

Abstract
In mucopolysaccharidosis-I (MPS-I), α-L-iduronidase deficiency leads to progressive heparan sulfate (HS) and dermatan sulfate (DS) glycosaminoglycan (GAG) accumulation. The functional consequences of these accumulated molecules are unknown. HS critically influences tissue morphogenesis by binding to and modulating the activity of several cytokines (eg, fibroblast growth factors [FGFs]) involved in developmental patterning. We recently isolated a multipotent progenitor cell from postnatal human bone marrow, which differentiates into cells of all 3 embryonic lineages. The availability of multipotent progenitor cells from healthy volunteers and patients with MPS-I (Hurler syndrome) provides a unique opportunity to directly examine the functional effects of abnormal HS on cytokine-mediated stem-cell proliferation and survival. We demonstrate here that abnormally sulfated HS in Hurler multipotent progenitor cells perturb critical FGF-2–FGFR1-HS interactions, resulting in defective FGF-2–induced proliferation and survival of Hurler multipotent progenitor cells. Both the mitogenic and survival-promoting activities of FGF-2 were restored by substitution of Hurler HS by normal HS. This perturbation of critical HS–cytokine receptor interactions may represent a mechanism by which accumulated HS contributes to the developmental pathophysiology of Hurler syndrome. Similar mechanisms may operate in the pathogenesis of other diseases where structurally abnormal GAGs accumulate.
Introduction
Hurler syndrome (mucopolysaccharidosis [MPS] type I) is an inborn error of glycosaminoglycan (GAG) metabolism caused by deficiency of α-L-iduronidase required for heparan sulfate (HS) and dermatan sulfate (DS) degradation.1-3 HS and DS accumulation leads to cell and organ dysfunction. While progressive neuropsychologic dysfunction is a hallmark of Hurler and related storage diseases,4 its cellular and molecular mechanisms remain undefined.
The fibroblast growth factor (FGF) cytokine family affects cell growth, migration, differentiation, and neuroectodermal development.5 FGF-2, a prototypical member involved in tissue morphogenesis and neurogenesis,6 has 2 kinds of cell-surface receptors. Specific, high-affinity FGF receptors (FGFRs) possessing intrinsic tyrosine kinase activity mediate cellular responses, while low-affinity receptors composed of HS proteoglycans (HSPGs) act as extracellular FGF-2 reservoirs and coreceptors.7 Although its mechanism of formation is unclear, the FGF-2–FGFR-HSPG complex is necessary for mitogenesis and optimal biologic response to FGF-2.8,9
Cell-type–specific and developmentally regulated HSPG synthesis regulates cell growth and differentiation by modulating extracellular signaling molecule activity.10-12 Specificity of interactions between HSPG and signaling molecules depends on HSPG sequence, location, and 3-dimensional structure.11,13 The importance of HSPG in FGF signaling is underscored by their role in neurogenesis, axonal guidance, and synapse formation.12,14 FGF-2 also protects neurons against apoptosis and promotes neuronal survival.15
While GAGs synthesized by cultured Hurler fibroblasts are thought to be structurally normal,16 GAGs that accumulate in a tissue-specific manner17 consist of a combination of large, normally sized GAGs and partially degraded, smaller chains (oligosaccharides).18,19 How accumulated oligosaccharides cause abnormal tissue development and function remains uncertain. Our previous studies on normal hematopoiesis suggest that a high concentration of small, abnormally sulfated HS chains (such as may be present in Hurler syndrome) are detrimental to orderly stem-cell growth and differentiation.20-22 We hypothesize that abnormal size and sulfation of accumulated HS oligosaccharides leads to aberrant functional properties of HS, including their capability to modulate GAG-binding cytokine activity, which may contribute to Hurler syndrome pathophysiology.
Clinically, following allogeneic hematopoietic stem-cell (HSC) transplantation, donor-derived tissue macrophages secrete α-L-iduronidase, which ameliorates clinical manifestations of Hurler syndrome and prolongs life.23-25 However, the hematopoietic system itself is not affected in Hurler syndrome. In vitro, a major limitation to examining the pathophysiologic role of HS remains the inability to obtain primary cells representative of diverse nonhematopoietic tissues such as the brain and skeletal system that are particularly affected in Hurler syndrome and not optimally corrected by HSC transplantation. We recently identified a multipotent, nonhematopoietic stem cell in postnatal human and rodent bone marrow (multipotent adult progenitor cell [MAPC])26,27 that now provides a powerful model for studying the effect of HS on cytokine signaling and nonhematopoietic stem-cell growth and differentiation. For the current studies, we used MAPCs rather than HSCs since (1) MAPCs expand without senescence, yet can be induced to differentiate into multiple mesodermal, endodermal, and ectodermal lineages26-30; (2) MAPCs acquire neural and glial phenotypes when cultured with FGF-231; (3) MAPCs represent an in vitro model for defining the effect of abnormal HS on progenitors of nonhematopoietic tissues affected by Hurler syndrome; and (4) transplantation of nonhematopoietic progenitor/stem cells is being tested as a therapeutic modality for Hurler syndrome.32 We examined if FGF-2–FGFR-HS interactions are abnormal in Hurler syndrome, and if this affects MAPC proliferation and survival.
We demonstrate that Hurler MAPC HSs are small and abnormally sulfated and have an impaired capability to induce FGF-2 binding to receptor FGFR1 and to mediate its biologic activity. The presence of normal HS or normal MAPCs corrects these abnormalities. These studies identify a mechanism by which accumulated HS may contribute to the developmental pathophysiology of Hurler syndrome by impairing critical HS–cytokine receptor interactions.
Materials and methods
Isolation and in vitro expansion of human MAPCs
The University of Minnesota Human Subjects Committee approved these studies. Informed consent was provided in accordance with the Declaration of Helsinki. MAPCs selected from normal bone marrow (BM) were expanded in culture as described.26,29 Briefly, BM mononuclear cells obtained by Ficoll-Hypaque density gradient centrifugation were depleted of CD45- and glycophorin-A (GlyA)–expressing cells using micromagnetic beads (Miltenyi Biotec, Sunnyvale, CA). CD45-/GlyA- cells were plated at 5000 cells/well in 96-well plates coated with 5 ng/mL fibronectin (Becton Dickinson, Franklin Lakes, NJ) in MAPC medium containing 54% low-glucose DMEM (Invitrogen, Grand Island, NY), 40% MCDB-201 (Sigma-Aldrich, St Louis, MO), 2% fetal calf serum (FCS; Hyclone Laboratories, Logan, UT), 1x insulin-transferrin-selenium, 1x linoleic acid–bovine serum albumin (BSA), 5 × 10-8 M dexamethasone, 10-4 M ascorbic acid 2-phosphate (all from Sigma), 1x penicillin/streptomycin (Invitrogen), 10 ng/mL epidermal growth factor (EGF; Sigma), and 10 ng/mL platelet-derived growth factor–BB (PDGF-BB; R&D Systems, Minneapolis, MN). Medium was changed every 4 to 5 days. Cells were replated in larger fibronectin-coated vessels to maintain a density of 1500 to 2000 cells/cm2. All cultures were incubated at 37°C in humidified 5% CO2 atmosphere.
