

Abstract
C/EBPε, a member of the CCAAT/enhancer binding protein family, is a transcription factor important in neutrophil differentiation. We have determined that it is phosphorylated on multiple serine and threonine residues and can be a target for phosphorylation by a number of kinases. We identified a threonine at amino acid 75, part of a consensus mitogen-activated protein (MAP) kinase site within the transactivation domain of C/EBPε, as being phosphorylated only by p38 MAP kinase. Phosphorylation of this residue resulted in enhanced transcriptional activity on a myeloid-specific promoter in in vitro transient transfection reporter assays. We also determined that phosphorylation at Thr75 yielded a protein that was more effective at binding its cognate DNA sequence compared with the wild-type nonphosphorylated C/EBPε. Stable expression of C/EBPεT75A in interleukin 3 (IL-3)–dependent 32Dcl3 did not result in the up-regulation of expression of secondary granule genes compared with wild-type C/EBPε or C/EBPεT75D. Therefore we suggest that C/EBPε is a target for p38 MAP kinase activity.
Introduction
Gene expression in response to external stimuli is regulated by the activity of transcription factors. The activity of these transcription factors is tightly regulated by a variety of posttranslational modifications, including acetylation and phosphorylation. Phosphorylation is one of the major, and most extensively studied, mechanisms for modulating transcription factor activity.
Many studies on transcription factors have demonstrated them to be modular proteins with separate domains for DNA binding and either transactivation or repression. C/EBPε is a member of the CCAAT/enhancer binding protein family of transcription factors, which we previously demonstrated had both transactivation and repression domains.1,2 The CCAAT/enhancer binding protein family also includes C/EBPα, C/EBPβ, C/EBPδ, and CHOP/GADD153 (C/EBP homology protein/growth arrest and DNA damage-inducible gene 153).3-6 The members of the C/EBP family of transcription factors are phosphoproteins and the phosphorylation of these proteins has been shown to affect their function. C/EBPβ is the most extensively studied member of this transcription factor family with respect to phosphorylation.
C/EBPβ is phosphorylated by a number of protein kinases including protein kinase A (PKA), protein kinase C (PKC), glycogen synthase kinase 3 (GSK3), mitogen-activated protein kinases (MAPKs), ribosomal S6 kinase, and calcium-calmodulin kinase II (CaMKII). Phosphorylation affects a number of the functions of C/EBPβ, including inhibition of DNA binding, intracellular relocalization to the nucleus, increased transcriptional activity, and inhibition of apoptosis.7-12
C/EBPα is a target for phosphorylation by PKC and GSK3.13,14 Although phosphorylation by PKC resulted in a decrease in DNA binding by C/EBPα, phosphorylation by GSK3 did not appear to have any effect on C/EBPα function.
In contrast to C/EBPα and C/EBPβ, phosphorylation in the DNA binding domain of C/EBPδ by casein kinase 2 actually increased DNA binding.15 C/EBPδ can also be phosphorylated by MAPKs resulting in the translocation of this protein to the nucleus.16 MAP kinases can phosphorylate another member of the C/EBP family, CHOP/GADD153. In this case CHOP/GADD153 showed increased transcriptional activity.
Members of the C/EBP family are expressed in myeloid cells but appear to play different roles in differentiating and mature cells. C/EBPε is expressed almost exclusively in myeloid cells with its expression reaching a peak in mature neutrophils and macrophages.17,18 Both C/EBPβ and C/EBPδ are regulators of cytokine gene expression in macrophages, whereas C/EBPα is critical for granulocytic differentiation.19-21 As well as being expressed in hematopoietic cells, C/EBPα, C/EBPβ, and CHOP/GADD153 also play an important role in adipocyte differentiation. The p38 MAP kinase can phosphorylate both C/EBPβ and CHOP/GADD153 with opposing effects on adipocyte differentiation. Phosphorylation of C/EBPβ by p38 MAPK is essential for adipocyte differentiation, whereas phosphorylation of CHOP/GADD153 by p38 MAPK results in an inhibition of adipocyte differentiation.22,23 Thus phosphorylation of transcription factors involved in the same pathway by the same kinase allows for tight regulation of that differentiation pathway. Also, phosphorylation of a specific transcription factor by a specific kinase may be required for the differentiation of a specific cell type.
Therefore, we studied C/EBPε in order to determine the sites of phosphorylation, the kinases responsible for these phosphorylations, and the effect, if any, on C/EBPε function. This study demonstrates that C/EBPε is phosphorylated on multiple serine and threonine residues by a number of different kinases. We identified a phosphoacceptor site within the N-terminal transactivation domain of C/EBPε that was specifically phosphorylated by p38 MAP kinase. Phosphorylation of this residue results in increased transactivation on a myeloid-specific promoter accompanied by increased binding of C/EBPε to its cognate DNA sequence. C/EBPε phosphorylated on this residue was also able to up-regulate the expression of myeloid-specific secondary granule genes in vivo.
Materials and methods
CMV-C/EBPε constructs
Single point mutations were introduced into wild-type C/EBPε by polymerase chain reaction (PCR) using overlapping mutated oligonucleotides, followed by a second round of PCR using external primers containing appropriate restriction sites.24 The resulting PCR products were digested with the appropriate restriction enzymes and ligated into a cytomegalovirus (CMV)–expression vector, pCMV-SPORT (Invitrogen, Carlsbad, CA). The presence of these point mutations within C/EBPε was confirmed by sequencing.
