

Abstract
Patients with early HIV-1 infection develop an autoimmune thrombocytopenia in which antibody is directed against an immunodominant epitope of the β3 (glycoprotein IIIa [GPIIIa]) integrin, GPIIIa49-66. This antibody induces thrombocytopenia by a novel complement-independent mechanism in which platelets are fragmented by antibody-induced generation of H2O2 derived from the interaction of platelet nicotinamide adenine dinucleotide phosphate (NADPH) oxidase and 12-lipoxygenase. To examine whether sharing of epitope between host and parasite may be responsible for this immunodominant epitope, we screened for antibody-reactive peptides capable of inhibiting platelet lysis and oxidation in vitro, using a filamentous phage display 7-mer peptide library. Fourteen of these phage-peptide clones were identified. Five shared close sequence similarity with GPIIIa49-66, as expected. Ten were molecular mimics with close sequence similarity to HIV-1 proteins nef, gag, env, and pol. Seven were synthesized as 10-mers from their known HIV-1 sequence and found to inhibit anti–GPIIIa49-66–induced platelet oxidation/fragmentation in vitro. Three rabbit antibodies raised against these peptides induced platelet oxidation/fragmentation in vitro and thrombocytopenia in vivo when passively transferred into mice. One of the peptides shared a known epitope region with HIV-1 protein nef and was derived from a variant region of the protein. These data provide strong support for molecular mimicry in HIV-1-immunologic thrombocytopenia within polymorphic regions of HIV-1 proteins. A known epitope of nef is particularly incriminated.
Introduction
Patients with early HIV-1 infection commonly develop an immunologic thrombocytopenia (HIV-1-ITP)1 with shortened platelet survival.2 Early onset HIV-1-ITP is clinically indistinguishable from classic autoimmune idiopathic thrombocytopenia (AITP) and responsive to the same therapeutic agents used to treat AITP.1,3-6 However, HIV-1-ITP is different from AITP with respect to markedly elevated platelet-associated IgG, IgM, C3, and C4 and the presence of serum circulating immune complexes (CICs) composed of the same.3,4 These CICs also contain specific high-affinity IgG1 antibody (Ab) against the platelet glycoprotein IIIa (GPIIIa) integrin,7 its anti-idiotype blocking Ab,8 and platelet fragments.9 Unlike classic AITP in which multiple Abs have been described with different specificities for platelet GPIIb, GPIIIa, and GPIb,6 patients with HIV-1-ITP have an IgG1 Ab against an immunodominant epitope, GPIIIa49-66 (CAPESIEFPVSEAREVLED). This Ab induces thrombocytopenia in mice (mouse GPIIIa has 83% sequence similarity with human GPIIIa and macrophages with Fc receptors capable of clearing human IgG1) and correlates inversely with platelet count in HIV-1-ITP patients (r =–0.71).7,10 Platelet affinity-purified antiplatelet Ab reacts predominantly with platelet GPIIIa following immunoprecipitation of 125I-labeled surface antigens from a platelet lysate, and more than 85% of platelet-reactive Ab is absorbed onto and eluted from a GPIIIa49-66 affinity column.10 This Ab is particularly unique in that it induces complement-independent platelet fragmentation in vitro and in vivo by the generation of peroxide through a newly described platelet nicotinamide adenine dinucleotide phosphate (NADPH) oxidase,9 interacting with platelet 12-lipoxygenase to produce 12(S)-hydroxyeicosatetraenoic acid (HETE).11 It is therefore of particular interest to determine how this Ab is induced in HIV-1 infection.
Molecular mimicry describes antigen sharing between host and parasite. Because of the immunodominant specificity of this Ab we chose to examine whether molecular mimicry might be the mechanism for HIV-1-ITP. Using a filamentous phage surface display 7-mer peptide library we screened for peptides reactive with a rabbit as well as human anti–GPIIIa49-66 Ab. The 10-mer HIV-1 peptides were synthesized from knowledge of the positive-reactive phage peptides. These peptides were then examined for their ability to inhibit human anti–GPIIIa49-66–induced platelet oxidation/fragmentation and compared to Ab inhibition by its immunodominant epitope, GPIIIa49-66. They were also examined for their ability to raise rabbit Ab capable of inducing platelet fragmentation as well as passive Ab-induced transfer of thrombocytopenia in mice.