Hurler MAPCs were similarly selected and expanded from a BM aliquot taken from patients with Hurler syndrome undergoing autologous BM harvesting prior to HSC transplantation. We confirmed that functional α-L-iduronidase enzyme activity was present in normal MAPCs but not in Hurler MAPCs (data not shown). Sera used for all studies in this paper were heat-inactivated (2 hours at 55°C) to inactivate bovine α-L-iduronidase.33
Metabolic labeling and purification of proteoglycans and HS
MAPCs grown to 80% confluence in MAPC medium were irradiated to 1600 rads. After 1 week, medium was changed to labeling medium (Ham F-12 Medium [Gibco BRL] plus 5% FCS and 10-6 M hydrocortisone) for 2 hours. Proteoglycans were metabolically labeled by adding 2.59 MBq (70 μCi)/mL to 2.77 MBq (75 μCi)/mL Na235SO4 (to label sulfate groups) and 740 KBq (20 μCi)/mL 3H-glucosamine (to label glucosamine monosaccharides in the polysaccharide backbone) in fresh Ham F-12 for 24 hours. After collecting supernatants, cell-surface plus extracellular matrix (ECM) PGs were obtained by treating cell layers with trypsin/EDTA (ethylenediaminetetraacetic acid), and intracellular PGs obtained by lysing cell pellets with 1% Triton X-100. Samples were dialyzed extensively, and proteoglycans enriched by diethylaminoethyl (DEAE)–Sephacel anion exchange HPLC (Beckman, CA), as described.20,21 Each proteoglycan peak resolved on HPLC was separately processed to purify HS for further analysis. To remove residual proteins, nucleic acids, and CS/DS GAGs, proteoglycans from each peak were sequentially digested with proteinase K (Sigma), chondroitinase ABC (Seikagaku America, Falmouth, MA), DNAse (Sigma), and RNAse (Sigma). HS chains were released from core proteins by alkaline sodium borohydride (NaBH4).21 Purified HS chains were separated from proteins, peptides, and mono- and disaccharides using Sephadex G-50 gel filtration chromatography followed by repeat anion exchange HPLC. Purity of HS preparations was consistently more than 95%, as determined by deaminative cleavage using low pH nitrous acid followed by Sephadex G50 gel filtration chromatography. HS purified from each HPLC peak was analyzed separately.
Size distribution of HS
The size of HS was estimated by gel filtration chromatography on a 0.75 × 95 cm Sepharose CL-6B column equilibrated in 4 M guanidine hydrochloride and 0.05 M sodium acetate, pH 5.8. The column was eluted at 3 mL/hour, and 500-μL fractions were collected. The approximate size (Mr) of HS was estimated by the method of Wasteson,34 as previously described.21
Preparation and HPLC analysis of HS disaccharides
The disaccharide composition of HS in normal and Hurler MAPCs was determined as recently described by us.35 A major advantage of this method is that it can be used to study HS from small numbers of cultured cells.
Samples of at least 3 × 106 cells were washed in PBS, scraped from the flasks, pelleted, and resuspended in 0.5 mL 50 mM Tris/HCl pH 8, 1 mM CaCl2, 1% Triton X-100, and sonicated 3 times for 30 seconds at 3 W to 4 W before protein estimation (Pierce BCA Assay). They were digested with 0.8 mg/mL protease for 16 hours at 55°C, heat inactivated at 96°C for 5 minutes, then incubated for 2 hours at 37°C with 1 μL 1 M MgCl2 and 0.5 μL (168.5 U) benzonase. NaCl (0.1 M) was added and insoluble material removed by microcentrifugation. The solution was applied on an Ultrafree-MC DEAE membrane equilibrated with sodium phosphate buffer (pH 6.0) containing 0.15 M NaCl. Fractions were eluted with 1.0 M NaCl in the same buffer, desalted with Biomax-5, lyophilized, and resuspended in 15 μL 0.03 M acetate buffer (pH 7.0) with 3.33 mM calcium acetate, 0.33 mIU heparinase, 0.33 mIU heparitinase II, and 0.33 mIU heparitinase I. The mixture was incubated at 37°C for 16 hours, lyophilized, and resuspended in 12 μL water to load onto the HPLC. HPLC conditions were as in Toyoda et al.35 Normal and Hurler HSs each comprised 6 major disaccharide types, assigned according to the elution positions of HS disaccharide standards. The disaccharide composition of each HS type was determined from the area under the peaks.
Binding of 125I-FGF2 to FGFR1 in presence of cell-surface HS
Heterodimerization of 125I-FGF-2 to cell-surface FGFR1 in the presence of normal or Hurler HS (formation of the FGF-2–FGFR1-HS complex) was determined as previously described.36 Normal and Hurler MAPCs were grown in 6-well plates until 80% confluent. After washing twice with serum-free, low sulfate Ham F-12 medium without penicillin/streptomycin, cells were incubated for 2 hours at 4°C with or without 25 ng/mL 125I-FGF2 (Perkin Elmer, Boston, MA), washed twice with PBS, then incubated with or without 0.25 mM disuccinimidyl suberate (DSS) in PBS for 30 minutes at room temperature (to cross-link the 125I-FGF–2-FGFR1 complex), and lysed with 100 μL lysis buffer. Proteins in the lysate were separated by 7.5% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to a polyvinylidene difluoride (PVDF) membrane. The membrane was first autoradiographed on a Kodak film to visualize 125I-FGF2, then immunoblotted with an anti-FGFR1 polyclonal antibody (flg [H-76], cat no. SC-7945, Santa Cruz; 1:500 dilution). FGFR1 was detected using ECF (Amersham Biosciences, Piscataway, NJ). Densitometric analysis was performed using NIH Image 1.60b7. Equivalent loading of all lanes was confirmed by reprobing the membrane for β-actin (cat no. SC-1616, Santa Cruz; 1:1000 dilution; not shown).
Binding of 125I-FGF-2 to normal and Hurler MAPCs
Normal and Hurler MAPCs were seeded at 105 cells/well in 24-well plates. After overnight adherence, cells were placed on ice for 5 minutes, then washed twice with binding buffer (low sulfate Ham F-12 medium plus 0.1% BSA) and blocked with 3% BSA in Ham F-12 for 30 minutes at room temperature. Then 62.5 pM (2 × 105 cpm) 125I-FGF-2 (specific activity, 87.5 μCi/μg) was added in 500 μL binding buffer and plates incubated at 4°C for 2 hours on a rotary shaker. Cells were washed thrice with binding buffer and then 4 times with 2 M NaCl at pH 7.4, to remove nonspecifically bound 125I-FGF-2.7 Specifically bound 125I-FGF-2 was quantified by washing the cells with 2 M NaCl at low pH (4.0) twice and counting radioactivity released in the washes on a γ-counter.7
To examine the effect of MAPC conditioned medium (CM) on 125I-FGF-2 binding, the binding buffer was incubated with normal or Hurler MAPCs for 24 hours. This CM was then used at 50% concentration in some wells during the 125I-FGF-2 binding step (normal CM on Hurler MAPCs, and Hurler CM on normal MAPCs).
For removal of cell-surface and ECM HS prior to the binding assay, cells were plated in nonfibronectin-coated wells in MAPC medium, allowed to adhere overnight, then washed with heparitinase buffer (50 mM Hepes, 50 mM sodium acetate, 150 mM NaCl, 0.1% BSA, pH 6.58) 3 times and digested with 0.4 μU/mL heparitinase I (EC 4.2.2.8; ICN Biomedicals, Aurora, OH) at 37°C for 1 hour in the same buffer with 10 μM CaCl2. Heparitinase III (EC 4.2.2.7; Seikagaku America) was then added at the same concentration and calcium increased to 1 mM. After 1 hour incubation, the cell layer was washed and the 125I-FGF-2 binding assay performed as described above in this section.
MAPC-cell proliferation assay
Normal or Hurler MAPCs were seeded (3000 cells/well) in 96-well plates in MAPC medium without EGF and PDGF. Different concentrations of FGF-2 were added to each well. Cells were cultured for 24 hours at 37°C, then CellTiter 96 Aqueous One Solution (Promega, Madison, WI) was added to quantify relative cell numbers. To examine the effect of Hurler CM, normal MAPCs were also cultured in presence of 50% Hurler MAPC CM and identical concentrations of FGF-2.