Transient transfections and reporter assays
Transient transfection assays were performed in Jurkat cells using lipofectin (Invitrogen) with 3 μg promoter-luciferase reporter and 0.3 μg of the C/EBPε expression vectors, wild-type or mutants, as previously described.25 The internal control for transfection efficiency was 1 μg pCMV–β-galactosidase. At 24 hours after transfection, the Jurkat cells were activated with TPA (12-O-tetradecanoyl phorbol 13-acetate; 2 ng/mL) and the calcium ionophore A23187 (250 ng/mL) for 16 hours. After this time the transfections were harvested and the luciferase and β-galactosidase activities were measured according to the manufacturer's instructions (Promega, Madison, WI). In some assays the following inhibitors (Calbiochem, San Diego, CA) were added for 60 minutes prior to activation: SB203580 (5 μM) and SB202190 (2.5 μM) for inhibition of p38 MAP kinase; H-89 (50 nM), an inhibitor of PKA; and KN-62 (1.5 μM), an inhibitor of CaMKII.
GST-C/EBPε fusion proteins
C/EBPε, C/EBPεT75A, and C/EBPεT75D were subcloned into pGEX-5X-1 and the resulting constructs transformed into BL-21 cells. The glutathione-S–transferase (GST)–C/EBPε fusion proteins were isolated according to the manufacturer's instructions (Amersham-Pharmacia, Piscataway, NJ), with specific modifications. Following overnight growth, the culture was divided 1:10 and then grown for a further 60 minutes. The GST-C/EBPε fusion protein was induced by 1 mM IPTG (isopropyl-B-D glucopyranoside) for 4 hours. The GST fusion proteins were then isolated by sonication and stored bound to the glutathione-sepharose beads in 50% glycerol. Expression of the GST fusion proteins was monitored by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) and Western immunoblotting with a specific C/EBPε antibody (Santa Cruz Biotechnology, Santa Cruz, CA).
In vitro kinase assays
Equal amounts (5 μg) of the GST-C/EBPε fusion proteins were incubated with the activated kinase (1-10 U) in kinase-specific reaction buffer containing 10 μCi (0.37 MBq) γ-32P–adenosine triphosphate. The kinases used for these assays were PKA (Roche, Indianapolis, IN), PKC (Roche), extracellular signal-related kinase 2 (ERK2; UBI, Lake Placid, NY), p38α MAP kinase (UBI), c-Jun N-terminal kinase (JNK2; UBI), casein kinase 2 (Roche), and CaMKII (UBI). The kinase reactions were incubated in appropriate kinase buffer at 30°C for 10 to 30 minutes and then stopped by the addition of Laemmli sample buffer (BioRad, Hercules, CA). The reactions were boiled and separated by SDS-PAGE. The gels were either dried for further analysis as described in the next section or the 32P-labeled protein was transferred to polyvinylidene fluoride (PVDF) membrane (Millipore, Bedford, MA) for immunoblotting with a C/EBPε-specific antibody. 32P-labeled protein was detected by autoradiography.
Two-dimensional (2D) phospho–amino acid analysis
The in vitro–kinased GST-C/EBPε fusion proteins were eluted overnight from the dried polyacrylamide gel in buffer P (25 mM N-ethylmorpholine, pH 7.7) and then precipitated. For 2D phospho–amino acid analysis the precipitated protein was treated with 6 M HCl for 60 minutes at 100°C and then lyophilized. The pellet was washed thoroughly and finally resuspended in pH 1.9 buffer (formic acid–acetic acid–water ratio, 1:3.1:35.9) and the liberated phospho–amino acids were separated on 100 μM thin-layer chromatography (TLC) plates (CBS Scientific, Del Mar, CA) on the Hunter thin layer peptide mapping electrophoresis system (HTLE-7000; CBS Scientific), with 1 μg cold phospho–amino acid standards. For 2D separation, the first dimension is electrophoresis in pH 1.9 buffer (formic acid–acetic acid–water ratio, 1:3.1:35.9) at 1.5 kV for 20 minutes and the second dimension is electrophoresis in pH 3.5 buffer (acetic acid–pyridine–water ratio, 10:1:189) at 1.3 kV for 16 minutes. Unlabeled phospho–amino acids were detected by ninhydrin staining of the TLC plate and the 32P-labeled phospho–amino acids were visualized by autoradiography.
Nuclear protein analysis
Jurkat cells were transfected with 5 μg of the CMV-C/EBPε expression plasmids, wild-type and mutated, as described above. The empty vector pCMV-SPORT was used as the negative control for these experiments. Nuclear proteins were prepared as previously described.25 The nuclear extracts were initially separated by SDS-PAGE and analyzed by Western immunoblot to confirm expression of the various C/EBPε proteins. DNA binding by the C/EBPε proteins was determined by electrophoretic mobility shift assay (EMSA) using double-stranded oligonucleotides containing the C/EBP consensus site and adjacent sequences of the mim promoter.25 A standard reaction for the EMSA contained 2.5 ng 32P-labeled oligonucleotide, 10 μg Jurkat nuclear extract expressing either wild-type or mutated C/EBPε, 1.5 μg poly dIdC (poly-deoxyinosinic-deoxycytidylic acid), and 4.5 μg bovine serum albumin in a 20-μL volume. Competing cold oligonucleotides at 100-fold molar excess or antibodies (1 μg) were added where indicated. Electrophoresis was performed on a 4% polyacrylamide gel at 34 mA for 2 to 3 hours. The gel was dried and the results visualized by autoradiography.