Materials and methods
Materials
All reagents were obtained from Sigma-Aldrich (St Louis, MO), unless otherwise indicated. 2′,7′-Dichlorodihydrofluorescein diacetate (DCFH-DA) was obtained from Molecular Probes (Eugene, OR). The anti–HIV-1 sera immunoblot kit was obtained from Immunetics (Boston, MA).
Mice
Female Balb/C mice were obtained from Taconic Farms (Germantown, NY). Animal work was approved by the New York University Medical Center Institutional Review Board.
Antibodies
Patient sera were obtained from early onset HIV-1–infected thrombocytopenic patients without AIDS and control subjects (healthy laboratory personnel). These studies were approved by the New York University Medical Center Institutional Review Board. Rabbit Ab against GPIIIa49-66 was produced as described previously. Human Ab was isolated from polyethylene glycol (PEG)–precipitable immune complexes,7 and affinity purified on a GPIIIa49-66 affinity column.9 Rabbit Ab was raised against mimicry HIV-1 peptides coupled to KLH or the phage itself. The sera were purified and affinity purified, as described.
Peptides
Peptides were synthesized by Bio-synthesis (Lewisville, TX). Peptides not cited in Table 1 are a variant nef (60-71) peptide H19R3 (QEGEEVSFPVK) (accession no. CAA13487) and a variant gag (25-30) peptide PHC39 (GKNHYMLKHL) (GenBank accession no. AAL34661) as well as a relatively conserved irrelevant env peptide (517-526) Env (GIGALFLGFL) (GenBank accession no. AAQ98126).
Table 1.
Panning of PhD 7-phage peptide library
| Clones | Peptide | HIV sequence | HIV gene |
|---|---|---|---|
| PHB22 | SIEFPTS | None | None |
| PHC31 | SFDPGLF | SKLFDEGLFN | Envelope glycoprotein |
| PHC32 | YAPEFPL | GEAPEFPSKQ | pol |
| PHC33 | RHTMIIP | None | None |
| PHC34 | VEFPVSD | QEEEEVGFPVT | nef protein |
| PHC35 | SFDPGLF | Same as PHC31 | Same as PHC31 |
| PHC36 | TIIFPVE | ELFNKTIIFP | Envelope glycoprotein |
| PHC37 | SFDPGLF | Same as PHC31 | Same as PHC31 |
| PHC38 | KWHWSDS | None | None |
| PHC39 | RHTMINP | GKTHYMINPL | gag protein |
| HR1 | VEFPVSD | QEEEEVGFPVT | nef protein |
| H19R3 | GIIFPVE | EDEGIGFPVR | nef protein |
| H20R1 | LVYPVSE | FKLVPVSEAE | nef protein |
| H20R4 | TPTTNLH | SSNTPTTNAA | nef protein |
GPIIIa49-66: CAPESIEFPVSEARVLED.
Boldface type indicates close sequence similarity with GPIIIa49-66; italics, similar amino acid sequence among different clones; and underlining, homology with HIV-1 proteins.