In parallel experiments, normal and Hurler MAPCs were depleted of cell-surface and ECM HS using heparitinases I and III (as described above, under “Binding of 125I-FGF-2 to normal and Hurler MAPCs”). The proliferation assay was then performed in the absence or presence of normal or Hurler MAPC CM (as a source of normal or Hurler HS). During the proliferation assay, the medium was supplemented with 0.1 μU/mL each of heparitinases I and III.
MAPC apoptosis assay
Hurler MAPCs were plated onto fibronectin-coated sterile coverslips placed in 6-well plates in MAPC medium. Separately, Hurler or normal MAPCs were plated onto fibronectin-coated microporous membranes of Transwell inserts, which were then placed in wells containing Hurler MAPCs on coverslips in the bottom chamber. The entire medium in the wells was changed to serum-free MAPC media (without EGF or PDGF-BB) and containing 100 ng/mL FGF-2 and 1% BSA. After 1 to 7 days, the coverslips (with Hurler MAPCs) were removed and examined for cell morphology and apoptosis.
Apoptosis was detected with the Annexin V-Cy3 Apoptosis Detection Kit (Sigma), according to the manufacturer's instructions. Annexin-Cy3.18 (AnnCy3; red) binds to the outer leaflet of apoptotic and dead cells, while 6-carboxyfluorescein diacetate (6-CFDA) is converted to fluorescent 6-CF (green) only in living cells. Thus, live cells appear green, cells in early apoptosis label with both AnnCy3 (red) and 6-CF (green) producing a yellow color on merged images, while dead cells label only with AnnCy3 (red) but not with 6-CF (green) since they cannot convert 6-CFDA to 6-CF, and thus appear red. The numbers of live, apoptotic, and dead cells were calculated by counting 100 to 250 cells in 4 to 8 fields photographed from different coverslips.
FGF-2 induced proliferation of F32 cells on fixed MAPCs
F32 cells (a kind gift from Dr Alan Rapraeger, University of Wisconsin) are FGFR1 transfected lymphoid cells that express no endogenous cell-surface HSPG, and therefore require exogenous HS/heparin to bind FGF-2 to FGFR1 and stimulate cell proliferation.9,37 To prepare a fixed MAPC layer, 5000 MAPCs/well were allowed to adhere as a confluent monolayer in 96-well flat-bottom plates for 24 hours at 37°C in MAPC medium, fixed for 1 hour in 0.5% glutaradehyde in PBS at room temperature followed by 3 washes in PBS containing 0.2 M glycine, then incubated overnight in RPMI 1640 containing 10% FBS, 4 mM l-glutamine, and 1% penicillin/streptomycin. To examine the effect of cell-surface plus ECM HS of normal and Hurler MAPCs on FGF-2–induced F32-cell proliferation, F32 cells were seeded on fixed MAPC monolayers at 105 cells/mL in cytokine- and serum-deficient medium (RPMI with 0.1% BSA, 4 mM l-glutamine, and penicillin/streptomycin). Various concentrations of FGF-2 were added and plates incubated at 37°C for 24 hours. CellTiter 96 Aqueous One Solution was added to quantify the relative number of cells in each well, as a measure of cell proliferation.
For assessment of proliferation of F32 cells on MAPCs depleted of cell-surface and ECM HS, the fixed normal and Hurler MAPCs in selected wells were pretreated with heparitinases I and III (as described above, under “Binding of 125I-FGF-2 to normal and Hurler MAPCs”).
Results
Structure of HS from normal and Hurler MAPCs
Anion-exchange HPLC profile. Proteoglycans deposited in the ECM plus cell surface, and those present in intracellular storage sites, were separately examined. Proteoglycans from Hurler MAPCs were markedly abnormal. On anion-exchange HPLC, there was an early eluting PG peak in both the ECM and intracellular pools from Hurler cells. This abnormally large peak (peak no. 1), comprised 60% of the total intracellular PGs in Hurler MAPCs, versus 19% in normal MAPCs (Figure 1). This peak comprised 6% of the PGs in the ECM of Hurler MAPCs, but was completely absent in the ECM of normal MAPCs. HSs were obtained separately from each of the various HPLC-resolved peaks and analyzed as follows.
Figure 1.

HPLC profile of proteoglycans. Metabolically radiolabeled proteoglycans from normal and Hurler MAPCs were subjected to DEAE-Sephacel anion-exchange HPLC. The column was eluted at 1 mL/minute using an increasing NaCl gradient, and 1-mL fractions were collected. The 35S and 3H radioactivities were measured and plotted for each fraction. The proportion of radioactivity in each peak was calculated as a percentage of the total radioactivity in the proteoglycan-containing peaks. DPM indicates disintegrations per minute.
Size distribution of HS. HS in the abnormal peak no. 1 of the intracellular pool of Hurler MAPCs was largely comprised of small oligosaccharides (Mr 5.5 kDa; Figure 2). Similarly, peak no. 1 of the ECM of Hurler MAPCs was comprised mostly of small- (Mr 5.5 kDa) and intermediate-sized (Mr 25 kDa) oligosaccharides. These data are consistent with accumulation of oligosaccharides whose complete degradation is blocked by lack of α-L-iduronidase in Hurler MAPCs. In contrast, intracellular HS in peak no. 1 from normal MAPCs were comprised of 3 groups of large (Mr 55 kDa), intermediate (Mr 25 kDa), and small (Mr 7 kDa) molecules. The size distribution of HS in peak no. 2 of the intracellular pool and the ECM (Figure 2) of Hurler MAPCs was comparable to that of normal MAPCs.
Figure 2.

Size distribution of HS. HSs were purified separately from each proteoglycan peak that was resolved by anion-exchange HPLC, and subjected to gel filtration chromatography on a CL-6B column, as described in “Materials and methods.” Three regions containing large-, intermediate-, and small-sized molecules were identified, based on the elution profile of normal intracellular HPLC peak no. 1 HS (Kav 0.1-0.4, 0.4-0.55, and 0.55-0.9, respectively [Kav = (elution volume - void volume)/(total volume - void volume)]). The proportion of HS present in each of these regions was calculated as a percentage of the total, for HS from intracellular peak no. 2 as well as HS from HPLC peaks no. 1 and no. 2 from the ECM.
Disaccharide composition of normal and Hurler MAPC HS
The aggregate disaccharide composition of HS chains in Hurler MAPCs was consistently and significantly different from that of normal MAPCs (Figure 3). The total amount of HS (= 5 kDa) was greater in Hurler compared with normal MAPCs (2670 pg/μg vs 1554 pg/μg protein, respectively), a consequence of glycosaminoglycan accumulation in Hurler cells. A small proportion of less than 5 kDa HS was also isolated, with 4-fold higher levels of this in the Hurler MAPCs (not analyzed further). Normal and Hurler HS each comprised 6 major disaccharide types (Figure 3A-B) including 3 6-O-sulfated disaccharides, UAGlcNAc6S (peak no. 3), UAGlcNS6S (peak no. 4), and UA2SGlcNS6S (peak no. 6). The levels of all 3 6-O-sulfated disaccharides were reduced in Hurler (= 5 kDa) HS, with the greatest reduction (a 52% decrease) in UAGlcNS6S (Figure 3C). Overall there was a 26% decrease in 6-O-sulfation in Hurler MAPC (= 5 kDa) HS (P < .05; Figure 3D). In contrast, the proportion of UA2SGlcNS was slightly higher in Hurler MAPCs whereas the proportion of UAGlcNAc and UAGlcNS was equivalent in the 2 cell types (Figure 3B). The extent of reduction in 6-O-sulfated disaccharides was highly correlated with progressive accumulation of GAGs in MAPCs, most evident for UAGlcNS6S (correlation coefficient R = 0.99), which was reduced by 84% in Hurler MAPCs with the highest total accumulation of HS (Figure 3E). These data indicate that there are both quantitative as well as structural differences between Hurler and normal MAPC HS.
Figure 3.
