

Abstract
Previous studies demonstrated that ataxia telangiectasia mutated– and Rad3-related (ATR) kinase and its downstream target checkpoint kinase 1 (Chk1) facilitate survival of cells treated with nucleoside analogs and other replication inhibitors. Recent results also demonstrated that Chk1 is depleted when cells are treated with heat shock protein 90 (Hsp90) inhibitor 17-allylamino-17-demethoxygeldanamycin (17-AAG). The present study examined the effects of 17-AAG and its major metabolite, 17-aminogeldanamycin (17-AG), on Chk1 levels and cellular responses to cytarabine in human acute myelogenous leukemia (AML) cell lines and clinical isolates. Cytarabine, at concentrations as low as 30 nM, caused activating phosphorylation of Chk1, loss of the phosphatase Cdc25A, and S-phase slowing. Conversely, treatment with 100 to 300 nM 17-AAG for 24 hours caused Chk1 depletion that was accompanied by diminished cytarabine-induced S-phase accumulation, decreased Cdc25A degradation, and enhanced cytotoxicity as measured by inhibition of colony formation and induction of apoptosis. Additional studies demonstrated that small inhibitory RNA (siRNA) depletion of Chk1 also sensitized cells to cytarabine, whereas disruption of the phosphatidylinositol 3-kinase (PI3k) signaling pathway, which is also blocked by Hsp90 inhibition, did not. Collectively, these results suggest that treatment with 17-AAG might represent a means of reversing checkpoint-mediated cytarabine resistance in AML.
Introduction
The nucleoside analog cytarabine, an effective and widely used agent for the treatment of acute myelogenous leukemia (AML),1 induces remissions when administered on various schedules either as a single agent or in combination with other antileukemic drugs.2,3 Unfortunately, despite the inclusion of cytarabine in a variety of induction and consolidation regimens, most patients with AML ultimately relapse and die with drug-resistant disease.3-6 Accordingly, there is considerable interest in understanding the mechanisms of resistance to cytarabine and devising strategies for overcoming them.6,7
Earlier studies identified a number of mechanisms of cytarabine resistance, including diminished uptake on nucleoside transporters,8 increased degradation of cytarabine to uracil arabinoside,9 diminished formation or retention of cytosine arabinoside triphosphate,10-12 and reduced incorporation into DNA resulting from decreased passage of cells through S phase.13 Strategies for overcoming several of these mechanisms have been successfully implemented in clinical trials.14-16
Recent observations suggest that signaling by checkpoint kinase Chk1 might also contribute to cytarabine resistance. Chk1 is activated by a number of replication inhibitors.17-21 According to current understanding, these inhibitors cause DNA polymerases to stall but allow DNA helicases to continue advancing.22,23 The resulting single-stranded DNA then binds replication protein A, which recruits 2 protein complexes, one consisting of the ataxia telangiectasia mutated– and Rad3-related (ATR) kinase and its binding partner ATR-interacting protein (ATRIP) and another consisting of the Rad9-Rad1-Hus1 clamp. The Rad9-Rad1-Hus1 complex facilitates ATR-mediated phosphorylation and activation of Chk1. Once activated, Chk1 phosphorylates the phosphatase Cdc25A.24-27 The resulting protease-mediated degradation of Cdc25A contributes to S-phase slowing by preventing phosphatase-mediated activation of cyclin E/cyclin dependent kinase 2 complexes (reviewed by Sagata28). In addition, Chk1-mediated phosphorylation stabilizes stalled replication forks until replication can resume.18,21 The potential importance of these events in drug resistance is highlighted by the observation that Chk1 gene deletion or pharmacologic Chk1 inhibition sensitizes cells to replication inhibitors.21,29-31 Collectively, these observations have raised the possibility that disrupting Chk1 signaling might enhance nucleoside analog cytotoxicity and overcome Chk1-mediated drug resistance.
Heat shock protein 90 (Hsp90) is currently receiving considerable attention as a potential anticancer drug target.32 The Hsp90 complex is a chaperone that facilitates the initial folding and/or stabilization of a variety of polypeptides, which are known as clients.33,34 Several Hsp90 clients, including c-Kit,35 FLT3,36 Akt,37,38 and Bcr/abl,39,40 play important roles in leukocyte biology and leukemogenesis. The ability of the Hsp90 complex to stabilize these clients is inhibited by the benzoquinone ansamycin antibiotic geldanamycin and its derivatives,41,42 which occupy the adenosine triphosphate (ATP) binding site on Hsp9043 and trap the chaperone complex in a conformation that targets client proteins for proteasome-mediated degradation.44,45
Differences between chaperoning complexes in neoplastic and normal cells make the Hsp90 complex a potentially useful target for cancer chemotherapy. In particular, Hsp90 in neoplastic cells exists in multimolecular complexes with high adenosine triphosphatase (ATPase) activity and a high affinity for the geldanamycin derivative 17-allylamino-17-demethoxygeldanamycin (17-AAG), whereas Hsp90 in normal cells exists in a latent form that has low ATPase activity and a low affinity for 17-AAG.46 These observations provide a potential explanation for the enhanced accumulation and selective cytotoxicity of 17-AAG in neoplastic cells.41,46,47
Based on this selective toxicity, the geldanamycin analog 17-AAG is currently in phase 1 and phase 2 trials in patients with solid tumors.42,47 These studies have shown that 17-AAG is reasonably well tolerated, with reversible elevation of serum transaminases, nausea, vomiting, and diarrhea being dose-limiting when this agent is administered as a 60-minute infusion on a weekly schedule.48 Pharmacokinetic analysis has demonstrated that serum levels of 17-AAG exceed 1 μM for 8 hours and 300 nM for 16 hours at the maximum tolerated dose. Levels of its principal metabolite 17-aminogeldanamycin (17-AG) also exceed 1 μM for 14 hours and 300 nM for 24 hours.48 Although the effects of 17-AG on chaperone function have not been well characterized, the net result of these 17-AAG and 17-AG exposures is an interruption of Hsp90 function, as evidenced by client protein down-regulation and Hsp70 up-regulation in peripheral blood mononuclear cells.48,49 These observations provide the drug concentrations used to guide the present experiments.
Recent studies showing that Chk1 is an Hsp90 client50 identified a potential link between chaperone complex function and S-phase checkpoint signaling. These studies showed that 17-AAG sensitizes cells to the nucleoside analog gemcitabine as it diminishes Chk1 levels.50 In view of these results as well as the tolerable side effects of 17-AAG in patients,48 we investigated the role of Chk1 activation in cytarabine-treated AML cells. In particular, the present studies examined whether cytarabine-induced S-phase arrest is accompanied by Chk1 activation and assessed the effect of 17-AAG–induced Chk1 depletion on the action of cytarabine in AML cells lines and clinical samples.
Materials and methods
Materials
17-AAG and 17-AG were kindly provided by R. Schultz of the Developmental Therapeutics Program, National Cancer Institute (Bethesda, MD) or T. Mueller, Kosan Biosciences (Hayward, CA). Reagents were purchased as follows: LY294002 from Calbiochem (San Diego, CA) or Alexis Biochemicals (San Diego, CA), cytarabine from Sigma (St Louis, MO), allophycocyanin (APC)–conjugated annexin V from BD Pharmingen (San Diego, CA), Apo-ONE homogeneous caspase 3/7 reagents from Promega (Madison, WI), and OptiMEM medium and Lipofectamine 2000 from Invitrogen (Carlsbad, CA). Antibodies to the following antigens were obtained as indicated: Akt, phosphatidylinositol-dependent kinase 1 (PDK1), procaspase-3, phospho-Thr308-Akt, phospho-Ser345-Chk1, and phospho-Ser21/9-glycogen synthase kinase (GSK)–3α/β from Cell Signaling Technology (Beverly, MA); total GSK3β from Oncogene Research (San Diego, CA); Akt1 from Upstate Biotechnology (Lake Placid, NY); cleaved poly(adenosine diphosphate-ribose) polymerase (PARP) from Promega (Madison, WI); Hsp70 from Stressgen (San Diego, CA); and Chk1 and Cdc25A from Santa Cruz Biotechnology (Santa Cruz, CA). Murine antibodies against Chk2, Hsp90, and PARP were generous gifts from Junjie Chen and David Toft (both at Mayo Clinic, Rochester, MN) and Guy Poirier (Laval University, Ste-Foy, QC, Canada), respectively.
Leukemia samples
After informed consent was obtained under the aegis of protocols approved by the review boards of both participating institutions, marrow or blood samples were acquired from patients with newly diagnosed AML prior to induction therapy. Mononuclear cells were isolated on Ficoll-Hypaque gradients as described,51 washed with RPMI 1640 medium, and resuspended at 1.5 × 106/mL in Iscove modified Dulbeccco medium (IMDM) containing 20% (vol/vol) heat-inactivated fetal bovine serum (FBS), 100 U/mL penicillin G, 100 μg/mL streptomycin, and 2 mM glutamine (medium A).
