

Abstract
Proliferative retinopathies, such as those complicating prematurity and diabetes, are major causes of blindness. A prominent feature of these retinopathies is excessive neovascularization, which is orchestrated by the hypoxia-induced vascular endothelial growth factor (VEGF) stimulating endothelial cells and the integrin-mediated adhesive interactions of endothelial cells with extracellular matrix components such as fibronectin (FN). Recently, we demonstrated that α-defensins interfere with α5β1–FN interactions and dependent endothelial cell functions. Here, α-defensins were studied in hypoxia-induced proliferative retinopathy. In vitro, α-defensins specifically inhibited α5β1-integrin–dependent migration of bovine retinal endothelial cells (BRECs) to FN, attenuated the VEGF-stimulated increase in endothelial permeability, and blocked BREC proliferation and capillary sprout formation in 3-dimensional fibrin-matrices. An up-regulation of β1-integrin and FN was observed in the retinal vessels in the mouse model of hypoxia-induced retinal angiogenesis. Systemic and local administration of α-defensins reduced retinal neovascularization by 45% and 60%, respectively, and this effect was comparable to the inhibitory effect of α5β1-blocking antibody. α-Defensins were detected in human diabetic retinas associated with normal retinal vessels but were absent from proliferative lesions. Together, these data show that α-defensins inhibit pathologic retinal neovascularization in vivo and may provide a clinically efficient strategy against proliferative retinopathies.
Introduction
Retinopathy of prematurity and proliferative diabetic retinopathy are the major causes of neonatal and adult blindness, respectively.1 Pathologic retinal neovascularization is ectopic—that is, the new vessels invade the vitreous cavity, resulting in vision-threatening complications, such as vitreous hemorrhage and retinal detachment. During proliferative retinopathy, neovascularization is regulated by several growth factors, especially the hypoxia-induced vascular endothelial growth factor (VEGF) that regulates endothelial cell proliferation, migration, and permeability and by the adhesive contacts of endothelial cells with the extracellular matrix.2-4 Proteins of the extracellular matrix, such as fibronectin (FN), vitronectin (VN), and fibrinogen (FBG), are deposited into an adhesive fibrillar network and control endothelial growth, differentiation, and migration by transmitting signals to the cells through specific integrins.5-8 Accordingly, we and others9,10 have previously shown that interference with integrin-extracellular matrix interactions and blockade of VEGF are effective in inhibiting retinal neovascularization in a mouse model of hypoxia-induced retinal angiogenesis.
Emerging evidence indicates that inflammatory cells may regulate endothelial cell functions related to angiogenesis in proliferative retinopathies. In retinopathy of prematurity and in diabetic retinopathy, leukocyte adhesion to retinal structures may lead to blood-retinal barrier breakdown, enhanced endothelial cell damage, and impaired capillary perfusion.2,11-13 Interestingly, VEGF regulates the recruitment of inflammatory cells in the context of retinopathies.1 Although the net effect of inflammatory cells in proliferative retinopathies is generally perceived to favor angiogenesis, inflammatory cells also bear antiangiogenic potential as a source of antiangiogenic compounds such as angiostatin14 and α-defensins.15
α-Defensins, also known as human neutrophil peptides, consist of a family of 4 closely related, small, cationic antimicrobial peptides comprising approximately 5% of the total neutrophil protein content, mainly found in the azurophilic granules.16 When neutrophils are activated during the phagocytosis of microorganisms or by exogenous inflammatory agonists, α-defensins are secreted intracellularly to provoke the lysis of prokaryotic organisms.16-19 During infection, α-defensins are released into the plasma at concentrations approaching 30 μM in some settings (normally less than 15 nM).20 α-Defensins accumulate in the vessel wall by binding to FN and can modify lipoprotein metabolism21 and inhibit plasminogen activation.22 Recently, we reported that α-defensins form a ternary complex with FN and α5β1-integrin and transform the FN–α5β1-integrin interaction to one that does not promote endothelial cell adhesion.15 α-Defensins were found to be an atypical ligand of α5β1, binding to the integrin in an Arg-Gly-Asp (RGD)–independent manner. Although α-defensins enhanced the binding of FN to α5β1, the FN–α5β1 interaction became RGD insensitive. α-Defensins thereby interfered with endothelial cell functions related to angiogenesis.15
Because the interaction between FN and α5β1 regulates retinal endothelial cell proliferation,8 we were prompted to investigate whether α-defensins could be used to affect proliferative retinopathy in vivo. Our present findings indicate that α-defensins specifically block retinal endothelial cell functions in vitro and pathologic retinal neovascularization in vivo. α-Defensins might thereby provide a platform to enable the development of novel approaches to treat proliferative retinopathies.
Materials and methods
Reagents and cells
α-Defensins (mixture of human neutrophil peptides [HNPs] 1-3) were isolated from human neutrophils and from the sputum of patients with cystic fibrosis and were characterized as described.18,23 Briefly, extracts were prepared from cells or from cystic fibrosis sputum incubated in 5% acetic acid for 16 hours at 4°C. Extracts were loaded on a Bio-gel P-10 polyacrylamide column (Bio-Rad, Hercules, CA). Defensins were eluted at 4 column volumes of 1% acetic acid, as described.18,23 Fractions that contained defensins were clearly identified by optical density at 280 nm and were characterized for their antibacterial activity using the radial diffusion assay as described.18 Positive fractions were buffer exchanged and separated on sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE). Three bands very close to each other with a mean molecular weight of 3.5 kDa were identified as HNPs 1 to 3 by Western blot analysis using rabbit antiserum (kindly provided by Dr T. Ganz, University of California, Los Angeles, CA). All preparations were free of endotoxin in the Limulus Amebocyte Lysate (E-toxate kit; Sigma, Munich, Germany; sensitivity less than 1.5 pg/mL) and were further diluted at least 1:100 before use. Synthetic α-defensin (HNP-1) was synthesized as previously described.24 Recombinant human β-defensin-2 (HBD-2) was prepared as described.25 Human FBG and FN were from Sigma. Blocking monoclonal antibody (mAb) against β1-integrin, 6S6, and mAb against αvβ3, LM609, were from Chemicon (Hofheim, Germany). mAb 5H10-27, which blocks mouse α5-integrin,26 was from BD Biosciences (San Jose, CA). Basic fibroblast growth factor (bFGF) and VEGF were from R&D Systems (Wiesbaden, Germany). Bovine retinal endothelial cells (BRECs) were kindly provided by Dr Sigrid Zink (University of Düsseldorf, Germany) and cultivated as described.27 Culture media were from Gibco (Eggenstein, Germany).
Cell proliferation
Endothelial cell proliferation was determined by measuring total cell number. Briefly, BRECs were plated onto 96-well plates and incubated for 12 hours, after which the medium was changed to MCDB-131 containing 0.05% fetal calf serum (FCS). Cells were incubated for 72 hours in the absence or presence of stimuli or competitors. Cells were then trypsinized, and the total cell number was counted with a Casy-Counter (Schärfe System, Reutlingen, Germany).