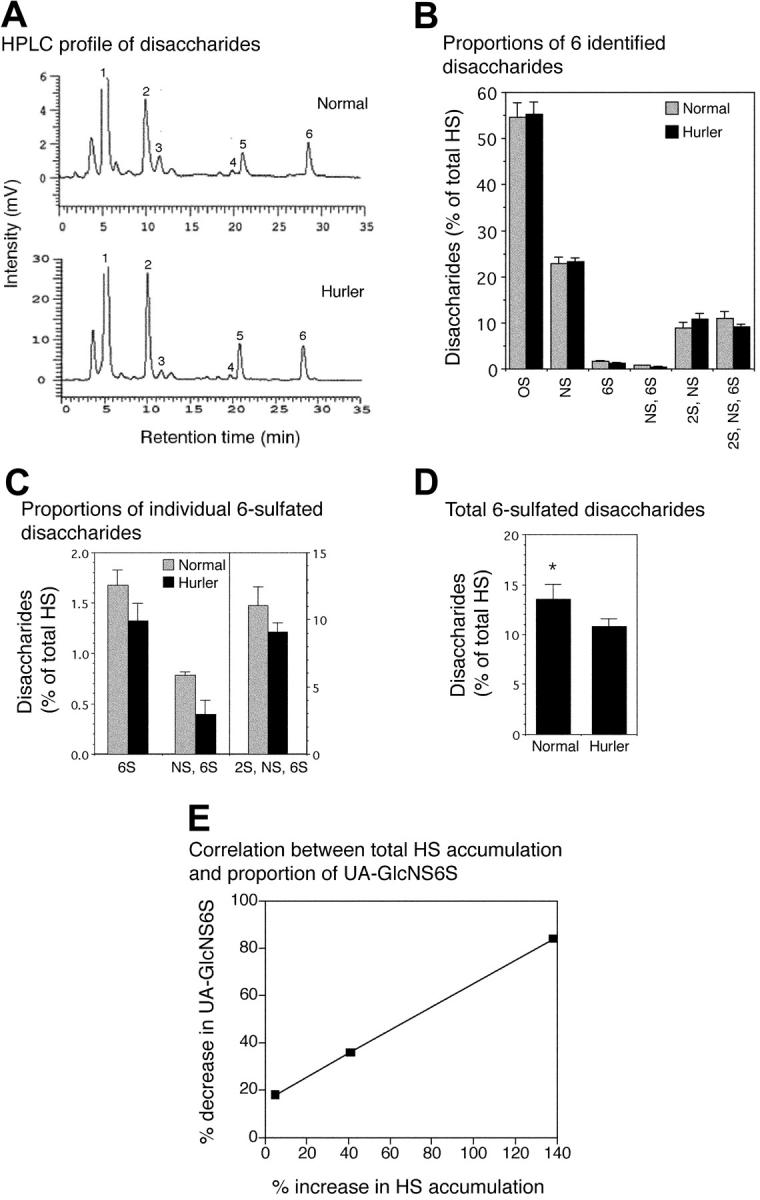
Analysis of disaccharide composition of normal and Hurler MAPC HS. HS from normal and Hurler MAPCs was enzymatically digested and the resulting disaccharides separated by reverse-phase ion pair HPLC. Individual disaccharides were quantified by determining the area under the peaks. n = 3 separate experiments. (A) High-performance liquid chromatography (HPLC) tracing of disaccharides from normal and Hurler MAPCs from one representative experiment. Peak no. 1 (0S): UAGlCNAc; peak no. 2 (NS): UAGlcNS; peak no. 3 (6S): UAGlcNAc6S; peak no. 4 (NS, 6S): UAGlcNS6S; peak no. 5 (2S, NS): UA2SGlcNS; peak no. 6 (2S, NS, 6S): UA2SGlcNS6S. (B) Disaccharide composition of normal and Hurler HS, expressed as percent of total HS. (C) Proportions of the 3 6-sulfated disaccharides in normal and Hurler HS. (D) Total 6-sulfation in normal and Hurler HS. *P < .05. (E) Progressive decrease in proportion of UA-GlcNS6S with increasing total HS accumulation. Correlation coefficient (R) = 0.99. Data in panels B-D are shown as the mean ± standard error (SE).
Binding of 125I-FGF2 to FGFR1 in presence of cell-surface HS
Both normal and Hurler MAPCs expressed FGFR1 (Figure 4A). However, the binding of 125I-FGF-2 to FGFR1 on Hurler MAPCs was less as compared with normal MAPCs (Figure 4B-C). Colocalization of FGFR1 and 125I-FGF-2, and the lack of the 125I-FGF-2 band in absence of cross-linking by DSS, indicate that the results represent specific ligand-receptor binding. These data suggest that the capability of Hurler HS to facilitate FGF-2 binding to FGFR1 (formation of the FGF-2–FGFR1-HS complex) is defective.
Figure 4.

(A-C) Binding of 125I-FGF-2 to MAPC cell-surface receptors. Binding of 125I-FGF-2 to FGFR1 in presence of cell-surface HS. The binding of 125I-FGF-2 to FGFR1 on normal and Hurler MAPCs was determined as described in “Materials and methods.” Briefly, normal or Hurler MAPCs were incubated with 125I-FGF-2 to induce binding to cell-surface receptors. Cell lysates were prepared and subjected to SDS-PAGE. FGFR1 was visualized by Western immunoblotting, and 125I-FGF-2 by autoradiography. In the control lanes, either 25I-FGF-2, DSS, or both were omitted, as shown. The densitometric intensity of the band in each lane is shown below the respective lanes. (A) Western immunoblotting with anti-FGFR1 antibody, demonstrating comparable expression of FGFR1 by normal and Hurler MAPCs. (B) Autoradiograph of the same membrane for 125I-FGF-2, demonstrating colocalization of FGFR1 and 125I-FGF-2. (C) The extent of binding of 125I-FGF-2 to FGFR1 determined by densitometry was expressed as a ratio of 125I-FGF-2 to FGFR1 (B/A). (D) 125I-FGF-2 binding to cell-surface receptors on normal and Hurler MAPCs. The binding of 125I-FGF-2 to normal and Hurler MAPC monolayers was examined as described in “Materials and methods.” The counts per minute (cpm) bound (specific binding) were corrected for the number of cells in each well, and then expressed as the proportion of cpm bound to normal MAPC/105 cells/well. Data are from 2 to 3 independent experiments with replicates. N: normal; H: Hurler; hep'ase: heparitinases I and III. Significance of differences between conditions is described in “Results.”
125I-FGF-2 binding to normal and Hurler MAPCs
Binding of 125I-FGF-2 to Hurler MAPCs was only 55% ± 3% of that compared with normal MAPCs (Figure 4D; P < .001). In presence of Hurler MAPC CM, binding of 125I-FGF-2 to normal MAPCs was reduced to 51% ± 4% of binding to normal MAPCs alone (P < .001). In contrast, 125I-FGF-2 binding to Hurler MAPCs was not improved by normal MAPC CM. Removal of cell-surface and ECM HS by heparitinase treatment further reduced 125I-FGF-2 binding to Hurler MAPCs (P < .001 compared with untreated cells). CM from normal MAPCs but not that from Hurler MAPCs (used as a source of HS) markedly improved binding to heparitinase-treated Hurler MAPCs (P < .001). Interestingly, binding of 125I-FGF-2 to heparitinase-treated Hurler MAPCs in presence of normal CM was significantly higher than binding to untreated Hurler MAPCs in the absence or presence of normal CM (P < .005 and P < .001 respectively). These data indicate that substances (likely HS) in the Hurler MAPC microenvironment interfere with FGF-2 binding to cell-surface receptors.
