

Abstract
Mantle-cell lymphoma (MCL) is a unique subtype of B-cell non-Hodgkin lymphoma (NHL) that behaves aggressively and remains incurable. In order to understand the pathogenesis of MCL and design new therapies, it is important to accurately analyze molecular changes in pathways dysregulated in MCL. We used antibody microarrays to compare patterns of protein expression between CD19+ purified B lymphocytes from normal tonsil and 7 cases of histologically confirmed MCL. Protein overexpression was defined as a higher than 1.3-fold or 2-fold increase in at least 67% of tumor samples compared with normal B-cell control. Of the polypeptides, 77 were overexpressed using the higher than 1.3-fold cutoff, and 13 were overexpressed using the 2-fold cutoff. These included cell cycle regulators (regulator of chromosome condensation 1 [RCC1], murine double minute 2 [MDM2]), a kinase (citron Rho-interacting kinase [CRIK]), chaperone proteins (heat shock 90-kDa protein [Hsp90], Hsp10), and phosphatase regulators (A-kinase anchor protein 1 [AKAP149], protein phosphatase 5 [PP5], and inhibitor 2). The elevated expression of some of these polypeptides was confirmed by immunoblotting and immunohistochemistry, whereas elevated expression of others could not be confirmed, illustrating the importance of confirmatory studies. This study describes a novel technique that identifies proteins dysregulated in MCL.
Introduction
Mantle-cell lymphoma (MCL) represents approximately 8% of all B-cell non-Hodgkin lymphomas (NHLs).1 The immunophenotype of MCL is characteristic and the cells are strongly positive for CD20 and CD5, but are negative for CD10 and CD23.2 Overexpression of cyclin D1 (CCND-1), which molecularly defines MCL,3 results from the t(11;14)(q13;q32) translocation that juxtaposes a portion of the CCND1 gene (11q13) and the immunoglobulin heavy-chain promoter (14q32).4
Despite advances in understanding the molecular pathogenesis of MCL, it remains the NHL subtype with the poorest prognosis.5 Most patients are diagnosed at an advanced stage, and extranodal sites are often involved.1 Even though patients with MCL often respond to therapy, the responses are usually partial and most patients eventually relapse.6 Aggressive therapeutic regimens yield complete responses in only 38% of patients with a median overall survival of only 3 to 4 years.5,7 New treatments based on the knowledge of what pathways are dysregulated in MCL cells are needed for this disease.
Although cyclin D1 overexpression is characteristic of MCL, overexpression of cyclin D1 alone cannot induce lymphoma.8 Instead, other oncogenic changes such as c-myc overexpression,8 loss of the ataxia-telangiectasia (ATM) gene, and p53 gene deregulation9 are required for the development and progression of MCL. The underlying molecular alterations in MCL have been studied with gene expression profiling. A recent study that compared the molecular profile of 38 MCLs with normal tonsillar mantle-zone B cells demonstrated dysregulation of 22 genes known to be involved in apoptosis (such as BCL2 and TOSO), cell-cycle control, and signaling.10 Cell-cycle genes, with the exception of CCND1, were all down-regulated, consistent with the relatively low rate of proliferation that characterizes MCL.10
Because changes in messenger RNA (mRNA) levels are not absolute indicators of alterations in the corresponding levels of cellular proteins, proteomic analysis provides an alternate approach. Antonucci et al11 compared 2-dimensional (2D) gels of proteins isolated from 2 MCL cases and 3 reactive lymph nodes. This analysis identified about 20 differentially expressed polypeptides. Of these proteins, T-cell leukemia/lymphoma protein 1A (TCL1) was found to be overexpressed 10-fold in samples of MCL. Other up-regulated proteins included the 78-kDA glucose-regulated protein (GRP 78), histone H2B2, α-1 antitrypsin precursor, and heat shock 27-kDA protein (Hsp27).11
Conventional methods for quantitation of proteins have relied on techniques such as 1D and 2D gel electrophoresis, immunoblotting, enzyme-linked immunosorbent assays (ELISAs), and radioim-munoassays (RIAs). Most of these established methods for protein characterization in cell extracts are not amenable to high-throughput applications. The 2D gel electrophoresis is time consuming and expensive. In addition, reproducibility is problematic.12 Recent advances in high-throughput screening have led to the development of biochip-based assays for detecting changes in protein expression. These novel techniques offer the potential of a comprehensive proteomic analysis with very low sample amounts, relatively low cost, and reasonable time consumption.13 The antibody-based protein microarray is a new technology that assesses polypeptide differences directly by binding fluorescently labeled protein mixtures from cell extracts onto glass slides spotted with different monoclonal antibodies (mAbs) specific for various human proteins. The goal of this study was to investigate the differential expression of proteins in tumor samples from patients with MCL compared with nonmalignant tonsillar B cells using the high-throughput technology of protein microarrays. This technology suggested that MCL overexpresses 13 proteins, which were further examined by immunoblotting and immunohistochemistry. The dysregulated expression of these proteins in MCL may represent future targets for novel therapeutic agents.
Patients, materials, and methods
Patients and tissue samples
This study was approved by the Mayo Foundation institutional review board and conducted in accordance with the Declaration of Helsinki. All patients gave written informed consent for their tissue to be used for research and for review of their clinical records. Frozen tissue samples were obtained from 7 patients diagnosed with MCL between August 1999 and February 2003. Tissue was obtained from lymph node biopsies (6 patients) or spleen (1 patient). The diagnosis of MCL was based on characteristic tumor cell morphology and immunophenotype as well as the demonstration of cyclin D1 expression and/or the presence of t(11;14) chromosomal translocation. Normal B cells isolated from a tonsillectomy specimen were used as nondiseased controls for all microarray and immunoblotting experiments to provide a constant reference point for arrays. Tonsillar B cells have been used as normal controls in cDNA microarray experiments10 and are preferred to circulating B cells because they represent B cells in a tissue context.
A total of 11 separate experiments with antibody-based protein microarrays were performed. Of these, 7 were performed with MCL extracts from the 7 patient samples versus control tonsil B-cell extracts. One experiment was performed with a normal B-cell extract versus itself to detect any nonspecific binding to the antibodies imprinted on the microarray slides, and to provide a normalized baseline ratio for comparison of all MCL samples to control. In addition, 2 experiments with patient MCL samples were repeated twice to confirm reproducibility of the data. In order to determine how the pattern of protein expression observed in primary human MCL samples correlates with MCL cell lines, one experiment was performed with the MCL cell line M02058.
Cell culture
The MO2058 human cell line is an Epstein-Barr virus–positive cell line containing a t(11;14)(q13;q32) chromosomal translocation.14 The cells were grown at 37°C in a 5% CO2 atmosphere in RPMI-1640 medium (Gibco/Life Technologies, Grand Island, NY) supplemented with 10% fetal calf serum, 2 mM l-glutamine, 100 U/mL penicillin, and 100 μg/mL streptomycin. On the day of the analysis, cells were pelleted and washed twice with 1 × phosphate-buffered saline (PBS).