Cell culture
HL-60 cells (American Type Culture Collection, Manassas, VA) and ML-1 cells (a kind gift from Michael Kastan, Saint Jude Children's Hospital, Memphis, TN) were maintained in RPMI 1640 containing 10% FBS, 100 U/mL penicillin G, 100 μg/mL streptomycin, and 2 mM glutamine (medium B) at a concentration of less than 1 × 106/mL at all times. HeLa cells were cultured in RPMI 1640 containing 10% FBS and 2 mM glutamine.
Assays of apoptosis
After treatment with the indicated concentrations of cytarabine and/or 17-AAG, fixation in 3:1 (vol/vol) methanol–acetic acid, and staining with 1 μg/mL Hoechst 33258, cells (at least 500 per sample) were examined by fluorescence microscopy for apoptotic changes in nuclear morphology.52 In parallel experiments, cells were washed, lysed at 4°C in buffer C (0.1% [wt/vol] Triton X-100 and 50 μg/mL propidium iodide in 0.1% [wt/vol] sodium citrate), and subjected to flow cytometry as previously described53,54 to assess DNA fragmentation. In further experiments, cells were stained with APC-conjugated annexin V and 0.1 μg/mL propidium iodide in 140 mM NaCl, 2.5 mM CaCl2, and 10 mM HEPES (N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid), pH 7.4, as instructed by the supplier. For this assay, 30 000 events were collected from the FL2 (excitation 488 nm, emission 585 ± 21 nm) and FL4 (excitation 635 nm, emission 661 ± 8 nm) channels of a Becton Dickinson (Mountain View, CA) FACSCalibur flow cytometer and analyzed using Becton Dickinson CellQuest software. Whole-cell lysates were harvested from the same experiments for immunoblotting to detect procaspases and caspase substrates52,55,56 as described in “Immunoblotting.” Finally, duplicate 100 μL aliquots of drug-treated cells were assayed for ability to cleave the substrate N-(Nα-aspartylglutamylvalinyl)aspartyl–rhodamine 110 according to instructions of the supplier using purified rhodamine 110 (Molecular Probes, Eugene, OR) to construct a standard curve. Additional aliquots were lysed in buffer C and analyzed in a SpectraMax Gemini EM fluorescent plate reader (Molecular Devices, Sunnyvale, CA) using excitation and emission wavelengths of 536 and 617 nm, respectively, to analyze DNA content, which was used to correct the caspase assay for variations in cell number.
Clonogenic assays
Aliquots containing 0.25 × 106 HL-60 cells in 1 mL medium B were incubated with diluent or cytarabine for 24 hours. Diluent or 17-AAG was added for an additional 24 hours. Cells were then sedimented at 80g for 5 minutes, resuspended in fresh medium B, diluted, and plated in gridded 35-mm plates in the medium of Pike and Robinson57 containing 0.3% (wt/vol) Bacto agar. After incubation for 14 days at 37°C, colonies containing 50 or more cells were counted on an inverted microscope. The same approach was used for clinical AML samples except that cells were treated with drug in medium A, washed, and plated in Methocult methylcellulose (StemCell Technologies, Vancouver, BC, Canada), which contains stem-cell factor, granulocyte-macrophage colony-stimulating factor, granulocyte colony-stimulating factor, erythropoietin, interleukin-3, and interleukin-6.
HeLa cells were transfected twice at 24-hour intervals with siRNA no. 1 (Dharmacon, Lafayette, CO), Chk1 siRNA,25 and/or PDK1 siRNA58 as described.59 Beginning 48 hours after the second transfection, cells were trypsinized, plated in 60-mm dishes, allowed to adhere for 4 hours, and treated with the indicated cytarabine concentrations for 24 hours. Alternatively, where indicated, nontransfected HeLa cells were treated with cytarabine for 24 hours followed by LY294002 for another 24 hours in the continued presence of cytarabine. Following drug treatment, cells were washed with RPMI 1640 and cultured for 7 days in medium B. After staining with Coomassie brilliant blue, colonies containing 50 or more cells were counted.
Cell-cycle analysis
Cells in medium B were incubated for 24 hours with diluent or cytarabine. Diluent or 17-AAG was then added for an additional 24 hours. Samples were then fixed in 50% ethanol, rehydrated, treated with RNase A, stained with 50 μg/mL propidium iodide in 0.1% sodium citrate, and analyzed (at least 20 000 events per sample) using a Becton Dickinson FACScan flow cytometer. Very small debris (forward scatter less than 10% of intact cells) was excluded from analysis by gating, but apoptotic cells were specifically included. Cell-cycle profiles were determined using Modfit Software (Verity, Topsham, ME) as previously described.51,60
Immunoblotting
After treatment with drug or diluent as indicated in the figure legends, cells were sedimented at 200g for 10 minutes, washed once with ice-cold RPMI 1640 medium containing 10 mM HEPES (pH 7.4 at 4°C), solubilized in buffered 6 M guanidine hydrochloride under reducing conditions, and prepared for electrophoresis as previously described.61 Aliquots containing 50 μg protein (determined by the bicinchoninic acid method62) were separated on sodium dodecyl sulfate (SDS)–polyacrylamide gels containing 5% to 15% acrylamide gradients, electrophoretically transferred to nitrocellulose, and probed as described.63 Alternatively, cells were lysed for 10 minutes on ice in buffer consisting of 50 mM HEPES (pH 7.4), 1% (wt/vol) Triton X-100, 10 mM NaF, 30 mM Na4P207, 150 mM NaCl, and 1 mM EDTA (ethylenediaminetetraacetic acid) supplemented with freshly added 10 mM β-glycerophosphate, 1 mM Na3VO4, 10 μg/mL pepstatin, 5 μg/mL aprotinin, 10 μg/mL leupeptin, and 20 nM microcystin-LR as described previously.64 After protein was estimated by the Bradford method,65 cell lysates were mixed with an equal volume of 2 × SDS-polyacrylamide gel electrophoresis sample buffer, boiled for 5 minutes, resolved on 10% SDS-polyacrylamide gels, transferred to Immobilon-P membranes, and probed with antiphospho-Ser345-Chk1, anti-PDK1, antiphospho-Thr308-Akt, antiphospho-Ser9/21-GSK3α/β, or anti-Chk1 antibodies as described.64
Statistical analysis
All experiments in tissue-culture cell lines were performed 3 to 5 times. Differences between groups were analyzed using the paired or unpaired Student t test as indicated. Several approaches outlined by Berenbaum66 were employed as indicated to determine whether effects of the cytarabine–17-AAG combination were synergistic.
Results
Cytarabine activates Chk1
Recent studies have demonstrated that the nucleoside analogs fludarabine30 and gemcitabine50 activate Chk1. To determine whether similar activation occurs in human leukemia cells treated with the clinically sustainable concentrations of 30 to 300 nM cytarabine,10,67 HL-60 cells were incubated with varying concentrations of this agent for 3 hours and probed with antibodies that recognize Chk1 phosphorylated on Ser345, a site modified by ATR and required for Chk1 activation.20 Results of this experiment demonstrated readily detectable Chk1 phosphorylation at cytarabine concentrations as low as 30 nM (Figure 1). Chk1 activation was accompanied by degradation of the phosphatase Cdc25A (Figure 2A), which is required for S-phase progression, and by S-phase slowing, as evidenced by the increased accumulation of HL-60 cells in S phase after cytarabine exposure (Figure 2B-D). Indistinguishable results were observed with a 48-hour cytarabine exposure (D.L. and S.H.K., unpublished observations, March 2003), suggesting that the S-phase checkpoint was not appreciably adapting in these cells. Because the flow cytometry assay used in these experiments cannot readily distinguish between cells in late S phase and G2, the results in Figure 2D represent a minimum estimate of the S-phase accumulation after cytarabine treatment.
Figure 1.

Cytarabine induces Chk1 activation in HL-60 cells. HL-60 cells were treated with diluent or the indicated concentrations of cytarabine for 3 hours. After cells were washed, postnuclear lysates were prepared. Equal amounts of protein were electrophoresed and sequentially immunoblotted for antiphospho-Ser345-Chk1 and total Chk1.
Figure 2.

Cytarabine-induced S-phase accumulation is abrogated by 17-AAG. (A) HL-60 cells treated with diluent (lane 1), 300 nM cytarabine (lane 2), 300 nM cytarabine and 1000 nM 17-AAG (lane 3), or 1000 nM 17-AAG alone (lane 4) were assayed for Cdc25A levels by immunoblotting as previously described.68 The asterisk indicates immunoglobulin G heavy chain. (B-C,E-F) HL-60 cells were treated for 24 hours with diluent (B), 100 nM cytarabine (C), or 300 nM 17-AAG (E). Alternatively, cells pretreated for 24 hours with 100 nM cytarabine (as in panel C) were then treated for an additional 24 hours with 300 nM 17-AAG (F) in the continued presence of cytarabine. At the completion of the incubation, the cell-cycle distributions were determined by flow microfluorimetry. Arrows denote cells with 2N and 4N DNA content determined by Modfit software analysis. (D) HL-60 cells were treated for 24 hours with diluent or the indicated concentrations of cytarabine and analyzed by flow microfluorimetry. Very similar results were obtained when the cytarabine incubation was continued for 48 hours (data not shown). (G) The percentage of S-phase cells detected after treatment with diluent or cytarabine and various concentrations of 17-AAG as illustrated in panels E and F.  indicates 300 nM cytarabine; ○, no cytarabine.
indicates 300 nM cytarabine; ○, no cytarabine.