FGF-2 induced proliferation of MAPCs
To investigate the mitogenic response of normal and Hurler MAPCs to FGF-2, MAPCs were cultured with various concentrations of FGF-2 alone, in the absence of other cytokines. Normal MAPCs showed dose-dependent proliferation at lower concentrations of FGF-2, and inhibition of proliferation at higher concentrations (Figure 5A). In contrast, Hurler MAPCs failed to show either stimulation or inhibition of proliferation over the range of concentrations of FGF-2 tested. When normal MAPCs were cultured in presence of Hurler CM, their proliferative response was markedly inhibited in an FGF-2 dose-dependent manner.
Figure 5.
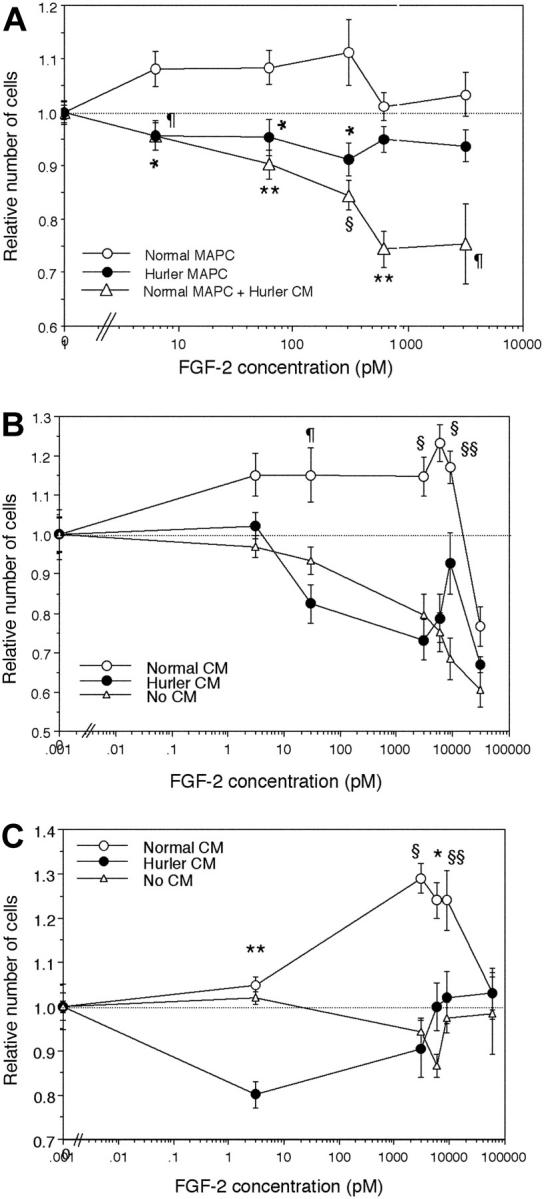
FGF-2–induced proliferation of normal and Hurler MAPCs. Normal or Hurler MAPCs were cultured for 24 hours in absence or presence of a range of concentrations of FGF-2. The numbers of cells present in each well were determined as detailed in “Materials and methods.” Cell numbers are expressed as a ratio (number of cell in wells supplemented with FGF-2–number of cells in absence of FGF-2). (A) Proliferation of normal and Hurler MAPCs in the absence or presence of the indicated concentrations of FGF-2. In separate wells in the same experiments, normal MAPCs were cultured in presence of 50% CM from Hurler MAPCs. n = 4 to 6 independent experiments with replicates. Comparison between normal MAPCs and the other conditions: *P < .01, ¶P < .005, §P < .002, **P < .001. (B) Proliferation of heparitinase I– and III–treated normal MAPCs, in the absence or presence of 50% CM from normal or Hurler MAPCs, was examined as in panel A. n = 3 independent experiments with replicates. Comparison between proliferation seen in presence of normal versus Hurler CM: §§P < .05, ¶P < .005, §P < .002. (C) Proliferation of heparitinase I– and III–treated Hurler MAPCs, in the absence or presence of 50% CM from normal or Hurler MAPCs, was examined as in panel A. n = 3 independent experiments with replicates. Comparison between proliferation seen in presence of normal versus Hurler CM: §§P < .05, *P < .01, §P < .002, **P < .001.
To examine if the observed differences were due to HS, proliferation of heparitinase I– and III–treated normal (Figure 5B) and Hurler MAPCs (Figure 5C) depleted of cell-surface and ECM HS was examined in absence or presence of normal or Hurler CM (as a source of exogenous HS). Following heparitinase treatment, neither normal nor Hurler MAPCs could proliferate in response to FGF-2 (Figure 5B-C). The FGF-2 dose-dependent proliferation of both normal and Hurler MAPCs was restored by normal but not by Hurler CM. These results indicate that factors in the Hurler-cell microenvironment (likely HS) interfere with FGF-2 mitogenic signaling, and are in keeping with the effect of Hurler CM on FGF-2 binding.
Apoptosis of Hurler MAPCs
We examined if the defective biologic activity of FGF-2 on Hurler MAPCs would lead to their apoptosis when deprived of other cytokines and serum, and if the presence of normal MAPCs (separated from the Hurler MAPCs by a Transwell membrane) would prevent apoptosis. When cultured in standard MAPC medium, both normal and Hurler MAPCs remained alive (Figure 6Ai-ii). However, when cultured in serum-free medium containing FGF-2 as the only cytokine (Figure 6Aiii-iv), a larger proportion of Hurler MAPCs on coverslips in the lower chamber of the well became apoptotic or died by 5 days when Hurler MAPCs were present in the Transwell inserts above the coverslips than when normal MAPCs were present in the Transwell inserts (39% ± 7% vs 17% ± 2%, respectively; P < .05). This difference was mainly due to a larger proportion of apoptotic cells (29% ± 4% vs 12% ± 2%, respectively; P < .02; Figure 6B). These data indicate that the survival-promoting activity of FGF-2 on Hurler MAPCs is impaired, but can be restored following cross-correction by normal MAPCs across a microporous Transwell membrane.
Figure 6.
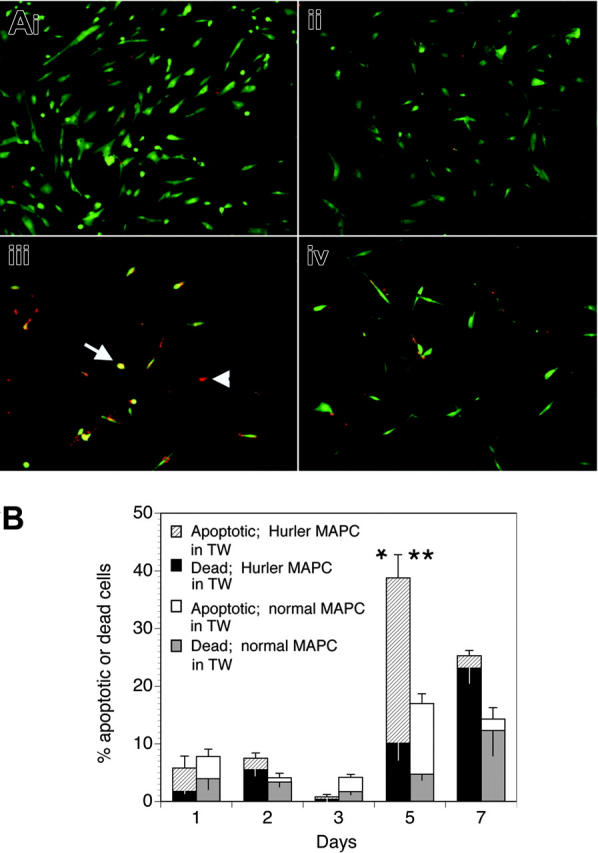
Apoptosis of Hurler MAPCs in presence of FGF-2. (A) Apoptosis was determined in Hurler MAPCs grown on coverslips placed in 6-well plates. Color definitions are as follows: green indicates viable cells; yellow, apoptotic cells (arrow); red, dead cells (arrowhead). Images were acquired on an Olympus BX60 fluorescent microscope using an UPlanApo 10×/0.4 dry objective lens at room temperature (final magnification, × 100). Fluorochromes were Cy3 (red) and 6-CF (green). Multiple fields were photographed using an Olympus U-ULS100HG digital camera system and SPOT 2.12 software (Diagnostics Instruments). Red and green images were merged and the composite figure made using Adobe Photoshop CS software. (i) Normal MAPCs grown on coverslips in standard MAPC medium (containing serum, EGF, and PDGF-BB). (ii) Hurler MAPCs grown on coverslips in standard MAPC medium (as in panel i). (iii) Hurler MAPCs grown on coverslips in FGF-2–supplemented serum-free medium, in presence of Hurler MAPCs in Transwells (TW) for 5 days. (iv) Hurler MAPCs grown on coverslips in FGF-2–supplemented serum-free medium, in presence of normal MAPCs in Transwells for 5 days. (B) Apoptotic and dead cells on the coverslips in the lower chambers of wells were counted on the indicated days after changing to FGF-2–supplemented serum-free medium and placement of Transwells with either Hurler or normal MAPCs. Data in the columns show the percentages of apoptotic and dead cells (upper and lower sections of the columns, respectively). The total height of the columns indicates the percentages of apoptotic and dead cells. The upper and lower error bars indicate the standard error (SE) of the mean for apoptotic and dead cells, respectively. Comparison between wells with Hurler MAPCs in the Transwells versus wells with normal MAPCs in Transwells: *P < .02 for apoptotic cells; **P < .05 for apoptotic and dead cells.
