der Heijden RT, van der Kaaij RM, Klis FM, Kools HJ, Kubicek CP, van Kuyk PA, Lauber J, Lu X, van der Maarel MJ, Meulenberg R, Menke H, Mortimer MA, Nielsen J, Oliver SG, Olsthoorn M, Pal K, van Peij NN, Ram AF, Rinas U, Roubos JA, Sagt CM, Schmoll M, Sun J, Ussery D, Varga J, Vervecken W, van de Vondervoort PJ, Wedler H, Wosten HA, Zeng AP, van Ooyen AJ, Visser J, Stam H. Genome sequencing and analysis of the versatile cell factory Aspergillus niger CBS 513.88. Nat Biotechnol. 2007;25:221–231. doi: 10.1038/nbt1282. [DOI] [PubMed] [Google Scholar]
- Elbein AD. Inhibitors of the biosynthesis and processing of N-linked oligosaccharide chains. Annu Rev Biochem. 1987;56:497–534. doi: 10.1146/annurev.bi.56.070187.002433. [DOI] [PubMed] [Google Scholar]
- Wiebe MG, Karandikar A, Robson GD, Trinci AP, Candia JL, Trappe S, Wallis G, Rinas U, Derkx PM, Madrid SM, Sisniega H, Faus I, Montijn R, van den Hondel CA, Punt PJ. Production of tissue plasminogen activator (t-PA) in Aspergillus niger. Biotechnol Bioeng. 2001;76:164–174. doi: 10.1002/bit.1156. [DOI] [PubMed] [Google Scholar]
- Mulder HJ, Saloheimo M, Penttila M, Madrid SM. The transcription factor HACA mediates the unfolded protein response in Aspergillus niger, and up-regulates its own transcription. Mol Genet Genomics. 2004;271:130–140. doi: 10.1007/s00438-003-0965-5. [DOI] [PubMed] [Google Scholar]
- Saloheimo M, Valkonen M, Penttila M. Activation mechanisms of the HAC1-mediated unfolded protein response in filamentous fungi. Mol Microbiol. 2003;47:1149–1161. doi: 10.1046/j.1365-2958.2003.03363.x. [DOI] [PubMed] [Google Scholar]
- MacKenzie DA, Guillemette T, Al-Sheikh H, Watson AJ, Jeenes DJ, Wongwathanarat P, Dunn-Coleman NS, van Peij N, Archer DB. UPR-independent dithiothreitol stress-induced genes in Aspergillus niger. Mol Genet Genomics. 2005;274:410–418. doi: 10.1007/s00438-005-0034-3. [DOI] [PubMed] [Google Scholar]
- Rand JD, Grant CM. The thioredoxin system protects ribosomes against stress-induced aggregation. Mol Biol Cell. 2006;17:387–401. doi: 10.1091/mbc.E05-06-0520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruegsegger U, Leber JH, Walter P. Block of HAC1 mRNA translation by long-range base pairing is released by cytoplasmic splicing upon induction of the unfolded protein response. Cell. 2001;107:103–114. doi: 10.1016/S0092-8674(01)00505-0. [DOI] [PubMed] [Google Scholar]
- Arava Y, Wang Y, Storey JD, Liu CL, Brown PO, Herschlag D. Genome-wide analysis of mRNA translation profiles in Saccharomyces cerevisiae. Proc Natl Acad Sci U S A. 2003;100:3889–3894. doi: 10.1073/pnas.0635171100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith CW, Klaasmeyer JG, Edeal JB, Woods TL, Jones SJ. Effects of serum deprivation, insulin and dexamethasone on polysome percentages in C2C12 myoblasts and differentiating myoblasts. Tissue Cell. 1999;31:451–458. doi: 10.1054/tice.1999.0053. [DOI] [PubMed] [Google Scholar]
- McCarthy JE. Posttranscriptional control of gene expression in yeast. Microbiol Mol Biol Rev. 1998;62:1492–1553. doi: 10.1128/mmbr.62.4.1492-1553.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pradet-Balade B, Boulme F, Beug H, Mullner EW, Garcia-Sanz JA. Translation control: bridging the gap between genomics and proteomics? Trends Biochem Sci. 2001;26:225–229. doi: 10.1016/S0968-0004(00)01776-X. [DOI] [PubMed] [Google Scholar]
- Arvas M, Pakula T, Lanthaler K, Saloheimo M, Valkonen M, Suortti T, Robson G, Penttila M. Common features and interesting differences in transcriptional responses to secretion stress in the fungi Trichoderma reesei and Saccharomyces cerevisiae. BMC Genomics. 2006;7:32. doi: 10.1186/1471-2164-7-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Archer DB, Turner G. Genomics of protein secretion and hyphal growth in Aspergillus. In: Brown AJP, editor. The Mycota XIII. New York , Springer; 2006. pp. 75–96. [Google Scholar]
- Watson AJ, Worley J, Elliott RM, Jeenes DJ, Archer DB. Cloning stress-induced genes from aspergillus niger using polymerase chain reaction-augmented subtractive hybridization. Anal Biochem. 2000;277:162–165. doi: 10.1006/abio.1999.4401. [DOI] [PubMed] [Google Scholar]