Assay of platelet particle formation
Whole blood was collected into 5.2 mM EDTA (ethylenediaminetetraacetic acid) and platelet-rich plasma (PRP) prepared by centrifugation at 200g for 5 minutes. Gel-filtered platelets were obtained by adding PRP to a Sepharose 2B column preincubated with Tyrode buffer, pH 7.4. Platelets (108/mL) were labeled with 10 μg/mL of an anti-CD61–fluorescein isothiocyanate (FITC) monoclonal antibody (mAb) for 30 minutes at 4°C. Cells were then centrifuged at 1000g for 6 minutes at room temperature and resuspended in Tyrode buffer. Then 10 μL of platelet suspension (106 platelets) was incubated with 10 μL anti–GPIIIa49-66 Ab (containing 2.5 μg) and 80 μL Tyrode buffer for 4 hours at 37°C and stored in an ice bucket prior to measurement of platelet particles. Fluorescent-labeled platelets/particles were measured by flow cytometry using a FACScan (Becton Dickinson Immunocytometry Systems, Mountain View, CA). Gates were adjusted for platelets by exclusion of other blood cells. Fluorescent-labeled intact platelets were monitored in the right upper quadrant with the y-axis measuring forward scatter and the x-axis measuring fluorescence. A shift in fluorescent particles from right upper quadrant to left upper quadrant and left lower quadrant reflected the percentage of platelet particle induction of 10 000 enumerated platelets/particles.
Assay of platelet oxidation
Gel-filtered platelets were loaded with 10 μM DCFH-DA for 30 minutes at 37°C as described and challenged with platelet Ab. Oxidation was quantified by measuring the increase in mean fluorescence by flow cytometry.9
Screening of the phage display peptide library
The PhD-7 phage library was obtained from New England Biolabs (Beverly, MA). This is a combinatorial library of random 7-mers fused to a minor coat protein (pIII) of M13 phage and capable of detecting 1.3 × 109 random 7-mer peptide sequences. Anti–GPIIIa49-66 Ab was incubated with the phage in solution and Ab complexes coupled with alternating protein A- or G-conjugated agarose beads to avoid fortuitous selection of peptide sequences specifically binding to protein A or G. Protein A beads were washed with TBST buffer (Tris [tris(hydroxymethyl)aminomethane] buffer 50 mM, NaCl 150 mM, Tween 0.1%, pH 7.5) and blocked with TBST + 1% bovine serum albumin (BSA). Then 2 × 1011 phage virions (10 μL of supplied library) and 300 ng anti–GPIIIa49-66 Ab were diluted in a final volume of 200 μL with TBST and incubated at room temperature for 20 minutes. The phage Ab mixture was then transferred to a tube containing washed agarose beads for 15 minutes. The agarose beads were washed 10 times with TBST and bound phage eluted with 0.2 M glycine-HCl, pH 2.2, and 1 mg/mL BSA for 10 minutes at room temperature. The eluate was immediately neutralized with Tris-HCl (pH 7.4). The eluted phage was then amplified in Escherichia coli ER2738. The amplified phage eluates were titered on LB/IPTG/Xgal plates and the titer, derived from the number of blue clones, used to calculate an input volume corresponding to 2 × 1011 pfu for the next round of panning. In the next round, protein G-agarose beads were used instead of protein A, and the Tween concentration raised in the binding and wash steps to 0.5% (vol/vol). The third round of panning was with protein A agarose beads with the Tween concentration kept at 0.5% (vol/vol) in the binding and wash steps. After the third round of panning, 20 clones were randomly selected for sequencing. Phage peptide sequences were analyzed for sequence similarity to other proteins using the BLAST algorithm of the Blast program and the database of the National Center for Biotechnology Information (NCBI). Antigen sequence similarities of HIV-1 peptide sequences available in the database were analyzed.
Results
Defining the epitope
To better determine the GPIIIa49-66 epitope, we synthesized a peptide within the middle of this GPIIIa fragment, 52-60 (ESIEF-PVSE), and tested for its ability to block anti–GPIIIa49-66–induced platelet fragmentation. Peptide GPIIIa52-60 was as effective as GPIIIa49-66, further defining the limits of the epitope (data not shown).