The lower level of apoptosis at early time points (days 1-3) upon serum starvation (Figure 6B) might possibly be due to the fact that MAPCs represent a cell type that is initially selected on the basis of its ability to survive and grow in very low or no serum. Further, it is likely that the biologic activity of FGF-2 is reduced but not absent on Hurler MAPCs, as suggested by the binding studies, providing some protection from early apoptosis. As expected, by a later time point (day 7), the proportion of dead cells continued to increase, whereas the proportion of apoptotic cells declined.
FGF-2 induced proliferation of F32 cells on fixed MAPCs
We also directly examined the capability of normal and Hurler-MAPC–cell surface and ECM HS to mediate the biologic activity of FGF-2 on an independent, HS-deficient, FGFR1-expressing cell line. To establish controls, we confirmed that F32 cells cultured with FGF-2 in absence of exogenous HS/heparin failed to proliferate (Figure 7), but did proliferate actively when heparin was added to the medium (not shown). F32 cells cultured on fixed normal MAPCs (which served as a source of normal HS) showed dose-dependent proliferation with increasing concentrations of FGF-2 till 1000 pM. In contrast, FGF-2–induced F32 proliferation was lower when fixed Hurler MAPCs served as a source of HS. These differences in F32 proliferation on normal versus Hurler MAPCs were larger at lower FGF-2 concentrations (3 pM-30 pM). At intermediate FGF-2 concentrations (1000 pM), there was a trend toward lower proliferation on Hurler MAPCs. However, at the highest FGF-2 concentrations (3000 pM-30 000 pM), F32 proliferation on Hurler MAPCs became comparable to that on normal MAPCs. That the HS presented by normal and Hurler MAPCs was responsible for the observed differences in FGF-2–mediated F32 proliferation was confirmed by enzymatic removal of HS (using heparitinases I and III) from MAPCs prior to the proliferation assay.
Figure 7.

FGF-2 induced proliferation of F32 cells on fixed MAPCs. F32 cells were cultured for 24 hours on fixed monolayers of normal and Hurler MAPCs in the presence of the indicated concentrations of FGF-2. The numbers of cells present in each well were determined as detailed in “Materials and methods.” Cell numbers are expressed as a ratio (number of cells in wells supplemented with FGF-2/number of cells in absence of FGF-2). Proliferation of F32 cells plated on heparitinase I and III (hep'ase)–treated normal and Hurler MAPCs was also examined in parallel wells. n = 2 independent experiments with replicates. ○ indicates Normal MAPC; •, Hurler MAPC; □, No cell layer; ▴, Hep'ase treated Hurler MAPC; and ▵, Hep'ase treated normal MAPC.
Discussion
We demonstrate that structurally and functionally abnormal HSs that accumulate in Hurler syndrome perturb critical FGF-2–FGFR1-HS interactions. Further, FGF-2–induced proliferation and survival of Hurler MAPCs is defective. This may be one mechanism by which accumulated HSs contribute to the developmental pathophysiology of Hurler syndrome.
Previous studies on Hurler syndrome examined structural features of GAGs obtained from the urine, autopsy tissue homogenates, or cultured fibroblasts.16-18,33,38-40 Overall, our findings are consistent with these studies. A unique aspect of our study is that we examined metabolically radiolabeled GAGs obtained from the progeny of primary, multipotent stem-cell populations from the BM of patients with Hurler syndrome and healthy volunteers. This enabled examination of the structure and functional properties of HS deposited in the ECM/cell surface and which accumulated intracellularly, and the association between these abnormalities and the activity of an important GAG-binding cytokine.
6-O-sulfation of HS is required for mediating FGF-2–FGFR1-HS complex formation and FGF-2 activity.41,42 We found that reduction in 6-O-sulfation of Hurler HS is associated with impaired FGF-2–FGFR1-HS complex formation and FGF-2 activity on Hurler MAPCs.
There was progressive reduction in the disaccharide UAGlcNS6S with increasing total accumulation of HS in Hurler MAPCs. It is possible that this may at least partly be due to a feedback mechanism from accumulated GAGs that downregulates HS6ST enzymes at a transcriptional or translational level. It also leads us to speculate that the progressive postnatal GAG accumulation in the brain (2- to 4-fold higher than normal brain),43-46 which is comparable to that in Hurler MAPCs, may lead to progressive structural abnormalities in HS and functional abnormalities in the biologic activity of critical cytokines.
At the cellular level, Hurler MAPCs demonstrated reduced 125I-FGF-2 binding, not corrected by normal MAPC CM unless Hurler MAPCs were pretreated with heparitinase. This indicates that removal of abnormal HS from Hurler-MAPC–cell surface and ECM and provision of normal HS restores 125I-FGF-2 binding. That Hurler MAPC CM was unable to improve FGF-2 binding to heparitinase-treated Hurler MAPCs is further evidence that Hurler HS is functionally abnormal.
This impairment of FGF-2 binding to Hurler MAPCs was associated with defective mitogenic activity of FGF-2 (MAPC proliferation experiments). Abnormal HS in Hurler syndrome thus impairs the ability of primitive progenitors to respond to mitogenic signaling from developmentally important cytokines. These results also suggest a mechanism for the recent observation that proliferation of neural progenitors (which depends on FGF-2)47 is reduced in a mouse model of MPS IIIB, another disease where partially degraded HSs accumulate.48
FGF-2–mediated proliferation of independent, FGFR1-expressing, HS-dependent F32 cells seeded on Hurler MAPCs was lower than on normal MAPCs. Since MAPCs served as a source of HS for F32 cells, this further indicates that HS on Hurler-MAPC–cell surface and ECM is abnormal, and directly impairs the FGF-2 mitogenic signaling. In contrast to the complete lack of FGF-2–induced proliferation of Hurler MAPCs themselves, proliferation of F32 cells on fixed Hurler MAPCs still occurred. This may be a result of alteration HS-FGF-2–FGFR1 complex formation dynamics because of large numbers of FGFR1 receptors on F32 cells.
FGF-2, like other cytokines, protects target cells, including neurons, from apoptosis.15 We show that FGF-2–mediated protection of serum-starved MAPCs (which can differentiate into neuronal cells)26,27,31 from apoptosis is defective in Hurler syndrome.