B-cell isolation
Cells were isolated from fresh MCL tumor samples and fresh tonsillar tissue by mincing over a wire mesh screen to achieve a single cell suspension in ice-cold culture medium (RPMI 1640). Mononuclear cells were isolated by Ficoll-Paque Plus (Amersham Pharmacia Biotech, Uppsala, Sweden). The MCL tumor cell suspensions were mixed with 10% dimethyl sulfoxide (DMSO) and frozen in liquid nitrogen until used for these experiments. On the day of protein extraction, the MCL samples were thawed, washed in RPMI 1640, and subjected to CD19+ immunomagnetic bead selection (Miltenyi Biotech, Auburn, CA). Tonsillar cells were enriched for B cells by performing CD19+ immunomagnetic bead selection. Purification of B cells to more than 90% homogeneity was confirmed by flow cytometry using CD20 monoclonal antibodies. Purified B-cell preparations were then stored in 10% DMSO and frozen in liquid nitrogen until needed for protein extraction.
Protein microarray procedure
The Ab Microarray (BD Clontech, Palo Alto, CA) detects a wide variety of proteins (both cytosolic and membrane bound) representing a broad range of biologic functions, including signal transduction, cell-cycle regulation, gene transcription, and apoptosis. The microarray contains 512 highly specific and sensitive mAbs against human polypeptides. The complete list of the arrayed antibodies, including Swiss-Prot ID numbers of the target antigens, is available elsewhere.15
Protein extraction
After CD19 enrichment, 6 to 30 × 106 cells were sedimented. Total protein was extracted by a single freeze-thaw cycle of the cell pellet in liquid nitrogen followed by homogenization in the manufacturer's extraction/labeling buffer, which contains nondenaturing detergents to maintain protein solubility and emulsify membrane-bound proteins. After measurement of the total protein concentration by the bicinchoninic acid method (Pierce, Rockford, IL), each sample was diluted to a final total protein concentration of 1.1 mg/mL.
Fluorescent labeling with green fluorescent (Cy3) and red fluorescent (Cy5) dyes
For each sample and control, 90 μL total protein was labeled with 10 μL labeling dyes, either cyanin 3 (Cy3) or Cy5 (PA25002 and PA23001, respectively; Amersham Biosciences, Piscataway, NJ). The dual-fluorescence detection method is designed so that inherent variations in dye labeling do not affect the outcome of the experiment. After 90 minutes of incubation and 30 minutes of blocking, the unbound dye was removed using PD-10–desalting columns (Amersham Biosciences). Protein concentration was again determined using the bicinchoninic acid method with subtraction of the dye's contribution to the overall optical density at 562. The average number of dye molecules covalently coupled to each protein was measured as per the user manual, and usually ranged from 2 to 4 molecules of Cy3 or Cy5 per molecule of protein.
The Clontech microarray slides
The arrays are printed on standard-size (75 × 25 × 1 mm) glass slides with 512 antibodies printed in duplicate on each slide. There were 2 slides provided for reverse-color labeling to allow normalization of the samples. Protein samples from normal B cells labeled with Cy5 were mixed with protein samples from MCL B cells labeled with Cy3 protein and added to slide 1. For slide 2, the protein samples from normal B cells were labeled with Cy3 and the proteins from MCL B cells labeled with Cy5. Labeling the proteins from both the normal and malignant B cells with Cy3 and Cy5 allows the microarray to detect differences in specific protein abundance between the MCL sample and the control sample with each experiment and compensates for any potential difference in binding of the protein to the Cy3 or Cy5 dyes. Total protein (20 μg) was added to each slide and incubated at room temperature for 30 minutes before a series of washes. The slides were dried and scanned according to the instructions of the supplier using the Axon GenePix 4000B scanner set to 635 nm (Cy5 channel: photomultiplier tube (PMT), 670 V; power, 33%; and 532 nm; and Cy3 channel: PMT, 550 V; power, 33% to produce a text file with signal intensities). There were 2 ratios generated from the spot images, MCL-Cy5/normal-Cy3 (slide 1) and normal-Cy5/MCL-Cy3 (slide 2), for each protein target.
Data analysis and clustering
The means of the ratios of Cy5/Cy3 for each polypeptide of both slides were analyzed using Clontech Array-Specific Analysis workbook software developed specifically for each microarray lot by the manufacturers. The 2 ratios were used to calculate an internally normalized ratio (INR), or ratio of ratios, for each spot on the array. This calculation normalizes for differences due to labeling efficiency and antibody-antigen binding affinity, greatly enhancing the precision and accuracy of the assay. The replicate values within each slide were then averaged and an INR was calculated where INR = √ Ratio 1/Ratio 2 and ratios 1 and 2 correspond to slides 1 and 2. Ratio 1 equals normal-Cy5 relative fluorescent units/MCL-Cy3 relative fluorescent units, and Ratio 2 equals MCL-Cy5 relative fluorescent units/normal-Cy3 relative fluorescent units. The average INR was calculated for each antibody, and duplicate INR values that varied by more than 30% were discarded. INR values higher than 1.3 or less than 0.77 were considered as valid changes in protein abundance for this study. These thresholds were determined by taking the global INR value for each experiment (ie, calculated average INR of all spots) and multiplying by 1.3 and 0.77 to get the specific threshold INR values for each subsequent microarray analysis. Genespring software (Silicon Genetics, Redwood City, CA) was used for analysis of all 7 experiments and normalized to the control experiment. An unsupervised clustering analysis was performed, and changes that were 1.3- and 2-fold or higher in 67% of the MCL samples with similar patterns compared with the B-lymphocyte control were identified.
Western-blot analysis
To confirm the expression of selected polypeptides identified in protein microarrays, immunoblotting was performed using the remaining total protein extracted for the microarray procedure. Total protein (50 μg) from each cell extract was precipitated with trichloroacetic acid, washed with ethanol, dried, and then solubilized in sodium dodecyl sulfate (SDS) sample buffer. Each extract was boiled for 5 minutes before loading on a 5% to 15% SDS polyacrylamide gel. After separation, the polypeptides were transferred to a nitrocellulose. After blocking, blots were probed with monoclonal anti–regulator of chromosome condensation 1 (RCC1), p43/endothelial monocyte-activating polypeptide II (EMAPII) precursor, A-kinase anchor protein 1 (AKAP149), procaspase-7, procaspase-14, cyclin D1 (Santa Cruz Biotechnology, Santa Cruz, CA), retinoblastoma 2 (Rb2), and Hsp90 (David Toft, Mayo Clinic, Rochester, MN) or polyclonal anti–procaspase-8.16 All antibodies were obtained from BD Pharmingen unless otherwise indicated. To ensure equal loading, the blot was probed with polyclonal goat antiactin antibody (Santa Cruz Biotechnology).
