

Abstract
Effector-memory T cells expressing Fas (Apo-1/CD95) are switched to an apoptotic program by cross-linking with Fas-ligand (FasL). Consequently, tumors that express FasL can induce apoptosis of infiltrating Fas-positive T lymphocytes and subdue any antitumor host immune response. Since Epstein-Barr virus (EBV)-associated tumors such as Hodgkin lymphoma (HL) and nasopharyngeal carcinoma (NPC) express FasL, we determined whether EBV-specific cytotoxic T lymphocytes (EBV-CTLs) could be modified to resist this evasion strategy. We show that long-term down-modulation of Fas can be achieved in EBV-CTLs by transduction with small interfering RNA (siRNA) encoded in a retrovirus. Modified T cells resisted Fas/FasL-mediated apoptosis compared with control cells and showed minimal cleavage of the caspase3 substrate poly(ADP-ribose) polymerase (PARP) protein after Fas engagement. Prolonged Fas stimulation selected a uniformly Faslow and FasL resistant population. Removal of responsiveness to this single death signal had no other discernible effects on EBV-CTLs. In particular, it did not lead to their autonomous growth since the modified EBV-CTLs remained polyclonal, and their survival and proliferation retained dependence on antigen-specific stimulation and on the presence of other physiologic growth signals. EBV-CTLs with knocked down Fas should have a selective functional and survival advantage over unmodified EBV-CTLs in the presence of tumors expressing FasL and may be of value for adoptive cellular therapy. (Blood. 2005;105:4677-4684)
Introduction
Generation of tumor-specific cytotoxic T lymphocytes has increasingly been considered an attractive strategy to treat human cancers.1,2 However, it has become evident that T-cell therapies for patients with cancer are limited in effectiveness at least in part by a failure of tumor-specific cytotoxic T lymphocytes to expand or survive in vivo.2-4
Fas (Apo-1/CD95) is a type I receptor that belongs to the tumor necrosis factor (TNF)/nerve growth factor receptor family.5 Its natural ligand FasL is a type II transmembrane molecule also homologous to TNF.6 The Fas/FasL pathway is one of the most important mechanisms for the induction of apoptosis in memory-effector T cells after elimination of antigen.7,8 However, following the discovery that several tumors can express FasL,9-11 the activation of this pathway has been considered a mechanism by which tumors expressing FasL escape destruction by the immune response.9,10 Knock down of Fas expression may, therefore, improve the survival and the function of tumor-specific cytotoxic T lymphocytes.
Small interfering RNA (siRNA) can transiently mediate specific degradation of homologous messenger RNA (mRNA), silencing the expression of the targeted gene.12 The recent development of retroviral constructs in which the polymerase-III H1-RNA gene promoter synthesizes siRNA-like transcripts allows stable expression of siRNA, thus rendering this technology applicable as gene transfer.13 We have used retroviral siRNA directed against Fas mRNA to knock down this molecule in human Epstein-Barr virus-specific cytotoxic T lymphocytes (EBV-CTLs), since the apparent benefit of these cells in the treatment of EBV-associated malignancies (including Hodgkin lymphoma and nasopharyngeal carcinoma) may be circumscribed by tumor expression of FasL.14,15
Our data show that stable knock down of Fas can be achieved in EBV-CTLs by transduction with retroviral siRNA and that modified cells became resistant to Fas/FasL-mediated apoptosis, allowing selection of the Faslow population. Nonetheless, the growth and survival of these modified cells remained fully dependent on antigen-specific stimulation and the presence of other physiologic growth signals, suggesting responsiveness to this single death signal may be safely removed from an effector-memory population.
Materials and methods
Cell lines
COS-7 and 293T cells were obtained from the American Type Culture Collection (ATCC, Rockville, MD) and were maintained in culture with Dulbecco modified Eagle medium (DMEM; BioWhittaker, Walkersville, MD) supplemented with 10% heat-inactivated fetal calf serum (FCS), 2 mM l-glutamine, 25 IU/mL penicillin, and 25 mg/mL streptomycin (BioWhittaker). Jurkat cells (ATCC) were maintained in RPMI 1640 containing 10% FCS and 2 mM l-glutamine. All work with human cells was approved by the Baylor College of Medicine Institutional Review Board.
Plasmid construction, retrovirus production, and transduction of Jurkat cells
To produce siRNA we used the pSUPER.puro vector13 (OligoEngine, Seattle, WA). We also modified this vector to express the green fluorescence protein (GFP) as a selectable marker instead of the puromycin resistance gene (pSUPER.GFP). The templates for anti-Fas siRNA were designed, annealed, and ligated into the vectors as described by Brummelkamp et al.13 293T cells were cotransfected with 3 plasmids (pSUPER.puro or pSUPER. GFP, Peg-Pam-e, and pRDF encoding the gene for RD114 envelop),16 using the Fugene6 transfection reagent (Roche, Indianapolis, IN), and retrovirus supernatant was collected 48 and 72 hours later. For transduction, 0.5 × 106 Jurkat/mL were plated in 24-well plates precoated with recombinant fibronectin fragment (FN CH-296; Retronectin; Takara Shuzo, Otsu, Japan). A second transduction was repeated the next day. Seventy-two hours later, cells transduced with pSUPER.puro were selected in the presence of puromycin 8 μg/mL (Sigma, Chicago, IL). Control cells were transduced with the empty vector (EV) or a vector coding for an irrelevant siRNA [CTGGCATCGGTGTGGATGA] (CsiRNA).17
The full-length human FasL cDNA was obtained from Clone 4849770 (Invitrogen, Carlsbad, CA) and cloned into the PL-X-SP retrovirus to generate the PL-hFasL-SP. Retrovirus supernatant was produced as described for pSUPER vectors. COS-7 cells were transduced either with PL-X-SP empty (COS-7/EV) or PL-hFasL-SP (COS-7/hFasL) and selected with puromycin (8 μg/mL). Human FasL was detected in supernatants (800-1600 pg/mL) and cell lysates (200 pg/mL) by enzyme-linked immunosorbent assay (ELISA; R&D System, Minneapolis, MN).
Generation and transduction of EBV-CTLs
EBV-CTLs were prepared by stimulating mononuclear cells (MNCs) each week with gamma-irradiated (40 Gy) autologous EBV-transformed lymphoblastoid cells (LCLs) and recombinant human interleukin 2 (rhIL-2; 50 U/mL; Proleukin; Chiron, Emeryville, CA), as previously described.1 EBV-CTLs obtained after at least 3 stimulations were transduced as described for Jurkat cells. One week after transduction, EBV-CTLs transduced with pSUPER.puro vectors were selected in the presence of puromycin (1 μg/mL) and stimulated weekly with autologous LCLs and rhIL-2. In experiments in which EBV-CTLs were transduced with pSUPER. GFP vectors, cells were stimulated weekly but without any selection.