Finally, we show that HS provided by normal MAPCs can correct the impairment of FGF-2 biologic activity, since removal of Hurler HS from MAPC surface and provision of normal HS in CM (1) improved 125I-FGF-2 binding to Hurler MAPCs, and (2) restored FGF-2–mediated Hurler MAPC proliferation. Also, the impaired capability of FGF-2 to prevent apoptosis of serum-starved Hurler MAPCs was restored by coculture with normal MAPCs.
It is possible that some of the observed abnormalities may be due to accumulated DS in Hurler MAPCs. However, specific enzymatic removal of cell-surface plus ECM HS and provision of normal HS (in normal MAPC CM) largely restored 125I-FGF-2 binding to and proliferation of Hurler MAPCs, indicating that abnormal HSs are mainly responsible for impairment of FGF-2 activity.
Our observation that progressive GAG accumulation is associated with a progressive decline in 6-O-sulfated disaccharides of HS, together with the known clinical behavior of Hurler syndrome where progressive GAG accumulation and neuropsychologic decline occurs postnatally, lead us to speculate that part of the pathophysiology of this disease may be related to progressive impairment of FGF-2 activity postnatally. This notion is consistent with normal prenatal brain development in Hurler syndrome, when GAG accumulation has not yet occurred, and therefore the biologic activity of FGF-2 may not be affected.
In conclusion, our studies suggest a potential mechanism by which accumulated GAGs contribute to the pathophysiology of Hurler syndrome via perturbation of cytokine activity, and how normal GAGs may correct these abnormalities. Similar mechanisms may possibly be involved in the pathogenesis of other neurodegenerative diseases associated with GAG accumulation.
Acknowledgments
We thank Dr Catherine M. Verfaillie, Director, Stem Cell Institute and Professor, Department of Medicine, University of Minnesota, Minneapolis, MN, and Dr Theodore R. Oegema Jr, Professor and Chairman, Department of Biochemistry, Rush University, Chicago, IL, for their advice and help with this work.
Prepublished online as Blood First Edition Paper, June 9, 2005; DOI 10.1182/blood-2005-02-0657.
Supported by the U.S. Department of Veterans Affairs (P.G.), NIH 1-R03-HD-41 411-01 and NIH 1-R01-NS-48 606-01 (P.G.), the Children's Cancer Research Fund (P.G., C.P.), and the University of Minnesota Medical School. C. Pan and S.E.S. designed and performed research, analyzed data, and wrote the manuscript; M.S.N., J.J.B., and E.J.S. performed research and analyzed data; M.R. and L.K. isolated and cultured multipotent progenitor cells; R.Z. and S.B.S. designed research; C. Peters provided bone marrow samples and designed research; P.G. designed research, analyzed data, and wrote the manuscript.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 U.S.C. section 1734.
References
- 1.Neufeld EF, Muenzer J. The mucopolysaccharidoses. In: Scriver CR, Beaudet AL, Sly WS, et al, eds. The Metabolic and Molecular basis of Inherited Disease. Vol III. New York, NY: McGraw-Hill; 2001: 3421-3452. [Google Scholar]
- 2.Russell C, Hendson G, Jevon G, et al. Murine MPS I: insights into the pathogenesis of Hurler syndrome. Clin Genet. 1998;53: 349-361. [DOI] [PubMed] [Google Scholar]
- 3.Whitley CB. The mucopolysaccharidoses. In: Moser HW, ed. Neurodystrophies and Neurolipidoses. Vol 66 (22). Elsevier Science; 1996: 281-328. [Google Scholar]
- 4.Lyon G, Adams RD, Kolodny EH. Late infantile progressive genetic encephalopathies: neurology of hereditary metabolic diseases of children (Second Ed). New York, NY: McGraw Hill; 1996: 124-176.
- 5.Conrad HE. Fibroblast growth factors. Heparin binding proteins. San Diego, CA: Academic Press; 1998: 301-349.
- 6.Vaccarino FM, Schwartz ML, Raballo R, Rhee J, Lyn-Cook R. Fibroblast growth factor signaling regulates growth and morphogenesis at multiple steps during brain development. Curr Top Dev Biol. 1999;46: 179-200. [DOI] [PubMed] [Google Scholar]
- 7.Yayon A, Klagsbrun M, Esko JD, Leder P, Ornitz DM. Cell surface, heparin-like molecules are required for binding of basic fibroblast growth factor to its high affinity receptor. Cell. 1991;64: 841-848. [DOI] [PubMed] [Google Scholar]
- 8.Chang Z, Meyer K, Rapraeger AC, Friedl A. Differential ability of heparan sulfate proteoglycans to assemble the fibroblast growth factor receptor complex in situ. FASEB J. 2000;14: 137-144. [DOI] [PubMed] [Google Scholar]
- 9.Krufka A, Guimond S, Rapraeger AC. Two hierarchies of FGF-2 signaling in heparin: mitogenic stimulation and high-affinity binding/receptor transphosphorylation. Biochemistry. 1996;35: 11131-11141. [DOI] [PubMed] [Google Scholar]
- 10.Bernfield M, Gotte M, Park PY, et al. Functions of cell surface heparan sulfate proteoglycans. Annu Rev Biochem. 1999;68: 729-777. [DOI] [PubMed] [Google Scholar]
- 11.Gallagher JT, Lyon M. Heparan sulfate: molecular structure and interactions with growth factors and morphogens. In: Iozzo RV, ed. Proteoglycans. New York, NY: Marcel Dekker, Inc; 2000: 27-60.
- 12.Perrimon N, Bernfield M. Specificities of heparan sulphate proteoglycans in developmental processes. Nature. 2000;404: 725-728. [DOI] [PubMed] [Google Scholar]
- 13.Pye DA, Vives RR, Hyde P, Gallagher JT. Regulation of FGF-1 mitogenic activity by heparan sulfate oligosaccharides is dependent on specific structural features: differential requirements for the modulation of FGF-1 and FGF-2. Glycobiology. 2000;10: 1183-1192. [DOI] [PubMed] [Google Scholar]
- 14.Yamaguchi Y. Heparan sulfate proteoglycans in the nervous system: their diverse roles in neurogenesis, axon guidance, and synaptogenesis. Semin Cell Dev Biol. 2001;12: 99-106. [DOI] [PubMed] [Google Scholar]
- 15.Alzheimer C, Werner S. Fibroblast growth factors and neuroprotection. Adv Exp Med Biol. 2002; 513: 335-351. [DOI] [PubMed] [Google Scholar]
- 16.Matalon R, Dorfman A. The structure of acid mucopolysaccharides produced by Hurler fibroblasts in tissue culture. Proc Natl Acad Sci U S A. 1968; 60: 179-185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Byers S, Rozaklis T, Brumfield LK, Ranieri E, Hopwood JJ. Glycosaminoglycan accumulation and excretion in the mucopolysaccharidoses: characterization and basis of a diagnostic test for MPS. Mol Genet Metab. 1998;65: 282-290. [DOI] [PubMed] [Google Scholar]
- 18.Knecht J, Cifonelli JA, Dorfman A. Structural studies on heparitin sulfate of normal and Hurler tissues. J Biol Chem. 1967;242: 4652-4661. [PubMed] [Google Scholar]
- 19.Knecht J, Dorfman A. Structure of heparitin sulfate in tissues of the Hurler syndrome. Biochem Biophys Res Commun. 1965;21: 509-515. [DOI] [PubMed] [Google Scholar]
- 20.Gupta P, McCarthy JB, Verfaillie CM. Stromal fibroblast heparan sulfate is required for cytokine-mediated ex vivo maintenance of human long-term culture-initiating cells. Blood. 1996;87: 3229-3236. [PubMed] [Google Scholar]