Immunohistochemistry
To further confirm the expression of specific proteins detected by the protein microarray technique, paraffin-embedded tissue biopsies available from the same patients with the same dates of sample acquisition that were used for microarray experiments were analyzed by immunohistochemistry using monoclonal antibodies to human proteins: Hsp90 (Stressgen, San Diego, CA); murine double minute 2 (MDM2); Rb-2, KU-80; Paxillin, Bcl-x (BD Biosciences Pharmingen, San Diego, CA); and citron Rho-interacting kinase-L (CRIK-L) (Santa Cruz Biotechnology). Paraffin-embedded colon and breast carcinoma tissues were used as positive controls.
Results
Baseline patient characteristics
The median age at diagnosis was 71 years (range, 51-88 years; Table 1). The median time from diagnosis to sample collection was 214 days (range, 0-2063 days). Histologically, all cases displayed the immunophenotypic picture of MCL, with cells that were positive for CD20 and CD5 but lacked CD10, CD23, and CD3. All were cyclin D1 positive by immunohistochemistry or by fluorescent in situ hybridization (FISH) analysis. Blastoid features were present in 1 of the 7 cases.
Table 1.
Baseline patient characteristics
| Patient no. | Age, y | Sex | Time from Dx to Bx, d | Site of Bx | Prior therapy before Bx | Time from Dx to last FU, y | Response at last FU | Death |
|---|---|---|---|---|---|---|---|---|
| 1 | 88 | M | 48 | Spleen | N | 0.9 | Prog | Y |
| 2 | 80 | F | 0 | LN | N | 1.8 | PR | N |
| 3 | 71 | F | 559 | LN | Y | 3.4 | PR | N |
| 4 | 51 | M | 2263 | LN | Y | 9.8 | Secondary AML | Y |
| 5 | 59 | F | 214 | LN | N | 1.8 | CR | N |
| 6 | 64 | M | 0 | LN | N | 0.7 | Stable | N |
| 7 | 74 | M | 2063 | LN | Y | 6.6 | Prog | Y |
Dx indicates diagnosis; Bx, biopsy; FU, follow-up; Prog, progression; LN, lymph node; PR, partial remission; AML, acute myeloid leukemia; and CR, complete remission.
At the time of sample collection, 2 (29%) patients were newly diagnosed, 4 (57%) had no previous therapy, and 3 (42%) were previously treated. Of the 3 previously treated patients, 2 had received chemotherapy followed by autologous stem cell transplantation, and the tissue used in this analysis was obtained at the time of relapse 2 and 3 years after transplantation. Of the 3 patients, 1 received 2-chlorodeoxyadenosine (2-CDA) and rituximab 1 year prior to the specimen collection. The median time of follow-up of living patients was 2.6 years (range, 1.8-3.4 years). The median overall survival was 1.8 years (range, 0.9-9.8 years).
Protein expression
Of the 7 MCL samples, 6 had a common pattern of protein overexpression (Figures 1, 2). Because of the homogenous expression pattern in the 6 samples, we further analyzed the proteins that were 2-fold or higher up- or down-regulated compared with normal control. The results obtained from MCL patient 7 and the M02058 cell line differed from the patterns observed with the other 6 MCL samples and are reported separately in the next section. We used a cutoff of 2-fold or higher because it has been the standard used in the analysis of cDNA array data. Using this cutoff, the microarrays identified 13 dysregulated polypeptides in at least 4 (66%) of the 6 samples with similar expression patterns (Table 2). All 13 polypeptides were overexpressed in the MCL cells compared with the normal tonsillar B lymphocytes (Figure 3). These polypeptides included cell cycle regulators such as MDM2 and RCC1; chaperone proteins such as Hsp10 and Hsp90; and phosphatase/kinase regulators such as AKAP149, PP5, inhibitor 2, and CRIK.
Figure 1.

Heat map with unsupervised clustering analysis of the 6 MCL samples compared with normal control. The MCL samples are numbered 1 to 6. The control sample is at the far right of the heat map. All the samples were normalized to the control sample using the Genespring software. The data points are colored by expression with lower signal values colored blue and higher values, red. Yellow signifies expression values equal to the control sample.
Figure 2.

Protein expression in the 6 MCL samples compared with normal control. The y-axis is a log scale of the normalized intensity. The data points are colored by expression with lower signal values colored blue and higher values, red and presented by their relative level in each sample compared with the normal control. The central line is the midpoint with the lines above and below at log-rank increase or decrease in expression. MCL7, which had a reversed expression, is presented on the right side of the control and is repeated twice. Note the reversed expression of the proteins in MCL7 as marked by the color changes. Data were analyzed with Genespring software.
Table 2.
Higher than 2-fold change in at least 66% of the 6 MCL patients
| Protein | Corresponding gene | Function |
|---|---|---|
| RCC1 (regulator of chromosome condensation) | CHC1 | Cell cycle, DNA binding, Ran guanyl-nucleotide exchange factor activity |
| p43/EMAPII (multisynthetase complex auxiliary component p43) precursor | SCYE1 | Chemotaxis, cell-cell signaling, inflammatory response |
| Inhibitor 2 | PPP1R2 | Type 1 serine/threonine-specific protein phosphatase inhibitor activity |
| AKAP149 (A kinase anchor protein 1, mitochondrial precursor) | AAKAP149 | Protein kinase A anchoring activity |
| CHD3 (chromodomain helicase-DNA-binding protein 3) | CHD3 | ATP-dependent DNA helicase activity, ATP binding |
| PP5 (protein phosphatase 5) | PPP5C | Protein phosphatase type 2C activity, involved in p53 and Hsp90 regulation |
| Caspase-7 | CASP7 | Apoptosis |
| Hsp10 (heat shock protein 10) | HSPE1 | Protein folding, chaperone protein |
| MDM2 (murine double minute 2) | MDM2 | Negative regulation of cell proliferation, oncogenesis |
| Hsp90 (heat shock protein 90) | HSPCA | Protein folding, chaperone protein |
| SH2B (SH2-B homolog) | SH2B | Growth factor, cytokine, and immunoreceptor signaling |
| CRIK (citron Rho-interacting kinase) | CIT | Small GTPase regulatory/interacting protein activity |
| Per2 (period circadian protein 2) | PER2 | Regulator of circadian expression of VEGF mRNA in hypoxic tumors |
CHC1 indicates chromosome condensation 1; SCYE1, small inducible cytokine subfamily E, member 1 (endothelial monocyte-activating); PPP1, protein phosphatase 1, regulatory (inhibitor) subunit 2R2; AKAP1, A kinase (PRKA) anchor protein 1; CHD3, chromodomain helicase DNA binding protein 3; ATP, adenosine triphosphate; PPP5C, protein phosphatase 5, catalytic subunit; CASP7, caspase-7, apoptosis-related cysteine protease; HSPE1, heat shock 10-kDa protein 1 (chaperonin 10); MDM2, Mdm2, transformed 3T3 cell double minute 2, p53 binding protein (mouse); HSPCA, heat shock 90-kDa protein 1; SH2B, Src homology 2B; CRIK, citron (rho-interacting, serine/threonine kinase 21); PER2, period homolog 2; and VEGF, vascular endothelial growth factor.