- van Gemeren IA, Punt PJ, Drint-Kuyvenhoven A, Broekhuijsen MP, van't Hoog A, Beijersbergen A, Verrips CT, van den Hondel CA. The ER chaperone encoding bipA gene of black Aspergilli is induced by heat shock and unfolded proteins. Gene. 1997;198:43–52. doi: 10.1016/S0378-1119(97)00290-4. [DOI] [PubMed] [Google Scholar]
- Ngiam C, Jeenes DJ, Punt PJ, Van Den Hondel CA, Archer DB. Characterization of a foldase, protein disulfide isomerase A, in the protein secretory pathway of Aspergillus niger. Appl Environ Microbiol. 2000;66:775–782. doi: 10.1128/AEM.66.2.775-782.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leber JH, Bernales S, Walter P. IRE1-independent gain control of the unfolded protein response. PLoS Biol. 2004;2:E235. doi: 10.1371/journal.pbio.0020235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patil CK, Li H, Walter P. Gcn4p and novel upstream activating sequences regulate targets of the unfolded protein response. PLoS Biol. 2004;2:E246. doi: 10.1371/journal.pbio.0020246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroder M, Kaufman RJ. ER stress and the unfolded protein response. Mutat Res. 2005;569:29–63. doi: 10.1016/j.mrfmmm.2004.06.056. [DOI] [PubMed] [Google Scholar]
- Kimata Y, Ishiwata-Kimata Y, Yamada S, Kohno K. Yeast unfolded protein response pathway regulates expression of genes for anti-oxidative stress and for cell surface proteins. Genes Cells. 2006;11:59–69. doi: 10.1111/j.1365-2443.2005.00921.x. [DOI] [PubMed] [Google Scholar]
- Dave A, Jeenes DJ, Mackenzie DA, Archer DB. HacA-independent induction of chaperone-encoding gene bipA in Aspergillus niger strains overproducing membrane proteins. Appl Environ Microbiol. 2006;72:953–955. doi: 10.1128/AEM.72.1.953-955.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez IM, Chrispeels MJ. Genomic analysis of the unfolded protein response in Arabidopsis shows its connection to important cellular processes. Plant Cell. 2003;15:561–576. doi: 10.1105/tpc.007609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonilla M, Cunningham KW. Mitogen-activated protein kinase stimulation of Ca(2+) signaling is required for survival of endoplasmic reticulum stress in yeast. Mol Biol Cell. 2003;14:4296–4305. doi: 10.1091/mbc.E03-02-0113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis CD, Wang F, MacDiarmid CW, Clark S, Lyons T, Eide DJ. Zinc and the Msc2 zinc transporter protein are required for endoplasmic reticulum function. J Cell Biol. 2004;166:325–335. doi: 10.1083/jcb.200401157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Entwistle J, Morlon E, Archer DB, Peberdy JF, Ward M, Jeenes DJ. Isolation and characterisation of a calnexin homologue, clxA, from Aspergillus niger. Mol Genet Genomics. 2003;268:684–691. doi: 10.1007/s00438-002-0790-2. [DOI] [PubMed] [Google Scholar]
- Steel GJ, Fullerton DM, Tyson JR, Stirling CJ. Coordinated activation of Hsp70 chaperones. Science. 2004;303:98–101. doi: 10.1126/science.1092287. [DOI] [PubMed] [Google Scholar]
- van Huizen R, Martindale JL, Gorospe M, Holbrook NJ. P58IPK, a novel endoplasmic reticulum stress-inducible protein and potential negative regulator of eIF2alpha signaling. J Biol Chem. 2003;278:15558–15564. doi: 10.1074/jbc.M212074200. [DOI] [PubMed] [Google Scholar]
- Lombrana M, Moralejo FJ, Pinto R, Martin JF. Modulation of Aspergillus awamori thaumatin secretion by modification of bipA gene expression. Appl Environ Microbiol. 2004;70:5145–5152. doi: 10.1128/AEM.70.9.5145-5152.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moralejo FJ, Watson AJ, Jeenes DJ, Archer DB, Martin JF. A defined level of protein disulfide isomerase expression is required for optimal secretion of thaumatin by Aspegillus awamori. Mol Genet Genomics. 2001;266:246–253. doi: 10.1007/s004380100550. [DOI] [PubMed] [Google Scholar]
- Valkonen M, Ward M, Wang H, Penttila M, Saloheimo M. Improvement of foreign-protein production in Aspergillus niger var. awamori by constitutive induction of the unfolded-protein response. Appl Environ Microbiol. 2003;69:6979–6986. doi: 10.1128/AEM.69.12.6979-6986.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeenes DJ, Pfaller R, Archer DB. Isolation and characterisation of a novel stress-inducible PDI-family gene from Aspergillus niger. Gene. 1997;193:151–156. doi: 10.1016/S0378-1119(97)00098-X. [DOI] [PubMed] [Google Scholar]
