

Abstract
Macrophages play an essential role in defending against invading pathogens by migrating to the sites of infection, removing apoptotic cells, and secreting inflammatory cytokines. The molecular mechanisms whereby macrophages regulate these processes are poorly understood. Using bone marrow–derived macrophages (BMMs) deficient in the expression of p85α-subunit of class IA phosphatidylinositol 3 (PI-3) kinase, we demonstrate 50% reduction in proliferation in response to macrophage–colony-stimulating factor (M-CSF) as well as granulocyte macrophage–colony-stimulating factor (GM-CSF) compared with wild-type controls. Furthermore, p85α–/– BMMs demonstrate a significant reduction in migration in a wound-healing assay compared with wild-type controls. The reduction in migration due to p85α deficiency in BMMs is associated with reduced adhesion and directed migration on fibronectin and vascular cell adhesion molecule-1. In addition, deficiency of p85α in BMMs also results in defective phagocytosis of sheep red blood cells. Biochemically, loss of p85α in BMMs results in reduced activation of Akt and Rac, but not Erk (extracellular signal-related kinase) mitogen-activated protein (MAP) kinase. Taken together, our results provide genetic evidence for the importance of p85α in regulating both actin- and growth-based functions in macrophages, and provide a potential therapeutic target for the treatment of diseases involving macrophages, including inflammation.
Introduction
Macrophages play an essential role in defending against invading pathogens. Their function(s) include phagocytosis, release of cytokines, maintaining inflammation, and immune responses. Most of these functions require macrophages to migrate to the sites of invasion or tissue damage. Macrophage migration can be induced by chemokines released during inflammation, as well as by macrophage–colony-stimulating factor (M-CSF). Additionally, migration of macrophages can also be regulated by components of the extracellular matrix (ECM). In particular, the expression of vascular cell adhesion molecule-1 (VCAM-1), fibronectin (FN), collagen, and vitronectin on the surface of endothelial cells as well as ECM plays an important role in the migration of macrophages to sites of inflammation via their interaction with members of the β1 integrin family, including α4β1 and α5β1.1-3 Importantly, interfering with the function of β1 integrins impairs the ability of macrophages to be recruited to the sites of inflammation.4,5 Thus, although it is clear that soluble factors such as M-CSF and adhesion molecules such as β1 integrins play a significant role in regulating growth and migration of macrophages, the signaling pathways responsible for coordinating these processes downstream from these molecules are poorly defined.
Phosphatidylinositol 3 (PI-3) kinases are involved in regulating a number of cellular responses, including growth, survival, and migration.6,7 PI-3 kinases are a family of enzymes that phosphorylate phosphatidylinositol (PI) lipids at the 3′ position.7 They are divided into 3 classes, based on sequence similarity and biochemical properties. Among the PI-3 kinases, class I has been shown to regulate signals in response to receptor tyrosine kinase stimulation as well as G-protein–coupled receptor stimulation. Class IA PI-3 kinases consist of a catalytic subunit, p110 (α, β, δ), and an adaptor subunit, p85 (α or β). The p85 subunit binds to phosphorylated tyrosines via the Src homology-2 (SH-2) domains, leading to the recruitment and activation of the p110 subunits at the plasma membrane. In addition to the presence of SH2 domains, p85 also contains an SH3 domain, 2 proline-rich domains (PRDs), and a domain homologous to the breakpoint cluster region (BCR) gene product.8 These domains may be involved in targeting p85α to additional subcellular compartments and may result in the activation of distinct downstream pathways. To this end, the regulatory subunits of p85 (p55α, p50α, and p55γ) that do not contain these additional domains present in p85α appear to have distinct biologic activities in cells.9,10
M-CSF receptor plays a critical role in regulating M-CSF–induced functions in early as well as more mature monocytes and macrophages. Binding of M-CSF to its receptor results in the autophosphorylation of the M-CSF receptor on several intracellular tyrosine residues, thereby creating docking sites for various SH2- and SH3-containing signaling molecules. Several of these tyrosine sites have been mapped in the M-CSF receptor, including Tyr 697, Tyr 706, and Tyr 721 in the kinase insert region, Tyr 807 in the catalytic domain, and Tyr 559 in the juxtamembrane region. The p85α regulatory subunit of PI-3 kinase binds at Tyr 721,11 thereby bringing PI-3 kinase into proximity with its substrates in the plasma membrane. Although this appears to be one of the major mechanisms by which p85α regulates PI-3 kinase activation in response to M-CSF stimulation, recent studies have demonstrated additional mechanisms by which p85α may regulate the activation of PI-3 kinase in response to M-CSF stimulation.12 Thus, although it is clear that p85α plays a major role in the activation of PI-3 kinase in response to M-CSF stimulation, the physiologic role or roles of p85α in regulating M-CSF–induced macrophage functions is not known. To this end, studies have suggested specificity with regards to the function of various catalytic subunits of PI-3 kinase in regulating M-CSF–mediated responses. In one study, microinjection of an anti-p110α antibody in fibroblasts expressing the M-CSF receptor demonstrated no effect on mitogenesis; however, a separate study showed that microinjection of an anti-p110α antibody completely inhibits M-CSF–dependent DNA synthesis in a macrophage cell line, BAC1.13 Likewise, injecting antibodies against p110β or p110δ isoforms of PI-3 kinase impaired lamellipodia formation and macrophage migration, which was not the case when the function of p110α isoform was inhibited. Additional studies performed using porcine aortic endothelial cells demonstrated that p110α is required for growth factor–induced migration, whereas p110β is required for insulin-induced migration.14 Thus, the involvement of different catalytic subunits of PI-3 kinase in migration might be cell type–specific. Taken together, the physiologic role of PI-3 kinase isoforms in regulating growth and actin cytoskeletal functions in response to M-CSF and integrin stimulation requires further investigation.