Figure 3.

Expression levels of the 13 overexpressed proteins in the MCL samples compared with control. The data points are colored in red/orange indicating overexpression. The samples are numbered 1 to 7. MCL7 showed reversed expression pattern compared with the other 6 MCL samples. MCL7 was repeated twice and demonstrates consistent expression pattern. Data were analyzed with Genespring software.
However, since protein expression levels up-regulated by less than 2-fold may still constitute important functional changes, we analyzed the data using higher than 1.3-fold expression difference. The higher than 1.3 cutoff was determined by the manufacturer as a significant value for up-regulated proteins. Using this cutoff, 77 of 512 polypeptides were identified as up-regulated in MCL compared with normal control (Table 3). These included polypeptides involved in the phosphatidylinositol 3–kinase (PI3K) pathway, the proteosome/ubiquitin pathway, the modulation of apoptosis, cell cycle regulation (including cyclin D1), and other signaling pathways. MCL sample 7 demonstrated a relatively reversed pattern of expression, with proteins that are overexpressed in all other samples showing a lower expression in this sample on 2 separate occasions (Figures 2 and 4). For example, Hsp90, MDM2, and many other proteins that were identified as up-regulated in the 6 samples were underexpressed in sample 7. The cell line M02058 also exhibited down-regulation of Hsp90 and Hsp10 compared with normal B-cell control. These polypeptides were identified as up-regulated in the MCL patient samples.
Table 3.
Higher than 1.3-fold change in at least 66% of the 6 MCL patients identified 77 proteins
| Protein | Corresponding gene | Molecular function |
|---|---|---|
| RCC1 (regulator of chromosome condensation) | CHC1 | DNA binding, Ran guanyl-nucleotide exchange factor activity |
| p43/EMAPII (endothelial monocyte-activating polypeptide II) precursor | SCYE1 | Chemotaxis, cell-cell signaling, inflammatory response |
| Inhibitor 2 | PPP1R2 | Type 1 serine/threonine-specific protein phosphatase inhibitor activity |
| AKAP149 (A-kinase anchor protein 149) | AKAP1 | Protein kinase A anchoring activity |
| CHD3 (chromodomain helicase DNA binding protein 3) | CHD3 | ATP-dependent DNA helicase activity, ATP binding |
| PP5 (protein phosphatase 5) | PPP5C | Protein phosphatase type 2C activity, involved in p53 and Hsp90 regulation |
| Caspase-7/Mch3 (modifier of chinchilla 3) | CASP7 | Apoptosis |
| Hsp10 (heat shock protein 10) | HSPE1 | Protein folding, chaperone protein |
| SH2B (Src-homology-3) | SH2B | Growth factor, cytokine, and immunoreceptor signaling |
| MDM2 (murine double minute 2) | MDM2 | Negative regulation of cell proliferation, oncogenesis |
| Hsp90 (heat shock protein 90) | HSPCA | Protein folding, chaperone protein |
| PKA c (protein kinase A) | PRKACA | Protein-tyrosine kinase activity |
| CRIK (citron Rho-interacting kinase) | CIT | Small GTPase regulatory/interacting protein activity |
| Per2 (period circadian protein 2) | PER2 | Regulator of circadian expression of VEGF mRNA in hypoxic tumors |
| Neurexin I | NRXNI | Receptor activity |
| Crk (Proto-oncogene C-Crk) | CRK | Adaptor in tyrosine kinase signal transduction |
| CDC25C (M-phase inducer phosphatase 3) | CDC25C | Prenylated protein tyrosine phosphatase activity |
| CPG16/CaM kinase VI (calcium calmodulin dependent kinase CPG16) | DCAMKL1 | Protein serine/threonine kinase activity |
| G3BP (Ras-GTPase-activating protein binding protein 1) | G3BP | Ras protein signal transduction |
| CDC34 (ubiquitin-conjugating enzyme E2-32 kDa complementing) | CDC34 | Ubiquitin-protein ligase activity |
| BRUCE (baculoviral IAP repeat-containing 6) | BIRC6 | Cysteine protease inhibitor activity, ubiquitin conjugating enzyme activity, apoptosis inhibitor activity |
| p73a (p73 alpha) | TP73 | Transcription factor activity |
| PKC i (protein kinase C i) | PRKCI | Atypical protein kinase C activity |
| Neurotensin receptor 3 | NT | Induces migration |
| La protein (Sjogren syndrome antigen B) | SSB | Ribonuclear protein complex |
| Rag-1 (V(D)J recombination activating protein 1) | RAG1 | Endonuclease activity, DNA binding |
| Cadherin-5 | CDH5 | Calcium ion binding, cell adhesion molecule activity |
| FADD (Fas-associating protein with death domain) | FADD | Induction of apoptosis |
| TBP (TATA box-binding protein) | TBP | DNA binding, RNA polymerase II transcription factor activity |
| Maspin | SERPINB5 | Serine protease inhibitor activity |
| CTBP2 (C-terminal binding protein 2) | CTBP2 | 2-Hacid DH; oxidoreductase activity |