Immunophenotyping and detection of apoptosis
Cells were stained with phycoerythrin (PE)-, fluorescein isothiocyanate (FITC)-, or peridinin chlorophyll protein (PerCP)-conjugated antibodies (Abs). We used CD3, CD4, CD8, CD56, and CD95 (anti-hFas) from Becton Dickinson (Mountain View, CA) and Abs specific for the T-cell receptor variable beta region (TCR-Vβ) repertoire (IOTest βMark kit; Immunotech, Emeryville, CA). Tetramers targeting known major histocompatibility complex (MHC) class I epitopes of EBV-related antigens (MHC Tetramer Core Facility, Houston, TX) were also used. Cells were analyzed by a FACScan (Becton Dickinson) equipped with the filter set for triple fluorescence signals. To induce apoptosis, Jurkat cells and EBV-CTLs were cultured for 16 hours with an anti-Fas-activating Ab (clone CH-11; Upstate, Charlottesville, VA) at 20 ng/mL and 40 ng/mL, respectively. To evaluate the effect of the natural hFasL, EBV-CTLs were cocultured (1:1 ratio) for 16 hours with COS-7/hFasL or COS-7/EV. For these experiments, Jurkat and EBV-CTLs were washed and resuspended in RPMI 1640 supplemented with 2% FCS. For experiments with CTLs, gradient centrifugation with Lymphoprep was performed before incubation with CH-11 or COS-7 cells to reduce the background from dead cells. As additional controls, we used purified CD3+ T lymphocytes and concanavalin-A (Con-A) blasts. CD3+ cells were enriched from isolated MNCs by immunomagnetic depletion of CD19+, CD14+, and CD56+ cells (Milteny Biotec, Bergisch Gladbach, Germany). Con-A blasts were generated by culturing MNCs in the presence of 5 μg/mL Con-A (Sigma) and 30 U/mL rhIL-2 for 48 hours, followed by incubation in the presence of rhIL-2 alone for 5 days.18 Apoptosis was measured by staining cells with PE-Annexin-V (BD Pharmingen, San Diego, CA) antibody and 7-Amino-actinomycin (7-AAD).
Enzyme-linked immunospot (ELISPOT) assay
The interferon-γ (IFNγ) ELISPOT assay was performed as previously described.19 EBV-CTLs were plated in triplicate and serially diluted from 1 × 105 to 1 × 104 cells/well and then 100 μL autologous, irradiated LCLs (1 × 105 cells) or peptides (5 μM) were added. Negative controls included EBV-CTLs alone and EBV-CTLs loaded with irrelevant peptides. As positive controls, EBV-CTLs were stimulated with 25 ng/mL phorbol myristate acetate and 1 μg/mL ionomycin (Sigma).
Chromium release assay
The cytotoxic activity of EBV-CTLs was evaluated in a standard 4-hour 51Cr release assay, as previously described.1 The targets tested included autologous LCLs, HLA class I and II mismatched LCLs, and the HSB-2 cell line, which provide a measure of lymphokine-activated killer (LAK) activity. Target cells incubated in media alone or in 1% Triton X-100 were used to determine spontaneous and maximum 51Cr release, respectively. The mean percentage of specific lysis of triplicate wells was calculated as follows: [(test counts - spontaneous counts)/(maximum counts - spontaneous counts)] × 100.
Proliferation assay
EBV-CTLs were cultured at 1 × 105 cells/well with irradiated autologous LCLs at a 4:1 stimulator-to-responder ratio with or without rhIL-2 (50 U/mL). After 72 hours, the cells were pulsed with 1 μCi (0.037 MBq) methyl-3[H]thymidine (Amersham Pharmacia Biotech, Piscataway, NJ) and cultured for an additional 15 hours. The cells were then harvested onto filters and dried, and counts per minute were measured in a β-scintillation counter (TriCarb 2500 TR; Packard BioScience, Boston, MA). The experiments were performed in triplicate.
Western blot analysis
Jurkat cell lysates, obtained from the same number of cells that had been cultured with or without CH-11 Ab, were resolved on a 7.5% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). Poly(ADP-ribose) polymerase (PARP) protein was detected by immunoblot using a monoclonal Ab (Santa Cruz Biotechnologies, Santa Cruz, CA). Immunoblots were developed using enhanced chemiluminescence detection reagents (Amersham Biosciences, Piscataway, NJ). Membranes were reprobed using the monoclonal anti-β-actin Ab (Sigma).
Reverse transcription-polymerase chain reaction amplification (RT-PCR)
Total RNA, complementary DNA (cDNA), and RT-PCR were performed according to standard procedures. The primers for the human Fas amplification were 5′-TTCGGAGGATTGCTCAACA-3′ (sense, exon 1) and 5′-GGTGAGTGTGCATTCCTTG-3′ (antisense, exon 5). In each experiment, the integrity and loading of RNA extracted was evaluated by amplification of the β-actin using the primers 5′-GTGGGGCGCCCCAGGCACCA-3′ (sense, exon 1) and 5′-CTCCTTAATGTCACGCACGATTTC-3′ (antisense, exon 3). Twenty-five cycles, each consisting of 30 seconds at 94°C, 30 seconds at 56°C, and 30 seconds at 72°C, were performed. The reaction products were electrophoretically separated through a 1.2% agarose gel and stained with ethidium bromide. The expected products generated by PCR were 500 base pairs (bp) and 546 bp for FAS and β-actin, respectively.
Statistical analysis
All data are presented as mean plus or minus 1 SD. Student t test was used to determine the statistical significance of differences between samples, and P less than .05 was accepted as indicating a significant difference.
Results
EBV-CTLs but not primary T cells express high levels of Fas and are susceptible to Fas-mediated death
EBV-CTLs generated from 7 EBV seropositive healthy donors were predominantly CD3+/CD8+ T lymphocytes (89%; range, 81%-97%), whereas CD3+/CD4+ T lymphocytes were 9% (range, 3%-14%). More than 95% of the EBV-CTLs had an effector memory phenotype CD45RA-, CD45RO+, CD62L+/- (data not shown). As shown in Figure 1, EBV-CTLs had high expression of Fas, unlike freshly isolated CD3+ T lymphocytes. Fas could also be up-regulated in virtually all normal T cells after brief activation in vitro with Con-A and rhIL-2.18 However, Fas expression by Con-A blasts remained lower than by EBV-CTLs (Fas mean fluorescence intensity [MFI], 216 ± 47 versus 341 ± 76; P = .002). Fas expression in EBV-CTLs did not significantly change with additional in vitro stimulation with antigen and rhIL-2 and was not significantly different between CD8+ and CD4+ cells (data not shown).
Figure 1.

EBV-CTLs express high levels of Fas and are susceptible to Fas-mediated apoptosis. EBV-CTLs (A), freshly isolated CD3+ cells (B), and Con-A blasts (C) were stained with anti-CD95-PE (solid line) or isotype control (dashed lines). (D) The mean fluorescence intensity MFI for CD95 in EBV-CTLs, freshly isolated CD3+ cells, and Con-A blasts are shown. Data are representative of 4 different donors for MNCs and 7 donors for the CTL lines (*P < .001; **P = .002). (E) Fas-mediated apoptosis is shown in EBV-CTLs, Con-A blasts, and isolated CD3+ cells incubated with CH-11 antibody (40 ng/mL;  ) or in media alone (□). Data represent the mean ± SD of 6 different donors for EBV-CTLs and 4 different donors for isolated CD3+ cells and Con-A blasts (*P < .001).
) or in media alone (□). Data represent the mean ± SD of 6 different donors for EBV-CTLs and 4 different donors for isolated CD3+ cells and Con-A blasts (*P < .001).