Endothelial cell permeability
BREC permeability was studied as previously described.28 Briefly, cells were grown to confluent monolayers on gelatin-coated membranes in double-chamber tissue culture plates (Transwell membrane, 0.4-μM pore size; Corning Costar, Bodenheim, Germany). After 2 days, the medium was switched to serum-free conditions, and the incubation was continued for an additional day. At this time, chambers were examined microscopically for integrity and uniformity of BREC monolayers, medium was changed, and stimuli/competitors were added at designated times in quadruplicate cups for each time point. At the end of the incubation period, fluorescein isothiocyanate (FITC)–conjugated dextran (1 mg/mL; Mr 42 000; Sigma) was added to the upper chambers, and fluorescence in the lower chamber was measured 2 hours later with a fluorescence reader (Biotek, Woburn, MA).
Adhesion assay
Cell adhesion to multiwell plates precoated with FN, FBG, or bovine serum albumin (BSA) as a control (each 10 μg/mL) was examined as previously described.27,29 Briefly, confluent BRECs were detached with trypsin, which was subsequently neutralized with soybean trypsin inhibitor (Sigma), washed in serum-free Dulbecco modified Eagle medium (DMEM), fluorescently labeled with 2′,7′-bis-(2-carboxyethyl)-5-(and-6)-carboxyfluorescein (BCECF; Invitrogen, Carlsbad, CA), and incubated in the precoated wells (30 000 cells/well) at 37°C in the absence or presence of competitors. Thereafter, the wells were washed, and the number of adherent cells was quantified as the percentage of total cells added with a fluorescence reader (Biotek). Adhesion to BSA-coated wells was used as blank and was subtracted to calculate specific adhesion.
Migration assay
Migration of BRECs was tested using Transwell membranes (8-μm pore size and 6.5-mm diameter; Corning Costar), coated with FN, FBG, or BSA (5 μg/mL), as previously described.15 Briefly, after gentle trypsinization, BRECs were resuspended in DMEM containing 0.2% (vol/vol) FCS, and 200 μL cell suspension at a density of 5 × 105/mL was added to the top chamber. Each assay was performed in quadruplicate. After incubating at 37°C for 6 hours, the number of migrated cells was quantified.15
In vitro angiogenesis assay
A previously described protocol15,30 was modified to use BRECs grown on Cytodex-3 microcarriers (Sigma) at 37°C with 5% CO2 in complete MCDB-131 medium. Approximately 100 confluent microcarriers coated with BRECs were added per well to sterile-filtered solutions containing 1.8 mg/mL FBG in phosphate-buffered saline (PBS), and α-thrombin (0.65 NIH U/mL) was immediately added for 30 minutes to induce fibrin clot formation. Then, 1 mL medium containing 200 KIU/mL aprotinin alone or together with stimuli or competitors was added, and the plates were incubated for an additional 48 hours at 37°C. The number of capillary-like sprouts was evaluated microscopically and was expressed as sprouts per microcarrier.
Hypoxia-induced retinal vascularization (ROP model)
The hypoxia-induced retinal vascularization model, or retinopathy of prematurity (ROP) model, was described by Smith et al31 and was used in our previous studies.9,32 Briefly, 7-day-old (p7) C57BL/6J mice were exposed to 75% oxygen for 5 days in an incubator (BioSpherix, Redfield, NY) with their nursing mothers. At p12, mice were returned to room air. From p14 until p17, mice were given intraperitoneal injections of α-defensin (HNP-1), blocking mAb to α5β1-integrin, β-defensin (each 30 μg/mouse) or buffer alone once daily (5 mice/group). Alternatively, p14 mice were anesthetized, and 5 μg α-defensin (HNP-1) in 2.5 μL PBS or the same volume of buffer alone was injected subconjunctivally into the superotemporal space of the right eye with a 30-gauge syringe (Hamilton, Reno, NV). Subconjunctival injection has previously been shown to be an effective method to deliver substances to the retina.33,34 Mice were killed at p17, and eyes were processed for retinal fluorescein angiography quantification of preretinal neovascular nuclei, and immunohistochemistry, as described.9,31,32 To quantify retinal neovascularization, 6-μm paraffin-embedded sections were stained with periodic acid-Schiff (PAS) and hematoxylin, and 10 intact sections of equal length, each 18 μm apart, were evaluated. All retinal vascular cell nuclei anterior to the internal limiting membrane were counted in each section by an observer blinded to the protocol. The mean of the 10 counted sections represented the average neovascular nuclei per section per eye. No neovascular cell nuclei anterior to the internal limiting membrane were observed in normal “room air” mice.9,31,32
Quantification of vascular permeability in the retina of p14 mice subjected to the ROP model was performed by measuring the leakage of Evans blue dye, according to a previously described protocol.35
Immunohistochemistry
Vertical cryostat sections were used for β1-integrin, FN, and platelet endothelial cell adhesion molecule-1 (PECAM-1) immunohistochemistry. Six-micrometer sections were prepared from the eyes of p17 mice with or without exposure to hyperoxia. The sections were collected on slides and were fixed in ice-cold acetone for 10 minutes. After blocking and subsequent washing, sections were incubated with combinations of the following antibodies: hamster anti–mouse β1 (Biolegend, San Diego, CA), rabbit anti-FN (Sigma), or biotin-conjugated rat anti–mouse PECAM-1 (PharMingen Hamburg, Germany). To detect antibodies bound to β1-integrin and FN, goat anti–hamster FITC and goat anti–rabbit tetramethylrhodamine isothiocyanate (TRITC), respectively, were used. To detect antibody bound to PECAM-1, extravidin-TRITC, or streptavidin-FITC were used. Coverslipped slides were examined using a fluorescence microscope (Zeiss, Oberkochen, Germany), using a Plan-Neofluar (Zeiss) 20 ×/0.5 numeric aperture (NA) objective, and photographs were collected with an ORCA ER camera (Hamamatsu, Hamamatsu City, Japan). Images were acquired and analyzed with Metamorph software (Molecular Devices, Downingtown, PA).