| Calnexin | CANX | Calcium ion binding, chaperone activity |
| SMRT (silencing mediator of retinoic and thyroid hormone receptors) | NCOR2 | Myb_DNA-binding |
| MST3 (serine/threonine-protein kinase 3) | MST3 | Apoptosis regulation |
| FBP (fructose 1,6-bisphosphate) | FUBP1 | Transcription factor activity |
| Mena (enabled protein homolog) | ENAH | Regulation of cell motility and adhesion |
| Rap 2 (Ras-related protein Rap-2) | RAP2B | RAS small monomeric GTPase activity |
| Topoisomerase II a | TOP2A | DNA metabolism |
| PYK2 (pyruvate kinase 2) | PTK2B | Protein tyrosine kinase activity |
| Pericentrin | PCNT2 | Structural constituent of cytoskeleton |
| Karyopherin b | KPNB1 | Zinc ion binding, protein transporter activity |
| eIF-4g (eukaryotic translation initiation factor 4 gamma) | EIF4G1 | Translation factor activity |
| Erp72 (protein disulfide-isomerase A4 precursor) | ERp72 | Endoplasmic reticulum protein, chaperone protein |
| Neurogenin 3 | NEUROG1 | Transcription factor |
| IkBa/MAD-3 (NF-kappaB inhibitor alpha) | NFKB1A | Transcription factor binding |
| GSK-3B (Glycogen synthase kinase-3 beta) | GSK3B | Regulates atypical PKC regulation of cell polarity, apoptosis |
| DNA topoisomerase II b | TOP2B | DNA topoisomerase type II activity |
| MCM6 (DNA replication licensing factor MCM6) | MCM6 | DNA binding, DNA dependent ATPase activity |
| Nexilin | Nexilin | Actin binding protein |
| KIF3B (kinesin-like protein KIF3B) | KIF3B | Kinase, microtubule motor activity |
| PTPIC (protein-tyrosine phosphatase, non-receptor type 6) | PTPN6 | Protein tyrosine phosphatase activity |
| PTEN/MMAC1 (phosphatase and tensin homolog deleted on chromosome 10) | PTEN | Phosphatidylinositol-3,4,5-trisphosphate 3-phosphatase activity, protein tyrosine phosphatase activity |
| JAM1 (junctional adhesion molecule 1 precursor) | JAM1 | Intracellular junction |
| DP103/Gemin3 (probable ATP-dependent RNA helicase DDX20) | DDX20 | DNA binding, ATP-dependent RNA helicase activity |
| Sam68 (substrate of Src in mitosis) | SAM68 | Transcription, RNA splicing, cell cycle regulation |
| NABC1 (breast carcinoma amplified sequence 1) | NABC1 | Ion transporting protein |
| GRB14 (growth factor receptor-bound protein 14) | GRB14 | SH3/SH2 adaptor protein activity |
| MLH1 (MutL homolog 1) | MLH1 | ATP binding |
| GM130 (Golgi matrix protein of 130 kDa) | GOLGA2 | Peripheral membrane protein associated with the Golgi bodies |
| mEPHX (microsomal epoxide hydrolase) | EPHX1 | Epoxide hydrolase activity |
| b-tubulin | TUBB | Structural constituent of cytoskeleton |
| JAK 1 (Janus kinase 1) | JAK1 | Protein-tyrosine kinase activity |
| SMAC (second mitochondria-derived activator of caspases)/DIABLO (Diablo homolog, mitochondrial precursor) | SMAC | Apoptosis |
| IGFBP-3 (insulin-like growth factor binding protein 3 precursor) | IGBP3 | Insulin-like growth factor binding |
| 14-3-3e | 14-3-3 | Apoptosis |
| DP-1 (Dodeca-satellite-binding protein 1) | TFDP1 | Transcription factor activity, cell cycle regulation |
| Cyclin D1 | CCND1 | Cell cycle regulation |
| FYB (FYN-binding protein)/SLAP-130 (Src-like adaptor protein 130) | SLAP130 | SLP76-associated phosphoprotein |
| SRPK1 (SFRS protein kinase 1) | SRPK1 | Protein serine threonine kinase activity |
| Kalinin B1 | LAMB3 | Cell laminin |
| L1 (deferiprone) | L1CAM | Cell adhesion |
| XCCR4 (X-ray repair complementing defective repair in Chinese hamster cells 4) | XCCR4 | X-ray repair |
| P54nrb (54 kDa nuclear RNA-and DNA-binding protein) | P54NRB | Nuclear RNA binding protein |
| PKA R1 (cAMP-dependent protein kinase type I regulatory subunit) | PRKAR1B | c-AMP-dependent protein kinase |
| Carboxypeptidase E | CPE | Metallopeptidase activity, hydrolase activity, carboxypeptidase activity |
| MST1 (hepatocyte growth factor-like protein precursor) | MST1 | Serine threonine kinase |
| PTP1D/SHP2 (Protein-tyrosine phosphatase, non-receptor type 11) | PTP1D | PI3K signaling pathway |
GTPase indicates guanosine triphosphatase; SLP76, SH2-containing leukocyte protein; and c-AMP, cyclic adenosine monophosphate.
Figure 4.
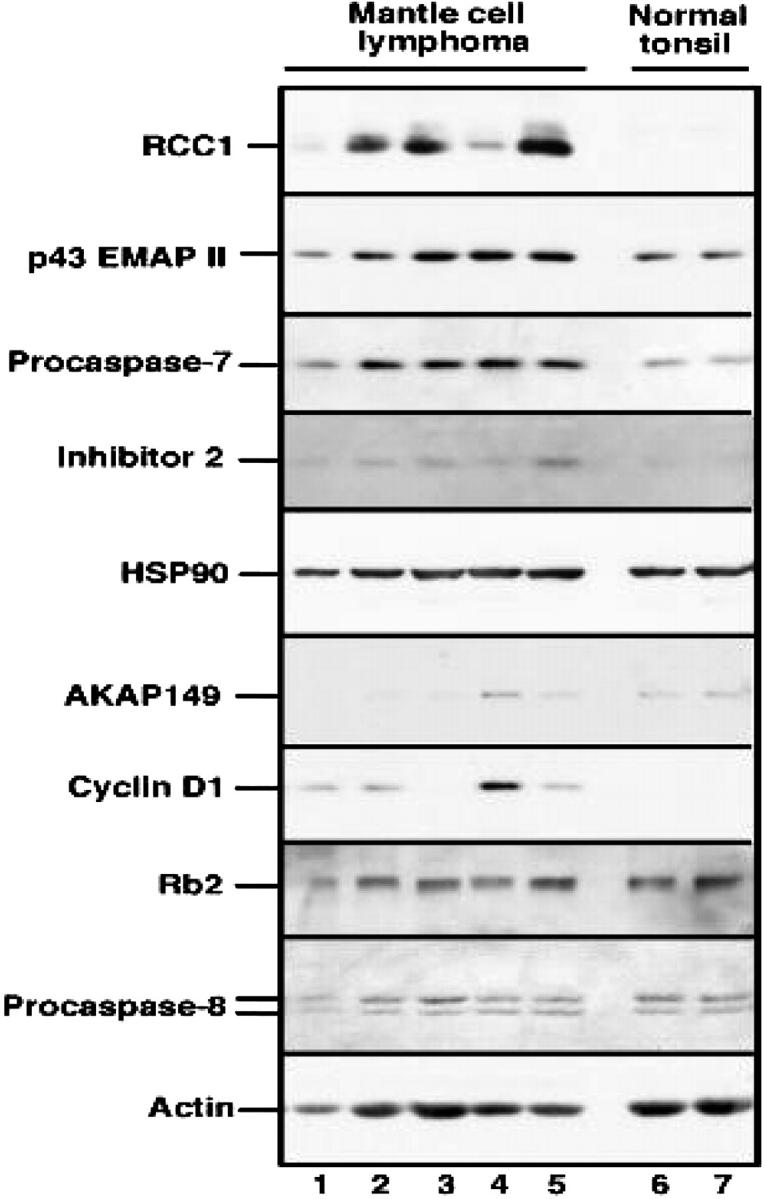
Immunoblotting analysis was performed to confirm the expression of several proteins identified in protein microarrays by using the rest of the total protein extracted for the microarray procedure. Protein from control B-lymphocyte extracts was loaded twice (normal tonsil) on each side of the gel. The nitrocellulose blots were probed with monoclonal anti-RCC1, p43/EMAPII precursor, procaspase-7, inhibitor 2, Hsp90, AKAP149, cyclin D1, Rb2, and procaspase-8. Loading control was performed with antiactin antibody (bottom row).