Transduction of C/EBPε
The 32Dcl3 cells were cultured in Iscoves modified Dulbecco medium containing 10% fetal calf serum and 10% Wehi-3–conditioned medium. C/EBPε, C/EBPεT75A, C/EBPεT75D, C/EBPεS181D, and C/EBPεS188D were ligated into the pBabePuro retroviral vector to allow for retroviral infection of 32Dcl3 cells. Bosc23 packaging cells were transfected with 5 μg of the pBabePuro-C/EBPε constructs using GenePorter (GTS, San Diego, CA) as recommended by the manufacturer's instructions. At 48 hours after transfection, 32Dcl3 cells were infected by incubation with medium removed from the Bosc23 cells for 48 hours in the presence of 4 μg/mL polybrene.23 After this time the 32Dcl3 cells were transferred to medium containing 2 μg/mL puromycin. Puromycin-resistant 32D-C/EBPε cells were expanded, and the expression of the wild-type and mutant C/EBPε proteins was examined by Western immunoblotting with an antibody specific for C/EBPε.
In order to demonstrate changes in 32Dcl3 cells occurring with the stable expression of C/EBPε proteins, total RNA was isolated from exponentially growing 32D-C/EBPε–expressing cells using Trizol according to the manufacturer's instructions (Invitrogen). Ten micrograms total RNA was separated on formaldehyde-agarose gels. The RNA was Northern blotted onto Hybond+ membrane with 10 × standard saline citrate (SSC), baked, and hybridized sequentially with 32P-α–dCTP (32P-α–deoxycytosine triphosphosphate)–labeled probes for the secondary granule genes NGAL and B9 and a probe for 18S as the control for equal sample loading of the gel.26 Results were visualized by autoradiography.
Results
C/EBPε can be phosphorylated in vitro by multiple kinases
Since other C/EBP family members have been shown to be the target of a number of kinases, we analyzed the protein sequence of C/EBPε and identified a number of potential phosphoacceptor sites. We then carried out a series of in vitro kinase assays to confirm that C/EBPε was a substrate for these kinases as suggested by the protein sequence. In vitro kinase assays demonstrated that GST-C/EBPε could be phosphorylated by ERK2, p38α MAP kinase, and PKA (Figure 1A-C) but was not phosphorylated in vitro by JNK2, casein kinase 2, or PKC (Figure 1D-E; data not shown). CaMKII phosphorylated C/EBPε weakly compared with the other kinases (Figure 1F). The appropriate positive controls were included in these kinase assays as verification of the activity of the kinase, and GST alone was not phosphorylated in any of these assays (Figure 1; data not shown).
Figure 1.

C/EBPε is phosphorylated in vitro by ERK2, PKA, and p38 MAP kinase. GST-C/EBPε was in vitro kinased by the individual protein kinases and the reaction separated on SDS-PAGE gel and transferred to PVDF membrane. The following enzymes were used: (A) ERK2; (B) p38α MAP kinase; (C) PKA; (D) JNK2; (E) casein kinase 2; (F) calcium-calmodulin kinase II. (i) Phosphorylation was visualized by autoradiography. (ii) The same membrane was subsequently probed with an antibody specific for C/EBPε and the results were visualized by enhanced chemiluminescence (ECL; Amersham).
Previously we had determined, by in vivo labeling, that C/EBPε was phosphorylated on multiple serine and threonine residues but not on tyrosine residues.2 Furthermore, in vivo labeling with a series of previously described1 C/EBPε deletion mutants demonstrated that only threonine residues were phosphorylated within the N-terminal transactivation domain of C/EBPε (data not shown). Potentially both serine and threonine residues in C/EBPε could be phosphorylated by PKA, ERK2, and p38α MAP kinase. Therefore we performed 2D phospho–amino acid analysis on the in vitro–kinased GST-C/EBPε to determine the identity of the residues phosphorylated by each of these 3 kinases. This experiment demonstrated that both ERK2 and PKA kinase activity resulted in the phosphorylation of serine residues only (Figure 2A-B). However, phosphorylation of both threonine and serine residues was observed when GST-C/EBPε was in vitro kinased by p38α MAP kinase (Figure 2C).
Figure 2.

C/EBPε is phosphorylated on Thr75 by p38 MAP kinase. In vitro–kinased GST-C/EBPε was treated with 6M HCl to liberate the phosphorylated residues that were resolved by 2D electrophoresis in buffers pH 1.9 and pH 3.5. (A) ERK2 and (B) PKA phosphorylated GST-C/EBPε. (C) p38 MAP kinase phosphorylated GST-C/EBPε. (D) p38 MAP kinase phosphorylated GST-C/EBPεT75A. (E) A diagrammatic representation of cold phospho–amino acid standards on ninhydrinstained TLC plate. Arrows indicate the location of phosphorylated amino acid residues.
Within C/EBPε, 4 potential MAP kinase phosphoacceptor sites occur, 1 threonine and 3 serine residues. The serine residues are at amino acids 109, 181, and 188 and are all minimal MAP kinase sites (SP). The threonine residue is at amino acid 75 and is part of a consensus MAP kinase site (PGTP) within the N-terminal transactivation domain of C/EBPε. Since this is the only potential site within C/EBPε for the threonine phosphorylation observed with p38 MAP kinase, we mutated this residue to the nonphosphorylatable alanine GST-C/EBPεT75A. This mutated GST-C/EBPε was then used as a substrate in an in vitro kinase assay with p38α MAP kinase. The 2D phospho–amino acid analysis of the kinased GST-C/EBPε confirmed that the mutation Thr75Ala did result in the loss of the threonine phosphorylation observed for the wild-type GST-C/EBPε and p38α MAP kinase (Figure 2D). Therefore, these results suggest that Thr75 within the N-terminal transactivation domain of C/EBPε is a target for the p38 MAP kinase signaling pathway.