Panning with a 7-mer combinatorial phage display peptide library
The first series of panning with rabbit anti–GPIIIa49-66 identified 3 peptide clones with close sequence similarity to GPIIIa49-66, as expected: SIEFPTS (Table 1, column 2, line 1) as well as 4 of the peptides not shown: IEWPVSD, TIEFPVA, VEFPVSD, NEFPVS. Nine others (Table 1, column 2) showed sequence similarity with HIV-1 proteins: envelope glycoprotein (clones PHC31/PHC35/PHC37, PHC36); polymerase protein (PHC32); nef protein (PHC34/HR1, H19R3, H20R1, H20R4); and gag protein (PHC39), as determined from the NCBI database. PHC33 and PHC38 had no sequence similarity with either HIV-1 or GPIIIa49-66. This is likely due to the use of a polyclonal Ab to screen the phage peptide library. Preliminary studies were performed with 3 intact phage peptides that had sequence similarity with HIV-1 peptides from nef (clones PHC34 and H20R1) and gag (PHC39). All 3 inhibited about 50% human platelet lysis at phage dilutions of 1:00 (Figure 1A), whereas 2 irrelevant phages showed no inhibition at dilutions of 1:3 (Figure 1B).
Figure 1.

Effect of panned phage 7-mer peptides on inhibition of anti–GPIIIa49-66–induced platelet fragmentation. (Left) First 3 bars refer to effect of patient Ab (Pt) on platelet fragmentation compared to buffer (C) or control IgG (Ci). Phage peptides were diluted at ratios of 1:3, 1:10, 1:30, and 1:100 and then preincubated with Ab for 15 minutes prior to the addition of the combined reagents to gel-filtered platelets. First bar is buffer diluent alone. Ab-induced platelet fragmentation was about 50% inhibited with a 1:100 dilution of the 3 phage peptides. (Right) Irrelevant peptide control phage (C1 and C2) as well as inhibitory phage peptides (H20R1, PHC39, PHC34) were diluted from 1:3 to 1000, preincubated with Ab as in the left panel, and then incubated with gel-filtered platelets. Data are representative of 2 different experiments.
Properties and specificity of mimicry HIV-1 peptides with GPIIIa49-66 peptides
The HIV-1 counterparts of the phage peptide are shown in Table 1. The 7 HIV-1 peptides were then tested for their ability to inhibit human Ab-induced platelet lysis in vitro and compared to inhibition by GPIIIa49-66 and GPIIIa52-60 peptides. All 7 HIV-1 mimicry peptides inhibited Ab-induced human platelet lysis significantly at about an inhibitory concentration of 50% (IC50) molar Ab/peptide ratio of 1:5, whereas irrelevant peptide (CVGKML-CKAK) had no effect (Figure 2). Similarly, no effect was observed with 2 other variant peptides of the active inhibitory peptides H19R3 (nef) or PHC39 (gag), nor an HIV-1 peptide derived from a relatively conserved envelope protein region (GIGALFLGFL), (514-523), as shown in Figure 3. Studies were next performed with 3 different human HIV-1-ITP anti–GPIIIa49-66 Abs to ensure further specificity. The phage clones obtained with rabbit anti–GPIIIa49-66 were repanned with the human anti–GPIIIa49-66. Four of 9 clones obtained showed close sequence similarity with wild-type nef (clones HR1, H20R1, H20R4, H19R3). Two were tested (clones H20R1, H19R3) and showed similar IC50 values as the previous group (Table 1).
Figure 2.

Effect of GPIIIa49-66 and wild-type HIV-1 peptides on Ab-induced platelet fragmentation. CAPE and ESIEF refer to positive control samples in which Ab was preincubated with peptide GPIIIa49-66 (CAPESIEFPVSEARVLED) or GPIIIa52-60 (SIEFPVSE). First set of bars refer to control, patient IgG, irrelevant peptide, or positive control inhibitory peptide (CAPE) prior to incubation with gel-filtered platelets. Second set of bars refers to positive control inhibitory peptide (ESIEF). The remaining bar panels refer to preincubation with various wild-type 10-mer HIV-1 peptides. □ indicates control IgG; ▪, patient Ig;  , irrelevant peptide. The horizontal line, dark gray, white, and black bars refer to molecular Ab/peptide ratios of 1:100, 1:10, 1:5, and 1:2, respectively. Numbers refer to PHC or H clones of Table 1. HR1 refers to H20R1; HR3 refers to H19R3.