Although a large body of evidence has demonstrated that PI-3 kinase is intimately associated with different phases of macrophage function, the exact role or roles this enzyme plays in macrophage functions in vivo, and which isoforms of PI-3 kinase are important for particular signaling events, remains poorly understood. The conclusions from most studies with regards to the role of PI-3 kinase are based on pharmacologic inhibitors of PI-3 kinase (wortmannin and LY294 002) or microinjection studies in cell lines. Although informative, these data are limited with regards to specificity. If, in the future, the PI-3 kinase pathway is going to be targeted for the treatment of diseases involving macrophages, including inflammation, then a better understanding of the relative contribution of various subunits of PI-3 kinase in regulating macrophage functions is necessary. To this end, we analyzed the function of macrophages deficient in the expression of the p85α subunit of class IA PI-3 kinase using p85α gene knockout mice.
Materials and methods
Mice
p85α+/– mice have been previously described.10,15 p85α–/– mice were obtained by mating of p85α+/– mice. The genotype of the p85α–/– mice was determined by polymerase chain reaction (PCR) analysis as previously described.15 The targeting strategy allowed selective disruption of p85α expression, while leaving p55α and p50α isoforms intact.15 These mice were maintained under specific pathogen-free conditions in the Indiana University Laboratory Animal Research Center, Indianapolis, IN.
Generation of bone marrow–derived macrophages
Bone marrow–derived macrophages (BMMs) were generated from 6- to 8-week-old wild-type and p85α–/– mouse femurs, tibias, and iliac crests as previously described.16 This method yields about 90% pure macrophages as described.16 Briefly, bone marrow was flushed into a 50-mL falcon tube using a syringe-needle and Iscoves modified Dulbecco medium (IMDM; Invitrogen, Carlsbad, CA). Cells were collected by centrifugation at 800g for 5 minutes and red blood cells (RBCs) were lysed with RBC lysis solution (155 mM ammonium chloride, 10 mM potassium bicarbonate containing 0.1 mM EDTA [ethylenediaminetetraacetic acid]) for 5 minutes at room temperature. The cells were centrifuged and resuspended in IMDM containing 20% fetal bovine serum (FBS) and 1% penicillin/streptomycin and plated in a 10-cm2 tissue-culture plate supplemented with 100 ng/mL M-CSF (Pepro Tech, Rocky Hill, NJ). After 24 hours, the nonadherent cells were collected and cultured in IMDM/20% FBS/100 ng/mL M-CSF on tissue-culture dishes for 8 to 10 days.
Proliferation assay
Bone marrow–harvested nonadherent cells (1 × 106) were collected after 24 hours and plated in 24-well plates in complete medium supplemented with 100 ng/mL M-CSF. After 3 days, the cells were starved by replacing the complete medium with IMDM without serum or growth factors overnight. The next day, starved cells were stimulated with M-CSF (100 ng/mL) or granulocyte macrophage–colony-stimulating factor (GM-CSF; 50 ng/mL) for 24 hours and pulsed with 0.037 MBq (1.0 μCi) of [3H]-thymidine (Amersham) for 6 hours at 37°C prior to harvesting. The cells were washed 4 to 5 times with cold phosphate-buffered saline (PBS) and lysed using 1 mL of 0.1 N sodium hydroxide (NaOH). Thymidine incorporation was measured using a Beckman Coulter LS 6500 Multipurpose Scintillation counter (Beckman Coulter, Fullerton, CA).
Adhesion on fibronectin
BMMs from wild-type and p85α–/– mice were generated as described in “Generation of bone marrow–derived macrophages.” Adhesion assay was performed as described.17 Briefly, flat-bottom 96-well polystyrene plates (BD Biosciences) were coated with 20 μg/mL fibronectin, retronectin, or fragments of fibronectin (H296 or CH271) and VCAM-1 in PBS for 1 hour at 37°C. Wells were washed once with PBS, incubated with 20 mg/mL bovine serum albumin (BSA) for 1 hour at 37°C for blocking nonspecific sites, and again washed twice with PBS. To examine cell adhesion on the coated surface, 1 × 105 cells were added to each well and incubated at 37°C for 15 minutes, 30 minutes, 1 hour, and 4 hours. At the end of the incubation, unbound cells were removed by aspiration carefully, and wells were washed twice with cold PBS. Adherent cells were fixed with 3.5% formaldehyde and stained with 0.1% crystal violet and photographs were taken. The stain was eluted with 10% acetic acid, and absorbance was determined at 600 nm using a microplate reader (Spectramax 250; Molecular Device, Sunnyvale, CA).