Mutation of Thr75 alters the capacity of C/EBPε to activate gene transcription
Although Thr75 appears to be a target for p38 MAP kinase in vitro, this does not necessarily mean that phosphorylation or lack of it at this site will affect the function of C/EBPε in vivo. Studies of other C/EBP family members showed that some residues can be phosphorylated in vitro but not in vivo and vice versa.9,11 Therefore we wanted to determine whether mutation of this amino acid could affect the ability of C/EBPε to transcriptionally activate a myeloid-specific promoter. Thus 2 C/EBPε mutants, C/EBPεT75A (nonphosphorylated) and C/EBPεT75D (threonine to aspartate to mimic a phosphorylation event), were generated and subcloned into a CMV-driven expression vector. These 2 constructs were then used in transient transfection assays with a myeloid-specific promoter-luciferase construct and compared with the wild-type C/EBPε activity on this promoter. The Jurkat cell line was used for these transient reporter assays since it does not express detectable levels of endogenous C/EBPε.
In the transient reporter assays, the wild-type C/EBPε activated the promoter-luciferase reporter construct by 7.5-fold, whereas the C/EBPεT75A mutant activated the promoter only 3.7-fold (Figure 3). In contrast, C/EBPεT75D was even more effective at activating the promoter, giving a 10.5-fold increase in activity on the promoter-luciferase reporter (Figure 3). These results suggest that phosphorylation (or lack of it) at Thr75 does have an effect on the ability of C/EBPε to activate gene transcription. Furthermore, when a series of protein kinase inhibitors was used in these transient reporter assays, only inhibitors of p38 MAP kinase inhibited the ability of wild-type C/EBPε to transactivate the promoter-luciferase reporter (Figure 3).
Figure 3.

Mutation of Thr75 or addition of p38 MAP kinase inhibitors affects the transactivation ability of C/EBPε on a myeloid-specific promoter. Jurkat cells were transfected with the C/EBPε constructs and the mim promoter in a luciferase vector. The cells were harvested 48 hours after transfection and luciferase (LUC) and β-galactosidase activities were measured. These results are shown as relative luciferase units (RLUs) representing the mean of 3 experiments.
Mutation of minimal MAP kinase sites can also affect C/EBPε function
We were also interested to examine the effect of phosphorylation on the 3 minimal MAP kinase sites; however, only 2 are conserved between human and murine C/EBPε. These are Ser181 and Ser188, which are located in the C-terminal region of the repression domain of C/EBPε. Therefore we generated mutations of these 2 serine residues converting each to either alanine or aspartate as we had done previously for Thr75. We then used these new constructs in transient transfection reporter assays using the mim promoter-luciferase construct.
For these 2 sites, C/EBPεS181A and C/EBPεS188D were able to activate the mim promoter-luciferase construct in these assays but were not as effective as the wild-type C/EBPε (Figure 3). However, the other mutations C/EBPεS181D and C/EBPεS188A did not significantly activate the mim promoter (Figure 3).
Phosphorylation of other C/EBP family members has been shown to affect intracellular localization.8,27 Therefore, the expression of these mutant C/EBPε proteins was examined (Figure 4). Western immunoblot analysis determined that all C/EBPε proteins were expressed at similar levels but the C/EBPεS188A mutant protein was the only one not localized to the nucleus (Figure 4). Subsequent analysis demonstrated that this mutant C/EBPε protein appeared to now be localized to the cytoplasm, hence its lack of activity in the reporter assays (Figure 3).
Figure 4.

Expression of wild-type and mutant C/EBPε proteins. Jurkat cells were transfected with either wild-type or mutant C/EBPε expression vectors alone. Forty-eight hours after transfection cells were harvested for total and nuclear protein extraction. Western immunoblot analysis was carried out using an antibody specific for C/EBPε. (A) Total protein extract. (B) Nuclear protein extract.
Phosphorylation alters DNA binding affinity of C/EBPε
Since all but 1 of our mutant C/EBPε proteins are correctly localized to the nucleus and expressed at similar levels, we were interested to examine their ability to bind their cognate DNA sequence. Therefore gel shift analysis was performed using nuclear extracts from Jurkat cells transfected with either wild-type or mutant forms of C/EBPε and an oligonucleotide from the mim promoter containing a cognate C/EBPε binding site and surrounding sequences.
The gel shift analysis demonstrated that wild-type C/EBPε, C/EBPεT75A, C/EBPεT75D, C/EBPεS181A, C/EBPεS181D, and C/EBPεS188D all bound the oligonucleotide containing the C/EBPε binding site from the mim promoter (Figure 5A). In these gel shift experiments, the DNA-protein complex could be competed away by the inclusion of the same unlabeled oligonucleotide and it could be supershifted with a C/EBPε-specific antibody (Figure 5A).
Figure 5.

DNA binding affinity of C/EBPε is altered by phosphorylation. Gel shift analysis was carried out on nuclear extracts derived from Jurkat cells transfected with wild-type and mutant C/EBPε expression plasmids and an oligonucleotide containing the C/EBP site from the mim promoter. (A) Nuclear extracts from transfected Jurkat cells give a slow migrating band with this oligonucleotide. This retarded band can be competed by 100-fold excess of cold double-stranded (ds) oligonucleotide and supershifted by C/EBPε-specific antisera. (B) EMSA was carried out using the same amount of nuclear extract with decreasing concentrations of hot oligonucleotides. The gel shifts were analyzed on a Phosphoimager (Molecular Dynamics, Piscataway, NJ) to obtain the radioactive counts of bound and free probe. Scatchard analysis determined the affinity of these C/EBPε proteins for the C/EBP site in the mim promoter as follows: C/EBPε 7.69 nM; C/EBPεT75D 0.48 nM.