Validation of the results
To validate the results of the protein microarray, studies on reproducibility and protein expression were performed by 2 other standard techniques—Western blotting (immunoblotting) and immunohistochemistry. To assess reproducibility, protein from the normal B cells was labeled with Cy3 and Cy5 on the same slide and no difference was found between the 2 samples (data not shown), indicating equal labeling of the proteins by the 2 fluorescent dyes. There were 2 MCL samples analyzed on multiple occasions and the results were highly reproducible (data not shown).
In an attempt to further validate the results of the antibody arrays, Western blot analysis and immunohistochemistry were performed on the same samples used for the protein microarray analysis. Figure 4 demonstrates the expression of RCC1, p43/EMAPII precursor, procaspase-7, inhibitor 2, Hsp90, AKAP149, cyclin D1, Rb2, procaspase-8, and actin by Western blot analysis using additional aliquots of the same protein extracts used for the antibody arrays. These aliquots were available for 5 of the 7 MCL patients. As a control, 2 separately processed aliquots of protein from the normal tonsil were included on the same blot. All lanes were loaded with 50 μg protein. Results of this analysis demonstrated that RCC-1, p43/EMAPII precursor, caspase-7, and inhibitor 2 were all overexpressed in a majority of the MCL samples compared with control B cells. In contrast Rb2 and procaspase-8, which were not identified as overexpressed proteins by the antibody arrays, were also not overexpressed on the Western blots.
Although some of the predictions of the arrays were confirmed, immunoblotting also demonstrated that the antibody arrays had both false positives and false negatives. While the antibody arrays indicated that Hsp90 was up-regulated 2-fold in the MCL samples, this was not evident on the immunoblots. A similar discrepancy was observed with AKAP149. These results demonstrate the importance of confirming predictions of the arrays by an independent method. As an alternative approach to validating results of the antibody arrays, protein expression in MCL and normal tonsil was also examined by immunohistochemistry. Figure 5 demonstrates the expression of CRIK, Hsp90, and MDM2 in the paraffin-embedded tissue from the same biopsies used for the protein array analysis. Consistent with the results of the antibody arrays, increased staining for CRIK and MDM2 was observed in MCL samples. As was the case with immunoblotting, immunohistochemistry failed to demonstrate elevated Hsp90. Consistent with the results of the antibody arrays, immunohistochemistry failed to detect up-regulation of Bcl-xL, Paxillin, Rb-2, and KU-80. Further analysis (Figure 6) demonstrated that the levels of Hsp90, MDM2, cyclin D1, and CRIK in the control tonsil sample used for the protein arrays are similar to those observed in other tonsillar tissues, indicating that these protein levels are confirmed in multiple control samples.
Figure 5.

Immunohistochemistry analysis to confirm the presence of the expression of the proteins in the MCL samples and confirm the absence of expression differences in other proteins not detected by the protein microarray. Paraffin-embedded tissue biopsies available from the same patients were analyzed using antibodies to human proteins: CRIK-L, Hsp90, and MDM2 (A); Rb2, KU80, Paxillin, and Bcl-x (B). Images were visualized under an Olympus IX70 inverted microscope equipped with objective lenses from 20 ×/0.40 and 60 ×/0.70 (Olympus, Tokyo, Japan). A MagnaFire digital camera (model 599806) and MagnaFire software version 4.1 were used to capture images (Olympus).
Figure 6.

Immunohistochemistry analysis of RCC-1, Hsp90, and MDM2 in several tonsillar tissues. This indicates that the expression of proteins detected in the control sample used in the array analysis is similar to other tonsillar tissues. Image acquisition was performed as in Figure 5.
Discussion
Despite the advances in understanding of the molecular pathogenesis of MCL, treatment remains inadequate.5 Accordingly, there is considerable interest in elucidating the molecular pathways that contribute to resistance to apoptosis and resistance to therapeutic interventions in MCL. A large body of information has been recently generated using transcriptional profiling in B-cell lymphomas, including MCL. However, not all changes in the mRNA expression lead to similar changes at the protein level.17 As an alternative, high-throughput proteomic profiling of cancer cells might yield more direct answers to the functional differences between malignant and normal cells. With this in mind, our objective was to use antibody-based protein microarrays to characterize the expression of polypeptides in MCL that might contribute to the biology of this neoplasm.
When a 2-fold or higher change in 4 of the 6 samples analyzed was used as a cutoff, the microarrays predicted 13 overexpressed proteins in MCL (Table 2). These included RCC-1, procaspase-7, CRIK, MDM2, inhibitor 2, and p43/EMAP. Overexpression of each of these in MCL compared with normal B cells was confirmed by immunoblotting or immunohistochemistry (Figures 5, 6). The potential contributions of these polypeptides to MCL are discussed in the next section.
Because antibody arrays represent a new technology for the study of lymphoid malignancies, it is important to understand their possible limitations. Our results suggest that antibody microarrays predict both false negatives and false positives. Cyclin D1 might be considered a false negative. According to the microarrays, cyclin D1 was increased in the MCL samples at a mean INR value of 1.66 higher than control cells (range, 0.9-3.3). Therefore, cyclin D1 was not identified as up-regulated when a 2-fold or higher elevation was used as a cutoff, but was identified using a higher than 1.3-fold cutoff. In contrast, cyclin D1 was readily detected by immunoblotting in 4 of the 5 MCL specimens examined but not in tonsillar B cells. Although, the 2-fold cutoff may increase the specificity of the microarray technique, it failed to identify polypeptides that are moderately up-regulated but are important in the pathogenesis of the disease, such as cyclin D1. On the other hand, Hsp90 appears to represent a false positive. Neither immunohistochemistry nor immunoblotting detected changes in Hsp90. Likewise, immunoblotting failed to detect up-regulation of AKAP149. While it would be convenient to attribute this discrepancy to lack of sensitivity of blotting to quantitative changes, the techniques used to probe these blots have recently yielded correlation coefficients of up to 0.99 when signal intensity and relative protein amount of a series of diluted standards are compared.18
These results are consistent with previous reports indicating deficiencies of sensitivity and specificity of protein microarray techniques.13 Therefore, we believe that confirmatory studies with conventional methodologies such as immunoblotting, immunohistochemistry, or ELISA are essential to validate the results of protein microarrays.
Because the analysis of the data depends on comparing ratios of malignant cells to normal control cells, we used B cells derived from a single control to allow comparison between the level of proteins across the different MCL samples. Immunohistochemistry confirmed that the levels of polypeptides detected in the control sample used in the microarray analysis are comparable with several other control tonsils. Tonsillar cells have been used in previous gene expression analyses and represent one of the best control cells available for mantle zone B cells. They are preferable to circulating B cells because they contain B cells in a tissue context with all of the signaling events that are inherent in cellular interactions. Nonetheless, it is important to acknowledge that some proteins may have been up-regulated in the tonsillar tissue due to chronic inflammation, obscuring their up-regulation in the MCL samples compared with the tonsillar B cells.