To determine their susceptibility to Fas-induced apoptosis, CTL lines were incubated with CH-11 Ab. As shown in Figure 1, after 16 hours, EBV-CTLs underwent significant apoptosis in the presence of CH-11 (40 ng/mL) compared with untreated EBV-CTLs (46% ± 9% versus 21% ± 5%, P < .001). The percentage of necrotic cells remained unchanged (16% ± 3% and 14% ± 4% for treated and untreated EBV-CTLs, respectively). Apoptosis of EBV-CTLs incubated with an irrelevant mouse immunoglobulin M (IgM) Ab was identical to control cells (data not shown). As previously reported,20 we observed that resting CD3+ cells were completely resistant to CH-11. Even after a brief activation of primary T cells with Con-A and rhIL-2, induction of apoptosis by CH-11 was minimal compared with EBV-CTLs.18
Stable expression of anti-Fas siRNA knocks down Fas in Jurkat cells
We prepared several pSUPER.puro and pSUPER.GFP vectors containing 10 different sequences predicted as siRNA for Fas mRNA. Two of 10 sequences tested (GGACATTACTAGTGACTCA [designed as siRNA10 and GFP-siRNA10] and GTTCAACTGCTTCGTAATT [designed as siRNA8 and GFP-siRNA8]) substantially knocked down Fas expression in Jurkat cells (Figure 2). The MFIs of Fas expression were 83 plus or minus 15 (nontransduced Jurkat), 83 plus or minus 8 (empty vector transduced), 75 plus or minus 5 (CsiRNA transduced), 27 plus or minus 15 (siRNA8 transduced), and 15 plus or minus 5 (siRNA10 transduced). Both siRNA8 and siRNA10 were significantly different from control vector values (P = .009 and P < .001, respectively). When Jurkat cells transduced with siRNA10 and selected with puromycin were transduced with GFP-siRNA8, Fas was almost completely knocked down in GFP+ cells (Figure 2). Knock-down expression of Fas in Jurkat cells transduced with siRNA8 and siRNA10 paralleled the reduced amount of specific mRNA, as demonstrated by RT-PCR amplification (Figure 2). Fas knock down was stable in cells maintained in culture for more than 3 months.
Figure 2.
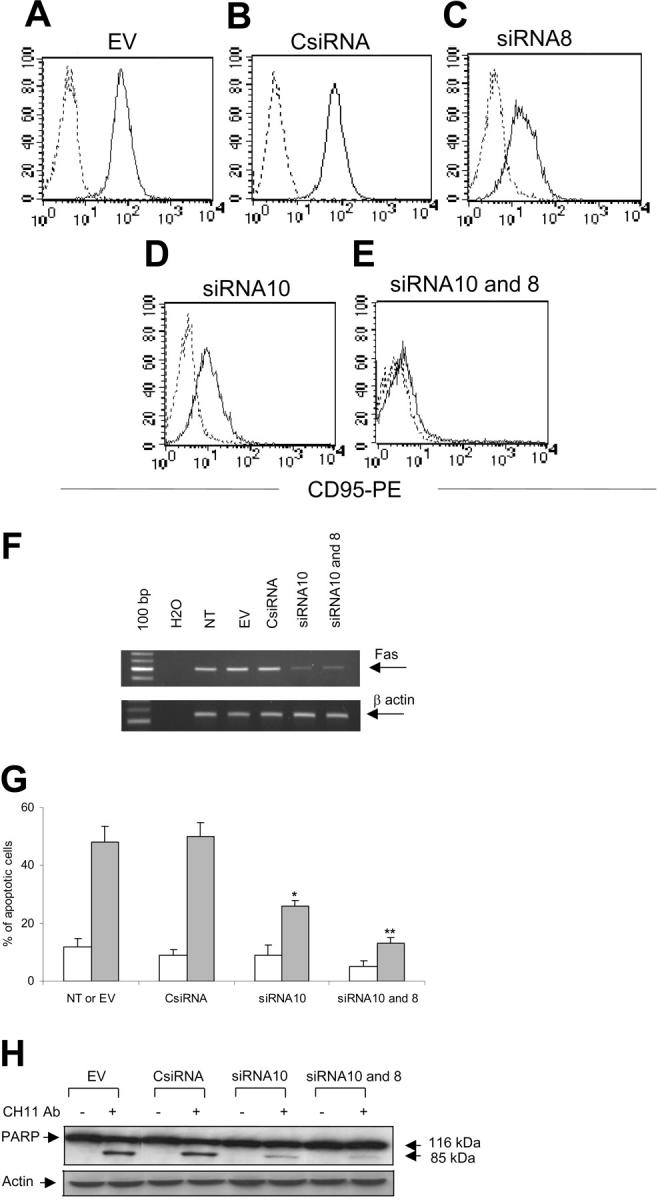
SiRNA knocks down Fas in Jurkat cells and reduces their susceptibility to Fas-mediated death. Jurkat cells were transduced with the EV (A), CsiRNA (B), siRNA8 (C), siRNA10 (D), or sequentially with siRNA10 and GFP-siRNA8 (siRNA10 and 8) (E). After selection, cells were stained with anti-CD95-PE (solid line) or isotype control (dashed lines). (F) The RT-PCR is shown for Fas (top) and β-actin (bottom) using the total RNA extracted from Jurkat cells. (G) Fas mediated-apoptosis is shown in Jurkat cells incubated with () or without (□) CH-11 antibody (20 ng/mL). Data represent the mean ± SD of 3 independent experiments (*P = .01; **P = .001). (H) The PARP protein cleavage in Jurkat cells is shown after incubation with or without CH-11 antibody (top gel). The bottom gel shows the membrane reprobed with anti-β-actin antibody.
Fas knock down reduces the susceptibility to Fas-mediated apoptosis in Jurkat cells
We next evaluated whether siRNA-mediated knock down of Fas reduced the susceptibility to Fas-mediated apoptosis. As shown in Figure 2, Jurkat cells transduced with siRNA10 were more resistant to apoptosis induced by CH-11 Ab (20 ng/mL) compared with control cells transduced with the EV (26% ± 2% versus 48% ± 6%, P < .001) or CsiRNA (26% ± 2% versus 50% ± 5%, P < .001). We observed no significant difference in protection from apoptosis between Jurkat cells transduced with siRNA8 and Jurkat cells transduced with siRNA10 (data not shown). However, protection from death correlated with the level of Fas expression, since Jurkat cells transduced with both vectors (siRNA10 and GFP-siRNA8) were almost completely resistant to death compared with cells transduced with 1 single vector (13% ± 2% versus 26% ± 2%, P = .001). Fas/FasL binding results in activation of cysteine proteases, or caspases, which induces proteolytic cleavage of numerous substrates. The PARP, a nuclear enzyme involved in DNA repair, is a well-known substrate for caspase-3 cleavage during Fas-induced apoptosis.21 To further demonstrate that knock down of Fas reduces Fas-mediated apoptosis in Jurkat cells, we evaluated the cleavage of the nuclear enzyme PARP in cells treated with CH-11 Ab. As shown in Figure 2, in Jurkat cells transduced with EV or CsiRNA, the 116-kDa PARP protein was cleaved into a stable 85-kDa fragment after incubation with CH-11 Ab (20 ng/mL) for 16 hours. However, the cleavage process was significantly reduced in cells where Fas was stably knocked down by anti-Fas siRNA.