, irrelevant peptide. The horizontal line, dark gray, white, and black bars refer to molecular Ab/peptide ratios of 1:100, 1:10, 1:5, and 1:2, respectively. Numbers refer to PHC or H clones of Table 1. HR1 refers to H20R1; HR3 refers to H19R3.
Figure 3.

Effect of wild-type polymorphic HIV-1 peptides derived from their inhibitory HIV-1 peptide regions on the inhibition of Ab-induced platelet fragmentation. Peptides were preincubated with Ab for 15 minutes at 37°C prior to addition of the combined reagents to gel-filtered platelets (as in Figure 1). □ and ▪refer to preincubation with molar peptide/Ab ratios of 1:100 and 1:10, respectively. C indicates control IgG; P, patient IgG; Ph, phage peptide PHC39; HR3, HIV-1 wild-type peptide HI9R3; PHR3 and P39, reported polymorphic peptides of HI9R3 and PHC39, respectively (Table 1); Env, irrelevant conserved region of env (514-523). N=4. SEM is given.
Properties of rabbit Ab raised against the HIV-1 peptides
To establish proof of concept, it was necessary to demonstrate that Ab raised against these molecular mimicry HIV-1 peptides could induce platelet fragmentation and oxidation in vitro and thrombocytopenia in vivo. Rabbit Abs were raised against a series of mimicry HIV-1 peptides as well as their intact phage peptide counterparts. Because of the numerous peptides, batches of 4 were immunized together: batch 1096 HIV-1 10-mer peptides (PHC31, 32, 34, and 39), batch 1098 HIV-1 10-mer peptides (PHC31, 32, 34, and H20R1), and batch 1099 phage 7-mer peptides (PHC 36, 39, H19R3, and H20R1). All 3 rabbit Abs strongly induced fragmentation (Figure 4 top panel) and oxidation (Figure 4 bottom panel) of gel-filtered human platelets, in a similar manner as human anti– GPIIIa49-66 Ab (28% versus control IgG of 2%). This activity could be removed (86% ± 1%) by absorbing the Abs with the peptides used to raise the Ab, whereas no activity was removed with envelope protein peptide Env (GIGALFLGFL), indicating specificity.