Apoptosis assay
Apoptosis in retinal cells was detected by terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL) using a cell apoptosis detection kit (Roche, Mannheim, Germany). Six-micrometer cryostat sections were obtained at p17 from the eyes of 4 mice treated with α-defensin and 4 buffer-treated mice. All sections included the optic nerve. Sections were fixed in 4% paraformaldehyde in PBS for 20 minutes at room temperature, rinsed 3 times in PBS and treated with 0.1% Triton X-100/0.1% sodium citrate at 4°C for 2 minutes, rinsed 3 times in PBS and incubated with TUNEL reaction mixture (50 mL/slide) in a humidified chamber at 37°C for 1 hour, washed 3 times in PBS and incubated with 50 mL Converter-AP in a humidified chamber at 37°C for 30 minutes, rinsed 3 times with PBS and exposed to nitroblue tetrazolium chloride/5-bromo-4-chloro-3′-indoyl-phosphatase (NBT/BCIP) substrate solution for color development in a dark chamber at room temperature for 10 minutes, and washed in PBS. As a negative control, 50 mL label solution without terminal transferase was substituted for TUNEL reaction mixture. Sections were observed under a light microscope, and the number of apoptotic cells was quantified in 15 sections from each eye.36
Retina whole-mounts
Eyes from α-defensin–treated and control mice were fixed in 4% paraformaldehyde for 2 hours, and the retinas were carefully removed. After the retinas were rinsed twice with PBS for 20 minutes, they were blocked with 1% BSA for 30 minutes and washed once more. Thereafter, they were allowed to incubate overnight with Griffonia simplicifolia isolectin (Sigma) at 4°C. Retinas were then washed and fixed once more in paraformaldehyde overnight, flat-mounted, and coverslipped. Photographs were obtained with a fluorescence microscope (Zeiss), using a Plan-Neofluar (Zeiss) 5 ×/0.15 NA (Figure 5), 10 ×/0.3 NA (Figure 6C, E, G), 20 ×/0.5 NA (Figure 6F), or 40 ×/0.75 NA (Figure 6A, B, D), objective lens, or a Plan-Apochromat (Zeiss) 63 ×/1.4 oil objective lens, as described in “Immunohistochemistry.”
Figure 5.

Inhibition of retinal angiogenesis by α-defensins. After 5 days of hyperoxia, mice were brought back to room air on p12 and were injected intraperitoneally or subconjunctivally with different treatments at different time points. (A) Typical photographs from lectin staining of retina whole-mount from a mouse that was treated with buffer alone or a mouse treated with synthetic α-defensin (HNP-1) intraperitoneally from p14 to p16 are shown. Arrowheads indicate the neovascular tufts. Neovascular regions are far more extensive in retinas from buffer-treated mice than in retinas from α-defensin–treated mice. (B) Retinal neovascularization was quantified on p17, as described in “Materials and methods.” Mice were treated from p14 until p16 once daily intraperitoneally with buffer, synthetic α-defensin (30 μg/mouse), blocking antibody to α5-integrin (30 μg/mouse), combination α-defensin and antibody to α5-integrin (each 30 μg/mouse), or β-defensin (30 μg/mouse). Retinal neovascularization is presented as percentage of control, defined as neovascularization in the presence of buffer only. Data are mean ± SD (n = 5 mice). (C) Retinal neovascularization was quantified on p17. Mice were treated on p14 with a subconjuctival injection into one eye of buffer or synthetic α-defensin (5 μg/eye). Retinal neovascularization is presented as percentage of control, defined as neovascularization in the presence of buffer only. Data are mean ± SD (n = 5 mice). * P < .05 compared with buffer; ns indicates not significant. (D) Induction of apoptosis by α-defensins in the ROP. Apoptosis in the retinas of 4 mice treated with buffer or 4 mice treated with α-defensin (30 μg/mouse) intraperitoneally from p14 until p17 was quantified. Apoptosis was determined by TUNEL by counting 15 sections per eye and is shown as number of apoptotic nuclei per section. *P < .05 compared with buffer.
Figure 6.

α-Defensins in human diabetic retinas. Ocular tissue from 5 patients with diabetes mellitus and 5 cases of traumatic injury in persons without diabetes mellitus were stained for α-defensins. Sections were stained with a rabbit antiserum that recognizes HNP1-3 or serum from unimmunized rabbits at the same dilution as a control. Normal-appearing regions of the retina in samples from patients with diabetes mellitus stained for α-defensin (A-C). In contrast, α-defensins were not detected in diabetic retinas when the normal rabbit serum control was used as first antibody (D). Little or no expression of α-defensins was noted in proliferative retinal vessels (E) or in proliferative vessels at the rubeotic membrane (F). α-Defensins were not detected in retinal vessels from eyes removed because of trauma (G). Hematoxylin and eosin staining identified neutrophils (H).
Immunohistochemistry for α-defensins in human retinas
Five cases of ocular globes from patients with diabetes mellitus and 5 cases of eyes removed because of trauma from patients without diabetes mellitus were retrieved from the archives of the Department of Ophthalmic Pathology, Scheie Eye Institute, University of Pennsylvania. Sections were previously fixed in 10% buffered formalin, paraffin embedded, cut in 5-μm sections, and mounted on microprobe slides. For immunohistochemistry, a modification of the streptavidin-biotin peroxidase method was performed using the Vectastain ABC kit (Vector Laboratories, Burlingame, CA) and capillary gap technology, as previously described by us.37,38 Briefly, tissue sections were blocked with 10% goat serum after blocking of endogenous peroxidase by 2.2% H2O2 in methanol, incubated with rabbit antiserum that recognizes HNP-1 to -3 but not HNP-4 to -6 (kindly provided by Dr Tomas Ganz, University of Los Angeles at California) at a dilution of 1:5000 as the primary antibody or serum from unimmunized rabbits at the same dilution as a control. Sections were incubated with biotinylated goat anti–rabbit immunoglobulin G (IgG) 1:200 as the secondary antibody followed by peroxidase-coupled streptavidin (DAKO) and were developed by adding 0.05% (vol/vol) 3,3′-diaminobenzidine solution (DAB) (Sigma, St Louis, MO) and 0.03% H2O2. Image acquisition and analysis was performed as described in “Immunohistochemistry.”
Statistical analysis
Data were compared using the Student t test and analysis of variance (ANOVA) with Bonferroni adjustment as appropriate; P less than .05 was regarded as significant.
Results
Inhibition of VEGF-induced retinal endothelial cell proliferation and permeability by α-defensins
VEGF-stimulated increase in endothelial cell proliferation and permeability plays a crucial role during pathologic retinal neovascularization. We therefore examined the influence of α-defensins on VEGF-stimulated BREC proliferation and permeability. VEGF-stimulated BREC proliferation was inhibited in the presence of synthetic α-defensin (HNP-1) (Figure 1) and natural α-defensins (not shown) in a dose-dependent manner. Early (1-hour) and long-term (24-hour) stimulating effects of VEGF on BREC permeability were also reduced by α-defensins and synthetic HNP-1 (Figure 2). VEGF-induced proliferation and permeability of BRECs were unaffected by identical concentrations of β-defensin (Figures 1, 2).
Figure 1.

Influence of α-defensins on BREC proliferation. BRECs were incubated in the absence (open bar) or presence (filled bars) of 10 ng/mL VEGF alone (–) or in the presence of increasing concentrations of synthetic α-defensin (HNP-1) or β-defensin (10 μM), as indicated. Proliferation of human umbilical vein endothelial cells (HUVECs) is expressed as a percentage of control, defined as cell proliferation in the absence of any stimulus or competitor. Data are mean ± SD (n = 3) of 1 experiment typical of 3 separate experiments so performed.