Integrin-mediated migration assay
In vitro haptotactic migration assay was conducted as previously described.16 Briefly, the transwell filters (8 μM pore filter; Costar, Boston, MA) were coated on the underside with 20 μg/mL fibronectin (FN) peptides (H-296, CH-271, and CH-296; Takara, Shiga, Japan) or vascular cell adhesion molecule-1 (VCAM-1; R&D Systems, Minneapolis, MN) for 2 hours at 37°C, and rinsed twice with PBS containing 2% BSA. The FN peptide-coated filters were placed in the lower chamber containing 500 μL complete medium with or without M-CSF (100 ng/mL). BMMs (2.5 × 105) were resuspended in 100 μL IMDM and allowed to migrate toward the underside of the top chamber. After 20 hours of incubation at 37°C, nonmigrated cells on the upper side of the chamber were removed and the migrated cells attached to the bottom surface of the membrane were stained with 0.1% crystal violet, 0.1 M borate, pH 9.0, and 2% ethanol for 5 minutes at room temperature. The numbers of migrated cells per membrane were counted in 10 random fields with an inverted microscope using ×20 objective lens. As a control, cell migration on BSA was also conducted, which resulted in about only 2 cells on average.
Flow cytometric analysis
Flow cytometry was used to examine the level of integrin expression on wild-type (WT) and p85α–/– BMM cells. Phycoerythrin (PE)–conjugated CD11b (Mac-1) and fluorescein isothiocyanate (FITC)–conjugated CD16/32 (FcγIII/II) antibodies were purchased from BD Pharmingen (San Diego, CA). WT or p85α–/– BMMs (0.5 × 106-1 × 106) were stained by incubating the cells at 4°C for 30 minutes with 1 μg of the above-mentioned antibodies. Cells were washed twice with PBS containing 0.1% BSA (Sigma, St Louis, MO), and analyzed by fluorescence-activated cell sorter (FACS; Becton Dickinson, San Jose, CA).
Wound-healing assay
Wound-healing assay was performed as described previously.16 The assay was conducted in either 24-well plates or 6-well tissue-culture plates. WT or p85α–/– BMMs were cultured as a confluent monolayer for 8 days, an artificial wound was created by scraping with a pipette tip, and cells migrating into open space were monitored microscopically. Photographs were taken immediately and at various intervals.
Phagocytosis
Phagocytosis assay was conducted as described previously.18 Briefly, 2 × 105 BMMs were plated overnight in a 6-well plate (Corning, Corning, NY). Sheep red blood cells (sRBCs) (1 mL; ICN Biomedicals, Irvine, CA) were added to 10 mL PBS, mixed, and centrifuged for 5 minutes at 1500 rpm. The sRBC pellet was resuspended in 10 mL Dulbecco modified Eagle medium (DMEM–high glucose). The sRBC solution (1 mL) was mixed with 10 μL immunoglobulin G (IgG) (ICN Biomedicals) and incubated at room temperature for 1 hour. BMMs were subjected to sRBCs coated with IgG at subagglutinating concentration (1:100), and incubated for 15 minutes. To determine the phagocytic index, uningested sRBCs were subjected to water shock to induce lysis. Diff-Quik stain (Dade Behring, Newark, DE) was used to stain the ingested sRBCs cells in macrophages. Number of sRBCs ingested by 100 random macrophage cells was counted. The phagocytic index was calculated as the percent of phagocytic cells × average number of sRBCs internalized by each BMM. Analysis of IgG-sRBC binding to macrophages was performed as described.19 IgG-sRBCs were allowed to bind BMMs for 5 minutes at 37°C. Unbound IgG-sRBCs were washed using cold PBS, and the percentage of macrophages that attached to IgG-sRBCs were counted microscopically.
Western blot analysis
WT or p85α–/– BMMs were starved overnight of serum and growth factors and stimulated with 50 ng/mL M-CSF for 5 minutes. The reaction was stopped by adding cold PBS and cells were lysed in previously described lysis buffer.20 Equal amount of protein was subjected to sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) and separated protein was transferred onto a nitrocellulose membrane. Western blot analysis was performed using an anti–phospho-Akt and an anti–phospho-Erk antibody (Cell Signaling, Beverly, MA). Rac activation was determined by means of a Rac activation assay kit (Upstate Biotechnology, Lake Placid, NY) according to the manufacturer's protocol.
Results
Deficiency of p85α in BMMs results in reduced proliferation
GM-CSF promotes the growth and differentiation of myelomonocytic progenitors. M-CSF is required for the proliferation, survival, differentiation, and activation of monocyte/macrophage lineage cells. Previous studies have shown that the p85α subunit of PI-3 kinase binds to the phosphorylated M-CSF receptor at tyrosine 723, thereby increasing the catalytic activity of the p110 subunit.11 Recent findings have suggested that phospholipase C γ 2 (PLC-γ2) also binds to this site.21 In a recent study,12 a 32D myeloid cell line engineered to express a truncated M-CSF receptor incapable of binding the p85α subunit of PI-3 kinase was shown to posses PI-3 kinase activity. Thus, although it is clear that PI-3 kinase gets activated in response to M-CSF stimulation, the physiologic role of the p85α subunit of class IAPI-3 kinase in regulating M-CSF–induced growth is not known. Here, we sought to evaluate the effect of deficiency of the p85α subunit of class IA PI-3 kinase in macrophages in M-CSF– and GM-CSF–induced proliferation. Macrophages from wild-type and p85α–/– mice were derived as previously described16 and stimulated with M-CSF or GM-CSF for 48 hours. As shown in Figure 1, deficiency of p85α in BMMs results in a significant decrease in proliferation compared with WT BMMs over 48 hours in response to M-CSF stimulation. A similar decrease in proliferation of p85α–/– BMMs was observed in response to GM-CSF stimulation (Figure 1). These results suggest that the p85α subunit of class IA PI-3 kinase plays an essential role in regulating growth via 2 distinct families of cytokine receptors.
Figure 1.