SiRNA-mediated knock down of Fas in EBV-CTLs reduces their susceptibility to Fas-mediated cell death
On the basis of the results obtained in Jurkat cells, we transduced EBV-CTLs either with the EV, CsiRNA, or siRNA10. EBV-CTLs were then selected in the presence of puromycin (1 μg/mL) and stimulated weekly with autologous LCLs and rhIL-2. As shown in Figure 3, Fas was knocked down in EBV-CTLs transduced with siRNA10 compared with EBV-CTLs transduced with EV (Fas MFI, 107 ± 47 versus 402 ± 47; P < .001) or CsiRNA (Fas MFI, 107 ± 47 versus 361 ± 61; P < .001). No significant differences in Fas expression was observed between nontransduced CTLs (NT) and those transduced with the EV (Fas MFI, 341 ± 76 versus 402 ± 47) or CsiRNA (Fas MFI, 341 ± 76 versus 361 ± 61). We next sequentially transduced EBV-CTLs with siRNA10 and GFP-siRNA8, as described for Jurkat cells. Although there was an additive reduction of Fas expression in double-transduced EBV-CTLs compared with single transduced EBV-CTLs, this was not statistically significant (MFI, 107 ± 47 versus 73 ± 23; P = .08). EBV-CTLs maintained in culture for 5 to 10 weeks after transduction were then incubated with CH-11 Ab (40 ng/mL) to induce apoptosis. As shown in Figure 3, EBV-CTLs transduced with siRNA10 were significantly resistant to apoptosis induced by CH-11 compared with EBV-CTLs transduced with the EV (23% ± 3% versus 44% ± 7%, P = .006) or CsiRNA (23% ± 3% versus 44% ± 3%, P = .006). We observed no significant difference in susceptibility to apoptosis between EBV-CTLs transduced with a single or both knock-down vectors (23% ± 3% versus 22% ± 4%). The percentage of necrotic EBV-CTLs was less than 20% for both treated and untreated EBV-CTLs and was not significantly different between transduced or EBV-CTLs NT.
Figure 3.

SiRNA knocks down the surface expression of Fas in EBV-CTLs. EBV-CTLs transduced with the EV (A), CsiRNA (B), siRNA10 (C), or with both siRNA10 and GFP-siRNA8 (siRNA10 and 8) (D) after selection were stained with anti-CD95-PE (solid lines) or isotype control (dashed lines). Bold solid lines and thin solid lines indicate CD95 expression in EBV-CTLs nontransduced (NT) and transduced, respectively. (E) The CD95 MFI in EBV-CTLs NT and transduced with the different vectors. Data represent the mean ± SD of 6 different donors (*P < .001). (F) The percentage of apoptotic EBV-CTLs is shown after incubation with () or without (□) CH-11 antibody (40 ng/mL). Data represent the mean ± SD of 5 different donors (**P = .006).
Knock down of Fas mediated by anti-Fas siRNA in EBV-CTLs is stable and increases CTL survival in presence of aberrant activation of the Fas/FasL pathway
To discover whether the Fas knock down mediated by siRNA was stable and sufficient to favor the survival of the modified cells, EBV-CTLs were transduced with GFP-siRNA10 or GFP-CsiRNA and stimulated weekly with LCLs and rhIL-2 without any selection. EBV-CTLs were counted and stained weekly to evaluate the percentage of GFP+ and GFP- cells as well as the expression of Fas in both GFP+ and GFP- EBV-CTLs. For CTLs transduced with GFP-CsiRNA and GFP-siRNA10, the percentage of GFP+ EBV-CTLs after transduction was 58% (range, 42%-77%) and 60% (range, 54%-77%) and remained stable during in vitro expansion for more than 8 stimulations (Figure 4). We confirmed in LCLs made from 10 different donors that these EBV-transformed normal B cells do not express or release FasL. It is likely for this reason that CTLs with knocked down Fas maintain the same proliferative capacity of control CTLs, since there is no selective advantage in the absence of Fas activation. However, the addition of CH-11 Ab (40 ng/mL) twice a week to the CTLs transduced with GFP-siRNA10, increased the absolute number and the percentage of GFP+ EBV-CTLs to 88% (range, 81%-97%; P = .001) (Figure 4), suggesting that these cells were protected from CH-11-induced cell death. In contrast, when the CH-11 Ab was added to CTLs transduced with GFP-CsiRNA, we observed reduced CTL expansion and no selection for GFP+ CTLs (Figure 4). Measurement of Fas expression over time showed that Fas was significantly and constantly knocked down in GFP+ cells transduced with GFP-siRNA10 compared with CTLs transduced with GFP-CsiRNA (Figure 4). As anticipated, addition of CH-11 Ab to the CTLs transduced with GFP-siRNA10 also selected for the survival of CTLs with low Fas expression (Faslow): the MFI for CD95 was 226 ± 99 in EBV-CTLs cultured without the addition of CH-11 and 79 ± 29 in those surviving to the prolonged exposure to CH-11 (P = .01). CTLs transduced with GFP-CsiRNA and surviving exposure to CH-11 showed the same low expression of Fas as those transduced with GFP-siRNA10.
Figure 4.

Fas knock down in EBV-CTLs is stable and increases their survival in the presence of aberrant activation of the Fas/FasL pathway. EBV-CTLs transduced with GFP-siRNA10 or GFP-CsiRNA were stimulated weekly with autologous LCLs and rhIL-2 without selection. The percentage of GFP+ () and GFP- (□) cells was stable over time for both CTLs transduced with GFP-CsiRNA (A) and GFP-siRNA10 (B). However, the addition of CH-11 antibody (40 ng/mL) to the culture induced selective growth advantage for GFP+ CTLs transduced with GFP-siRNA10 (D) compared with those transduced with GFP-CsiRNA (C). (E-G) The expression is shown of Fas in GFP+ CTLs transduced with GFP-siRNA10 (bold solid lines) and GFP-CsiRNA (thin solid lines), respectively, over time with or without the addition of CH-11 to the culture. Dashed lines indicate the profile of the isotype control. The analysis was performed on gated GFP+ cells. Data illustrate a representative donor of 5 independent experiments.
To mimic activation of the Fas pathway by the natural aberrantly expressed FasL by tumor cells, we cocultured EBV-CTLs (ratio 1:1) for 16 hours with COS-7/hFasL cells or control COS-7/EV. As shown in Figure 5, the percentage of dead cells in the presence of hFasL was 44% ± 12% for EBV-CTLs transduced with the EV or CsiRNA but 25% ± 10% for EBV-CTLs transduced with siRNA10 (P = .006). When EBV-CTLs transduced with GFP-siRNA10 were stimulated with LCLs and rhIL-2 and cultured in the presence of COS-7/hFasL cells, there was progressive selection of CTLs with knocked down Fas, increasing the percentage of GFP+ cells from 46% (range, 42%-50%) to 81% (range, 78%-84%) (P = .001) by day 4. These cultures also selected a Faslow CTL population (Fas MFI, 116 ± 5). When the same experiment was performed with control CTLs transduced with GFP-CsiRNA, we did not observe any selection in the few surviving cells (Figure 5).
Figure 5.