Figure 2.

Influence of α-defensins on BREC permeability. BRECs were incubated for 1 hour or for 24 hours in the absence (100% control) or presence of VEGF (filled bars; 10 ng/mL), VEGF +α-defensins (10 μM; open bars), VEGF + synthetic HNP-1 (10 μM; gray bars), or VEGF + β-defensin (10 μM; dotted bars). FITC-conjugated dextran (1 mg/mL) was then added to the upper chambers, and fluorescence in the lower chamber was measured with a fluorescence reader. Permeability is expressed as percentage of control, defined as permeability in the absence of any stimulus or competitor. Data are mean ± SD (n = 3) of 1 experiment typical of 3 separate experiments so performed.
Inhibition of α5β1-integrin–dependent retinal endothelial cell adhesion, migration, and capillary sprouting by α-defensins
Recently, we demonstrated that α-defensins blocked FN–α5β1-integrin interactions.15 In accordance with these findings, α5β1-integrin–dependent adhesion of BRECs to FN was inhibited by α-defensin in a dose-dependent manner. In contrast, αv-integrin–dependent adhesion of BRECs to FBG was not affected by α-defensin (Figure 3A). α-Defensin also specifically inhibited VEGF-induced migration of BRECs toward FN (Figure 3B) in a dose-dependent manner (not shown), whereas migration toward FBG was unaffected. The effect of purified α-defensins and synthetic HNP-1 on BREC adhesion and migration were comparable. In contrast, β-defensin did not inhibit BREC adhesion or migration (Figure 3). Thus, α-defensins block retinal endothelial cell adhesion and migration in an FN–α5β1-integrin–specific manner.
Figure 3.

Influence of α-defensins on BREC adhesion, migration, and capillary sprout formation. (A) Adhesion of BRECs to immobilized FN or FBG (each at 5 μg/mL) is shown in the absence (–; filled bars) or presence of the blocking antibody against β1-integrin, 6S6 (for FN), or the blocking antibody against αvβ3-integrin, LM609 (for FBG) (hatched bars; antibody concentration, 20 μg/mL), or in the presence of increasing concentrations of synthetic HNP-1 (gray bars), or in the presence of β-defensin (dotted bars; 10 μM). Cell adhesion is expressed as percentage of adherent cells to total added cells. Adhesion was performed in triplicate, and data are shown as the mean ± SD of a typical experiment; similar results were obtained in at least 3 separate experiments. (B) VEGF-stimulated migration of BRECs toward FN or FBG (each 5 μg/mL) is shown in the absence (filled bars) or presence of the blocking antibody against β1-integrin, 6S6 (for FN), or the blocking antibody against αvβ3-integrin, LM609 (for FBG) (hatched bars; antibody concentration, 20 μg/mL), in the presence of α-defensins (open bars; 5 μM), in the presence of synthetic HNP-1 (gray bars; 5 μM), or in the presence of β-defensin (dotted bars; 10 μM). Cell migration is expressed as percentage of control, which is represented as cell migration in the absence of any stimulus or competitor. Data are mean ± SD (n = 3) of a typical experiment; similar results were obtained in 3 separate experiments. (C) BRECs were incubated for 48 hours in the absence (–; open bar) or presence of bFGF (5 ng/mL; gray bars) or of VEGF (5 ng/mL; filled bars) alone or in the presence of α-defensins (10 μM), as indicated. Capillary-like sprout formation is expressed as percentage of control, defined as sprouts per microcarrier in the absence of any stimulus or competitor. Data are mean ± SD (n = 3) of a typical experiment; similar results were obtained in 3 separate experiments.
We next examined the role of α-defensins in sprouting angiogenesis exhibited by BRECs cultivated on microcarrier beads. VEGF- and bFGF-stimulated capillary-like sprout formation in 3-dimensional fibrin gels was reduced by α-defensins (Figure 3C). The extent of inhibition was also dose dependent and, at its peak, was comparable to the inhibitory effect of blocking antibody to β1-integrins (not shown). Again, no inhibition was observed with β-defensins (not shown).
Expression of FN and α5β1-integrin during pathologic retinal angiogenesis
To study the expression of FN and α5β1-integrin in pathologic retinal neovascularization, we compared the patterns of the retinal vasculature in control mice and mice that underwent the ROP model of vascularization. By immunofluorescence, FN and α5β1-integrin were predominantly expressed in the vasculature in which each colocalized with the endothelial cell marker PECAM-1 and with each other (Figure 4). β1-Integrin and FN localized primarily to the vascular areas between the inner and outer nuclear layers. In mice subjected to the ROP model, β1-integrin and FN were also found in proliferating vessels above the internal limiting membrane. Staining for both proteins was strongly enhanced in the retinal neovasculature compared with that for control mice, indicating that both are up-regulated in proliferative retinopathy (Figure 4).
Figure 4.
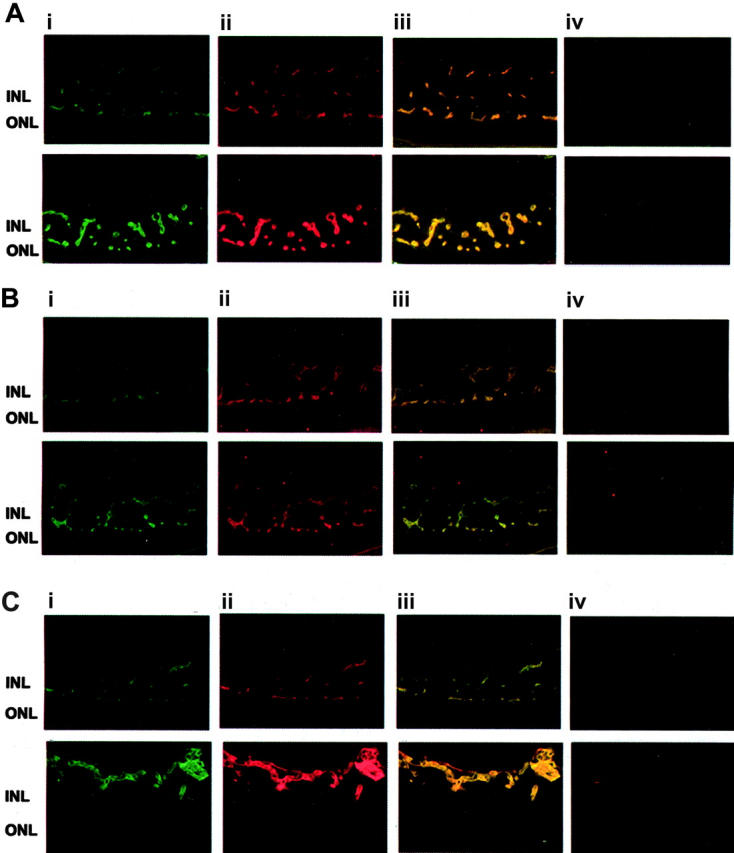
Up-regulation of β1-integrin and FN in the ROP model. (A) Typical fluorescence staining patterns for β1-integrin (i; green) and FN (ii; red) and the overlay picture (iii) and appropriate negative control (first antibody omitted) (iv) of retinas from control mice (top row) or mice subjected to the ROP model (bottom row) are shown. (B) Typical fluorescence staining patterns for β1-integrin (i; green) and PECAM-1 (ii; red) and the overlay picture (iii) and appropriate negative control (first antibody omitted) (iv) of retinas from control mice (top row) or mice subjected to the ROP model (bottom row) are shown. (C) Typical fluorescence staining patterns for PECAM-1 (i; green) and FN (ii; red) and the overlay picture (iii) and appropriate negative control (first antibody omitted) (iv) of retinas from control mice (top row) or mice that were subjected to the ROP model (bottom row) are shown. INL indicates inner nuclear layer; ONL, outer nuclear layer.