Although all C/EBPε proteins bound to the cognate DNA sequence, there appeared to be an increase DNA binding by the nuclear extract containing C/EBPεT75D compared with the wild-type C/EBPε (Figure 5A). Therefore we wanted to determine whether there was a real change in DNA binding by the C/EBPεT75D mutant protein. Therefore, further EMSAs were carried out using a constant amount of the C/EBPε proteins and increasing dilutions of the oligonucleotide (Figure 5B). Wild-type C/EBPε demonstrated good binding for its cognate DNA sequence at 10 nM of oligonucleotide (Figure 5B). However, this DNA-protein complex was very weak even when the oligonucleotide was diluted to only 5 nM (Figure 5B). In contrast, binding of C/EBPεT75D was strong even when the oligonucleotide had been diluted to 0.25 nM (Figure 5B). Scatchard analysis determined that the DNA binding affinity for wild-type C/EBPε is 7.69 nM. This value is about the same as the DNA binding affinity of C/EBPε previously determined by our group.25 In contrast, the DNA binding affinity of C/EBPεT75D is 0.48 nM. This demonstrates that C/EBPεT75D binds its cognate DNA with 16-fold more affinity than wild-type C/EBPε. Therefore, phosphorylation of Thr75 dramatically increases the DNA binding ability of the C/EBPε protein.
In vivo effects of C/EBPεT75D expression
The previous experiments are all in vitro analyses for effects on C/EBPε function after phosphorylation. Therefore we were interested in investigating the biologic effects of expression of phosphorylation mutants of C/EBPε in a cell line capable of granulocytic differentiation. The 32Dcl3 cells were chosen for stable transfection. Following retroviral infection of the 32Dcl3 cells, a series of clones were isolated for each C/EBPε protein. Protein lysates were made from each clone and analyzed by Western immunoblotting with an antibody specific for C/EBPε. Clones were selected that expressed similar levels of the C/EBPε proteins (Figure 6A).
Figure 6.
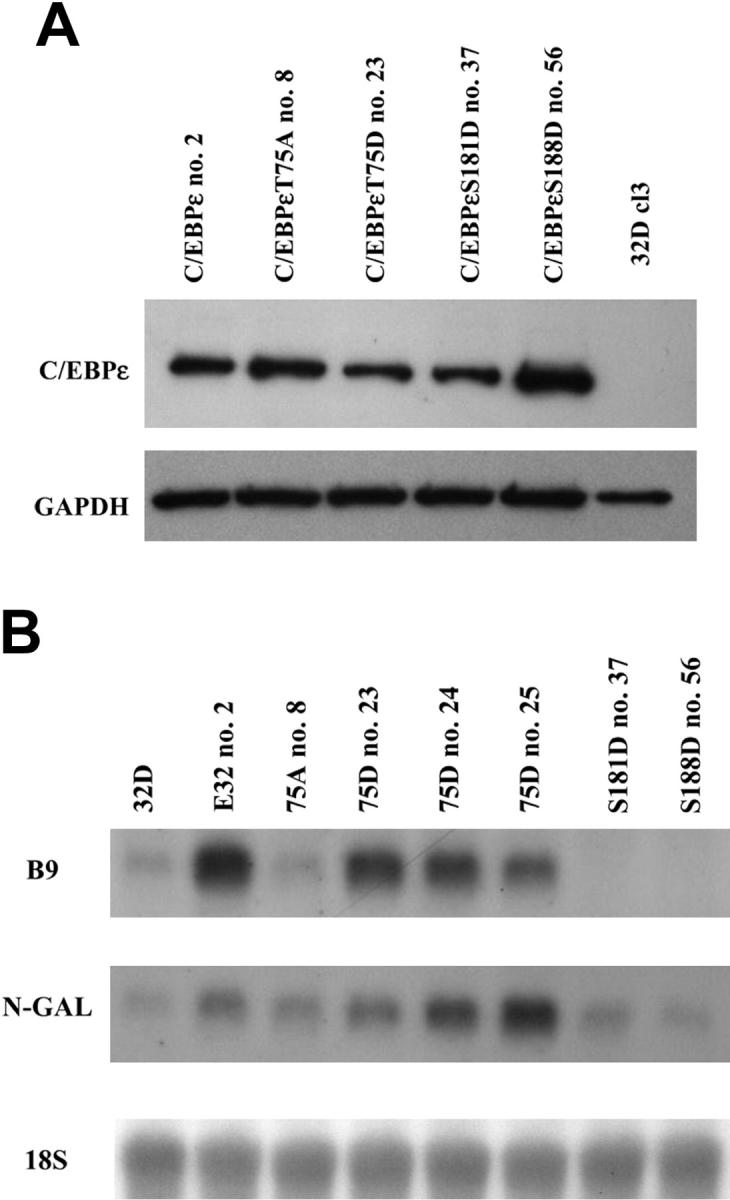
In vivo effects of constitutive expression of C/EBPε on endogenous gene expression. (A) Expression of the C/EBPε proteins from 32Dcl3 stably transfected cells was analyzed by Western immunoblot on total cell lysate with an antibody specific for C/EBPε. Results were visualized by ECL. GAPDH indicates glyceraldehyde 6-phosphate dehydrogenase. (B) Ten micrograms total RNA was examined by Northern blot analysis with probes for the secondary granule genes NGAL and B9.Aprobe specific for 18S was used as the loading control.