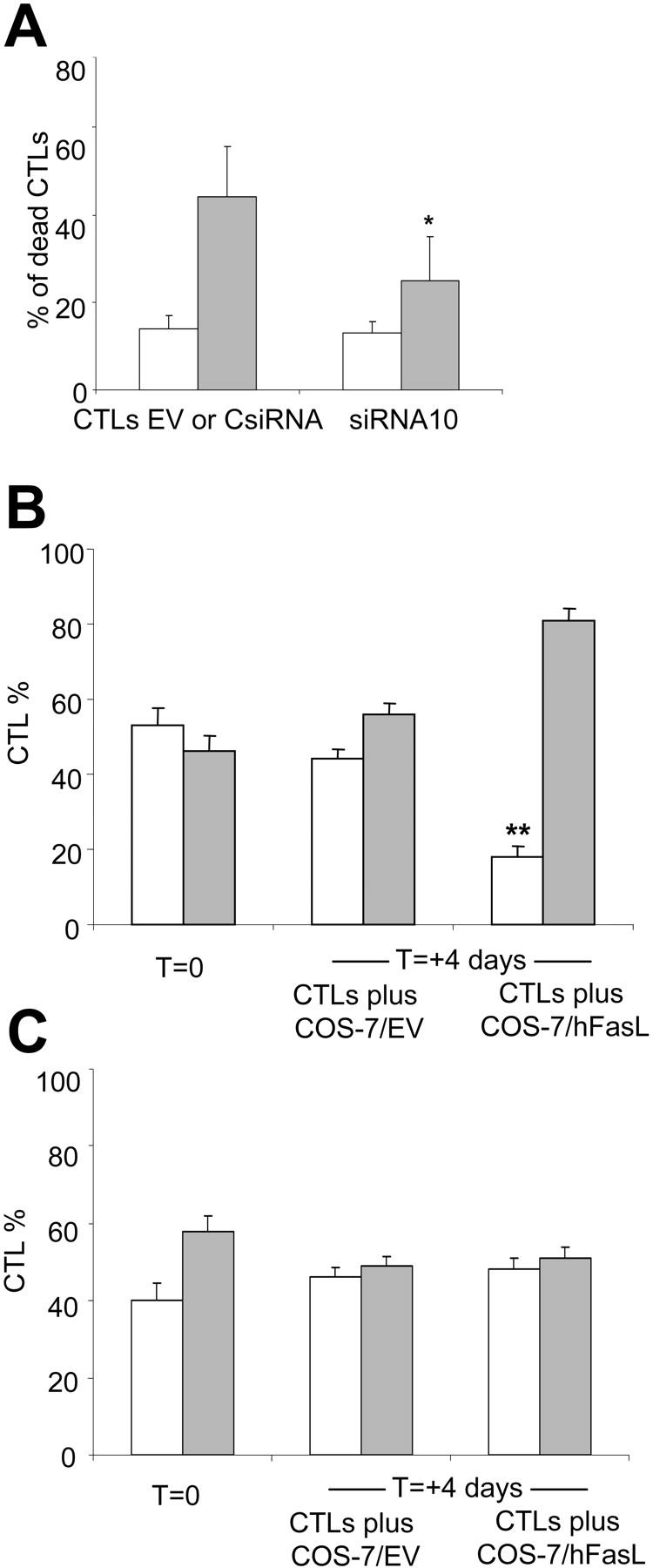
Fas knock down in EBV-CTLs increases CTL survival in the presence of cells producing hFasL. (A) The percentage is shown of dead EBV-CTLs transduced with siRNA10, EV, or CsiRNA cocultured (ratio 1:1) with COS-7/hFasL () or COS-7/EV (□). EBV-CTLs transduced with siRNA10 were more resistant to FasL-induced death compared with CsiRNA-transduced CTLs. Data represent the mean ± SD of 4 different donors (*P = .008). EBV-CTLs were then transduced with GFP-siRNA10 (B) or GFP-CsiRNA (C) and cultured for 4 days with COS-7/EV or COS-7/hFasL (ratio 10:1). A selective increase of GFP+ () compared with GFP- (□) cells was observed in CTLs with knocked down Fas (B) (**P = .001), while no selection was observed in control CTLs transduced with GFP-CsiRNA. Data represent the mean of 3 different donors.
EBV-CTLs with Fas knock down retain their dependency on antigen and growth factors for expansion and survival
Germ line deletions as well as point mutations of Fas and FasL can induce T-lymphoproliferation and autoimmune diseases.22 To discover whether similar problems could be anticipated when Fas function was modified in EBV-CTLs, we compared the growth characteristics and the antigen specificity of unmodified and Fas knock-down EBV-CTLs, looking in particular for evidence of clonal lymphoproiliferation.
We first used the methyl-3[H]thymidine incorporation assay to evaluate whether EBV-CTLs maintain their IL-2 dependency for in vitro proliferation. As shown in Figure 6, EBV-CTLs transduced with siRNA10, and selected with puromycin, proliferated only in the presence of both specific antigen (autologous LCLs) and exogenous rhIL-2. Moreover, their level of proliferation was similar to that observed in EBV-CTLs NT or EBV-CTLs transduced with the EV or CsiRNA. Both EBV-CTLs transduced with siRNA10 and control cells maintained in culture without any stimulation with LCLs and rhIL-2 died within 10 to 14 days (data not shown). Similarly, knock down of Fas in EBV-CTLs did not alter their phenotype: EBV-CTLs transduced with siRNA10 were 86% plus or minus 14% CD3+/CD8+, while EBV-CTLs NT or EBV-CTLs transduced with EV or CsiRNA were 83% plus or minus 16%, 84% plus or minus 15%, and 85% plus or minus 15% positive for these markers, respectively. The percentages of CD3+/CD4+ cells were also unchanged at 6% plus or minus 4%, 6% plus or minus 4%, 7% plus or minus 5%, and 5% plus or minus 4% for the same groups. 51Cr release assays demonstrated that the percentage of autologous LCLs lysed by the EBV-CTLs transduced with siRNA10 was 64% plus or minus 19%, the same as observed for control EBV-CTLs. The MHC-restricted killing by the CTL lines was maintained, since the reactivity detected against allogeneic LCLs was 15% plus or minus 7% and the reactivity against the HSB-2 cell line was 26% plus or minus 17% (Figure 6).
Figure 6.

Fas down-modulation does not modify growth or antigen specificity of EBV-CTLs. EBV-CTLs nontransduced (□) and EBV-CTLs transduced with the EV (▪), CsiRNA ( ) or siRNA10 () after selection were cocultured with autologous LCLs with or without the addition of rIL-2 (50 U/mL). After 3 days, cells were pulsed with methyl-3H-thymidine (A). (B) A standard 51Cr release assay is shown in which CTL killing of autologous LCLs, allogeneic LCLs, and the HBS-2 cell line was tested at an effector-target ratio of 20:1. Data represent the mean ± SD of 3 and 5 different donors, respectively. (C) A representative staining is shown of EBV-CTLs using the tetramers HLA-B8-RAKFQLL and HLA-B8-QAKWRLQTL tetramers targeting BZLF1 and EBNA3A, respectively.