Inhibition of retinal angiogenesis in vivo by α-defensins
We then examined the role of α-defensins in pathologic retinal neovascularization in vivo in the ROP model in mice. Mice were given intraperitoneal injections of synthetic α-defensin (HNP-1; 30 μg), blocking mAb against mouse α5-integrin (30 μg), a combination thereof, β-defensin (30 μg), or the same volume of buffer alone once daily from p14 to p16. We chose p14 to begin treatment because this was when the retinal vessels started to invade the vitreous cavity. Immunohistochemical analysis of retinal whole-mounts demonstrated a significant reduction in the extent of neovascularization in mice given α-defensin (Figure 5A). Specifically, p17 retinas of control-treated animals had prominent neovascular tufts extending into the vitreous body. In contrast, p17 retinas from mice treated with α-defensins demonstrated a marked reduction in the number of neovascular tufts (Figure 5A). Angiogenesis was reduced by 45% in mice given α-defensin compared with control mice (Figure 4B). Intraperitoneal injection of the blocking mAb against mouse α5-integrin inhibited retinal neovascularization to the same extent (45%-50% inhibition; Figure 5B). When this antibody was combined with α-defensin, no additive effect was observed. We then examined the effect of α-defensin given locally. A subconjunctival injection of α-defensins on p14 inhibited retinal neovascularization by 60% compared with mice that received similar injections of buffer (Figure 5C). In contrast, β-defensin had no effect on retinal angiogenesis, whether given systemically (Figure 5B) or locally (not shown).
We then asked whether the inhibition of VEGF-induced proliferation of retinal endothelial cells by α-defensin was associated with the induction of apoptosis. As demonstrated in Figure 5D, increased numbers of apoptotic nuclei were found in retinas from mice treated with α-defensin, indicating that the proapoptotic/antiproliferative and the antiadhesive effects of α-defensins on retinal endothelial cells that had been observed in vitro might have contributed to the inhibitory effect of α-defensins on retinal neovascularization in vivo. In addition, α-defensin inhibited the increase in retinal vascular permeability that develops during the course of the ROP model. Leakage of Evans blue dye in retinas of p14 mice was reduced by 42% in animals treated with α-defensin, whereas β-defensins had no effect (not shown). Taken together, these data demonstrate that the administration of α-defensin inhibits pathologic retinal neovascularization and hyperpermeability in vivo.
α-Defensins in human diabetic retinas
These data demonstrated that α-defensins interfere with biologic functions of retinal endothelial cells and inhibit retinal neovascularization in vivo in the mouse model of ROP. As in the ROP model, exuberant hypoxia-driven neovascularization was also the most prominent feature in the pathophysiology of proliferative diabetic retinopathy. Thus, to begin to explore the role that α-defensins may play in regulating pathologic angiogenesis in humans, we studied their expression and distribution in human diabetic retinas. Ocular globes from 5 patients with diabetes mellitus and 5 cases of eyes removed as a result of trauma from patients without diabetes mellitus were immunostained for α-defensins. In all patients with clinical history of diabetes mellitus, a subpopulation of retinal vessels showed expression of α-defensins (Figure 6A-C). α-Defensin was detected in neutrophils (Figure 6A-B, solid arrows), as expected, but was also diffusely distributed within vascular walls in association with and beneath the endothelium (Figure 6A-B, dashed arrows). Neutrophils were recognized by their size and morphology in the immunohistochemical sections, and identification was confirmed in hematoxylin and eosin–stained sections (Figure 6H). α-Defensin was not detected in retinal vessels in the nondiabetic controls (traumatic eyes) and in sections from diabetic eyes incubated with normal rabbit serum as a control (Figure 6G, D, respectively). Interestingly, only retinal vessels from relatively normal-appearing sections of the diabetic retinas stained positive for α-defensins, whereas proliferative retinal vessels and proliferative vessels of the rubeotic membrane were typically negative (Figure 6E-F).
Discussion
Pathologic retinal neovascularization is the most prominent feature of retinopathy of prematurity and proliferative diabetic retinopathy, the major causes of neonatal and adult blindness, respectively.1 The development of pathologic retinal neovascularization depends on integrin-mediated adhesive contacts of the endothelial cells with the extracellular matrix and hypoxia-induced VEGF, which regulate endothelial cell functions.1-4 In the present study, α-defensins were found to inhibit aspects of retinal endothelial cell function that play crucial roles in retinal neovascularization, such as VEGF-dependent migration, proliferation, and increased vascular permeability. We also show that α-defensins block pathologic retinal neovascularization in vivo, thereby providing a platform for developing a novel and potentially feasible strategy to treat proliferative retinopathies.
We recently reported that α-defensins, the most abundant proteins released from activated neutrophils, interfere with several endothelial cell functions that are involved in angiogenesis. α-Defensins function as atypical ligands of α5β1-integrin, transforming the interaction with one of its prominent ligands, FN, to one that does not promote cell adhesion.15 Consistent with these findings, we found that α-defensins specifically blocked α5β1-mediated adhesion of retinal endothelial cells to FN and their migration toward FN under control conditions and on stimulation by VEGF, whereas adhesion and migration in response to other extracellular matrix (ECM) proteins, such as fibrinogen/fibrin, were unaffected, excluding a nonspecific cytotoxic effect. Moreover, α-defensins blocked VEGF-induced proliferation and VEGF- and bFGF-induced capillary sprout formation of retinal endothelial cells in 3-dimensional fibrin gels. Interference with the interaction between FN and α5β1 by α-defensins may account for their inhibitory effect on retinal endothelial cell proliferation because this ligand-receptor pair has been shown convincingly to regulate retinal endothelial cell proliferation.8 These effects of α-defensins on retinal endothelial cell function cannot be attributed solely to a nonspecific effect of their cationic charge or their hydrophobic properties because comparable concentrations of the more highly cationic and hydrophobic β-defensin, HBD-2, had no effect on either process.