- Wang H, Ward M. Molecular characterization of a PDI-related gene prpA in Aspergillus niger var. awamori. Curr Genet. 2000;37:57–64. doi: 10.1007/s002940050009. [DOI] [PubMed] [Google Scholar]
- Kimura T, Hosoda Y, Sato Y, Kitamura Y, Ikeda T, Horibe T, Kikuchi M. Interactions among yeast protein-disulfide isomerase proteins and endoplasmic reticulum chaperone proteins influence their activities. J Biol Chem. 2005;280:31438–31441. doi: 10.1074/jbc.M503377200. [DOI] [PubMed] [Google Scholar]
- Pagani M, Pilati S, Bertoli G, Valsasina B, Sitia R. The C-terminal domain of yeast Ero1p mediates membrane localization and is essential for function. FEBS Lett. 2001;508:117–120. doi: 10.1016/S0014-5793(01)03034-4. [DOI] [PubMed] [Google Scholar]
- Helenius J, Ng DT, Marolda CL, Walter P, Valvano MA, Aebi M. Translocation of lipid-linked oligosaccharides across the ER membrane requires Rft1 protein. Nature. 2002;415:447–450. doi: 10.1038/415447a. [DOI] [PubMed] [Google Scholar]
- Kuhn KM, DeRisi JL, Brown PO, Sarnow P. Global and specific translational regulation in the genomic response of Saccharomyces cerevisiae to a rapid transfer from a fermentable to a nonfermentable carbon source. Mol Cell Biol. 2001;21:916–927. doi: 10.1128/MCB.21.3.916-927.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Warner JR. Mutation of the Rab6 homologue of Saccharomyces cerevisiae, YPT6, inhibits both early Golgi function and ribosome biosynthesis. J Biol Chem. 1996;271:16813–16819. doi: 10.1074/jbc.271.28.16813. [DOI] [PubMed] [Google Scholar]
- Mizuta K, Warner JR. Continued functioning of the secretory pathway is essential for ribosome synthesis. Mol Cell Biol. 1994;14:2493–2502. doi: 10.1128/mcb.14.4.2493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Hartingsveldt W, Mattern IE, van Zeijl CM, Pouwels PH, van den Hondel CA. Development of a homologous transformation system for Aspergillus niger based on the pyrG gene. Mol Gen Genet. 1987;206:71–75. doi: 10.1007/BF00326538. [DOI] [PubMed] [Google Scholar]
- Archer DB, Jeenes DJ, MacKenzie DA, Brightwell G, Lambert N, Lowe G, Radford SE, Dobson CM. Hen egg white lysozyme expressed in, and secreted from, Aspergillus niger is correctly processed and folded. Biotechnology (N Y) 1990;8:741–745. doi: 10.1038/nbt0890-741. [DOI] [PubMed] [Google Scholar]
- Wiebe MG, Trinci AP. Dilution rate as a determinant of mycelial morphology in continuous culture. Biotechnol Bioeng. 1991;38:75–81. doi: 10.1002/bit.260380110. [DOI] [PubMed] [Google Scholar]
- Aspergillus niger genome Database . http://www.ebi.ac.uk/genomes/eukaryota.html; [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Scaling (normalization) factors for the GeneChips used in the stress studies. The figure provided show that overall signal intensities were comparable throughout all GeneChips.
Overall variations in gene expression levels induced by tunicamycin treatment. In panel A, signals of tunicamycin treatment (y-axis) vs DMSO (x-axis). In panel B, distribution of signal log ratio's (2 log).
Overall variations in gene expression levels induced by t-PA expression. In panel A, signals of t-PA treatment (y-axis) vs control strain (x-axis). In panel B, distribution of signal log ratio's (2log).
Overall variations in gene expression levels induced by DTT treatment. In panel A, signals of DTT (y-axis) vs control strain (x-axis). In panel B, distribution of signal log ratio's (2 log).
Genes induced by tPA and/or tunicamycin treatments. The fold changes in expression are indicated for all the treatments (Tun = tunicamycin, DTT = dithiothreitol, tPA = production of t-PA). Similarities are expressed in comparison with the Saccharomyces cerevisiae genome except when indicated. The symbol* indicates that the value do not meet the defined restrictive criteria. The genes induced by the three treatments are marked in bold.
Genes repressed by tPA and/or tunicamycin treatments. The fold changes in expression are indicated for all the treatments (Tun = tunicamycin, DTT = dithiothreitol, tPA = production of t-PA). Similarities are expressed in comparison with the Saccharomyces cerevisiae genome except when indicated. The symbol* indicates that the value do not meet the defined restrictive criteria.