) or siRNA10 () after selection were cocultured with autologous LCLs with or without the addition of rIL-2 (50 U/mL). After 3 days, cells were pulsed with methyl-3H-thymidine (A). (B) A standard 51Cr release assay is shown in which CTL killing of autologous LCLs, allogeneic LCLs, and the HBS-2 cell line was tested at an effector-target ratio of 20:1. Data represent the mean ± SD of 3 and 5 different donors, respectively. (C) A representative staining is shown of EBV-CTLs using the tetramers HLA-B8-RAKFQLL and HLA-B8-QAKWRLQTL tetramers targeting BZLF1 and EBNA3A, respectively.
To further characterize the effects of Fas knock down on the maintenance of polyclonal antigen specificity of EBV-CTLs, we evaluated the frequency of EBV-CTLs responding to available HLA class I-restricted EBV peptides using specific tetramers. We also measured the distribution of Vβ-TcR repertoire. As shown in Figure 6 and Table 1 the frequency of tetramer-positive EBV-CTLs as well as the Vβ TcR repertoire were not significantly modified in EBV-CTLs transduced with siRNA10 compared with controls. Finally, we determined whether the modified EBV-CTLs selected by prolonged exposure to CH-11 Ab retained functional reactivity to identical EBV-derived peptide epitopes. As shown in Table 2, EBV-CTLs transduced with siRNA10 and maintained in culture with the addition of CH-11 twice a week retained the same capacity to release IFNγ in response to HLA class I-restricted EBV peptides as unselected or control EBV-CTLs.
Table 1.
Repertoire analysis of CTL lines generated from 3 representative donors
|
CTLs, donor 1
|
CTLs, donor 2
|
CTLs, donor 3
|
|||||||
|---|---|---|---|---|---|---|---|---|---|
| Vβ | NT | EV or CsiRNA | SiRNA10 | NT | EV or CsiRNA | SiRNA10 | NT | EV or CsiRNA | SiRNA10 |
| Vβ1 | 1.66 | 0.98 | 1.43 | 5.71 | 4.38 | 4.66 | 7.48 | 9.67 | 5.14 |
| Vβ2 | 1.54 | 1.81 | 1.54 | 4.55 | 4.59 | 4.59 | 8.05 | 7.61 | 8.24 |
| Vβ3 | 2.49 | 2.02 | 2.2 | 0.58 | 1.15 | 0.83 | 7.67 | 11.18 | 8.21 |
| Vβ4 | 0.01 | 0.06 | 0.06 | 0.11 | 0.32 | 0.16 | 0.03 | 0.06 | 0.02 |
| Vβ5.1 | 3.37 | 2.99 | 3.17 | 5.55 | 5.07 | 5.47 | 6.42 | 9.45 | 5.11 |
| Vβ5.2 | 0.92 | 0.22 | 0.53 | 0.19 | 0.29 | 0.25 | 0.55 | 0.32 | 0.58 |
| Vβ5.3 | 2.17 | 2.42 | 2.25 | 0.49 | 0.38 | 0.5 | 0.15 | 0.14 | 0.2 |
| Vβ7.1 | 2.73 | 3.03 | 2.66 | 4.25 | 3.71 | 3.51 | 4.43 | 3.89 | 5.93 |
| Vβ7.2 | 0.04 | 0.06 | 0.01 | 1.11 | 0.7 | 0.85 | 0.32 | 0.28 | 0.4 |
| Vβ8 | 1.51 | 1.43 | 1.08 | 4.79 | 4.88 | 4.79 | 0.78 | 0.51 | 0.41 |
| Vβ9 | 2.05 | 1.39 | 3.1 | 0.82 | 0.67 | 0.49 | 0.9 | 0.87 | 0.57 |
| Vβ11 | 0.06 | 0.35 | 0.48 | 3.99 | 2.41 | 3.24 | 0.07 | 0.06 | 0.02 |
| Vβ12 | 0.51 | 1.14 | 0.58 | 3.55 | 3.97 | 3.11 | 0.7 | 0.96 | 0.38 |
| Vβ13.1 | 1.98 | 1.1 | 1.66 | 1.8 | 2.38 | 1.72 | 0.63 | 0.44 | 0.53 |
| Vβ13.2 | 8.52 | 1.53 | 13.24 | 1.83 | 1.49 | 1.66 | 1.78 | 1.24 | 1.26 |
| Vβ13.6 | 1.24 | 1.0 | 0.89 | 0.59 | 0.84 | 0.65 | 0.11 | 0.09 | 0.15 |
| Vβ14 | 13.97 | 19.4 | 17.73 | 5.93 | 7.48 | 4.97 | 6.73 | 8.55 | 6.45 |
| Vβ16 | 0.46 | 0.62 | 0.57 | 2.71 | 4.97 | 2.36 | 0.45 | 0.33 | 0.35 |
| Vβ17 | 0.38 | 0.25 | 0.33 | 2.65 | 1.78 | 1.6 | 0.13 | 0.35 | 0.21 |
| Vβ18 | 0.06 | 0.18 | 0.05 | 1.07 | 1.67 | 1.39 | 0.06 | 0.19 | 0.09 |
| Vβ20 | 0.5 | 0.39 | 0.47 | 1.25 | 1.28 | 0.75 | 0.46 | 0.41 | 0.28 |
| Vβ21.3 | 1.22 | 0.84 | 0.9 | 0.71 | 0.77 | 0.77 | 3.54 | 4.11 | 3.23 |
| Vβ22 | 3.19 | 2.12 | 2.9 | 1.78 | 1.9 | 1.97 | 1.33 | 0.81 | 0.9 |
| Vβ23 | 0.71 | 1.2 | 0.94 | 1.97 | 2.44 | 1.48 | 0.23 | 0.19 | 0.37 |
Data represent the percentage of T cells recognized by the specific antibodies.
NT indicates nontransduced CTLs; EV, CTLs transduced with the pSUPER.puro empty; CsiRNA, CTLs transduced with pSUPER.puroCsiRNA; siRNA10, CTLs transduced with pSUPER.puro.siRNA10.
Table 2.
IFNγ release of CTLs transduced with siRNA10 and maintained in culture with or without CH-11 antibody
| CTLs | HLA*typing | A2-CLG | A2-GLC | B8-RAK | B8-QAK | B35-AVL | B35-HPV | CTL alone | A11†DEP |
|---|---|---|---|---|---|---|---|---|---|
| 1 | A1,24;B8,18 | — | — | 543 ± 83‡ | 1199 ± 25 | — | — | 0 | NT |
| 1 + CH-11 | A1,24;B8,18 | — | — | 615 ± 64 | 1022 ± 185 | — | — | 0 | NT |
| 2 | A1,2,;B7,B8 | 499 ± 54 | 227 ± 21 | NT | NT | — | — | 0 | 0 |
| 2 + CH-11 | A1,2,;B7,B8 | 433 ± 2 | 296 ± 20 | NT | NT | — | — | 0 | 0 |
| 3 | A2,3;B35,57 | 208 ± 22 | NT | — | — | 310 ± 61 | 533 ± 87 | 0 | 0 |
| 3 + CH-11 | A2,3;B35,57 | 203 ± 34 | NT | — | — | 245 ± 76 | 553 ± 26 | 0 | 0 |
| 4 | A1,2;B7,8 | 12 ± 4 | NT | 338 ± 34 | 576 ± 100 | — | — | 0 | NT |
| 4 + CH-11 | A1,2;B7,8 | 32 ± 7 | NT | 326 ± 26 | 458 ± 50 | — | — | 0 | NT |
NT indicates not tested; —, not applicable.