17-AAG–induced Chk1 depletion is accompanied by S-phase exit and enhanced cytotoxicity in HL-60 cells
Recent results from our laboratory demonstrated that Chk1 is a client of the Hsp90 chaperone complex.50 Consequently, treatment of several cell lines with Hsp90 inhibitors led to Chk1 depletion. To confirm that Hsp90 inhibitors also promote Chk1 depletion in HL-60 cells, cells were treated for up to 24 hours with increasing concentrations of 17-AAG or its major metabolite, 17-AG, harvested, and analyzed by immunoblotting with anti-Chk1 antibodies. This analysis demonstrated a readily detectable decrease in Chk1 levels after a 24-hour treatment with 100 to 300 nM 17-AAG (Figure 3A, lanes 7 and 8). This decrease occurred at somewhat lower 17-AAG concentrations than were required to diminish the kinase Akt1 (Figure 3A, lanes 9 and 10), another Hsp90 client. 17-AG induced similar Chk1 and Akt1 down-regulation but at 3-fold lower concentrations (Figure 3B). Of importance, these changes occurred before detectable caspase activation, as evidenced by the lack of PARP cleavage (Figure 3A, lane 10). More prolonged 17-AAG treatment, however, induced PARP cleavage (not shown) and other stigmata of apoptosis (Figures 4, 5), as has been reported in other cell lines.36,39,73,74
Figure 3.

Effect of 17-AAG and 17-AG on Chk1 and other polypeptides in HL-60 cells. (A) Log-phase HL-60 cells were treated for 6 hours (lanes 1 to 5) or 24 hours (lanes 6 to 10) with diluent (lanes 1 and 6), 100 nM 17-AAG (lanes 2 and 7), 300 nM 17-AAG (lanes 3 and 8), 1000 nM 17-AAG (lanes 4 and 9), or 3000 nM 17-AAG (lanes 5 and 10). After cells were washed in serum-free medium, whole-cell lysates were subjected to SDS-polyacrylamide gel electrophoresis, transferred to nitrocellulose, and immunoblotted for Chk1 and Akt1. PARP, a well-recognized substrate of caspases 3, 7, and 9,69-72 served as a marker of caspase activation. Chk2 served as a loading control. (B) Log-phase HL-60 cells were treated for 24 hours with diluent (lanes 1 and 6), 17-AAG (lanes 2 to 5), or 17-AG (lanes 7 to 10) at 30 nM (lanes 2 and 7), 100 nM (lanes 3 and 8), 300 nM (lanes 4 and 9), and 1000 nM (lanes 5 and 10). At the completion of the incubation, sample preparation and immunoblotting were performed as described in panel A.
Figure 4.
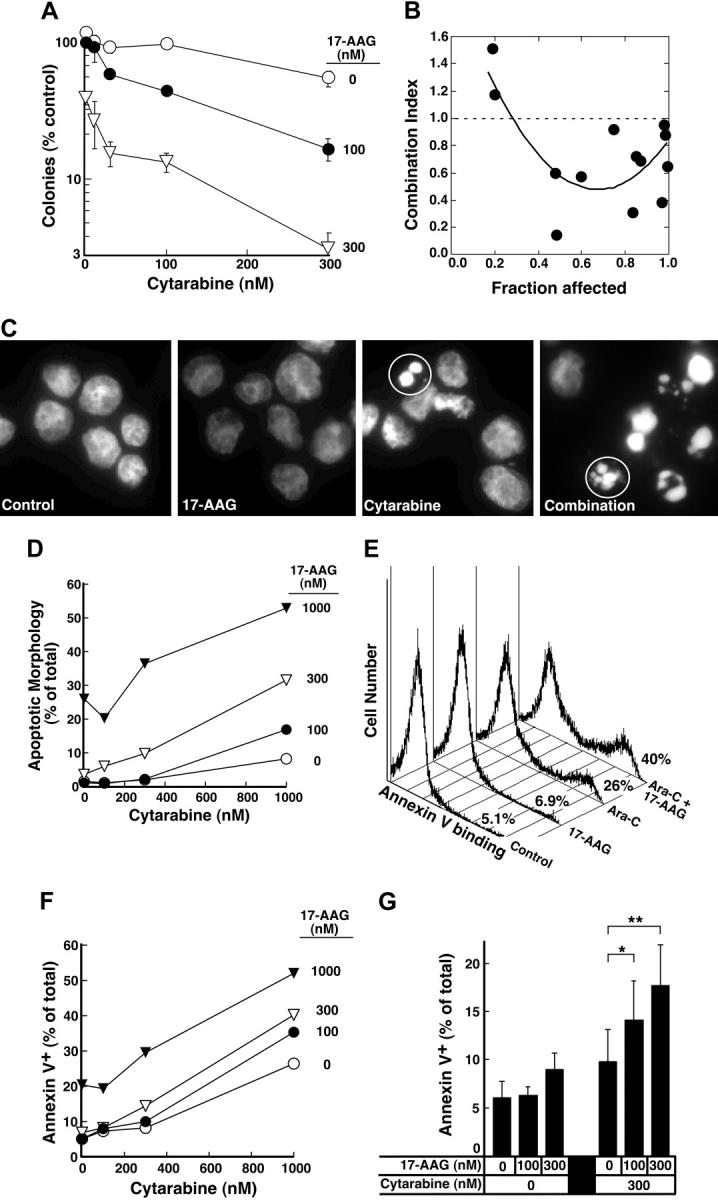
Effect of 17-AAG on cytarabine-induced cytotoxicity. (A) Log-phase HL-60 cells were treated with 0.1% dimethyl sulfoxide (DMSO) containing 0, 10, 30, 100, or 300 nM cytarabine for 24 hours. The indicated concentration of 17-AAG was then added for an additional 24 hours in the continued presence of cytarabine. At the completion of the incubation, cells were sedimented, washed, plated in drug-free medium, and allowed to form colonies for 14 days. Cloning efficiency of diluent-treated cells in multiple experiments ranged from 25% to 35%. Error bars: mean ± standard deviation of quadruplicate samples. (B) Dots represent combination index values calculated from the data in panel A under the assumption that effects of cytarabine and 17-AAG were mutually exclusive.82 Solid line is second-order regression line calculated from the individual data points. (C-D) Log-phase HL-60 cells were treated with 0.1% DMSO containing 0, 100, 300, or 1000 nM cytarabine for 24 hours. The indicated concentration of 17-AAG was then added for an additional 24 hours in the continued presence of cytarabine. At the completion of the incubation, cells were sedimented, incubated in drug-free medium for 72 hours, and examined for apoptotic morphologic changes. (C) Micrographs contain representative fields of cells harvested 72 hours after completion of treatment with diluent (control), 300 nM 17-AAG for 24 hours, 1000 nM cytarabine for 48 hours, or 1000 nM cytarabine for 24 hours followed by the addition of 300 nM 17-AAG for the second 24 hours in the continued presence of the cytarabine (combination). Cells were then incubated for 72 hours and analyzed. (D) Results obtained when more than 500 cells per treatment were examined from the experiment in panel C. (E) Log-phase HL-60 cells were treated with diluent (control or 17-AAG) or 1000 nM cytarabine (Ara-C) for 24 hours. Diluent or 300 nM 17-AAG was then added for 24 hours. At the completion of the drug treatment, cells were washed and cultured in drug-free medium for 3 days prior to staining with APC–annexin V. For these assays, both propidium iodide–negative (early apoptotic) and propidium iodide–positive (late apoptotic) cells were quantitated, a common practice in these prolonged assays.83,84 (F) Log-phase HL-60 cells were treated with 0.1% DMSO containing 0, 100, 300, or 1000 nM cytarabine for 24 hours. The indicated concentration of 17-AAG was then added for an additional 24 hours in the continued presence of cytarabine. At the completion of the incubation, cells were sedimented, incubated in drug-free medium for 72 hours, and stained with APC–annexin V as illustrated in panel E. (G) Combined results obtained in 4 independent experiments performed as described for panels E and F. Error bars: ± 1 standard deviation. *P = .03 and **P = .002 by paired t test.
Figure 5.

Effect of 17-AAG on cytarabine-induced cell-cycle arrest and cytotoxicity in ML-1 cells. (A-B) ML-1 cells were treated for 24 hours with diluent or 30 nM cytarabine (Ara-C). Either 300 nM 17-AAG (A) or the indicated concentration of 17-AAG (B) was then added for an additional 24 hours in the continued presence of cytarabine. At the completion of the drug treatment, cells were analyzed by flow microfluorimetry as illustrated in Figure 2. Arrows in panel A indicate subdiploid (apoptotic) cells. (C) ML-1 cells were treated with diluent or 30 nM cytarabine for 24 hours, washed, and incubated with diluent, 300 nM 17-AAG, 30 nM cytarabine, or 30 nM cytarabine plus 300 nM 17-AAG for an additional 24 hours. Morphologic apoptotic changes were then assessed as illustrated in Figure 4C. (D-F) ML-1 cells were treated with diluent or 100 nM cytarabine for 24 hours, sedimented, and incubated with diluent, 300 nM 17-AAG, 100 nM cytarabine, or 100 nM cytarabine plus 300 nM 17-AAG for an additional 24 hours. At the completion of the incubation, whole-cell lysates were assayed for ability to cleave DEVD–rhodamine 110 and for DNA content (D). Alternatively, whole-cell lysates were subjected to immunoblotting using sera that recognize the indicated antigen (E); dashed line indicates where nonadjacent lanes on each blot were juxtaposed; arrow, 89 kDa PARP fragment that results from caspase-mediated cleavage.86 At the completion of the incubation, cells were also examined for APC–annexin V binding (F) as illustrated in Figure 4E. Error bars: ± 1 standard deviation of 5 (C) or 3 (F) independent experiments. *P = .02 and **P < .001 by paired t test.