One sample in this study (MCL7) expressed a protein pattern that was reversed compared with the other 6 samples. For example, Hsp90, MDM2, and many other proteins up-regulated in the 6 samples were underexpressed in sample 7. The experiments were repeated on 2 separate occasions with the same results. This patient was heavily pretreated with multiple chemotherapies and autologous stem cell transplantation 3 years prior to the collection of the sample, which was confirmed to be a relapse of MCL. The results from patient 7 indicate that there will likely be heterogeneity in protein expression when larger numbers of MCL patients are studied. Whether this heterogeneity will correlate with disease activity or provide insights into the mechanisms of disease progression in MCL remains to be determined.
The expression data obtained from the MCL cell line MO2058 also failed to correlate with the patient samples. This lack of correlation might reflect both the effect of the microenvironment on the malignant cells in clinical specimens and the molecular changes that occur during derivation of a continuously cycling immortal cell line. These observations suggest that it is important to analyze primary cells obtained from tumor samples rather than cell lines when proteomic profiling of malignant cells is being performed.
Among the polypeptides correctly identified by the antibody microarrays as being dysregulated in MCL compared with normal B cells are several that might contribute to the pathogenesis of MCL. These include cell cycle regulators such as RCC1 and MDM2, the cytokine p43/EMAPII, and signal transduction regulators such as inhibitor 2 and CRIK.
The MDM2 protein is an E3 ubiquitin ligase that regulates levels of the tumor suppressor protein p53. Up-regulation of MDM2 has been shown to contribute to myc-induced lymphomagenesis in a mouse model.19,20 In MCL, MDM2 overexpression might not only participate in tumorigenesis, but also diminish the ability of DNA damage to up-regulate p53 and its downstream proapoptotic transcriptional targets, thereby contributing to drug resistance. Consistent with this hypothesis, MDM2 overexpression has been detected in a variety of NHLs,21-25 including MCL,25 with particularly high levels in relapsed24 or poor prognosis cases.22 Results of the present study are consistent with these previous observations, providing some assurance that the up-regulated polypeptides described in the present study are truly up-regulated.
In light of these observations, it is interesting to speculate whether the other polypeptides that are most highly up-regulated in MCL might contribute to the pathogenesis of this disorder. RCC1 is a 45-kDa guanine nucleotide exchange factor that contributes to nuclear localization and activation of the small GTP-binding factor Ran.26,27 Because expression of RCC1 is required for DNA synthesis,28,29 the up-regulation of RCC1 observed in the MCL samples might reflect entry into G1 as a consequence of cyclin D1 overexpression. p43/EMAPII is a cytokine30 that has recently been reported to induce lymphocyte apoptosis.31,32 Because recent studies have implicated EMAPII in tumor-induced immunosuppression,32 it is possible that p43/EMAPII overexpression helps MCL evade the immune system and contributes to resistance of MCL to immunotherapeutic approaches. Protein phosphatase inhibitor-2 serves dual functions in signal transduction pathways. It was initially identified as an inhibitor of type 1 protein phosphatases (PP1),33 which dephosphorylate and inactivate a variety of signal transduction components, including the antiapoptotic kinase Akt.34 More recently inhibitor-2 has also been identified as an allosteric activator of Aurora A, a kinase required for cell cycle progression through late G2 and M phases of the cell cycle.35 Accordingly, inhibitor-2 overexpression would be predicted to enhance antiapoptotic signaling through the Akt pathway as well as promote cell cycle progression. CRIK is a recently identified Rho-A–dependent kinase.36 The Rho subfamily GTPases mediate actin cytoskeleton organization, motility, and cell cycle progression. Activated mutants of Rho-A family proteins have been implicated in carcinogenesis.37 Thus, several of the up-regulated polypeptides could conceivably contribute to the pathogenesis of MCL.
In summary, high-throughput antibody-based protein microarrays represent a new and potentially useful technology for analyzing differences between malignant cells and normal controls. As illustrated in the present study, confirmation with other methods of protein detection such as immunohistochemistry and immunoblotting is required. This study shows overexpression of polypeptides involved in cell-cycle regulation (RCC1), p53 regulation (MDM2), phosphatase regulators (inhibitor 2), and kinases such as CRIK in MCL. Aside from MDM2, overexpression of these polypeptides in MCL was previously unknown. Whether these observations can be exploited for therapeutic benefit or predicting prognosis remains to be determined in future studies.
Prepublished online as Blood First Edition Paper, January 13, 2005; DOI 10.1182/blood-2004-10-3999.
Supported in part by the National Cancer Institute (NCI) Cancer Center CA15083-29C1 and CA97274. I.M.G. is supported in part by CA97274, an American Society of Clinical Oncology (ASCO) Young Investigator award, the Multiple Myeloma Research Foundation, the American Society of Hematology (ASH) Scholar award, and the Research Fund for Waldenstrom.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 U.S.C. section 1734.
References
- 1.Argatoff LH, Connors JM, Klasa RJ, Horsman DE, Gascoyne RD. Mantle cell lymphoma: a clinicopathologic study of 80 cases. Blood. 1997;89: 2067-2078. [PubMed] [Google Scholar]
- 2.Kurtin, PM. Mantle cell lymphoma. Adv Anat Pathol. 1998;5: 376-398. [DOI] [PubMed] [Google Scholar]
- 3.Yatabe Y, Suzuki R, Matsuno Y, et al. Morphological spectrum of cyclin D1-positive mantle cell lymphoma: study of 168 cases. Pathol Int. 2001;51: 747-761. [DOI] [PubMed] [Google Scholar]
- 4.Katz RL Wojcik EM, el-Naggar AK, Ordonez NG, Johnston DA. Proliferation markers in non-Hodgkin's lymphoma: a comparative study between cytophotometric quantitation of Ki-67 and flow cytometric proliferation index on fine needle aspirates. Anal Quant Cytol Histol. 1993;15: 179-186. [PubMed] [Google Scholar]
- 5.Weisenburger D, Vose JM, Greiner TC, et al. Mantle cell lymphoma: a clinicopathologic study of 68 cases from the Nebraska Lymphoma Study Group. Amer J Hematol. 2000;64: 190-196. [DOI] [PubMed] [Google Scholar]
- 6.Oinonen R, Franssila K, Teerenhovi L, Lappalainen K, Elonen E. Mantle cell lymphoma: clinical features, treatment and prognosis of 94 patients. Eur J Cancer. 1998;34: 329-336. [DOI] [PubMed] [Google Scholar]
- 7.Hiddemann W, Unterhalt M, Herrmann R, et al. Mantle-cell lymphomas have more widespread disease and a slower response to chemotherapy compared with follicle-center lymphomas: results of a prospective comparative analysis of the German Low-Grade Lymphoma Study Group. J Clin Oncol. 1998;16: 1922-1930. [DOI] [PubMed] [Google Scholar]
- 8.Bodrug SEW, Bath ML, Lindeman GJ, Harris AW, Adams JM. Cyclin D1 transgene impedes lymphocyte maturation and collaborates in lymphomagenesis with the myc gene. EMBO J. 1994;13: 2124-2130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stilgenbauer S, Schaffner C, Winkler D, et al. The ATM gene in the pathogenesis of mantle-cell lymphoma. Ann Oncol. 2000;11(suppl 1): 127-130. [PubMed] [Google Scholar]
- 10.Martinez NC, Algara P, Rodriguez A, et al. The molecular signature of mantle cell lymphoma reveals multiple signals favoring cell survival. Cancer Res. 2003;63: 8226-8232. [PubMed] [Google Scholar]
- 11.Antonucci FC, Parolini C, Hamdan M, Astner H, Righetti PG. Two-dimensional molecular profiling of mantle cell lymphoma. Electrophoresis. 2003;24: 2376-2385. [DOI] [PubMed] [Google Scholar]
- 12.Hamdan M, Righetti PG. Assessment of protein expression by means of 2-D gel electrophoresis with and without mass spectrometry. Mass Spectrom Rev. 2003;22: 272-284. [DOI] [PubMed] [Google Scholar]
- 13.Zhu H, Snyder M. Protein chip technology. Curr Opin Chem Biol. 2003;7: 55-63. [DOI] [PubMed] [Google Scholar]
- 14.Meeker T, Sellers W, Harvey R, et al. Cloning of the t(11;14)(q13;q32) translocation breakpoints from two human leukemia cell lines. Leukemia. 1991;5: 733-737. [PubMed] [Google Scholar]
- 15.BD Biosciences. Antibody microarray analysis workbook. http://bioinfo2.clontech.com/abinfo/array-list-action.do. Accessed January 7, 2005.