Deficiency of p85α in BMMs results in reduced proliferation. Nonadherent bone marrow cells (1 × 106) from WT and p85α–/– mice were cultured in a 24-well tissue-culture plate in the presence of M-CSF (100 ng/mL) for 3 days. Cells were starved overnight using IMDM and stimulated with either M-CSF (100 ng/mL) or GM-CSF (50 ng/mL) for 24 hours. Proliferation was measured by thymidine incorporation assay. Bars represent the mean thymidine incorporation in BMMs in response to either M-CSF or GM-CSF stimulation (counts per minute [cpm] ± SD) of 2 experiments performed in duplicate. Similar results were observed in 3 independent experiments. *P<.05 for WT versus p85α–/–.
Deficiency of p85α in BMMs results in reduced adhesion, migration, and phagocytosis
Macrophages play a significant role in wound healing. Furthermore, during the process of inflammation, macrophages migrate and extravagate into numerous tissues. Previous studies have suggested a role for fibronectin and VCAM-1 in the migration of macrophages to sites of inflammation. However, the signaling pathways that regulate the migration of macrophages are poorly understood. Studies using macrophage cell lines, as well as pharmacologic inhibitors of PI-3 kinase and injection of antibodies to inhibit the function of the catalytic subunits of PI-3 kinase, have suggested a role for the PI-3 kinase pathway in migration. To determine if the p85α subunit of PI-3 kinase plays an essential role in adhesion and migration of macrophages and to determine if integrin-directed migration cooperates with the M-CSF receptor, we performed an in vitro adhesion and migration assay. Figure 2A shows similar level of Mac-1 expression on both wild-type and p85α–/– BMMs as measured by flow cytometry. As seen in Figure 2B, loss of p85α results in a profound reduction in the adhesion of BMMs to different fibronectin fragments and VCAM-1 compared with wild-type BMMs. The decrease in adhesion of p85α–/– BMMs to FN is associated with a significant reduction in directed migration of these cells on different fibronectin fragments via both α5β1 and α4β1 as well as VCAM-1 (Figure 2C-D). Consistent with reduced migration on fibronectin and VCAM-1, deficiency of p85α in BMMs also resulted in defective migration of BMMs in an in vitro wound-healing assay (Figure 3A-B).
Figure 2.

Deficiency of p85α in BMMs results in reduced adhesion and migration. (A) Expression of Mac-1 on WT and p85α–/– BMMs. BMMs were stained with an anti–Mac-1 antibody or an isotype control antibody and subjected to flow cytometric analysis. Filled histograms indicate the level of Mac-1 staining on the surface of both WT and p85α–/– BMMs; open histograms, the level of staining using an isotype control antibody. (B) WT or p85α–/– BMMs (5 × 105) were subjected to an in vitro adhesion assay on fibronectin fragments CH271 (which contains a binding site for α5β1), H296 (which contains binding sites for α4β1), and CH-296 (which contains binding sites for both α4β1 and α5β1) and VCAM-1. Adhesion was assessed by measuring absorbance at indicated times. Shown is the optical density of the adherent cells at 600 nm. *P<.05 for WT versus p85α–/–.WTor p85α–/– BMMs (2.5 × 105) were subjected to an in vitro migration assay on fibronectin fragment CH271 (which contains a binding site for α5β1), H296 (which contains binding sites for α4β1), and CH-296 (which contains binding sites for both α4β1 and α5β1) as well as VCAM-1–coated transwell membranes. Cell migration was performed either in the absence (C) or presence (D) of M-CSF. Cell migration is expressed as the average number of cells migrated. Figure shows the average number of cells migrated ± SD. Ten fields were scored from one representative experiment. Similar results were observed in 3 other experiments. *P<.05 for WT versus p85α–/–.
Figure 3.

Impaired migration of p85α–/– BMMs in a wound-healing assay. (A) WT and p85α–/– BMMs were cultured for 8 days in 24-well plates in IMDM/20% FBS/100 ng/mL M-CSF. An artificial wound was created in the macrophage monolayer using a pipette tip. Photographs were taken immediately and again at indicated time periods after creating the wound. Data are from one representative experiment. Similar results were obtained in 2 independent experiments. Images were acquired through an Olympus TH3 microscope equipped with a 20×/0.4 objective lens (Olympus, Melville, NY), and were captured with a Nikon HRD100 camera (Nikon, Melville, NY). (B) Quantification of the wound-healing assay. The figure shows the mean number of cells migrated ± SD. Data are from one representative experiment. *P<.05 for WT versus p85α–/–.
Since PI-3 kinase plays a critical role in producing polyphosphoinositides, and since recent studies have suggested a role for these phospholipids in mediating phagocytosis,22,23 we next investigated the role of the p85α subunit of PI-3 kinase in phagocytosis in BMMs. We conducted a phagocytosis assay using IgG-sensitized sRBCs. As shown in Figure 4A-B, a significant decrease in phagocytosis was observed in p85α–/– BMMs compared with WT controls. The decrease in phagocytosis observed in p85α–/– BMMs was not due to impaired binding of sRBCs to BMMs, nor was it due to differences in the expression of FcγIII/II (Figure 4C-D).
Figure 4.