Because 17-AAG treatment results in Chk1 depletion and Chk1 is required for activation of the S-phase checkpoint, we reasoned that 17-AAG would disrupt cytarabine-induced Cdc25A degradation and S-phase accumulation. However, in agreement with previous results,75-77 we also observed that 17-AAG administered as a single agent arrested HL-60 cells in G1 and G2 (Figure 2E), which would prevent cells from reaching S phase, where cytarabine is most effective.1 To circumvent this problem, we first arrested cells in S phase by treating for 24 hours with cytarabine and then added 17-AAG for the subsequent 24 hours. Immunoblotting demonstrated that 17-AAG–induced Chk1 depletion under these conditions was indistinguishable from that shown in Figure 3 (data not shown). Consistent with the loss of Chk1, 17-AAG attenuated cytarabine-induced Cdc25A loss (Figure 2A) and overcame cytarabine-induced S-phase slowing, as indicated by the fact that cells accumulated in G1 and G2 phases despite the continued presence of cytarabine (Figure 2F-G).
The depletion of S-phase cells observed in Figure 2F-G was not due to cell death. Few cells with sub-G1 DNA content or apoptotic morphology were observed at the completion of the combined drug treatment (Figure 2F; D.L. and S.H.K., unpublished observations, January 2004). Nonetheless, examination of the long-term consequences of the sequential treatment indicated that 17-AAG enhanced the antiproliferative effects of cytarabine. For these studies, we initially used colony-forming assays, which have been widely employed to determine the effects of cytokines and drugs on normal and leukemic myeloid cells in the past.57,78-81 These assays demonstrated that 17-AAG diminished the ability of cytarabine-treated HL-60 cells to form colonies (Figure 4A). The ability of one agent to enhance the activity of another without having any activity itself is one definition of synergy.66 Based on this definition, 100 nM 17-AAG clearly synergized with cytarabine (Figure 4A). Formal analysis of these data by the median effect method of Chou and Talalay82 under the assumption that the combined effects of the 2 agents were mutually exclusive (equivalent to isobologram analysis)66 showed that most combination index values were below 1 (Figure 4B), confirming that the effects were synergistic.
Further experiments investigated whether 17-AAG also enhanced cytarabine-induced apoptosis. Cells treated with cytarabine, 17-AAG, or the combination appeared healthy at the end of the combined drug treatment (data not shown). By 72 hours after drug withdrawal, however, a substantial number of cells appeared apoptotic (Figure 4C-D). Analysis by the median effect method again demonstrated that the effects were synergistic, consistent with the results of the clonogenic assays. Similar results were observed when the induction of apoptosis was examined using APC-conjugated annexin V (Figure 4E-F), a phosphatidylserine-binding polypeptide that preferentially recognizes cells undergoing apoptosis.85 Combined results of 4 independent experiments demonstrated that 17-AAG concentrations as low as 100 to 300 nM significantly enhanced cytarabine-induced killing. As shown in Figure 4G, the percentage of annexin V–positive cells increased from 9.8% ± 3.3% after treatment with 300 nM cytarabine alone to 14.1% ± 4.1% when we added 100 nM 17-AAG, a concentration that exhibited no effect by itself, and to 17.7% ± 4.2% when we added 300 nM 17-AAG, a concentration that induced only 3% more apoptosis than diluent.
Effects of the cytarabine–17-AAG combination in ML-1 cells
To rule out the possibility that these effects occurred only in HL-60 cells, ML-1 cells were treated with cytarabine alone or cytarabine followed by 17-AAG. Previous results from our laboratory demonstrated that 17-AAG treatment decreases Chk1 levels in this AML cell line.50 Flow cytometry confirmed that cytarabine-induced S-phase slowing was also diminished by addition of 17-AAG (Figure 5A-B). Although ML-1 cells do not form colonies, analysis of apoptotic changes by several different assays demonstrated that 17-AAG enhanced cytarabine-induced cell death. In contrast to HL-60 cells, which did not show a sub-G1 peak following drug exposure, ML-1 cells exposed to cytarabine and 17-AAG displayed DNA fragmentation in this assay (Figure 5A, double-headed arrow). As indicated in Figure 5C, morphologic analysis also indicated that addition of 17-AAG enhanced cytarabine-induced apoptosis. These same experiments also explored whether continued cytarabine treatment was required for the enhanced apoptosis during the 17-AAG exposure (Figure 5C). Even though the amount of cell death was similar when cells were exposed to cytarabine for either 24 or 48 hours (3.25% ± 0.95% versus 7.5% ± 3.0% apoptosis, n = 5), significantly more cells died when cytarabine was present during the 17-AAG exposure compared with when cytarabine was removed (41% ± 13% versus 21% ± 9% apoptosis, n = 5; P = .02 by paired t test).
In a subsequent series of experiments, multiple independent techniques compared the induction of apoptosis by the individual drugs and the cytarabine–17-AAG combination. More caspase activity was detected in cells treated with the drug combination than in cells treated with either drug alone (Figure 5D). Consistent with these results, immunoblotting demonstrated that cleavage of the caspase substrate PARP was enhanced by treatment with the combination (Figure 5E, top panel). Because this cleaved PARP fragment was specifically detected by a serum that recognizes only PARP cleaved after Asp214 (Figure 5E, second panel from top), a known caspase cleavage site,86 this enhanced cleavage reflects increased caspase activity. Consistent with this interpretation, increased amounts of cleaved (activated)56 caspase 3 were also detected after treatment with the combination (Figure 5E, third panel from top). Analysis of these cells for apoptosis using an annexin V binding assay again demonstrated that the continued presence of cytarabine enhanced the effect of 17-AAG. In particular, 46.1% ± 6.3% of the cells bound annexin V when cytarabine was present during the 17-AAG exposure, but only 32.7% ± 6.7% stained positive when cytarabine was removed (Figure 5F, P < .001 by paired t test). These results suggest that the enhanced cytotoxicity observed when 17-AAG is added to cytarabine-containing cells reflects, at least in part, the effect of inducing additional replication stress when Chk1 is down-regulated by 17-AAG.
siRNA-mediated Chk1 depletion also sensitizes cells to cytarabine
Hsp90 inhibition depletes numerous Hsp90 clients associated with increased survival following genotoxin treatment.33,41,42 To independently assess the effect of Chk1 depletion on cytarabine cytotoxicity, we down-regulated Chk1 with siRNA. Despite multiple attempts, we could not deplete Chk1 by transfecting either HL-60 or ML-1 cells with synthetic siRNA or plasmid-produced hairpin siRNAs that were effective in other cell lines. Consequently, HeLa cells were used in these experiments.
As was seen in HL-60 cells, cytarabine induced Ser345 phosphorylation of Chk1 in HeLa cells, although somewhat higher cytarabine concentrations were required (compare Figures 6A and 1). Chk1 down-regulation by siRNA (Figure 6B, inset) was accompanied by enhanced sensitivity to single-agent cytarabine (Figure 6B). These results are consistent with the hypothesis that 17-AAG–induced Chk1 down-regulation contributes to the enhanced cytotoxicity of cytarabine.
Figure 6.

Effect of various treatments on cytarabine sensitivity in HeLa cells. (A) Cells were treated with diluent or the indicated concentrations of cytarabine (Ara-C) for 4 hours. After cells were washed, postnuclear lysates were prepared. Equal amounts of protein were electrophoresed and sequentially immunoblotted for phospho-Ser345-Chk1 (P-Chk1) and total Chk1. (B) Forty-eight hours after the second transfection with control or Chk1 siRNA, cells were harvested for immunoblotting with anti-Chk1 and antiactin antibodies (inset) or treated for 24 hours with the indicated concentration of cytarabine. At the completion of the drug incubation, colonies were allowed to form under drug-free conditions. (C) Forty-eight hours after the second transfection with control or PDK1 siRNA, cells were harvested for immunoblotting with anti-PDK1, anti-Thr308-phospho-Akt (P-Akt), and anti-Akt antibodies (inset) or treated for 24 hours with the indicated concentration of cytarabine. At the completion of the drug incubation, colonies were allowed to form under drug-free conditions. (D) In parallel experiments, cells were treated for 24 hours with diluent or the indicated concentration of cytarabine, and diluent or 20 μM LY294002 was then added for an additional 24 hours in the continued presence of diluent or cytarabine. At the completion of the drug incubation, cells were harvested for immunoblotting with antiphospho-Thr308-Akt, anti-Akt, antiphospho-Ser9/21-GSK3α/β, and anti-GSK3β antibodies (inset) or washed and allowed to form colonies under drug-free conditions. Error bars: mean ± standard deviation of triplicate aliquots. Each panel is representative of 3 or more independent experiments with similar results. HeLa-cell cloning efficiency ranged from 70% to 90%. The total period of cytarabine exposure was 48 hours in panel D but only 24 hours in panels B and C, explaining the enhanced sensitivity in panel D.