The following HLA class I-restricted peptides were used for stimulation in an ELISPOT assay: HLA-A2 LMP2 epitope CLGGLLTML (CLG), HLA-A2 BMLF1 epitope GLCTLVAML (GLC), HLA-B8 BZLF1 epitope RAKFQLL (RAK), HLA-B8 EBNA3A QAKWRLQTL (QAK), HLA-B35 EBNA3B epitope AVLLHEESL (AVL), and HLA-B35 EBNA1 epitope HPVGEADYF (HPV).
The A11-DEP peptide was used as irrelevant peptide.
Number of spot-forming cells/105CTL.
Discussion
We have used knock down of Fas expression to discover whether we could increase the resistance of tumor-specific cytotoxic T lymphocytes to immune evasion strategies that rely on constitutive expression of FasL by tumor cells. We also determined whether such resistance would result in unwanted T-cell proliferation or autonomy. Using EBV as our model system, we found that EBV-CTLs, unlike resting human T cells, are highly sensitive to apoptosis induced by the Fas/FasL system, but that sustained knock down of Fas expression can be obtained using retroviral-siRNA constructs. Transduced EBV-CTLs are highly resistant to FasL-induced apoptosis. Importantly, their growth and survival remain dependent on antigen and exogenous growth factors, suggesting autonomous growth or development of autoimmunity will not occur when antigen-specific effector-memory cells are the targets of Fas knock down. Thus, Fas knocked down EBV-CTLs may have improved efficacy for the treatment of FasL-expressing EBV-associated tumors such as Hodgkin lymphoma (HL) and nasopharyngeal carcinoma (NPC).
Potentially immunogenic tumors have evolved many different immune evasion strategies. While these often affect antigen presentation or modulate effector T-cell induction and function,23,24 several tumors, including those associated with EBV, also express FasL, which likely impedes the host immune response by accelerating the death of tumor-infiltrating T cells.9-11 Despite the high frequency of FasL expression by tumor cells, the importance of this molecule for tumor escape from host defenses has been questioned.25 Several in vivo experiments have shown that tumor cells expressing supraphysiologic levels of FasL were rejected by an inflammatory process mediated by neutrophils,26 suggesting that any potential benefit to be gained by accelerated killing of effector T cells would be more than offset by increased destruction by the innate immune system. However, it is now clear that apparently distinct tumor defense mechanisms may interact in a mutually supportive manner. For example, animal models show that the concomitant presence of transforming growth factor-beta (TGF-β) within the tumor microenvironment prevents the rejection of FasL-positive tumor cells by innate immunity.27 It is noteworthy that the malignant Reed-Sternberg cells of Hodgkin disease both express FasL and secrete TGFβ.15,28 The effects of FasL may further depend on its biologic form. FasL, like other members of the TNF family, is cleaved by matrix metalloproteinases from the cell surface to generate a soluble protein29 that retains apoptotic but not proinflammatory activity, at least in humans.18,30 Thus, a high level of metalloproteinase activity, which is frequently present in the tumor microenvironment,31 likely promotes elimination of infiltrating T cells while avoiding induction of an inflammatory response.
Degradation of mRNA mediated by siRNA is a powerful means of specifically knocking down the expression of a target gene.13,32 The ability to stably express siRNA from a retroviral vector allowed Fas mRNA and Fas protein to be knocked down for at least 3 months both in a T-cell line (Jurkat) and in EBV-CTLs. EBV-CTLs transduced with anti-Fas siRNA showed increased resistance to Fas/FasL-induced apoptosis and survival advantage in culture after prolonged Fas stimulation. This strategy might improve the persistence of EBV-CTLs in vivo, promoting their survival in a hostile tumor microenvironment expressing FasL as shown in our cocultures with hFasL-expressing cells. However, one of the major concerns about Fas knock down is that it would become associated with a loss in antigen-directed functionality of the T cells, the development of autonomous growth, or both. Certainly animal models in which Fas is mutated are associated with the development of T-lymphoproliferative disorders and a range of autoimmune conditions.22 A similar autoimmune lymphoproliferative condition has been observed in affected humans.33 However, in each of these examples, Fas function was diminished in all cells of the T lineage, including prethymic T cells and naive postthymic T lymphocytes, suggesting that activation of the Fas pathway is of particular importance in these cell types to eradicate autoreactive clones.34 While the Fas/FasL pathway remains important for the rapid control of the numbers of peripheral effector-memory T cells, the development of autoreactivity is now much less of a concern, and lymphocyte growth and survival can also be controlled by passive mechanisms, dependent on the attenuation of antigenic stimulation and availability of growth factors.35 Other researchers have proposed the transgenic overexpression of antiapoptotic downstream molecules such as bcl-xL to reduce susceptibility to apoptosis of T cells.36 Although this approach is effective, it is nonspecific, allowing protection from multiple proapoptotic stimuli, including antigen and cytokine deprivation. This may increase the possibility of losing control of the proliferation of genetically modified T cells. We suggest that a more selective genetic modification of antigen-specific CTLs such as the down-regulation of Fas that we propose allows the counterattack of a specific mechanism of tumor escape while leaving intact other proapoptotic signal pathways.
The data we report specifically support the use of Fas knocked down antigen-specific CTLs for EBV-related tumors and potentially for other malignant cell types expressing FasL, although the ultimate benefit of this genetic manipulation will receive confirmation in clinical study.
Acknowledgments
We thank Tatiana Goltsova for the technical assistance.
Prepublished online as Blood First Edition Paper, February 15, 2005; DOI 10.1182/blood-2004-08-3337.
Supported in part from the National Institutes of Health, National Cancer Institute (POI CA94234), Leukemia and Lymphoma Society Specialized Center of Research (SCOR) (grant no. 7018), The Shell Center for Cell and Gene Therapy, The Methodist Hospital Foundation award (G.D., B.S., and K.S.), and a Chao Scholar grant (G.D.). M.P. was supported by a fellowship from the British Society of Haematology.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 U.S.C. section 1734.