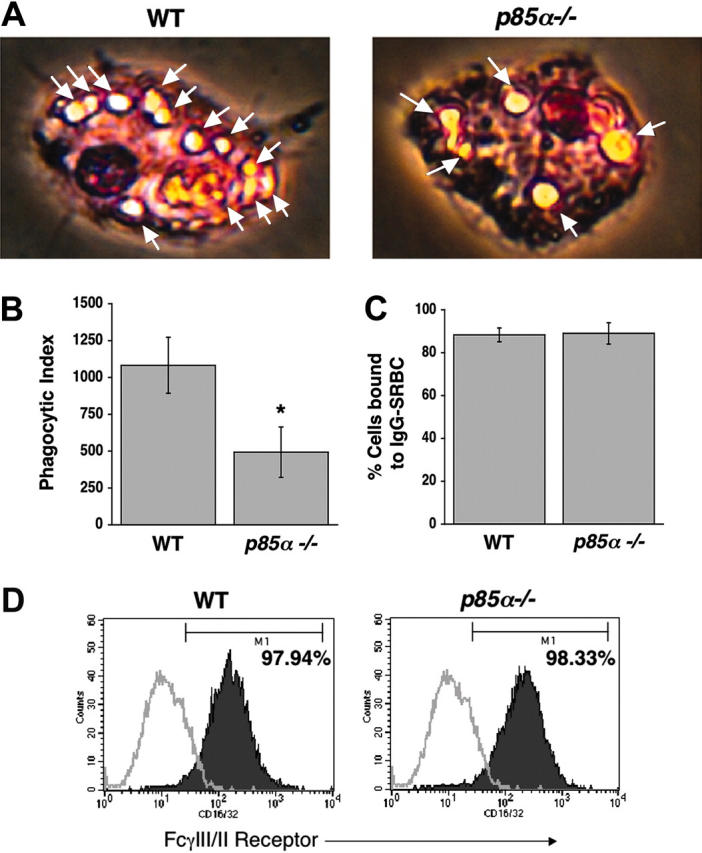
Reduced phagocytosis in p85α–/– BMMs. Phagocytosis assay was performed using WT and p85α–/– BMMs as described in “Materials and methods.” Phagocytosis assay was initiated by subjecting BMMs to IgG-sensitized sRBCs at a target-effector ratio of 100:1 for 15 minutes at 37°C. Nonengulfed sRBCs were lysed by water shock treatment and cells were fixed and stained using Diff-Quik stain before measuring the phagocytic index. (A) Arrows indicate engulfed cells. Images were acquired through a Zeiss Axioskop 2 Plus microscope equipped with a Plan-Neofluar 20×/0.5 objective lens, and were captured with an Axiocam MRC-5 camera and Axiovision 4 software (all from Zeiss, San Marcos, CA). (B) Bars indicate the mean phagocytic index ± SD. Data are from 4 independent experiments. *P<.05 for WT versus p85α–/–. The ability of WT and p85α–/– BMMs to bind IgG-sRBCs was assessed as described in “Materials and methods.” (C) Shown is the percent of BMMs bound to IgG sRBCs ± SD. (D) Expression of FcγIII/II receptor on WT and p85α–/– BMMs was analyzed by using an anti-FcγIII/II receptor (CD16/32) antibody or an isotype control antibody and flow cytometric analysis. Solid histograms indicate the level of FcγIII/II receptor staining on the surface of both WT (97.94% positivity) and p85α–/– (98.33% positivity) BMMs. Open histograms show the level of staining using an isotype control antibody.
Deficiency of p85α in BMMs results in reduced activation of Akt and Rac, but not Erk MAP kinase
Downstream effectors of PI-3 kinase include Akt and Rac. Other studies have suggested a role for PI-3 kinase in Ras-induced Erk activation. To determine the in vivo consequences of loss of p85α in BMMs on the activation of Akt, Rac, and Erk MAP kinase, we performed Western blot analysis using phospho-specific antibodies that recognize the activated forms of Akt and Erk MAP kinase. As seen in Figure 5A, deficiency of p85α in BMMs resulted in a significant decrease in the activation of Akt, but not Erk MAP kinase. Furthermore, loss of p85α in BMMs also resulted in reduced activation of Rac (Figure 5B).
Figure 5.

Reduced activation of Akt and Rac in p85α–/– BMMs. Wild-type and p85α–/– BMMs were starved overnight and stimulated with M-CSF. Equal amounts of cell lysates were subjected to Akt and Erk MAP kinase activation (A) by Western blot analysis using an anti–phospho Akt or Erk1/2 antibody or to a p21-activated kinase (PAK)–binding pull-down assay (B), which measures active, GTP-bound Rac. Shown are representative blots. Three independent experiments showed similar results. The bars indicate the band intensity of activated Akt and Rac, expressed as arbitrary units.