α-Defensins also inhibited early (1-hour) and long-term (24-hour) stimulation of retinal endothelial cell permeability by VEGF, a prerequisite step in the induction of neovascularization. Moreover, we found reduced vascular permeability in retinas of α-defensin–treated mice in the ROP model, in which a VEGF-induced increase in permeability is a prominent feature. Our data provide the first report, to our knowledge, of the effect of α-defensins on endothelial cell permeability. However, additional work is required to understand its underlying mechanism. Given that previous reports demonstrated the induction of permeability in epithelial cells by α-defensins, especially during inflammatory responses in the lung,39 it is likely that concentration, timing, ambient oxygen tension, and perhaps cell specificity may account for the distinct effects of α-defensins in the 2 experimental models.
Our in vitro findings were strengthened by the observation that systemic administration of α-defensins on p14 to p16 blocked pathologic neovascularization in a mouse model of hypoxia-induced proliferative retinopathy by 45% relative to buffer-treated mice. Subconjunctival application of α-defensin on p14 significantly inhibited retinal neovascularization by 60%. Thus, the local application of α-defensin represents one possible strategy to improve clinical efficacy. To evaluate α-defensins as potential therapeutic agents, more detailed pharmacokinetic studies, including comparison of different routes of administration, doses, and times of administration, are needed. Combining α-defensins with VEGF-inhibitory agents may provide additive or synergistic benefits in the treatment of hypoxia-induced retinopathies.
Our data suggest that the inhibition of neovascularization by α-defensins in vivo is mediated predominantly by the disruption of α5β1–FN interactions. Enhanced staining and colocalization of FN and the β1-integrin are seen in retinas from mice subjected to the ROP model. The inhibitory effect of α-defensins and neutralizing antibody to α5β1-integrin was indistinguishable. Moreover, when both inhibitors were combined, no significant additive effect was observed. However, α-defensins in vivo may regulate vascular proliferation/apoptosis through α5β1-integrin–independent pathways.
Expression of β1-integrins and FN has been shown previously to be up-regulated in hypoxia-driven diabetic proliferative retinopathy,40-42 and their interaction is important for retinal endothelial cell proliferation.8 Here we demonstrate that these 2 components are also up-regulated in the mouse model of hypoxia-induced retinopathy. Inhibition of angiogenesis in proliferative retinopathy by α-defensins and α5β1-integrin–neutralizing antibody also underscores the involvement of the α5β1–FN system in this pathologic process. Our data provide the first direct evidence that α5β1-integrin antagonism may retard pathologic retinal neovascularization. FN–α5β1-integrin interaction undoubtedly promotes angiogenesis, as evidenced by inhibition studies,7 and genetic ablation of either component leads to embryonic lethality with major vascular defects.43,44 However, only the effects of αv-integrin antagonists in pathologic retinal neovascularization have been studied to date. Recent data demonstrating that mice lacking αv-integrins exhibit extensive angiogenesis6 complicate our views concerning the role of these integrins in physiologic and pathologic angiogenesis in the retina and in other organs as the mechanism by which pharmacologic inhibition of these integrins prevents angiogenesis.
The inhibitory effect of α-defensins seems to be equipotent to the VEGF-inhibiting agents that have been used experimentally in the same mouse model. VEGF-neutralizing chimeric proteins consisting of the extracellular domain of human or mouse VEGF receptors injected locally inhibited retinal neovascularization by 47% and 37%, respectively,10 whereas systemic and intraocular injection of VEGF-neutralizing antibody inhibited neovascularization by 18% and 46%, respectively.45 In preclinical and phase 1A clinical study, 80% inhibition of neovascularization in the ROP model was achieved using an optimized anti-VEGF pegylated aptamer.46
Consistent with our in vitro and in vivo data, we found that α-defensins were present in human diabetic retinas, whereas they were minimally expressed or absent in nondiabetic control retinas from traumatized eyes. α-Defensins were most prevalent in nonproliferative regions of the diabetic retinas, whereas there was no staining of proliferative vessels. These results are preliminary because of the limited number of cases examined, and further studies are required to explore the localization and distribution of a potential endogenous antiangiogenic substance in human diabetic retinopathy. However, these findings do represent the first demonstration that α-defensins are present in diabetic retinas. Their presence in normal-appearing vessels and their absence from proliferative vessels are consistent with their antiangiogenic function activities described here. Thus, α-defensins may function as endogenous angiogenesis antagonists that counteract and limit the proangiogenic activity of inflammatory cells in proliferative retinopathies. The concentrations of endogenous α-defensins achieved in the retina may not suffice to completely counterbalance the proangiogenic activity of these cells, so that the net effect of inflammatory cells may be proangiogenic,2,11,12 depending on the extent of neutrophil activation and secretion. However, pharmacologic administration of α-defensins, as performed in the present study, may prove a promising approach to control proliferative retinopathies. Another advantage of α-defensins is that they are endogenous molecules; hence, fewer side effects would be expected after their pharmacologic application. Lastly, it may be possible to administer α-defensins intraocularly, further limiting potential systemic side effects and improving efficacy; our studies provide the first evidence in this direction.
It has recently been reported that β-defensin is proangiogenic.47 The proangiogenic effect of β-defensins was observed in tumor angiogenesis, whereas α-defensins were tested in retinal angiogenesis in the present study. The effect of β-defensins is mediated by the chemotactic recruitment of dendritic cell precursors to tumors where they may promote neovascularization, in part by undergoing endothelial-like transdifferentiation, whereas α-defensins are released from activated neutrophils The contribution of this subtype of dendritic cells in retinal angiogenesis (if any) is unknown. In addition, we have shown that the antiangiogenic effect of α-defensins is predominantly mediated by the disruption of α5β1–FN interactions, which is not affected by β-defensins. Thus, the proangiogenic effect of β-defensins is clearly distinct from the antiangiogenic effect of α-defensins. Nevertheless, it is an interesting observation that 2 similar small cationic antimicrobial peptides, α-defensins and β-defensins, have opposite actions on angiogenesis. α-Defensins may provide an endogenous balance to the effect of β-defensins, and vice versa, in different biologic settings associated with angiogenesis, depending on the extent to which recruitment of dendritic cells or neutrophils predominates.