Although it is not possible to examine the effects of all of the other Hsp90 clients, the same model system was used to evaluate the contribution of 17-AAG–induced Akt down-regulation. As a key signaling kinase of the phosphatidylinositol 3-kinase (PI3k) pathway, Akt plays critical roles in blocking apoptosis and promoting survival following cellular stress.87 Because Akt and the upstream kinase that activates it, PDK1, are both Hsp90 clients,33,41,42 inactivation of this pathway has been postulated to contribute to the ability of 17-AAG to sensitize cells to a variety of proapoptotic stimuli. To assess the effect of PDK1/Akt pathway inhibition in sensitizing cells to cytarabine, PDK1 was down-regulated with siRNA (Figure 6C, inset). Concomitant with PDK1 depletion, phosphorylation of Akt on Thr308 was decreased (Figure 6C, inset). In contrast to Chk1 down-regulation, however, PDK1 depletion did not enhance cytarabine sensitivity (Figure 6C). To the contrary, cells were more resistant after PDK1 down-regulation. To confirm that this was not a result of an off-target effect of the PDK1 siRNA, we inhibited the same pathway with LY294002, a small molecule that inhibits PI3k. LY294002 inhibited Akt phosphorylation and activity (Figure 6D, inset) but again did not sensitize cells to cytarabine (Figure 6D). These results suggest that Chk1 depletion, but not modulation of the PI3k/PDK1/ATR pathway, plays a predominant role in sensitizing HeLa cells to cytarabine.
Effect of cytarabine and 17-AAG in clinical AML isolates
In a final series of experiments, the effects of 17-AAG on primary AML isolates were examined. Immunoblotting demonstrated that a 24-hour 17-AAG treatment caused depletion of both Chk1 and Akt1, as illustrated in Figure 7 for samples from 3 separate AML patients. Consistent with previous observations, Hsp70 levels were up-regulated after 17-AAG exposure, and Hsp90 levels were unaltered. Similar effects of 17-AAG on Chk1, Hsp70, and Hsp90 were observed in all 5 AML marrow samples examined (Figure 7 and D.L., K.F, and S.H.K., unpublished observations, May 2003–August 2004).
Figure 7.

Effects of 17-AAG on client protein levels in primary AML isolates. Aliquots containing 1.5 × 106 to 2 × 106 AML cells were incubated for 24 hours with diluent (lane 1), 300 or 1000 nM cytarabine (lanes 2 and 3), or 300 or 1000 nM 17-AAG (lanes 4 and 5). Whole cell lysates containing 50 μg protein were then subjected to immunoblotting with reagents that detect the indicated polypeptides. Actin, which is not an Hsp90 client, and Hsp90, which does not change after 17-AAG treatment, served as loading controls. Samples in panels A-C came from 3 different AML patients.
Colony-forming assays were used to evaluate the antiproliferative effects of the cytarabine–17-AAG combination. For these studies, cells were treated with cytarabine for 24 hours followed by 17-AAG for an additional 24 hours in the continued presence of cytarabine (Figure 8). Replicate aliquots of the same cells were treated with the individual drugs at exactly the same time. Results of these assays were analyzed using 2 different approaches. First, the fractional product approach66 was used. According to this method, the fractional survival expected from the combination was assumed to be the product of the fractional survivals of the single agents. Effects of the combination predicted by this method are indicated by open circles in Figure 8A,C-D. In each case, the survival observed after treatment with the combination was much lower than would be expected if the combination were strictly additive. Similar results were observed in 2 additional AML samples incubated with cytarabine for 24 hours before addition of 17-AAG for 24 hours and in 7 separate AML samples incubated with cytarabine for 48 hours before addition of 17-AAG for 24 hours (H.L.P., R.A.M., and S.H.K., unpublished observations, March to July 2004). Because the limited number of cells in some samples precluded exposure to a large number of drug concentrations, data from only 6 samples were analyzed by the more formal method of Chou and Talalay.82 In 1 of the 6 samples the combination index was above 1.0, indicating that the antiproliferative effects of the combination were less than additive. In the remaining 5, the combination index was below 1 (Figure 8B), indicating that the antiproliferative effects of the combination were synergistic.
Figure 8.

Effect of 17-AAG on antiproliferative effects of cytarabine in colony-forming assays. (A,C-D) Freshly isolated mononuclear cells from 3 newly diagnosedAML patients were treated for 24 hours with diluent or 100 nM cytarabine (Ara-C) as indicated in the panels. Diluent or 17-AAG was then added to the indicated final concentration for an additional 24 hours. At the completion of the drug treatment, samples were washed and plated in Methocult medium to permit growth and quantitation of leukemic colonies. Bars indicate relative colony counts observed after the indicated treatment; ○, relative colony counts predicted from the effects of the individual agents using the fractional product method.66 (B) Combination index values calculated from the data in panel A and additional results from the same clonogenic assay under the assumption that effects of 17-AAG and cytarabine are mutually exclusive ().82 Other symbols indicate combination index values from clonogenic assays performed in 4 additional AML samples as described in the text. A combination index level below 1.0 indicates synergy.
Discussion
Results of the present study provide the first demonstration that the cytarabine-induced S-phase checkpoint can be abrogated by 17-AAG in myeloid leukemia cell lines. Treatment with therapeutically achievable cytarabine concentrations10,67 resulted in activating phosphorylation of Chk1 (Figure 1), Cdc25A degradation (Figure 2A), and diminished progression through S phase (Figures 2 and 5). Treatment with 17-AAG not only diminished Chk1 levels (Figure 3)50 but also reduced Cdc25A degradation and decreased the number of cells in S phase (Figures 2 and 5). This S-phase exit in the continued presence of cytarabine is consistent with the notion that accumulation in S phase represents activation of the replication checkpoint29 rather than physical blockage of replication complexes by cytidine arabinoside incorporation into DNA.
This 17-AAG–induced exit of cells from S phase in the continued presence of cytarabine is followed by increased apoptosis as detected by caspase activation (Figure 5D), cleavage of caspase substrates (Figure 5E), DNA fragmentation (Figure 5A), phosphatidylserine externalization (Figures 4E-G and 5F), and apoptotic morphologic changes (Figures 4C-D and 5C). The enhancement of cell death was observed in HL-60 cells, which lack p5388 and express high levels of Bcl-2,89,90 as well as ML-1 cells, which express wild-type p5391 and more modest levels of Bcl-2.90 The ability of 17-AAG to enhance cytarabine-induced killing is consistent with earlier observations demonstrating that the Chk1 inhibitor UCN-01 increases death induced by the nucleoside analogs gemcitabine,29 fludarabine,30 and cytarabine.31 Of importance, 17-AAG also depleted Chk1 in primary AML cells (Figure 7). In these samples, the lower percentage of blasts that are actively traversing the cell cycle92,93 likely limits the efficacy of the cytarabine plus 17-AAG combination during the arbitrary treatment time used in the present experiments. Nonetheless, 17-AAG also synergized with cytarabine in primary AML isolates (Figure 8). All of these results are consistent with previous studies showing that targeted Chk1 gene deletion enhances the cytotoxicity of the replication inhibitor aphidicolin in chicken B cells.21 The cytarabine–17-AAG combination described in the present study might be particularly effective because cytarabine stalls replication forks94,95 and 17-AAG depletes Chk1, which plays critical roles in facilitating survival when replication forks stall.21 In view of data showing that the incorporation and retention of cytarabine metabolites into nascent DNA is increased by proliferative cytokines,12,13,15,96,97 it is possible that the cytotoxicity of the cytarabine–17-AAG combination might be further enhanced by cytokine priming.
Recent pharmacokinetic analysis performed in conjunction with phase 1 clinical trials of 17-AAG demonstrated that 17-AAG is metabolized to 17-AG, which persists in the circulation longer than 17-AAG.48 The present study provides the first indication that 17-AG more potently down-regulates Hsp90 client proteins than 17-AAG (Figure 3B). These findings indicate that the contributions of both 17-AAG and 17-AG need to be taken into account when considering the implications of drug levels on client protein stability.