Discussion
A critical role for PI-3 kinase in regulating actin cytoskeleton rearrangements, membrane ruffling, and cell migration following integrin clustering has been demonstrated in several cell systems.6,7 Although the importance of PI-3 kinase in regulating actin-based functions has been derived using distinct cellular systems, in each system, integrin-mediated adhesion lead to the activation of PI-3 kinase. Furthermore, treatment of cells with the PI-3 kinase inhibitors resulted in the inhibition of integrin functions. Although informative, pharmacologic inhibitors of PI-3 kinase inhibit the function of all PI-3 kinase isoforms, thereby underscoring the importance of specific subunits. Identifying the function of specific subunits is crucial, as recent studies have begun to demonstrate specificity with regards to the function of distinct catalytic subunits of PI-3 kinase. To this end, actin cytoskeleton regulation by epidermal growth factor (EGF) requires the p85/p110α isoform of PI-3 kinase.24 Further, EGF-stimulated lamellipodia extension is blocked by microinjection of inhibitory antibodies to p110α, but not p110β.24 Thus, different PI-3 kinase isoforms appear to signal differently within the same cell. Our results in primary BMMs demonstrate an essential and nonredundant role for the p85α regulatory subunit of class IA PI-3 kinase in regulating multiple actin-based functions, including adhesion, migration, wound-healing, and phagocytosis. These defects are observed in spite of the presence of the p85β as well as the p50α and p55α subunits of class IA PI-3 kinase in these mice.10,15
The regulation of integrin-mediated functions in macrophages, including adhesion and migration, is largely controlled by tyrosine kinases. To this end, the role of PYK2 (proline-rich tyrosine kinase 2) and Src family kinases (SFKs) has been investigated in some detail in macrophages. Deficiency of PYK2 in BMMs results in reduced chemokine-induced macrophage migration, which is associated with reduced PI-3 kinase activity as well as Rho H activation.25 Likewise, the Src family kinase–Cbl-PI-3 kinase axis plays an essential role in regulating β1 integrin–directed migration and adhesion in macrophages.26 In this axis, Cbl participates in membrane targeting of PI-3 kinase by binding to the SH2 domain of the p85α subunit of class IA PI-3 kinase.26 Using Hck/Fgr/Lyn SFK–deficient macrophages, Meng and Lowell26 showed defective spreading and migration, which was associated with reduced membrane localized PI-3 kinase activity, but not total cellular PI-3 kinase activity, suggesting that the membrane-associated fraction of PI-3 kinase may be most critical in regulating actin cytoskeletal rearrangements that lead to cell spreading and migration in macrophages. Consistent with these observations, our results using p85α-deficient BMMs also demonstrate impaired adhesion and migration on ECM protein fibronectin.
Although the mechanism or mechanisms by which the p85α subunit of class IA PI-3 kinase affects actin- and growth-based functions in BMMs is not clear, several possibilities exist. To this end, Rho family GTPases, including Rac, have been shown to play a key role in actin reorganization27 and p85/p110 subunits have been shown to activate Rac in response to growth factor receptor stimulation.28 Furthermore, exchange factors for Rho GTPases, such as Vav, contain pleckstrin homology domains, which are likely to be activated by 3-phosphoinositides.29,30 Macrophages derived from mice deficient in the expression of Rac1 or Rac2 demonstrate different degrees of actin-based defects, including impaired spreading and migration.17,31
In addition to Rho family GTPases as potential downstream targets of p85α, several cytoskeletal proteins also interact with this subunit. These include proteins such as Cas and focal adhesion kinase, which bind to the SH2 domains of p85α32,33; Cbl, which binds to the SH3 domain of p8534; and ezrin, which also binds to the SH2 domains of p85α. The GTP exchange factor Pak interacting exchange factor and the actin-binding protein profilin also bind to p85 at unknown sites and could modulate macrophage functions.35,36 Thus, different regions of the p85 regulatory subunit can recruit distinct signaling proteins and modulate multiple functions in BMMs. Consistent with this notion, our results using p85α–/– BMMs demonstrate both cytoskeletal defects as well as defects in DNA synthesis via cytokine receptors as well as integrin receptors.
One of the most important functions of macrophages is to perform phagocytosis of virally infected cells. The mechanism of phagocytosis by macrophages involves the recognition of molecules on the surface of infected cells, including immunoglobulins (Igs) and the complement component C3b. Although previous studies have shown a role for PI-3 kinase in the phagocytic uptake of Ig-coated particles, the physiologic role of specific regulatory subunits of PI-3 kinase in controlling phagocytosis in primary BMMs has not been described. To this end, recent studies have confirmed that different catalytic subunits of PI-3 kinase play a unique role with respect to phagocytosis. Leverrier et al37 demonstrated that the p110β catalytic subunit is the major isoform required for apoptotic and FcγR-mediated phagocytosis. Other catalytic subunits, including p110γ, showed no effect on phagocytosis. Our results using p85α-deficient BMMs suggest an essential role for the p85α regulatory subunit of PI-3 kinase in regulating FcγR-mediated phagocytosis in primary BMMs. Our results demonstrating a 50% reduction in the phagocytosis of sRBCs in primary p85α–/– BMMs are consistent with those of Vieira et al38 who showed a complete loss of phagocytosis of sRBCs by embryonic fibroblasts deficient in the expression of both the p85α and p85β subunits of class IA PI-3 kinase engineered to express FcγR.38
Our results also demonstrate an essential role for p85α in regulating GM-CSF– and M-CSF–induced growth. Loss of p85α results in reduced PI-3 kinase activation as determined by Akt phosphorylation. Interestingly, the reduced growth and migration observed in p85α–/– BMMs was observed in spite of the presence of the p85β as well as the p55α and p50α isoforms of p85 in these cells.10,15 These results suggest that p85α functions with specificity in regulating macrophage functions. Alternatively, the residual growth and migration observed in p85α–/– BMMs may be due to the expression and activation of remaining regulatory subunits, including p85β.10,15 Unfortunately, eliminating all the members of p85 in mice results in embryonic lethality, thus precluding the analysis of these subunits in adult BMMs. Future studies using conditional knockouts of p85 regulatory subunits will shed light on the function of these additional subunits and how they cooperate with each other. In summary, this report provides the first direct genetic evidence of the effect of PI-3 kinase p85α deficiency on macrophage function(s) in vivo, and demonstrates that p85α plays an essential role in coordinating signals derived from growth factor receptors as well as integrins.