Figure 4.
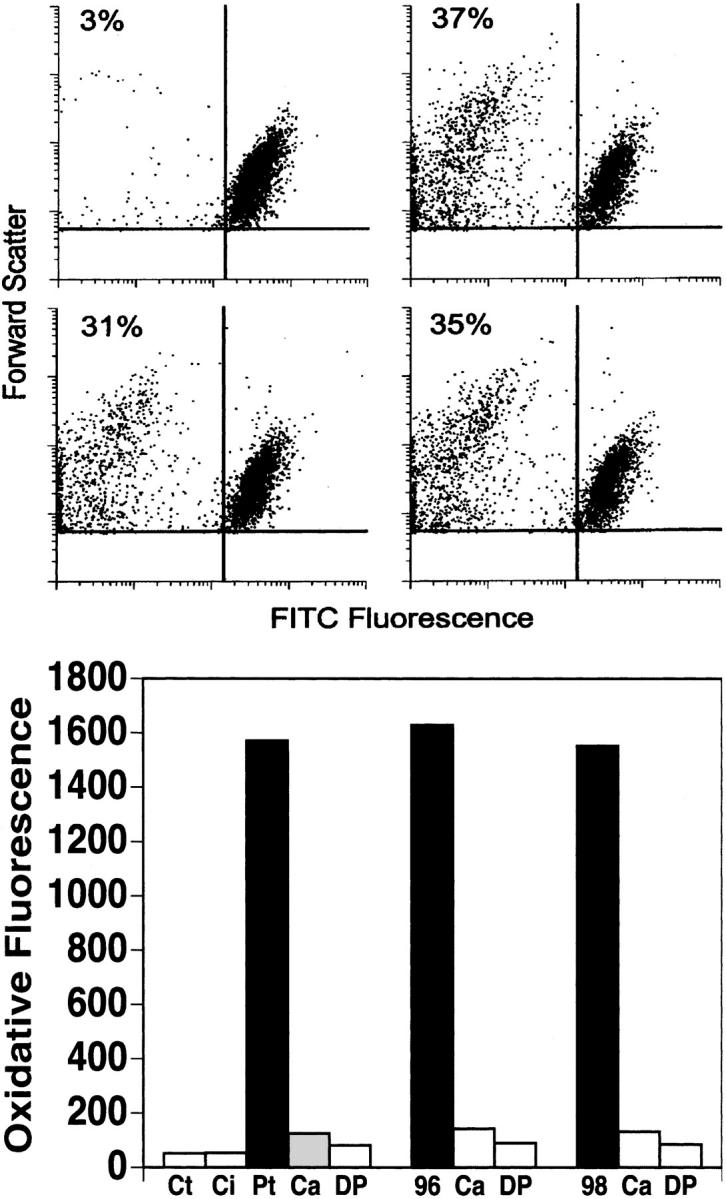
Effect of rabbit Abs raised against HIV-1 10-mer wild-type peptides or 7-mer phage peptides on platelet fragmentation and oxidation. (Top) Gel-filtered platelets were incubated with control rabbit IgG (upper left quadrant) or rabbit Ab against peptide batches 1096 (upper right quadrant), 1098 (lower left), and 1099 (lower right) at 37°C for 4 hours. The percentages refer to fragmented platelets per 10 000 events counted. (Bottom) Similar incubation with assay of Ab-induced platelet oxidation. Ct indicates control buffer; Ci, control IgG; and Pt, patient IgG. Ca and DP indicate preincubation with scavenger of reactive oxygen species and inhibitor of NADPH oxidase, catalase (60 μg/mL) and diphenylenediodonium (100 nM), respectively; 96 and 98 refer to rabbit Ab against peptide batches 1096 and 1098.
We hypothesized that we would find several mimicry HIV-1 peptides capable of inhibiting Ab-induced platelet oxidation/fragmentation because (1) they were derived from their similar sequence with panned phage peptides against the same Abs capable of inducing platelet oxidation/fragmentation and (2) HIV-1 proteins are continually mutating in the host. It was therefore necessary to determine whether these mimicry HIV-1 peptides were pathophysiologically relevant. This was achieved by demonstrating that the rabbit Ab derived from these mimicry HIV-1 peptides reacted with intact HIV-1 proteins capable of being detected in a similar manner as patient anti–HIV-1 sera as detected by immunoblot (Figure 5).
Figure 5.
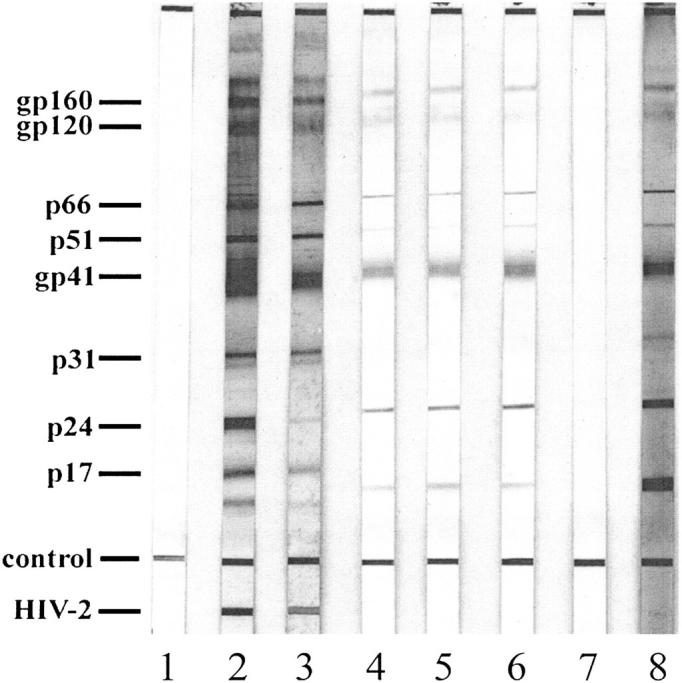
Reactivity of rabbit antimolecular mimicry Ab with HIV-1 proteins. Immunoblot of control sera (lane 1); 2 positive control sera (lanes 2 and 3); 3 rabbit antimolecular mimicry Ab sera, 1096, 1098, and 1099 (lanes 4, 5, and 6, respectively); preimmune rabbit sera (lane 7); and affinity-purified HIV-1-ITP patient sera (lane 8).