- 21.Gupta P, Oegema TR, Brazil JJ, Dudek AZ, Slungaard A, Verfaillie CM. Structurally specific heparan sulfates support primitive human hematopoiesis by formation of a multimolecular stem cell niche. Blood. 1998;92: 4641-4651. [PubMed] [Google Scholar]
- 22.Gupta P, Oegema TR, Brazil JJ, Dudek AZ, Slungaard A, Verfaillie CM. Human LTC-IC can be maintained for at least 5 weeks in vitro when interleukin-3 and a single chemokine are combined with O-sulfated heparan sulfates: requirement for optimal binding interactions of heparan sulfate with early-acting cytokines and matrix proteins. Blood. 2000;95: 147-155. [PubMed] [Google Scholar]
- 23.Peters C, Balthazor M, Shapiro EG, et al. Outcome of unrelated donor bone marrow transplantation in 40 children with Hurler syndrome. Blood. 1996;87: 4894-4902. [PubMed] [Google Scholar]
- 24.Peters C, Shapiro EG, Anderson J, et al. Hurler syndrome, II: outcome of HLA-genotypically identical sibling and HLA-haploidentical related donor bone marrow transplantation in fifty-four children. The Storage Disease Collaborative Study Group. Blood. 1998;91: 2601-2608. [PubMed] [Google Scholar]
- 25.Staba SL, Escolar ML, Poe M, et al. Cord-blood transplants from unrelated donors in patients with Hurler's syndrome. N Engl J Med. 2004;350: 1960-1969. [DOI] [PubMed] [Google Scholar]
- 26.Reyes M, Lund T, Lenvik T, Aguiar D, Koodie L, Verfaillie CM. Purification and ex vivo expansion of postnatal human marrow mesodermal progenitor cells. Blood. 2001;98: 2615-2625. [DOI] [PubMed] [Google Scholar]
- 27.Jiang Y, Jahagirdar BN, Reinhardt RL, et al. Pluripotency of mesenchymal stem cells derived from adult marrow. Nature. 2002;418: 41-49. [DOI] [PubMed] [Google Scholar]
- 28.Keene CD, Ortiz-Gonzalez XR, Jiang Y, Largaespada DA, Verfaillie CM, Low WC. Neural differentiation and incorporation of bone marrow-derived multipotent adult progenitor cells after single cell transplantation into blastocyst stage mouse embryos. Cell Transplant. 2003;12: 201-213. [DOI] [PubMed] [Google Scholar]
- 29.Reyes M, Dudek A, Jahagirdar B, Koodie L, Marker PH, Verfaillie CM. Origin of endothelial progenitors in human postnatal bone marrow. J Clin Invest. 2002;109: 337-346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schwartz RE, Reyes M, Koodie L, et al. Multipotent adult progenitor cells from bone marrow differentiate into functional hepatocyte-like cells. J Clin Invest. 2002;109: 1291-1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jiang Y, Henderson D, Blackstad M, Chen A, Miller RF, Verfaillie CM. Neuroectodermal differentiation from mouse multipotent adult progenitor cells. Proc Natl Acad Sci USA. 2003;100: 11854-11860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Koc ON, Day J, Nieder M, Gerson SL, Lazarus HM, Krivit W. Allogeneic mesenchymal stem cell infusion for treatment of metachromatic leukodystrophy (MLD) and Hurler syndrome (MPS-IH). Bone Marrow Transplant. 2002;30: 215-222. [DOI] [PubMed] [Google Scholar]
- 33.Bunge S, Clements PR, Byers S, Kleijer WJ, Brooks DA, Hopwood JJ. Genotype-phenotype correlations in mucopolysaccharidosis type I using enzyme kinetics, immunoquantification and in vitro turnover studies. Biochim Biophys Acta. 1998;1407: 249-256. [DOI] [PubMed] [Google Scholar]
- 34.Wasteson A. A method for the determination of the molecular weight and molecular weight distribution of chondroitin sulfate. J Chromatogr. 1971; 59: 87-97. [DOI] [PubMed] [Google Scholar]
- 35.Toyoda H, Kinoshita-Toyoda A, Selleck SB. Structural analysis of glycosaminoglycans in Drosophila and Caenorhabditis elegans and demonstration that tout-velu, a Drosophila gene related to EXT tumor suppressors, affects heparan sulfate in vivo. J Biol Chem. 2000;275: 2269-2275. [DOI] [PubMed] [Google Scholar]
- 36.Spivak-Kroizman T, Lemmon MA, Dikic I, et al. Heparin-induced oligomerization of FGF molecules is responsible for FGF receptor dimerization, activation, and cell proliferation. Cell. 1994; 79: 1015-1024. [DOI] [PubMed] [Google Scholar]
- 37.Ornitz DM, Yayon A, Flanagan JG, Svahn CM, Levi E, Leder P. Heparin is required for cell-free binding of basic fibroblast growth factor to a soluble receptor and for mitogenesis in whole cells. Mol Cell Biol. 1992;12: 240-247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Constantopoulos G. Hunter-Hurler syndrome: gel filtration and dialysis of urinary acid mucopolysaccharides. Nature. 1968;220: 583-585. [DOI] [PubMed] [Google Scholar]
- 39.Constantopoulos G, Iqbal K, Dekaban AS. Mucopolysaccharidosis types IH, IS, II, and IIIA: glycosaminoglycans and lipids of isolated brain cells and other fractions from autopsied tissues. J Neurochem. 1980;34: 1399-1411. [DOI] [PubMed] [Google Scholar]
- 40.Dekaban AS, Constantopoulos G. Mucopolysaccharidosis type I, II, IIIA and V: pathological and biochemical abnormalities in the neural and mesenchymal elements of the brain. Acta Neuropathol (Berl). 1977;39: 1-7. [DOI] [PubMed] [Google Scholar]
- 41.Guimond S, Maccarana M, Olwin BB, Lindahl U, Rapraeger AC. Activating and inhibitory heparin sequences for FGF-2 (basic FGF): distinct requirements for FGF-1, FGF-2, and FGF-4. J Biol Chem. 1993;268: 23906-23914. [PubMed] [Google Scholar]
- 42.Pye DA, Vives RR, Turnbull JE, Hyde P, Gallagher JT. Heparan sulfate oligosaccharides require 6-O-sulfation for promotion of basic fibroblast growth factor mitogenic activity. J Biol Chem. 1998;273: 22936-22942. [DOI] [PubMed] [Google Scholar]
- 43.Constantopoulos G, Dekaban AS. Content and distribution of molecular weights of mucopolysaccharides in the brain and other organs of Hurler's patients. J Neuropathol Exp Neurol. 1971;30: 144-145. [PubMed] [Google Scholar]
- 44.Constantopoulos G, Dekaban AS. Neurochemistry of the mucopolysaccharidoses: brain lipids and lysosomal enzymes in patients with four types of mucopolysaccharidosis and in normal controls. J Neurochem. 1978;30: 965-973. [DOI] [PubMed] [Google Scholar]
- 45.Constantopoulos G, McComb RD, Dekaban AS. Neurochemistry of the mucopolysaccharidoses: brain glycosaminoglycans in normals and four types of mucopolysaccharidoses. J Neurochem. 1976;26: 901-908. [DOI] [PubMed] [Google Scholar]
- 46.Dekaban AS, Constantopoulos G, Herman MM, Steusing JK. Mucopolysaccharidosis type V (Scheie syndrome): a postmortem study by multidisciplinary techniques with emphasis on the brain. Arch Pathol Lab Med. 1976;100: 237-245. [PubMed] [Google Scholar]
- 47.Gritti A, Parati EA, Cova L, et al. Multipotential stem cells from the adult mouse brain proliferate and self-renew in response to basic fibroblast growth factor. J Neurosci. 1996;16: 1091-1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li HH, Zhao HZ, Neufeld EF, Cai Y, Gomez-Pinilla F. Attenuated plasticity in neurons and astrocytes in the mouse model of Sanfilippo syndrome type B. J Neurosci Res. 2002;69: 30-38. [DOI] [PubMed] [Google Scholar]