The C/EBPε knockout mice are deficient in secondary granule gene expression.27 Therefore we were interested to examine whether these stably transfected 32Dcl3 cells were expressing secondary granule genes, even when not undergoing differentiation. For this analysis RNA was isolated from exponentially growing 32Dcl3 cells, parental and stably transfected, and Northern blot analysis was carried out (Figure 6B). The results from this experiment demonstrate that constitutive expression of C/EBPε in 32Dcl3 cells did affect endogenous secondary granule gene expression (Figure 6B). The secondary granule genes NGAL and B9 were both up-regulated in cells expressing wild-type C/EBPε and C/EBPεT75D (Figure 6B). However, more significantly, constitutive expression of C/EBPεT75A did not result in the up-regulation of endogenous secondary granule gene expression (Figure 6B). This result demonstrates that phosphorylation of this threonine in the consensus MAP kinase site in C/EBPε by p38 MAP kinase is important for the in vivo function of C/EBPε.
Analysis of 32Dcl3 cells expressing either C/EBPεS181D or C/EBPεS188D demonstrated that neither of these mutant proteins could up-regulate secondary granule gene expression. This lack of activity correlated with their level of activity in the luciferase-reporter assays (Figure 3).
Discussion
The activity of transcription factors is regulated in a rapid and reversible manner by specific phosphorylations. This study demonstrates that C/EBPε is phosphorylated in vivo on multiple serine and threonine residues and that it is a substrate in vitro for the protein kinases PKA, ERK2, and p38 MAP kinase. The combination of these in vivo and in vitro analyses suggested that p38 MAP kinase was the protein kinase responsible for the phosphorylation of a threonine residue within the N-terminal transactivation domain of C/EBPε.
Phosphorylation of this threonine residue resulted in an increase in the transactivation ability of C/EBPε as determined by transient transfection reporter assays. EMSA analysis showed that this increase in activity on a myeloid-specific promoter was accompanied by an increase in the binding affinity of C/EBPεT75D for its cognate DNA sequence compared with the wild-type C/EBPε protein.
Transcription factors are generally organized into discrete domains responsible for the functions of DNA binding and transactivation. However, phosphorylation within 1 domain may affect the function of other separate domains within the whole protein. This effect might occur due to a change in conformation of the transcription factor as a result of phosphorylation. Studies on the transcription factors Ets-like gene-1 (Elk-1) and sphingolipid activator protein-1 (SAP-1) have suggested that their activity is regulated in this manner.28-30 For both of these transcription factors, phosphorylation of the C-terminal transactivation domain increased DNA binding via the N-terminal DNA binding domain. This suggests that the conformation change upon phosphorylation may unmask or alter the DNA binding surface of the transcription factor.
Therefore, we postulate that phosphorylation of C/EBPε on Thr75 exerts such an effect on this transcription factor. It is possible that phosphorylation of Thr75 could change C/EBPε from a “closed” conformation where it cannot bind DNA efficiently to a more “open” conformation allowing for much more efficient binding to its cognate DNA sequence.
Although we focused mainly on Thr75, we also assessed the effect of other phosphorylations by MAP kinases on C/EBPε function. The effect of phosphorylation on 2 serine residues within the repression domain of C/EBPε was studied. For these, the effects were mixed and not as dramatic as for Thr75. Dephosphorylation of Ser188 appeared to relocalize the C/EBPε protein to the cytoplasm. Whereas phosphorylation of Ser181 gave a C/EBPε protein that could bind DNA as well as wild-type C/EBPε but was inactive on a myeloid-specific promoter.
We analyzed individually these 3 MAP kinase sites: Thr75, Ser181, and Ser188. However, in vivo distinct protein kinases and phosphatases exist that orchestrate the responses of a transcription factor to a variety of external stimuli. These transcription factors frequently contain both stimulatory and inhibitory phosphorylation sites, which allows for a dynamic process resulting in the fine tuning of the transactivation potential. Thus phosphorylation is a mechanism that provides for tight regulation of the activity of a transcription factor rather than being a simple switch for turning both on and off the activity of a transcription factor.
With respect to C/EBPε, we suggest that Thr75 is frequently phosphorylated under normal growth conditions but that this is elevated upon other external stimuli. In this case, a stoichiometric shift occurs toward phospho-Thr75 in any given population of C/EBPε molecules. Also, under these conditions the molar ratio between activating and repressing phosphorylation sites would determine the magnitude of transactivation function of C/EBPε.
We next investigated the in vivo biologic effects of phosphorylation of C/EBPε. The 32Dcl3 cells were stably transfected to constitutively express these C/EBPε proteins, both wild-type and phosphorylation mutants. C/EBPε plays a role in neutrophilic differentiation. Neutrophils are responsible for protecting the host against bacterial and fungal infections and are activated rapidly in response to inflammatory signals. In C/EBPε knockout mice it was found that the cells of the granulocytic lineage have an impaired ability to migrate, a reduced capacity to produce superoxide, and do not express secondary granule proteins.27 The p38 MAP kinase is involved in mediating intracellular signals resulting in superoxide production and cell migration. In human neutrophils, inhibition of p38 MAP kinase results in decreased superoxide anion production, reduction in adhesion and chemotaxis in response to fMLP (formyl-methionyl-leucyl-phenylalanine), and inhibition of release of interleukin-8 (IL-8) in response to lipopolysaccharide (LPS).31,32 Therefore, p38 MAP kinase appears to be important for neutrophil function.
The defects observed for the C/EBPε knockout mice are also observed in a number of diseases as well as other mouse knockout models. These include humans with specific granule disease, for which mutations of C/EBPε have been identified, and chronic granulomatous disease, as well as Rac2 knockout mice.33-36 More recently a human neutrophil immunodeficiency syndrome has been identified resulting from a mutation of Rac2 (ras-related C3 botulinum toxin substrate 2).37 Rac2 is a member of the Rho family of guanosine triphosphatases (GTPases) that is highly expressed in myeloid cells and is a regulator of the nicotinamide adenine dinucleotide phosphate (NADPH)–oxidase complex. Therefore, the phenotype observed in C/EBPε knockout mice represents a defect intrinsic to neutrophil function.