Effect of rabbit Ab against HIV-1 molecular mimicry peptides on induction of thrombocytopenia in mice
Figure 6 demonstrates passive transfer of thrombocytopenia into mice with the same rabbit antimolecular mimicry peptide Abs, in a similar manner as patient anti–GPIIIa49-66 Ab. No effect was noted with control IgG.
Figure 6.

Effect of rabbit Ab against HIV-1 peptides on induction of thrombocytopenia in mice. Purified control IgG (Ctl), patient IgG (Pt IgG) and rabbit Ab (25 μg) against wild-type HIV-1 peptide batches 1096, 1098, and 1099 were injected intraperitoneally into Balb/c mice, and platelet counts followed for 24 hours. N=4. SEM is given.
Database for HIV-1 epitopes
The epitope significance of the cross-reactive wild-type HIV-1 peptides with anti–GPIIIa49-66 Ab was further examined by searching the HIV-1 database at the Los Alamos National Library for human or mouse Abs that have been generated from patients or mice immunized with intact HIV-1 proteins.12 Three of the 7 HIV-1 peptides (PHC34/HR1, PHC39, H19R3) are located in known epitope regions of nef and gag HIV-1 proteins. This would be an underestimate because reported epitopes are from vaccine or other immunization studies that may not reflect all naturally occurring epitopes.
Discussion
Evidence indicates that molecular mimicry may contribute to autoimmune disease in HIV-1 infection. It has been suggested that this mechanism could protect invading microorganisms from the immune response of the host.13,14 Indeed it can be argued that HIV-1 is continuously undergoing mutation at its nonconservative region to mimic host protein. Autoantibodies with various specificities arise during early HIV-1 infection in association with persistent activation of the immune response. Protein sequences shared between humans and HIV-1 protein have been reported supporting the argument for molecular mimicry.15-19
Previous studies from our laboratory on the presence of an immunodominant epitope, GPIIIa49-66, on platelets have suggested a role for molecular mimicry in HIV-1–related thrombocytopenia.6,7 The presence of a relatively high-affinity Ab against GPIIIa49-667 suggested that the Ab was generated by antigen-driven B-cell clonal expansion. We therefore reasoned that mimicry epitopes from HIV-1 protein or other opportunistic bacterial, viral, or fungal protein could be responsible for the generation of such an Ab.
Cross-reactivity between viral antibodies and platelets has previously been reported. These include anti–HIV-GP160/12020 and anti–HIV p2421 for platelet GPIIb/IIIa and anti–varicella zoster virus (anti-VZV)22 for platelets. However, cross-reactive linear epitopes were neither defined nor found.23 Nevertheless, these observations support our present data that specifically define the cross-reactive platelet-HIV epitopes on GP120, as well as other HIV-1 proteins, gag and nef, and demonstrate that these cross-reactive Abs can induce thrombocytopenia in vivo through the oxidation and fragmentation of platelets.