While the present study focused on Chk1, other client proteins are also down-regulated by Hsp90 inhibitors. The antiapoptotic kinase Akt,87 for example, was down-regulated in AML cell lines (Figure 3) and clinical AML isolates (Figure 7). Although Akt clearly plays important roles in enhancing cell survival following certain cellular stresses, our results in HeLa cells (Figure 6) suggest that signals mediated by PI3k and downstream kinases are less important than Chk1 when replication forks are arrested by cytarabine. In particular, PDK1 siRNA diminishes Akt signaling but does not enhance cytarabine-induced antiproliferative effects (Figure 6), arguing against the possibility that 17-AAG–induced decreases in Akt1 phosphorylation on Thr308 are responsible for the synergy of cytarabine and 17-AAG. On the other hand, the tyrosine kinase receptor FLT3, a mutational target associated with a particularly poor prognosis in 20% to 40% of AML cases,98,99 is also an Hsp90 client that is depleted by 17-AAG.36 These observations, coupled with the finding that tumor-cell Hsp90 has a higher affinity for 17-AAG,46 raise the possibility that 17-AAG might selectively target leukemia cells, where it will inhibit multiple signaling pathways that contribute to drug resistance.
In summary, the present results indicate that 17-AAG depletes Chk1, disrupts the S-phase checkpoint, and enhances the antiproliferative effects of cytarabine in multiple human leukemia cell lines and clinical AML samples. In view of previous data suggesting that activation of the S-phase checkpoint contributes to nucleoside analog resistance,29,30 further preclinical and clinical investigation of the cytarabine–17-AAG combination appears warranted. Further study is also required to determine whether 17-AAG synergizes with other antileukemic agents.
Acknowledgments
We gratefully acknowledge generous gifts of antibodies from Junjie Chen, Guy Poirier, and David Toft; editorial assistance of Deb Strauss; and flow cytometric analysis by the Mayo Clinic Flow Cytometry Shared Resource.
Prepublished online as Blood First Edition Paper, March 22, 2005; DOI 10.1182/blood-2004-09-3523.
Supported in part by R01 CA73709 (S.H.K.), R01 CA104378 (L.M.K.), R01 CA90390 (C.E.), K23 CA96780 (R.A.M.), and a grant from the Jack Taylor Family Foundation.
R.A.M. and D.L. contributed equally to this study; S.H.K. and L.M.K. contributed equally to this study.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 U.S.C. section 1734.
References
- 1.Chabner BA. Cytidine analogues. In: Chabner BA, Longo DL, eds. Cancer Chemotherapy and Biotherapy. Philadelphia, PA: Lippincott-Raven; 1996: 213-233.
- 2.Rowe JM, Tallman MS. Intensifying induction therapy in acute myeloid leukemia: has a new standard of care emerged? Blood. 1997;90: 2121-2126. [PubMed] [Google Scholar]
- 3.Tallman MS. Therapy of acute myeloid leukemia. Cancer Control. 2001;8: 62-78. [DOI] [PubMed] [Google Scholar]
- 4.Cripe LD, Hinton S. Acute myeloid leukemia in adults. Curr Treat Options Oncol. 2000;1: 9-17. [DOI] [PubMed] [Google Scholar]
- 5.Estey EH, Thall PF, Cortes JE, et al. Comparison of idarubicin + ara-C-, fludarabine + ara-C-, and topotecan + ara-C-based regimens in treatment of newly diagnosed acute myeloid leukemia, refractory anemia with excess blasts in transformation, or refractory anemia with excess blasts. Blood. 2001;98: 3575-3583. [DOI] [PubMed] [Google Scholar]
- 6.Schiffer CA. Acute myeloid leukemia in adults: where do we go from here? Cancer Chemother Pharmacol. 2001;48(suppl 1): S45-S52. [DOI] [PubMed] [Google Scholar]
- 7.Stone RM. Treatment of acute myeloid leukemia: state-of-the-art and future directions. Semin Hematol. 2002;39: 4-10. [DOI] [PubMed] [Google Scholar]
- 8.Huang M, Wang Y, Collins M, Gu JJ, Mitchell BS, Graves LM. Inhibition of nucleoside transport by p38 MAPK inhibitors. J Biol Chem. 2002;277: 28364-28367. [DOI] [PubMed] [Google Scholar]
- 9.Steuart CD, Burke PJ. Cytidine deaminase and the development of resistance to arabinosyl cytosine. Nat New Biol. 1971;233: 109-110. [DOI] [PubMed] [Google Scholar]
- 10.Liliemark JO, Plunkett W, Dixon DO. Relationship of 1-beta-D-arabinofuranosylcytosine in plasma to 1-beta-D-arabinofuranosylcytosine 5′-triphosphate levels in leukemic cells during treatment with high-dose 1-beta-D-arabinofuranosylcytosine. Cancer Res. 1985;45: 5952-5957. [PubMed] [Google Scholar]
- 11.Karp JE, Donehower RC, Dole GB, Burke PJ. Correlation of drug-perturbed marrow cell growth kinetics and intracellular 1-B-D-arabinofuranosyl-cytosine metabolism with clinical response in adult acute myelogenous leukemia. Blood. 1987;69: 1134-1140. [PubMed] [Google Scholar]
- 12.Karp JE, Donehower RC, Enterline JP, Dole GB, Fox MG, Burke PJ. In vivo cell growth and pharmacologic determinants of clinical response in acute myelogenous leukemia. Blood. 1989;73: 24-30. [PubMed] [Google Scholar]
- 13.Karp JE, Donehower RC, Dole GB, Burke PJ. Direct relationship of marrow cell growth and 1-beta-D-arabinofuranosylcytosine metabolism. Cancer Res. 1984;44: 5046-5050. [PubMed] [Google Scholar]
- 14.Gandhi V, Estey E, Keating MJ, Plunkett W. Fludarabine potentiates metabolism of cytarabine in patients with acute myelogenous leukemia during therapy. J Clin Oncol. 1993;11: 116-124. [DOI] [PubMed] [Google Scholar]
- 15.Gandhi V, Estey E, Du M, Nowak B, Keating MJ, Plunkett W. Modulation of the cellular metabolism of cytarabine and fludarabine by granulocyte-colony-stimulating factor during therapy of acute myelogenous leukemia. Clin Cancer Res. 1995;1: 169-178. [PubMed] [Google Scholar]
- 16.Lowenberg B, van Putten W, Theobald M, et al. Effect of priming with granulocyte colony-stimulating factor on the outcome of chemotherapy for acute myeloid leukemia. N Engl J Med. 2003;349: 743-752. [DOI] [PubMed] [Google Scholar]
- 17.Hekmat-Nejad M, You Z, Yee MC, Newport JW, Cimprich KA. Xenopus ATR is a replication-dependent chromatin-binding protein required for the DNA replication checkpoint. Curr Biol. 2000;10: 1565-1573. [DOI] [PubMed] [Google Scholar]
- 18.Feijoo C, Hall-Jackson C, Wu R, et al. Activation of mammalian Chk1 during DNA replication arrest: a role for Chk1 in the intra-S phase checkpoint monitoring replication origin firing. J Cell Biol. 2001;154: 913-923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhao H, Piwnica-Worms H. ATR-mediated checkpoint pathways regulate phosphorylation and activation of human Chk1. Mol Cell Biol. 2001;21: 4129-4139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu Q, Guntuku S, Cui XS, et al. Chk1 is an essential kinase that is regulated by Atr and required for the G(2)/M DNA damage checkpoint. Genes Dev. 2000;14: 1448-1459. [PMC free article] [PubMed] [Google Scholar]
- 21.Zachos G, Rainey MD, Gillespie DA. Chk1-deficient tumour cells are viable but exhibit multiple checkpoint and survival defects. EMBO J. 2003;22: 713-723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kastan MB, Bartek J. Cell-cycle checkpoints and cancer. Nature. 2004;432: 316-323. [DOI] [PubMed] [Google Scholar]
- 23.O'Connell MJ, Cimprich KA. G2 damage checkpoints: what is the turn-on? J Cell Sci. 2005;118: 1-6. [DOI] [PubMed] [Google Scholar]
- 24.Mailand N, Falck J, Lukas C, et al. Rapid destruction of human Cdc25A in response to DNA damage. Science. 2000;288: 1425-1429. [DOI] [PubMed] [Google Scholar]
- 25.Zhao H, Watkins JL, Piwnica-Worms H. Disruption of the checkpoint kinase 1/cell division cycle 25A pathway abrogates ionizing radiation-induced S and G2 checkpoints. Proc Natl Acad Sci U S A. 2002;99: 14795-14800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shimuta K, Nakajo N, Uto K, Hayano Y, Okazaki K, Sagata N. Chk1 is activated transiently and targets Cdc25A for degradation at the Xenopus midblastula transition. EMBO J. 2002;21: 3694-3703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sorensen CS, Syljuasen RG, Falck J, et al. Chk1 regulates the S phase checkpoint by coupling the physiological turnover and ionizing radiation-induced accelerated proteolysis of Cdc25A. Cancer Cell. 2003;3: 247-258. [DOI] [PubMed] [Google Scholar]