Neutrophils are rapidly activated and short-lived. Therefore, C/EBPε has to be rapidly activated in response to an external stimulus. We hypothesize that C/EBPε may be phosphorylated in nonstimulated neutrophils and bound to its cognate DNA sequence. Activation of p38 MAP kinase signaling pathways could then result in the rapid phosphorylation of the threonine residue at amino acid 75 such that C/EBPε more efficiently binds DNA and can rapidly activate the transcription of genes necessary for neutrophil function.
Acknowledgments
Many thanks to members of the Koeffler laboratory for helpful suggestions and Tina Duong for excellent technical assistance.
Prepublished online as Blood First Edition Paper, January 27, 2005; DOI 10.1182/blood-2004-09-3708.
Supported by National Institutes of Health (NIH) grants (5 ROI CA26038-21 and 5 ROI CA26038-25), the Joseph Troy Fund, and the Parker Hughes Trust.
H.P.K. is a member of the Jonsson Comprehensive Cancer Center and the Molecular Biology Institute at UCLA.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 U.S.C. section 1734.
References
- 1.Williamson EA, Xu HN, Gombart AF, et al. Identification of transcriptional activation and repression domains in human CCAAT/enhancer-binding protein ε. J Biol Chem. 1998;273: 14796-14804. [DOI] [PubMed] [Google Scholar]
- 2.Chumakov AM, Grillier I, Chumakova E, Chih D, Slater J, Koeffler HP. Cloning of the novel human myeloid-cell-specific C/EBPε transcription factor. Mol Cell Biol. 1997;17: 1375-1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Akira S, Isshiki H, Sugita T, et al. IL-6 and NF-IL6 in acute-phase response and viral infection. EMBO J. 1990;9: 1897-1906.2112087 [Google Scholar]
- 4.Antonson P, Xanthanopoulos K. Molecular cloning, sequence and expression of the human gene encoding CCAAT/enhancer binding protein α (C/EBPα). Biochem Biophys Res Comm. 1995;215: 106-113. [DOI] [PubMed] [Google Scholar]
- 5.Cleutjens CBJM, Van Eekelen CCEM, Van Dekken H, et al. The human C/EBPδ (CRP3/CELF) gene: structure and chromosomal localization. Genomics. 1993;16: 520-523. [DOI] [PubMed] [Google Scholar]
- 6.Park JS, Luethy JD, Wang MG, et al. Isolation, characterization and chromosomal localization of the human GADD153 gene. Gene. 1992;116: 259-267. [DOI] [PubMed] [Google Scholar]
- 7.Buck M, Poli V, Hunter T, Chojkier M. C/EBPβ phosphorylation by RSK creates a functional XEXD caspase inhibitory box critical for cell survival. Mol Cell. 2001;8: 8807-8816. [DOI] [PubMed] [Google Scholar]
- 8.Chinery R, Brockman JA, Dransfield DT, Coffey RJ. Antioxidant-induced nuclear translocation of CCAAT/enhancer binding protein β. J Biol Chem. 1997;272: 30356-30361. [DOI] [PubMed] [Google Scholar]
- 9.Nakajima T, Kinoshita S, Sasagawa T, et al. Phosphorylation at threonine-235 by a ras-dependent mitogen-activated protein kinase cascade is essential for transcription factor NF-IL6. Proc Natl Acad Sci U S A. 1993;90: 2207-2211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Piwien-Pilipuk G, Van Mater D, Ross SE, MacDougald OA, Schwartz J. Growth hormone regulates phosphorylation and function of CCAAT/enhancer-binding protein β by modulating Akt and glycogen synthase kinase-3. J Biol Chem. 2001;276: 19664-19671. [DOI] [PubMed] [Google Scholar]
- 11.Trautwein C, Van Der Geer P, Karin M, Hunter T, Chojkier M. Protein kinase A and C site-specific phosphorylations of LAP (NF-IL6) modulate its binding affinity to DNA recognition elements. J Clin Invest. 1994;93: 2554-2561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wegner M, Cao Z, Rosenfeld MG. Calcium-regulated phosphorylation within the leucine zipper of C/EBPβ. Science. 1992;256: 370-373. [DOI] [PubMed] [Google Scholar]
- 13.Mahoney CW, Shuman J, McKnight SL, Chen H-C, Huang, K-P. Phosphorylation of CCAAT-enhancer binding protein by protein kinase C attenuates site-selective DNA binding. J Biol Chem. 1992;267: 19396-19403. [PubMed] [Google Scholar]
- 14.Ross SE, Erickson RL, Hemati N, MacDougald OA. Glycogen synthase kinase 3 is an insulin-regulated C/EBPα kinase. Mol Cell Biol. 1999;19: 8433-8441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Osada S, Yamamoto H, Nishihara T, Imagawa M. DNA binding specificity of the CCAAT/enhancer-binding protein transcription factor family. J Biol Chem. 1996;271: 3891-3896. [DOI] [PubMed] [Google Scholar]
- 16.Yamanaka R, Lekstrom-Himes J, Barlow C, Wynshaw-Boris A, Xanthanopoulos KG. CCAAT/enhancer binding proteins are critical components of the transcriptional regulation of hematopoiesis. Int J Mol Med. 1998;1: 213-221. [DOI] [PubMed] [Google Scholar]
- 17.Chih DY, Chumakov AM, Park DJ, Silla AJ, Koeffler HP. Modulation of mRNA expression of a novel human myeloid-selective CCAAT/enhancer binding protein gene (C/EBP epsilon). Blood. 1997;90: 2987-2994. [PubMed] [Google Scholar]