α-Defensins are found in extraordinarily high concentrations in neutrophil granules and are secreted locally in high concentrations when these cells are activated on severe bacterial infection.15 Our data indicate that α-defensins may act as endogenous antiangiogenic regulators modulating the balance between proangiogenesis and antiangiogenesis in several pathophysiologic processes, linking inflammation and angiogenesis. For example, α-defensins are abundant in the atherosclerotic vessel wall, where they may impede the development of a functional vasa vasorum and thereby modulate plaque stability.37 In addition, the presence of α-defensins in human tumors48 may help control tumor angiogenesis and, hence, tumor growth. Taken together, our findings demonstrate that α-defensins inhibit retinal endothelial cell functions and are potent regulators of pathologic retinal angiogenesis in vivo. α-Defensins may thus provide a platform for developing a novel class of antiangiogenesis compounds to treat the proliferative retinopathy that complicates diabetes and prematurity.
Acknowledgments
We thank Athanasios Athanasopoulos, Kaimei Song, and Uwe Schubert for their skillful technical assistance. We also thank M. Kruhlak for help with microscopy. M.E. thanks Prof H. Kaufmann (Department for Ophthalmology, Justus-Liebig-University, Giessen, Germany) for his continuing support.
Prepublished online as Blood First Edition Paper, August 25, 2005; DOI 10.1182/blood-2005-03-0889.
Supported in part by the Intramural Research Program of the National Institutes of Health (NIH), National Cancer Institute (T.C.), by NIH grant PO1 HL076406 (D.C.), by the University of Pennsylvania Research Foundation (K.H.B), by Phillip Morris USA, and by Phillip Morris International (K.H.B).
M.E., K.B., F.F., Y.B., K.T.P., H.P.H., and T.C performed the research. M.E., K.B., D.B.C., and T.C. wrote the manuscript. D.B.C., H.P.H., and T.C. participated in designing the research. J.L. and W.L. provided vital reagents.
M.E. and K.B. contributed equally to this study.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 U.S.C. section 1734.
References
- 1.Ishida S, Usui T, Yamashiro K, et al. VEGF164-mediated inflammation is required for pathological, but not physiological, ischemia-induced retinal neovascularization. J Exp Med. 2003;198: 483-489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stone J, Chan-Ling T, Pe'er T, Itin A, Gnessin H, Keshet E. Roles of vascular endothelial growth factor and astrocyte degeneration in the genesis of retinopathy of prematurity. Invest Ophthalmol Vis Sci. 1996;37: 290-299. [PubMed] [Google Scholar]
- 3.Campochiaro PA, Hackett SF. Ocular neovascularization: a valuable model system. Oncogene. 2003;22: 6537-6548. [DOI] [PubMed] [Google Scholar]
- 4.Eliceiri BP, Cheresh DA. Adhesion events in angiogenesis. Curr Opin Cell Biol. 2001;13: 563-568. [DOI] [PubMed] [Google Scholar]
- 5.Giancotti FG, Ruoslahti E. Integrin signaling. Science. 1999;285: 1028-1032. [DOI] [PubMed] [Google Scholar]
- 6.Hynes RO. A reevaluation of integrins as regulators of angiogenesis. Nat Med. 2002;8: 918-921. [DOI] [PubMed] [Google Scholar]
- 7.Kim S, Bell K, Mousa SA, Varner JA. Regulation of angiogenesis in vivo by ligation of integrin α5β1 with the central cell-binding domain of fibronectin. Am J Pathol. 2000;156: 1345-1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wilson SH, Ljubimov AV, Morla AO, et al. Fibronectin fragments promote human retinal endothelial cell adhesion and proliferation and ERK activation through α5β1 integrin and PI 3-kinase. Invest Ophthalmol Vis Sci. 2003;44: 1704-1715. [DOI] [PubMed] [Google Scholar]
- 9.Hammes HP, Brownlee M, Jonczyk A, Sutter A, Preissner KT. Subcutaneous injection of a cyclic peptide antagonist of vitronectin receptor-type integrins inhibits retinal neovascularization. Nat Med. 1996;2: 529-533. [DOI] [PubMed] [Google Scholar]
- 10.Aiello LP, Pierce EA, Foley ED, et al. Suppression of retinal neovascularozation in vivo by inhibition of vascular endothelial growth factor (VEGF) using soluble VEGF-receptor chimeric proteins. Proc Natl Acad Sci U S A. 1995;92: 10457-10461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Joussen AM, Murata T, Tsujikawa A, Kirchhof B, Bursell SE, Adamis AP. Leukocyte-mediated endothelial cell injury and death in the diabetic retina. Am J Pathol. 2001;158: 147-152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ishida S, Yamashiro K, Usui T, et al. Leukocytes mediate retinal vascular remodeling during development and vaso-obliteration in disease. Nat Med. 2003;9: 781-788. [DOI] [PubMed] [Google Scholar]
- 13.Miyamoto K, Khosrof S, Bursell S-E, et al. Vascular endothelial growth factor-induced retinal vascular permeability is mediated by intercellular adhesion molecule-1 (ICAM-1). Am J Pathol. 2000; 156: 1733-1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Scapini P, Nesi L, Morini M, et al. Generation of biologically active angiostatin kringle 1-3 by activated human neutrophils. J Immunol. 2002;168: 5798-5804. [DOI] [PubMed] [Google Scholar]
- 15.Chavakis T, Cines DB, Rhee JS, et al. Regulation of neovascularization by human neutrophil peptides (alpha-defensins): a link between inflammation and angiogenesis. FASEB J. 2004;18: 1306-1308. [DOI] [PubMed] [Google Scholar]
- 16.Ganz T. Antimicrobial polypeptides in host defense of the respiratory tract. J Clin Invest. 2002; 109: 693-697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kagan BL, Ganz T, Lehrer RI. Defensins: a family of antimicrobial and cytotoxic peptides. Toxicology. 1994;87: 131-149. [DOI] [PubMed] [Google Scholar]
- 18.Ganz T, Selsted ME, Szklarek D, et al. Natural peptide antibiotics of human neutrophils. J Clin Invest. 1985;76: 1427-1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Harwig SS, Ganz T, Lehrer RI. Neutrophil defensins: puification, characterization and antimicrobial testing. Methods Enzymol. 1994;236: 160-172. [DOI] [PubMed] [Google Scholar]
- 20.Panyutich AV, Panyutich EA, Krapivin VA, Baturevich EA, Ganz T. Plasma defensin concentrations are elevated in patients with sepsis or bacterial meningitis. J Lab Clin Med. 1993;122: 202-207. [PubMed] [Google Scholar]