- 28.Sagata N. Molecular biology. Untangling checkpoints. Science. 2002;298: 1905-1907. [DOI] [PubMed] [Google Scholar]
- 29.Shi Z, Azuma A, Sampath D, Li YX, Huang P, Plunkett W. S-phase arrest by nucleoside analogues and abrogation of survival without cell cycle progression by 7-hydroxystaurosporine. Cancer Res. 2001;61: 1065-1072. [PubMed] [Google Scholar]
- 30.Sampath D, Shi Z, Plunkett W. Inhibition of cyclin-dependent kinase 2 by the Chk1-Cdc25A pathway during the S-phase checkpoint activated by fludarabine: dysregulation by 7-hydroxystaurosporine. Mol Pharmacol. 2002;62: 680-688. [DOI] [PubMed] [Google Scholar]
- 31.Wang S, Wang Z, Grant S. Bryostatin 1 and UCN-01 potentiate 1-beta-D-arabinofuranosylcytosine-induced apoptosis in human myeloid leukemia cells through disparate mechanisms. Mol Pharmacol. 2003;63: 232-242. [DOI] [PubMed] [Google Scholar]
- 32.Neckers L, Ivy SP. Heat shock protein 90. Curr Opin Oncol. 2003;15: 419-424. [DOI] [PubMed] [Google Scholar]
- 33.Neckers L. Hsp90 inhibitors as novel cancer chemotherapeutic agents. Trends Mol Med. 2002;8: S55-S61. [DOI] [PubMed] [Google Scholar]
- 34.Pratt W, Toft D. Regulation of signaling protein function and trafficking by the hsp90/hsp70-based chaperone machinery. Exp Biol Med. 2003;228: 111-133. [DOI] [PubMed] [Google Scholar]
- 35.Fumo G, Akin C, Metcalfe DD, Neckers L. 17-allylamino-17-demethoxygeldanamycin (17-AAG) is effective in down-regulating mutated, constitutively activated KIT protein in human mast cells. Blood. 2004;103: 1078-1084. [DOI] [PubMed] [Google Scholar]
- 36.Yao Q, Nishiuchi R, Li Q, Kumar AR, Hudson WA, Kersey JH. FLT3 expressing leukemias are selectively sensitive to inhibitors of the molecular chaperone heat shock protein 90 through destabilization of signal transduction-associated kinases. Clin Cancer Res. 2003;9: 4483-4493. [PubMed] [Google Scholar]
- 37.Basso AD, Solit DB, Munster PN, Rosen N. Ansamycin antibiotics inhibit Akt activation and cyclin D expression in breast cancer cells that overexpress HER2. Oncogene. 2002;21: 1159-1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Basso AD, Solit DB, Chiosis G, Giri B, Tsichlis P, Rosen N. Akt forms an intracellular complex with heat shock protein 90 (Hsp90) and Cdc37 and is destabilized by inhibitors of Hsp90 function. J Biol Chem. 2002;277: 39858-39866. [DOI] [PubMed] [Google Scholar]
- 39.Nimmanapalli R, O'Bryan E, Bhalla K. Geldanamycin and its analogue 17-allylamino-17-demethoxygeldanamycin lowers Bcr-Abl levels and induces apoptosis and differentiation of Bcr-Abl-positive human leukemic blasts. Cancer Res. 2001;61: 1799-1804. [PubMed] [Google Scholar]
- 40.Blagosklonny MV, Fojo T, Bhalla KN, et al. The Hsp90 inhibitor geldanamycin selectively sensitizes Bcr-Abl-expressing leukemia cells to cytotoxic chemotherapy. Leukemia. 2001;15: 1537-1543. [DOI] [PubMed] [Google Scholar]
- 41.Isaacs JS, Xu W, Neckers L. Heat shock protein 90 as a molecular target for cancer therapeutics. Cancer Cell. 2003;3: 213-217. [DOI] [PubMed] [Google Scholar]
- 42.Goetz MP, Toft DO, Ames MM, Erlichman C. The Hsp90 chaperone complex as a novel target for cancer therapy. Ann Oncol. 2003;14: 1169-1176. [DOI] [PubMed] [Google Scholar]
- 43.Grenert JP, Sullivan WP, Faddan P, et al. The amino-terminal domain of heat shock protein 90 (hsp90) that binds geldanamycin is an ATP/ADP switch domain that regulates hsp90 conformation. J Biol Chem. 1997;272: 23843-23850. [DOI] [PubMed] [Google Scholar]
- 44.Schneider C, Sepp-Lorenzino L, Nimmesgern E, et al. Pharmacologic shifting of a balance between protein refolding and degradation mediated by Hsp90. Proc Natl Acad Sci U S A. 1996;93: 14536-14541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stebbins CE, Russo AA, Schneider C, Rosen N, Hartl FU, Pavletich NP. Crystal structure of an Hsp90-geldanamycin complex: targeting of a protein chaperone by an antitumor agent. Cell. 1997;89: 239-250. [DOI] [PubMed] [Google Scholar]
- 46.Kamal A, Thao L, Sensintaffar J, et al. A high-affinity conformation of Hsp90 confers tumour selectivity on Hsp90 inhibitors. Nature. 2003;425: 407-410. [DOI] [PubMed] [Google Scholar]
- 47.Workman P. Auditing the pharmacological accounts for Hsp90 molecular chaperone inhibitors: unfolding the relationship between pharmacokinetics and pharmacodynamics. Mol Cancer Ther. 2003;2: 131-138. [PubMed] [Google Scholar]
- 48.Goetz MP, Toft DO, Reid JM, et al. A phase I trial of 17-allylamino-17-demethoxygeldanamycin in patients with advanced cancer. J Clin Oncol. 2005;23: 1078-1087. [DOI] [PubMed] [Google Scholar]
- 49.Erlichman C, Toft DO, Reid JM, et al. A phase I trial of 17-allyl-amino-geldanamycin in patients with advanced cancer [abstract]. Proc Am Assoc Cancer Res. 2001;42: 833. [Google Scholar]
- 50.Arlander SJH, Eapen AK, Vroman BT, McDonald RJ, Toft DO, Karnitz LM. Hsp90 inhibition depletes Chk1 and sensitizes tumor cells to replication stress. J Biol Chem. 2003;278: 52572-52577. [DOI] [PubMed] [Google Scholar]
- 51.Mow BMF, Chandra J, Svingen PA, et al. Effects of the Bcr/abl kinase inhibitors STI571 and adaphostin (NSC 680410) on chronic myelogenous leukemia cells in vitro. Blood. 2002;99: 664-671. [DOI] [PubMed] [Google Scholar]
- 52.Martins LM, Mesner PW, Kottke TJ, et al. Comparison of caspase activation and subcellular localization in HL-60 and K562 cells undergoing etoposide-induced apoptosis. Blood. 1997;90: 4283-4296. [PubMed] [Google Scholar]
- 53.Nicoletti I, Migliorati G, Pagliacci MC, Grignani F, Riccardi C. A rapid and simple method for measuring thymocyte apoptosis by propidium iodide staining and flow cytometry. J Immunol Methods. 1991;139: 271-279. [DOI] [PubMed] [Google Scholar]
- 54.Meng XW, Chandra J, Loegering D, et al. Central role of Fas-associated death domain protein in apoptosis induction by the mitogen-activated protein kinase kinase inhibitor CI-1040 (PD184352) in acute lymphocytic leukemia cells in vitro. J Biol Chem. 2003;278: 47236-47339. [DOI] [PubMed] [Google Scholar]
- 55.Kaufmann SH. Induction of endonucleolytic DNA cleavage in human acute myelogenous leukemia cells by etoposide, camptothecin, and other cytotoxic anticancer drugs: a cautionary note. Cancer Res. 1989;49: 5870-5878. [PubMed] [Google Scholar]
- 56.Earnshaw WC, Martins LM, Kaufmann SH. Mammalian caspases: structure, activation, substrates and functions during apoptosis. Annu Rev Biochem. 1999;68: 383-424. [DOI] [PubMed] [Google Scholar]
- 57.Pike BL, Robinson WA. Human bone marrow colony growth in agar-gel. J Cell Physiol. 1970;76: 77-84. [DOI] [PubMed] [Google Scholar]
- 58.Sato S, Fujita N, Tsuruo T. Involvement of 3-phosphoinositide-dependent protein kinase-1 in the MEK/MAPK signal transduction pathway. J Biol Chem. 2004;279: 33759-33767. [DOI] [PubMed] [Google Scholar]
- 59.Flatten K, Dai NT, Vroman BT, et al. The role of checkpoint kinase 1 in sensitivity to topoisomerase I poisons. J Biol Chem. 2005;280: 14349-14355. [DOI] [PubMed] [Google Scholar]
- 60.Bible K, Kaufmann SH. Cytotoxic synergy between flavopiridol (NSC 649890, L86–8275) and various antineoplastic agents: importance of sequence of administration. Cancer Res. 1997;57: 3375-3380. [PubMed] [Google Scholar]
- 61.Kaufmann SH, Svingen PA, Gore SD, Armstrong DK, Cheng Y-C, Rowinsky EK. Altered formation of topotecan-stabilized topoisomerase I-DNA adducts in human leukemia cells. Blood. 1997;89: 2098-2104. [PubMed] [Google Scholar]