References
- 1.Rooney CM, Smith CA, Ng CY, et al. Infusion of cytotoxic T cells for the prevention and treatment of Epstein-Barr virus-induced lymphoma in allogeneic transplant recipients. Blood. 1998;92: 1549-1555. [PubMed] [Google Scholar]
- 2.Yee C, Thompson JA, Byrd D, et al. Adoptive T cell therapy using antigen-specific CD8+ T cell clones for the treatment of patients with metastatic melanoma: in vivo persistence, migration, and antitumor effect of transferred T cells. Proc Natl Acad Sci U S A. 2002;99: 16168-16173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Roskrow MA, Suzuki N, Gan Y, et al. Epstein-Barr virus (EBV)-specific cytotoxic T lymphocytes for the treatment of patients with EBV-positive relapsed Hodgkin's disease. Blood. 1998;91: 2925-2934. [PubMed] [Google Scholar]
- 4.Dudley ME, Wunderlich J, Nishimura MI, et al. Adoptive transfer of cloned melanoma-reactive T lymphocytes for the treatment of patients with metastatic melanoma. J Immunother. 2001;24: 363-373. [DOI] [PubMed] [Google Scholar]
- 5.Itoh N, Yonehara S, Ishii A, et al. The polypeptide encoded by the cDNA for human cell surface antigen Fas can mediate apoptosis. Cell. 1991;66: 233-243. [DOI] [PubMed] [Google Scholar]
- 6.Suda T, Takahashi T, Golstein P, Nagata S. Molecular cloning and expression of the Fas ligand, a novel member of the tumor necrosis factor family. Cell. 1993;75: 1169-1178. [DOI] [PubMed] [Google Scholar]
- 7.Nagata S, Golstein P. The Fas death factor. Science. 1995;267: 1449-1456. [DOI] [PubMed] [Google Scholar]
- 8.Bonfoco E, Stuart PM, Brunner T, et al. Inducible nonlymphoid expression of Fas ligand is responsible for superantigen-induced peripheral deletion of T cells. Immunity. 1998;9: 711-720. [DOI] [PubMed] [Google Scholar]
- 9.O'Connell J, O'Sullivan GC, Collins JK, Shanahan F. The Fas counterattack: Fas-mediated T cell killing by colon cancer cells expressing Fas ligand. J Exp Med. 1996;184: 1075-1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Strand S, Hofmann WJ, Hug H, et al. Lymphocyte apoptosis induced by CD95 (APO-1/Fas) ligand-expressing tumor cells—a mechanism of immune evasion? Nat Med. 1996;2: 1361-1366. [DOI] [PubMed] [Google Scholar]
- 11.Hahne M, Rimoldi D, Schroter M, et al. Melanoma cell expression of Fas(Apo-1/CD95) ligand: implications for tumor immune escape. Science. 1996;274: 1363-1366. [DOI] [PubMed] [Google Scholar]
- 12.Hunter T, Hunt T, Jackson RJ, Robertson HD. The characteristics of inhibition of protein synthesis by double-stranded ribonucleic acid in reticulocyte lysates. J Biol Chem. 1975;250: 409-417. [PubMed] [Google Scholar]
- 13.Brummelkamp TR, Bernards R, Agami R. A system for stable expression of short interfering RNAs in mammalian cells. Science. 2002;296: 550-553. [DOI] [PubMed] [Google Scholar]
- 14.Tsai ST, Fang SY, Jin YT, Su IJ, Yang BC. Analysis of the expression of Fas-L in nasopharyngeal carcinoma tissues. Oral Oncol. 1999;35: 421-424. [DOI] [PubMed] [Google Scholar]
- 15.Verbeke CS, Wenthe U, Grobholz R, Zentgraf H. Fas ligand expression in Hodgkin lymphoma. Am J Surg Pathol. 2001;25: 388-394. [DOI] [PubMed] [Google Scholar]
- 16.Kelly PF, Carrington J, Nathwani A, Vanin EF. RD114-pseudotyped oncoretroviral vectors. Biological and physical properties. Ann N Y Acad Sci. 2001;938: 262-276. [PubMed] [Google Scholar]
- 17.Schomber T, Kalberer CP, Wodnar-Filipowicz A, Skoda RC. Gene silencing by lentivirus-mediated delivery of siRNA in human CD34+ cells. Blood. 2004;103: 4511-4513. [DOI] [PubMed] [Google Scholar]
- 18.Suda T, Hashimoto H, Tanaka M, Ochi T, Nagata S. Membrane Fas ligand kills human peripheral blood T lymphocytes, and soluble Fas ligand blocks the killing. J Exp Med. 1997;186: 2045-2050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Savoldo B, Huls MH, Liu Z, et al. Autologous Epstein-Barr virus (EBV)-specific cytotoxic T cells for the treatment of persistent active EBV infection. Blood. 2002;100: 4059-4066. [DOI] [PubMed] [Google Scholar]
- 20.Miyawaki T, Uehara T, Nibu R, et al. Differential expression of apoptosis-related Fas antigen on lymphocyte subpopulations in human peripheral blood. J Immunol. 1992;149: 3753-3758. [PubMed] [Google Scholar]
- 21.Chinnaiyan AM, Orth K, O'Rourke K, et al. Molecular ordering of the cell death pathway. Bcl-2 and Bcl-xL function upstream of the CED-3-like apoptotic proteases. J Biol Chem. 1996;271: 4573-4576. [DOI] [PubMed] [Google Scholar]
- 22.Takahashi T, Tanaka M, Brannan CI, et al. Generalized lymphoproliferative disease in mice, caused by a point mutation in the Fas ligand. Cell. 1994;76: 969-976. [DOI] [PubMed] [Google Scholar]
- 23.Khong HT, Restifo NP. Natural selection of tumor variants in the generation of “tumor escape” phenotypes. Nat Immunol. 2002;3: 999-1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bollard CM, Rossig C, Calonge MJ, et al. Adapting a transforming growth factor beta-related tumor protection strategy to enhance antitumor immunity. Blood. 2002;99: 3179-3187. [DOI] [PubMed] [Google Scholar]
- 25.Restifo NP. Not so Fas: Re-evaluating the mechanisms of immune privilege and tumor escape. Nat Med. 2000;6: 493-495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Arai H, Gordon D, Nabel EG, Nabel GJ. Gene transfer of Fas ligand induces tumor regression in vivo. Proc Natl Acad Sci U S A. 1997;94: 13862-13867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen JJ, Sun Y, Nabel GJ. Regulation of the proinflammatory effects of Fas ligand (CD95L). Science. 1998;282: 1714-1717. [DOI] [PubMed] [Google Scholar]
- 28.Poppema S, Potters M, Visser L, van den Berg AM. Immune escape mechanisms in Hodgkin's disease. Ann Oncol. 1998;9(suppl 5): S21-S24. [DOI] [PubMed] [Google Scholar]
- 29.Kayagaki N, Kawasaki A, Ebata T, et al. Metalloproteinase-mediated release of human Fas ligand. J Exp Med. 1995;182: 1777-1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hohlbaum AM, Moe S, Marshak-Rothstein A. Opposing effects of transmembrane and soluble Fas ligand expression on inflammation and tumor cell survival. J Exp Med. 2000;191: 1209-1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bauvois B. Transmembrane proteases in cell growth and invasion: new contributors to angiogenesis? Oncogene. 2004;23: 317-329. [DOI] [PubMed] [Google Scholar]
- 32.Sharp PA. RNAi and double-strand RNA. Genes Dev. 1999;13: 139-141. [PubMed] [Google Scholar]
- 33.Rieux-Laucat F, Le Deist F, Hivroz C, et al. Mutations in Fas associated with human lymphoproliferative syndrome and autoimmunity. Science. 1995;268: 1347-1349. [DOI] [PubMed] [Google Scholar]
- 34.Castro JE, Listman JA, Jacobson BA, et al. Fas modulation of apoptosis during negative selection of thymocytes. Immunity. 1996;5: 617-627. [DOI] [PubMed] [Google Scholar]
- 35.Lenardo M, Chan KM, Hornung F, et al. Mature T lymphocyte apoptosis—immune regulation in a dynamic and unpredictable antigenic environment. Annu Rev Immunol. 1999;17: 221-253. [DOI] [PubMed] [Google Scholar]
- 36.Eaton D, Gilham DE, O'Neill A, Hawkins RE. Retroviral transduction of human peripheral blood lymphocytes with Bcl-X(L) promotes in vitro lymphocyte survival in pro-apoptotic conditions. Gene Ther. 2002;9: 527-535. [DOI] [PubMed] [Google Scholar]