- 21.Bdeir K, Cane W, Canziani G, et al. Defensin promotes the binding of lipoprotein(a) to vascular matrix. Blood. 1999;94: 2007-2019. [PubMed] [Google Scholar]
- 22.Higazi AA, Ganz T, Kariko K, Cines DB. Defensin modulates tissue-plasminogen activator and plasminogen binding to fibrin and endothelial cells. J Biol Chem. 1996;271: 17650-17655. [DOI] [PubMed] [Google Scholar]
- 23.Soong LB, Ganz T, Ellison A, Caughey GH. Purification and characterization of defensins from cystic fibrosis sputum. Inflamm. Res. 1997;46: 98-102. [DOI] [PubMed] [Google Scholar]
- 24.Wu Z, Prahl A, Powell R, Ericksen B, Lubkowski J, Lu W. From pro defensins to defensins: synthesis and characterization of human neutrophil pro alpha-defensin-1 and its mature domain. J Pept Res. 2003;62: 53-62. [DOI] [PubMed] [Google Scholar]
- 25.Hoover DM, Rajashankar KR, Blumenthal R, et al. The structure of human beta-defensin-2 shows evidence of higher order oligomerization. J Biol Chem. 2000;275: 32911-32918. [DOI] [PubMed] [Google Scholar]
- 26.Schultz JF, Armant DR. Beta 1- and beta 3-class integrins mediate fibronectin binding activity at the surface of developing mouse periimplantation blastocysts: regulation by ligand-induced mobilization of stored receptor. J Biol Chem. 1995;270: 11522-11531. [DOI] [PubMed] [Google Scholar]
- 27.Chavakis T, Kanse SM, Lupu F, et al. Different mechanisms define the antiadhesive function of high molecular weight kininogen in integrin- and urokinase receptor-dependent interactions. Blood. 2000;96: 514-522. [PubMed] [Google Scholar]
- 28.Behzadian MA, Windsor LJ, Ghaly N, Liou G, Tsai NT, Caldwell RB. VEGF-induced paracellular permeability in cultured endothelial cells involves urokinase and its receptor. FASEB J. 2003;17: 752-754. [DOI] [PubMed] [Google Scholar]
- 29.Chavakis T, Bierhaus A, Al-Fakhri N, et al. The pattern recognition receptor (RAGE) is a counter-receptor for leukocyte integrins: a novel pathway for inflammatory cell recruitment. J Exp Med. 2003;198: 1507-1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Koblizek TI, Weiss C, Yancopoulos GD, Deutsch U, Risau W. Angiopoietin-1 induces sprouting angiogenesis in vitro. Curr Biol. 1998;8: 529-532. [DOI] [PubMed] [Google Scholar]
- 31.Smith LE, Wesolowski E, McLellan A, et al. Oxygen-induced retinopathy in the mouse. Invest Ophthalmol Vis Sci. 1994;35: 101-111. [PubMed] [Google Scholar]
- 32.Rhee JS, Black M, Schubert U, et al. The functional role of blood platelet components in angiogenesis. Thromb Haemost. 2004;92: 394-402. [DOI] [PubMed] [Google Scholar]
- 33.Kim TW, Lindsey JD, Aihara M, Anthony TL, Weinreb RN. Intraocular distribution of 70-kDa dextran after subconjunctival injection in mice. Invest Ophthalmol Vis Sci. 2002;43: 1809-1816. [PubMed] [Google Scholar]
- 34.Escalona-Benz E, Jockovich ME, Murray TG, et al. Combretastatin A-4 prodrug in the treatment of a murine model of retinoblastoma. Invest Ophthalmol Vis Sci. 2005;46: 8-11. [DOI] [PubMed] [Google Scholar]
- 35.Brankin B, Campbell M, Canning P, Gardiner TA, Stitt AW. Endostatin modulates VEGF-mediated barrier dysfunction in the retinal microvascular endothelium. Exp Eye Res. 2005;81: 22-31. [DOI] [PubMed] [Google Scholar]
- 36.Alon T, Hemo I, Itin A, Pe'er J, Stone J, Keshet E. Vascular endothelial growth factor acts as a survival factor for newly formed retinal vessels and has implications for retinopathy of prematurity. Nat Med. 1995;1: 1024-1028. [DOI] [PubMed] [Google Scholar]
- 37.Barnathan ES, Raghunath PN, Tomaszewski JE, Ganz T, Cines DB, Higazi AA. Immunohistochemical localization of defensin in human coronary vessels. Am J Pathol. 1997;150: 1009-1020. [PMC free article] [PubMed] [Google Scholar]
- 38.Higazi AA, Lavi E, Bdeir K, et al. Defensin stimulates the binding of lipoprotein (a) to human vascular endothelial and smooth muscle cells. Blood. 1997;89: 4290-4298. [PubMed] [Google Scholar]
- 39.Zhang H, Porro G, Orzech N, Mullen B, Liu M, Slutsky AS. Neutrophil defensins mediate acute inflammatory response and lung dysfunction in dose-related fashion. Am J Physiol Lung Cell Mol Physiol. 2001;280: L947-L954. [DOI] [PubMed] [Google Scholar]
- 40.Roth T, Podesta F, Stepp MA, Boeri D, Lorenzi M. Integrin overexpression induced by high glucose and by human diabetes: potential pathway to cell dysfunction in diabetic microangiopathy. Proc Natl Acad Sci U S A. 1993;90: 9640-9644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Casaroli Marano RP, Preissner KT, Vilaro S. Fibronectin, laminin, vitronectin and their receptors at newly-formed capillaries in proliferative diabetic retinopathy. Exp Eye Res. 1995;60: 5-17. [DOI] [PubMed] [Google Scholar]
- 42.Roy S, Cagliero E, Lorenzi M. Fibronectin overexpression in retinal microvessels of patients with diabetes. Invest Ophthalmol Vis Sci. 1996;37: 258-266. [PubMed] [Google Scholar]
- 43.George EL, Georges-Labouesse EN, Patel-King RS, Rayburn H, Hynes RO. Defects in mesoderm, neural tube and vascular development in mouse embryos lacking fibronectin. Development. 1993;119: 1079-1091. [DOI] [PubMed] [Google Scholar]
- 44.Yang JT, Rayburn H, Hynes RO. Embryonic mesodermal defects in α5 integrin-deficient mice. Development. 1993;119: 1093-1105. [DOI] [PubMed] [Google Scholar]
- 45.Sone H, Kawakami Y, Segawa T, et al. Effects of intraocular or systemic administration of neutralizing antibody against vascular endothelial growth factor on the murine experimental model of retinopathy. Life Sci. 1999;65: 2573-2580. [DOI] [PubMed] [Google Scholar]
- 46.Eyetech Study Group. Preclinical and phase 1A clinical evaluation of an anti-VEGF pegylated aptamer (EYE001) for the treatment of exudative age-related macular degeneration. Retina. 2002; 22: 143-152. [DOI] [PubMed] [Google Scholar]
- 47.Conejo-Garcia JR, Benencia F, Courreges MC, et al. Tumor-infiltrating dendritic cell precursors recruited by a β-defensin contribute to vasculogenesis under the influence of VEGF-A. Nat Med. 2004;10: 950-958. [DOI] [PubMed] [Google Scholar]
- 48.Mizukawa N, Sugiyama K, Kamio M, et al. Immunohistochemical staining of human α-defensin-1 (HNP-1), in the submandibular glands of patients with oral carcinomas. Anticancer. Res. 2000;20: 1125-1127. [PubMed] [Google Scholar]