- 62.Smith PK, Krohn RI, Hermanson GT, et al. Measurement of protein using bicinchoninic acid. Anal Biochem. 1985;150: 76-85. [DOI] [PubMed] [Google Scholar]
- 63.Kaufmann SH. Reutilization of immunoblots after chemiluminescent detection. Anal Biochem. 2001;296: 283-286. [DOI] [PubMed] [Google Scholar]
- 64.Roos-Mattjus P, Hopkins KM, Vroman BT, et al. Phosphorylation of human Rad9 is required for genotoxin-activated checkpoint signaling. J Biol Chem. 2003;278: 24428-24437. [DOI] [PubMed] [Google Scholar]
- 65.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72: 248-254. [DOI] [PubMed] [Google Scholar]
- 66.Berenbaum MC. What is synergy? Pharmacol Rev. 1989;41: 93-141. [PubMed] [Google Scholar]
- 67.Donehower RC, Karp JE, Burke PJ. Pharmacology and toxicity of high-dose cytarabine by 72-hour continuous infusion. Cancer Treat Rep. 1986;70: 1059-1065. [PubMed] [Google Scholar]
- 68.Loegering D, Arlander SJH, Hackbarth J, et al. Rad9 protects cells from topoisomerase poison-induced cell death. J Biol Chem. 2004;279: 18641-18647. [DOI] [PubMed] [Google Scholar]
- 69.Nicholson DW, Ali A, Thornberry NA, et al. Identification and inhibition of the ICE/CED-3 protease necessary for mammalian apoptosis. Nature. 1995;376: 37-43. [DOI] [PubMed] [Google Scholar]
- 70.Takahashi A, Alnemri ES, Lazebnik YA, et al. Cleavage of lamin A by Mch2 alpha but not CPP32: multiple interleukin 1 beta-converting enzyme-related proteases with distinct substrate recognition properties are active in apoptosis. Proc Natl Acad Sci U S A. 1996;93: 8395-8400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Fernandes-Alnemri T, Takahashi A, Armstrong R, et al. Mch3, a novel human apoptotic cysteine protease highly related to CPP32. Cancer Res. 1995;55: 6045-6052. [PubMed] [Google Scholar]
- 72.Duan H, Orth K, Chinnaiyan AM, et al. ICE-LAP6, a novel member of the ICE/Ced-3 gene family, is activated by the cytotoxic T cell protease granzyme B. J Biol Chem. 1996;271: 16720-16724. [DOI] [PubMed] [Google Scholar]
- 73.Hostein I, Robertson D, DiStefano F, Workman P, Clarke PA. Inhibition of signal transduction by the Hsp90 inhibitor 17-allylamino-17-demethoxy-geldanamycin results in cytostasis and apoptosis. Cancer Res. 2001;61: 4003-4009. [PubMed] [Google Scholar]
- 74.Nimmanapalli R, O'Bryan E, Kuhn D, Yamaguchi H, Wang HG, Bhalla KN. Regulation of 17-AAG-induced apoptosis: role of Bcl-2, Bcl-XL, and Bax downstream of 17-AAG-mediated down-regulation of Akt, Raf-1, and Src kinases. Blood. 2003;102: 269-275. [DOI] [PubMed] [Google Scholar]
- 75.Yamaki H, Nakajima M, Seimiya H, Saya H, Sugita M, Tsuruo T. Inhibition of the association with nuclear matrix of pRB, p70 and p40 proteins along with the specific suppression of c-MYC expression by geldanamycin, an inhibitor of Src tyrosine kinase. J Antibiot. 1995;48: 1021-1026. [DOI] [PubMed] [Google Scholar]
- 76.McIlwrath AJ, Brunton VG, Brown R. Cell-cycle arrest and p53 accumulation induced by geldanamycin in human ovarian tumour cells. Cancer Chemother Pharmacol. 1996;37: 423-428. [DOI] [PubMed] [Google Scholar]
- 77.Munster PN, Srethapakdi M, Moasser MM, Rosen N. Inhibition of heat shock protein 90 function by ansamycins causes the morphological and functional differentiation of breast cancer cells. Cancer Res. 2001;61: 2945-2952. [PubMed] [Google Scholar]
- 78.Iscove NN, Senn JS, Till JE, McCulloch EA. Colony formation by normal and leukemic human marrow cells in culture: effect of conditioned medium from human leukocytes. Blood. 1971;37: 1-5. [PubMed] [Google Scholar]
- 79.Griffin JD, Lowenberg B. Clonogenic cells in acute myeloblastic leukemia. Blood. 1986;68: 1185-1195. [PubMed] [Google Scholar]
- 80.Hu ZB, Yang GS, Li M, Miyamoto N, Minden MD, McCulloch EA. Mechanism of cytosine arabinoside toxicity to the blast cells of acute myeloblastic leukemia: involvement of free radicals. Leukemia. 1995;9: 789-798. [PubMed] [Google Scholar]
- 81.Konopleva M, Tari AM, Estrov Z, et al. Liposomal Bcl-2 antisense oligonucleotides enhance proliferation, sensitize acute myeloid leukemia to cytosine-arabinoside, and induce apoptosis independent of other antiapoptotic proteins. Blood. 2000;95: 3929-3938. [PubMed] [Google Scholar]
- 82.Chou T-C, Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 1984;22: 27-55. [DOI] [PubMed] [Google Scholar]
- 83.Xiang J, Chao DT, Korsmeyer SJ. BAX-induced cell death may not require interleukin 1 beta-converting enzyme-like proteases. Proc Natl Acad Sci U S A. 1996;93: 14559-14563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Cheng EH, Wei MC, Weiler S, et al. BCL-2, BCL-X(L) sequester BH3 domain-only molecules preventing BAX- and BAK-mediated mitochondrial apoptosis. Mol Cell. 2001;8: 705-711. [DOI] [PubMed] [Google Scholar]
- 85.Koopman G, Reutelingsperger CPM, Kuijten GAM, Keehnen RMJ, Pals ST, van Oers MHJ. Annexin V for flow cytometric detection of phosphatidylserine expression on B cells undergoing apoptosis. Blood. 1994;84: 1415-1420. [PubMed] [Google Scholar]
- 86.Lazebnik YA, Kaufmann SH, Desnoyers S, Poirier GG, Earnshaw WC. Cleavage of poly(ADP-ribose) polymerase by a proteinase with properties like ICE. Nature. 1994;371: 346-347. [DOI] [PubMed] [Google Scholar]
- 87.Datta SR, Brunet A, Greenberg ME. Cellular survival: a play in three Akts. Genes Dev. 1999;13: 2905-2927. [DOI] [PubMed] [Google Scholar]
- 88.Wolf D, Rotter V. Major deletions in the gene encoding the p53 tumor antigen cause lack of p53 expression in HL-60 cells. Proc Natl Acad Sci U S A. 1985;82: 790-794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Delia D, Aiello A, Soligo D, et al. bcl-2 proto-oncogene expression in normal and neoplastic human myeloid cells. Blood. 1992;79: 1291-1298. [PubMed] [Google Scholar]
- 90.Kaufmann SH, Karp JE, Svingen PA, Krajewski S, Burke PJ, Gore SD. Elevated expression of the apoptotic regulator Mcl-1 at the time of leukemic relapse. Blood. 1998;91: 991-1000. [PubMed] [Google Scholar]
- 91.Kastan MB, Onyekwere O, Sidransky D, Vogelstein B, Craig RW. Participation of p53 protein in the cellular response to DNA damage. Cancer Res. 1991;51: 6304-6311. [PubMed] [Google Scholar]
- 92.Killmann S-A. Proliferative activity of blast cells in leukemia and myelofibrosis. Morphological differences between proliferating and non-proliferating blast cells. Acta Med Scand. 1965;178: 263-280. [DOI] [PubMed] [Google Scholar]
- 93.Mauer AM, Fisher V. Comparison of the proliferative capacity of acute leukaemia cells in bone marrow and blood. Nature. 1962;193: 1085-1086. [DOI] [PubMed] [Google Scholar]
- 94.Graham FL, Whitmore GF. Studies in mouse L-cells on the incorporation of 1-beta-D-arabinofuranosylcytosine into DNA and on inhibition of DNA polymerase by 1-beta-D-arabinofuranosylcytosine 5′-triphosphate. Cancer Res. 1970;30: 2636-2644. [PubMed] [Google Scholar]
- 95.Huang P, Chubb S, Hertel LW, Grindey GB, Plunkett W. Action of 2′,2′-difluorodeoxycytidine on DNA synthesis. Cancer Res. 1991;51: 6110-6117. [PubMed] [Google Scholar]
- 96.Bhalla K, Birkhofer M, Arlin Z, et al. Differential effect of interleukin 3 on the metabolism of high-dose cytosine arabinoside in normal versus leukemic human bone marrow cells. Exp Hematol. 1991;19: 669-673. [PubMed] [Google Scholar]
- 97.Hiddemann W, Kiehl M, Zuhlsdorf M, et al. Granulocyte-macrophage colony-stimulating factor and interleukin-3 enhance the incorporation of cytosine arabinoside into the DNA of leukemic blasts and the cytotoxic effect on clonogenic cells from patients with acute myeloid leukemia. Semin Oncol. 1992;19: 31-37. [PubMed] [Google Scholar]
- 98.Gilliland DG, Griffin JD. The roles of FLT3 in hematopoiesis and leukemia. Blood. 2002;100: 1532-1542. [DOI] [PubMed] [Google Scholar]
- 99.Levis M, Small D. FLT3: ITDoes matter in leukemia. Leukemia. 2003;17: 1738-1752. [DOI] [PubMed] [Google Scholar]