Acknowledgments
The authors wish to thank Arliene K. Britt and Janice Walls for assistance in manuscript preparation.
Prepublished online as Blood First Edition Paper, March 15, 2005; DOI 10.1182/blood-2004-10-4041.
Supported in part by the National Institutes of Health, National Heart, Lung, Blood Institute, R01 HL075 816 (R.K.).
An Inside Blood analysis of this article appears at the front of the issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 U.S.C. section 1734.
References
- 1.Chuluyan HE, Issekutz AC. VLA-4 integrin can mediate CD11/CD18-independent transendothelial migration of human monocytes. J Clin Invest. 1993;92: 2768-2777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Meerschaert J, Furie MB. Monocytes use either CD11/CD18 or VLA-4 to migrate across human endothelium in vitro. J Immunol. 1994;152: 1915-1926. [PubMed] [Google Scholar]
- 3.Weber C, Alon R, Moser B, Springer TA. Sequential regulation of alpha 4 beta 1 and alpha 5 beta 1 integrin avidity by CC chemokines in monocytes: implications for transendothelial chemotaxis. J Cell Biol. 1996;134: 1063-1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chuluyan HE, Schall TJ, Yoshimura T, Issekutz AC. IL-1 activation of endothelium supports VLA-4 (CD49d/CD29)-mediated monocyte transendothelial migration to C5a, MIP-1 alpha, RANTES, and PAF but inhibits migration to MCP-1: a regulatory role for endothelium-derived MCP-1. J Leukoc Biol. 1995;58: 71-79. [DOI] [PubMed] [Google Scholar]
- 5.Meerschaert J, Furie MB. The adhesion molecules used by monocytes for migration across endothelium include CD11a/CD18, CD11b/CD18, and VLA-4 on monocytes and ICAM-1, VCAM-1, and other ligands on endothelium. J Immunol. 1995;154: 4099-4112. [PubMed] [Google Scholar]
- 6.Vanhaesebroeck B, Leevers SJ, Ahmadi K, et al. Synthesis and function of 3-phosphorylated inositol lipids. Annu Rev Biochem. 2001;70: 535-602. [DOI] [PubMed] [Google Scholar]
- 7.Katso R, Okkenhaug K, Ahmadi K, White S, Timms J, Waterfield MD. Cellular function of phosphoinositide 3-kinases: implications for development, homeostasis, and cancer. Annu Rev Cell Dev Biol. 2001;17: 615-675. [DOI] [PubMed] [Google Scholar]
- 8.Vanhaesebroeck B, Leevers SJ, Panayotou G, Waterfield MD. Phosphoinositide 3-kinases: a conserved family of signal transducers. Trends Biochem Sci. 1997;22: 267-272. [DOI] [PubMed] [Google Scholar]
- 9.Shin BC, Suzuki M, Inukai K, Anai M, Asano T, Takata K. Multiple isoforms of the regulatory subunit for phosphatidylinositol 3-kinase (PI3-kinase) are expressed in neurons in the rat brain. Biochem Biophys Res Commun. 1998;246: 313-319. [DOI] [PubMed] [Google Scholar]
- 10.Terauchi Y, Tsuji Y, Satoh S, et al. Increased insulin sensitivity and hypoglycaemia in mice lacking the p85 alpha subunit of phosphoinositide 3-kinase. Nat Genet. 1999;21: 230-235. [DOI] [PubMed] [Google Scholar]
- 11.Reedijk M, Liu X, van der Geer P, et al. Tyr721 regulates specific binding of the CSF-1 receptor kinase insert to PI 3'-kinase SH2 domains: a model for SH2-mediated receptor-target interactions. Embo J. 1992;11: 1365-1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lee AW, States DJ. Both src-dependent and -independent mechanisms mediate phosphatidylinositol 3-kinase regulation of colony-stimulating factor 1-activated mitogen-activated protein kinases in myeloid progenitors. Mol Cell Biol. 2000; 20: 6779-6798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vanhaesebroeck B, Jones GE, Allen WE, et al. Distinct PI(3)Ks mediate mitogenic signalling and cell migration in macrophages. Nat Cell Biol. 1999;1: 69-71. [DOI] [PubMed] [Google Scholar]
- 14.Hooshmand-Rad R, Hajkova L, Klint P, et al. The PI 3-kinase isoforms p110(alpha) and p110(beta) have differential roles in PDGF- and insulin-mediated signaling. J Cell Sci. 2000;113(Pt 2): 207-214. [DOI] [PubMed] [Google Scholar]
- 15.Suzuki H, Terauchi Y, Fujiwara M, et al. Xid-like immunodeficiency in mice with disruption of the p85alpha subunit of phosphoinositide 3-kinase. Science. 1999;283: 390-392. [DOI] [PubMed] [Google Scholar]
- 16.Suen PW, Ilic D, Caveggion E, Berton G, Damsky CH, Lowell CA. Impaired integrin-mediated signal transduction, altered cytoskeletal structure and reduced motility in Hck/Fgr deficient macrophages. J Cell Sci. 1999;112(Pt 22): 4067-4078. [DOI] [PubMed] [Google Scholar]
- 17.Pradip D, Peng X, Durden DL. Rac2 specificity in macrophage integrin signaling: potential role for Syk kinase. J Biol Chem. 2003;278: 41661-41669. [DOI] [PubMed] [Google Scholar]