- 16.Svingen P, Karp JE, Krajewski S, et al. Evaluation of Apaf-1 and procaspases-2, -3, -7, -8, and -9 as potential prognostic markers in acute leukemia. Blood. 2000;96: 3922-3931. [PubMed] [Google Scholar]
- 17.Chen G, Gharib TG, Huang CC, et al. Discordant protein and mRNA expression in lung adenocarcinomas. Mol Cell Proteomics. 2002;1: 304-313. [DOI] [PubMed] [Google Scholar]
- 18.Svingen PA, Loegering DA, Meng XW, et al. Components of the cell death machine and drug sensitivity of the NCI cell line panel. Clinical Cancer Res. 2004;10: 6807-6820. [DOI] [PubMed] [Google Scholar]
- 19.Eischen C, Weber JD, Roussel MF, Sherr CJ, Cleveland JL. Disruption of the ARF-Mdm2-p53 tumor suppressor pathway in Myc-induced lymphomagenesis. Genes Dev. 1999;13: 2658-2669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Alt J, Greiner TC, Cleveland JL, Eischen CM. Mdm2 haplo-insufficiency profoundly inhibits Myc-induced lymphomagenesis. EMBO J. 2003;22: 1442-1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Watanabe T, Ichikawa A, Saito H, Hotta T. Overexpression of the MDM2 oncogene in leukemia and lymphoma. Leuk Lymphoma. 1996;21: 391-397. [DOI] [PubMed] [Google Scholar]
- 22.Pagnano K, Vassallo J, Lorand-Metze I, Costa FF, Saad ST. p53, Mdm2, and c-Myc overexpression is associated with a poor prognosis in aggressive non-Hodgkin's lymphomas [erratum published in: Am J Hematol. 2001;67:279]. Am J Hematol. 2001;67: 84-92. [DOI] [PubMed] [Google Scholar]
- 23.Lindstrom M, Klangby U, Wiman KG. p14ARF homozygous deletion or MDM2 overexpression in Burkitt lymphoma lines carrying wild type p53. Oncogene. 2001;20: 2171-2177. [DOI] [PubMed] [Google Scholar]
- 24.Moller M, Nielsen O, Pedersen NT. Frequent alteration of MDM2 and p53 in the molecular progression of recurring non-Hodgkin's lymphoma. Histopathology. 2002;41: 322-330. [DOI] [PubMed] [Google Scholar]
- 25.Solenthaler M, Matutes E, Brito-Babapulle V, Morilla R, Catovsky D. p53 and mdm2 in mantle cell lymphoma in leukemic phase. Haematologica. 2002;87: 1141-1150. [PubMed] [Google Scholar]
- 26.Bischoff F, Ponstingl H. Mitotic regulator protein RCC1 is complexed with a nuclear ras-related polypeptide. Proc Natl Acad Sci U S A. 1991;88: 10830-10834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bischoff F, Ponstingl H. Catalysis of guanine nucleotide exchange on Ran by the mitotic regulator RCC1. Nature. 1991;354: 80-82. [DOI] [PubMed] [Google Scholar]
- 28.Dasso M, Nishitani H, Kornbluth S, Nishimoto T, Newport JW. RCC1, a regulator of mitosis, is essential for DNA replication. Mol Cell Biol. 1992;12: 3337-3345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ren M, Drivas G, D'Eustachio P, Rush MG. Ran/TC4: a small nuclear GTP-binding protein that regulates DNA synthesis. J Cell Biol. 1993;120: 313-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Berger A, Tang G, Alexander HR, Libutti SK. Endothelial monocyte-activating polypeptide II, a tumor-derived cytokine that plays an important role in inflammation, apoptosis, and angiogenesis. J Immunother. 2000;23: 519-527. [DOI] [PubMed] [Google Scholar]
- 31.Murray J, Heng YM, Symonds P, et al. Endothelial monocyte-activating polypeptide-II (EMAP-II): a novel inducer of lymphocyte apoptosis. J Leukoc Biol. 2004;75: 772-776. [DOI] [PubMed] [Google Scholar]
- 32.Murray J, Symonds P, Ward W, et al. Colorectal cancer cells induce lymphocyte apoptosis by an endothelial monocyte-activating polypeptide-II-dependent mechanism. J Immunol. 2004;172: 274-281. [DOI] [PubMed] [Google Scholar]
- 33.Wera S, Hemmings BA. Serine/threonine protein phosphatases. Biochem J. 1995;311(pt 1): 17-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xu W, Yuan X, Jung YJ, et al. The heat shock protein 90 inhibitor geldanamycin and the ErbB inhibitor ZD1839 promote rapid PP1 phosphatase-dependent inactivation of AKT in ErbB2 overexpressing breast cancer cells. Cancer Res. 2003;63: 7777-7784. [PubMed] [Google Scholar]
- 35.Satinover D, Leach CA, Stukenberg PT, Brautigan DL. Activation of Aurora-A kinase by protein phosphatase inhibitor-2, a bifunctional signaling protein. Proc Natl Acad Sci U S A. 2004;101: 8625-8630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Di Cunto F, Calautti E, Hsiao J, et al. Citron rho-interacting kinase, a novel tissue-specific ser/thr kinase encompassing the Rho-Rac-binding protein Citron. J Biol Chem. 1998;273: 29706-29711. [DOI] [PubMed] [Google Scholar]
- 37.Ridley A. Rho proteins and cancer. Breast Cancer Res Treat. 2004;84: 13-19. [DOI] [PubMed] [Google Scholar]