- 18.Morosetti R, Park DJ, Chumakov AM, et al. A novel, myeloid transcription factor, C/EBP epsilon, is upregulated during granulocytic, but not monocytic differentiation. Blood. 1997;90: 2591-2600. [PubMed] [Google Scholar]
- 19.Akira S, Kishimoto T. IL-6 and NF-IL6 in acute-phase response and viral infection. Immunol Rev. 1992;127: 25-50. [DOI] [PubMed] [Google Scholar]
- 20.Juan TS, Wilson DR, Wilde MD, Darlington GJ. Participation of the transcription factor C/EBP delta in the acute-phase regulation of the human gene for complement component C3. Proc Natl Acad Sci U S A. 1993;90: 2584-2588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang DE, Hetherington CJ, Meyers S, et al. CCAAT enhancer binding protein (C/EBP) and AML1 (CBFα2) synergistically activate the macrophage colony-stimulating factor receptor promoter. Mol Cell Biol. 1996;16: 1231-1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Engelman JA, Lisanti MP, Scherer PE. Specific inhibitors of p38 mitogen-activated protein kinase blocks 3T3-L1 adipogenesis. J Biol Chem.. 1998; 273: 32111-32120. [DOI] [PubMed] [Google Scholar]
- 23.Wang XZ, Ron D. Stress-induced phosphorylation and activation of the transcription factor CHOP (GADD153) by p38 MAP kinase. Science. 1996;272: 1347-1349. [DOI] [PubMed] [Google Scholar]
- 24.Ito W, Ishiguro H, Kurosawa Y. A general method for introducing a series of mutations into cloned DNA using the polymerase chain reaction. Gene. 1991;102: 67-70. [DOI] [PubMed] [Google Scholar]
- 25.Verbeek W, Gombart AF, Chumakov AM, Muller C, Friedman AD, Koeffler HP. C/EBPε directly interacts with the DNA binding domain of c-myb and cooperatively activates transcription of myeloid promoters. Blood. 1999;93: 3327-3337. [PubMed] [Google Scholar]
- 26.Gombart AF, Kwok SH, Anderson, KL, Yamaguchi Y, Torbett BE, Koeffler HP. Regulation of neutrophil and eosinophil secondary granule gene expression by transcription factors C/EBPε and PU.1. Blood. 2003;101: 3265-3273. [DOI] [PubMed] [Google Scholar]
- 27.Yamanaka R, Barlow C, Lekstrom-Himes J, et al. Impaired granulopoiesis, myelodysplasia, and early lethality in CCAAT/enhancer binding protein ε-deficient mice. Proc Natl Acad Sci U S A. 1997; 94: 13187-13192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shore P, Whitmarsh AJ, Bhaskaran R, Davis, RJ, Waltho JP, Sharrocks AD. Determinants of DNA-binding specificity of ETS-domain transcription factors. Mol Cell Biol. 1996;16: 3338-3349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Strahl T, Gille H, Shaw PE. Selective response of ternary complex factor Sap-1a to different mitogen-activated protein kinase sub-groups. Proc Natl Acad Sci U S A. 1996;93: 11563-11568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yang S-H, Shore P, Willingham N, Lakey JH, Sharrocks AD. Targeting of p38 mitogen-activated protein kinase to MEF2 transcription factors. EMBO J. 2002;18: 5666-5674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nick JA, Avdi NJ, Young SK, et al. Selective activation and functional significance of p38α mitogen-activated protein kinase in lipopolysaccharide-stimulated neutrophils. J Clin Invest. 1999; 103: 851-858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Suzuki K, Hino M, Hato F, Tatsumi N, Kitigawa S. Cytokine-specific activation of distinct mitogen-activated protein kinase subtype cascades in human neutrophils stimulated by granulocyte colony-stimulating factor, granulocyte-macrophage colony-stimulating factor, and tumor necrosis factor-α. Blood. 1999;93: 341-349. [PubMed] [Google Scholar]
- 33.Allen L-AH, DeLeo FR, Gallois A, Toyoshima S, Suzuki K, Nauseef WM. Transient association of the nicotinamide adenine dinucleotide phosphate oxidase subunits p47phox and p67phox with phagosomes in neutrophils from patients with X-linked chronic granulomatous disease. Blood. 1999;93: 3521-3530. [PubMed] [Google Scholar]
- 34.Gombart AF, Shiohara M, Kwok SH, Agematsu K, Komiyama A, Koeffler HP. Neutrophil-specific granule deficiency: homozygous recessive inheritance of a frameshift mutation in the gene encoding the transcription factor CCAAT/enhancer binding protein-epsilon. Blood. 2001;97: 2561-2567. [DOI] [PubMed] [Google Scholar]
- 35.Lekstrom-Himes JA, Dorman SE, Kopar P, Holland SM, Gallin JI. Neutrophil-specific granule deficiency results from a novel mutation with loss of function of the transcription factor CCAAT/enhancer binding protein epsilon. J Exp Med. 1999; 189: 1847-1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Roberts A, Kim C, Zhen L, et al. Deficiency of the hematopoietic cell-specific Rho family GTPase Rac2 is characterized by abnormalities in neutrophil function and host defense. Immunity. 1999; 10: 183-196. [DOI] [PubMed] [Google Scholar]
- 37.Ambruso DR, Knall C, Abell AN, et al. Human neutrophil immunodeficiency syndrome is associated with an inhibitory Rac2 mutation. Proc Natl Acad Sci U S A. 2000;97: 4654-4659. [DOI] [PMC free article] [PubMed] [Google Scholar]