- 18.Kant AM, De P, Peng X, et al. SHP-1 regulates Fcgamma receptor-mediated phagocytosis and the activation of RAC. Blood. 2002;100: 1852-1859. [PubMed] [Google Scholar]
- 19.Yamauchi A, Kim C, Li S, et al. Rac2-deficient murine macrophages have selective defects in superoxide production and phagocytosis of opsonized particles. J Immunol. 2004;173: 5971-5979. [DOI] [PubMed] [Google Scholar]
- 20.Tan BL, Yazicioglu MN, Ingram D, et al. Genetic evidence for convergence of c-Kit- and alpha4 integrin-mediated signals on class IA PI-3kinase and the Rac pathway in regulating integrin-directed migration in mast cells. Blood. 2003;101: 4725-4732. [DOI] [PubMed] [Google Scholar]
- 21.Bourette RP, Myles GM, Choi JL, Rohrschneider LR. Sequential activation of phoshatidylinositol 3-kinase and phospholipase C-gamma2 by the M-CSF receptor is necessary for differentiation signaling. Embo J. 1997;16: 5880-5893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cox D, Tseng CC, Bjekic G, Greenberg S. A requirement for phosphatidylinositol 3-kinase in pseudopod extension. J Biol Chem. 1999;274: 1240-1247. [DOI] [PubMed] [Google Scholar]
- 23.Araki N, Johnson MT, Swanson JA. A role for phosphoinositide 3-kinase in the completion of macropinocytosis and phagocytosis by macrophages. J Cell Biol. 1996;135: 1249-1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hill K, Welti S, Yu J, et al. Specific requirement for the p85-p110alpha phosphatidylinositol 3-kinase during epidermal growth factor-stimulated actin nucleation in breast cancer cells. J Biol Chem. 2000;275: 3741-3744. [DOI] [PubMed] [Google Scholar]
- 25.Okigaki M, Davis C, Falasca M, et al. Pyk2 regulates multiple signaling events crucial for macrophage morphology and migration. Proc Natl Acad Sci U S A. 2003;100: 10740-10745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Meng F, Lowell CA. A beta 1 integrin signaling pathway involving Src-family kinases, Cbl and PI-3 kinase is required for macrophage spreading and migration. Embo J. 1998;17: 4391-4403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mackay DJ, Hall A. Rho GTPases. J Biol Chem. 1998;273: 20685-20688. [DOI] [PubMed] [Google Scholar]
- 28.Hawkins PT, Eguinoa A, Qiu RG, et al. PDGF stimulates an increase in GTP-Rac via activation of phosphoinositide 3-kinase. Curr Biol. 1995;5: 393-403. [DOI] [PubMed] [Google Scholar]
- 29.Han J, Luby-Phelps K, Das B, et al. Role of substrates and products of PI 3-kinase in regulating activation of Rac-related guanosine triphosphatases by Vav. Science. 1998;279: 558-560. [DOI] [PubMed] [Google Scholar]
- 30.Olson MF, Pasteris NG, Gorski JL, Hall A. Faciogenital dysplasia protein (FGD1) and Vav, two related proteins required for normal embryonic development, are upstream regulators of Rho GTPases. Curr Biol. 1996;6: 1628-1633. [DOI] [PubMed] [Google Scholar]
- 31.Wells CM, Walmsley M, Ooi S, Tybulewicz V, Ridley AJ. Rac1-deficient macrophages exhibit defects in cell spreading and membrane ruffling but not migration. J Cell Sci. 2004;117: 1259-1268. [DOI] [PubMed] [Google Scholar]
- 32.Reiske HR, Kao SC, Cary LA, Guan JL, Lai JF, Chen HC. Requirement of phosphatidylinositol 3-kinase in focal adhesion kinase-promoted cell migration. J Biol Chem. 1999;274: 12361-12366. [DOI] [PubMed] [Google Scholar]
- 33.Li E, Stupack DG, Brown SL, Klemke R, Schlaepfer DD, Nemerow GR. Association of p130CAS with phosphatidylinositol-3-OH kinase mediates adenovirus cell entry. J Biol Chem. 2000;275: 14729-14735. [DOI] [PubMed] [Google Scholar]
- 34.Hunter S, Koch BL, Anderson SM. Phosphorylation of cbl after stimulation of Nb2 cells with prolactin and its association with phosphatidylinositol 3-kinase. Mol Endocrinol. 1997;11: 1213-1222. [DOI] [PubMed] [Google Scholar]
- 35.Yoshii S, Tanaka M, Otsuki Y, et al. alphaPIX nucleotide exchange factor is activated by interaction with phosphatidylinositol 3-kinase. Oncogene. 1999;18: 5680-5690. [DOI] [PubMed] [Google Scholar]
- 36.Bhargavi V, Chari VB, Singh SS. Phosphatidylinositol 3-kinase binds to profilin through the p85 alpha subunit and regulates cytoskeletal assembly. Biochem Mol Biol Int. 1998;46: 241-248. [DOI] [PubMed] [Google Scholar]
- 37.Leverrier Y, Okkenhaug K, Sawyer C, Bilancio A, Vanhaesebroeck B, Ridley AJ. Class I phosphoinositide 3-kinase p110beta is required for apoptotic cell and Fcgamma receptor-mediated phagocytosis by macrophages. J Biol Chem. 2003;278: 38437-38442. [DOI] [PubMed] [Google Scholar]
- 38.Vieira OV, Botelho RJ, Rameh L, et al. Distinct roles of class I and class III phosphatidylinositol 3-kinases in phagosome formation and maturation. J Cell Biol. 2001;155: 19-25. [DOI] [PMC free article] [PubMed] [Google Scholar]