Our present results clearly make a strong case for molecular mimicry. (1) Affinity-purified rabbit as well as human anti–GPIIIa49-66 Abs are capable of panning high-affinity 7-mer phage peptides with similar sequence identity to HIV-1 peptides of HIV-1 proteins (env, pol, nef, gag). (2) The phage-peptides inhibit human anti–GPIIIa49-66 platelet lysis via oxidation of human platelets, whereas irrelevant phage peptides do not. (3) Synthesized HIV-1 mimicry 10-mer peptides inhibit Ab-induced platelet oxidation/fragmentation, whereas 2 variant peptides of the active HIV-1 peptides do not, nor does a relatively conserved HIV-1env peptide nor a random irrelevant peptide. (4) Rabbit Ab raised against the inhibitory HIV-1 peptides induces platelet oxidation/fragmentation in a similar manner and with a similar intensity as human anti–GPIIIa49-66. (5) The same rabbit anti–HIV-1 peptide Ab induces thrombocytopenia in mice in a similar manner and intensity as human anti–GPIIIa49-66. (6) Rabbit Ab against mimicry HIV-1 peptides binds to intact HIV-1 proteins on immunoblot. (7) At least 3 of the mimicry HIV-1 peptides are located within known immunogenic HIV-1-epitopes.
Our present results are also supported by the work of others. (1) Two of the 3 mimicry epitopes (PHC34/HR1 and H19R3) are located in variant regions of nef having no secondary structure and shown by Schneider et al17 to be distinct epitopes of nef following intact protein immunization and that 3 of 10 anti–HIV-1 human sera react with the same epitopes. This region maps in the predicted similar sequence region between nef and HLA class II.17 (2) A recent report by Kiepiela et al24 on the influence of HLA-B in the coevolution of HIV and HLA has revealed that the nef mimicry epitopes PHC34/HR1 and H19R3 are highly immunogenic and T-cell responsive. These observations were derived from a study of T-cell responsiveness in 375 HIV-infected subjects using a panel of 410 overlapping synthetic peptides spanning the HIV genome.
These data support the role of a variant mutation region of nef at amino acid position 60-73 as being responsible for the autoimmune thrombocytopenia in some patients with HIV-1-ITP. Our data also support the concept that other such variant mutations or nonconserved regions of HIV-1 may also be responsible in other patients. It is of interest that all 7 HIV-1 peptides are located in the highly mutated region of HIV-1 proteins. It has been suggested that HIV is continuously undergoing mutations in the nonconserved region to mimic host protein and escape host immune surveillance. It is likely that these mutations may contribute to the susceptibility of thrombocytopenia in patients infected with HIV-1.
Our data on Ab cross-reactivity between known epitope regions of HIV-1 proteins and platelet GPIIIa49-66 support our observation that anti–GPIIIa49-66 is an immunodominant Ab in HIV-1-ITP and provides proof for molecular mimicry being responsible for HIV-1-ITP.
Prepublished online as Blood First Edition Paper, March 17, 2005; DOI 10.1182/blood-2005-01-0243.
Supported by National Institutes of Health (NIH) grants HL13336 and DAO-04315 and the Dorothy and Seymour Weinstein Platelet Research Fund. Presented at the 45th Annual Meeting of the American Society of Hematology, San Diego, CA, December 8, 2003. Blood 102:125A, 2003.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 U.S.C. section 1734.
References
- 1.Morris L, Distenfeld A, Amorosi E, Karpatkin S. Autoimmune thrombocytopenic purpura in homosexual men. Ann Intern Med. 1982;96: 714-717. [DOI] [PubMed] [Google Scholar]
- 2.Najean Y, Rain JD. The mechanism of thrombocytopenia in patients with HIV infection. J Lab Clin Med. 1994;123: 415-420. [PubMed] [Google Scholar]
- 3.Walsh C, Nardi M, Karpatkin S. On the mechanism of thrombocytopenic purpura in sexually-active homosexual men. N Engl J Med. 1984;311: 635-639. [DOI] [PubMed] [Google Scholar]
- 4.Savona S, Nardi M, Karpatkin S. Thrombocytopenic purpura in narcotics addicts. Ann Intern Med. 1985;102: 737-741. [DOI] [PubMed] [Google Scholar]
- 5.Ratnoff O, Menitove J, Aster R, Lederman M. Coincident classic hemophilia and “idiopathic” thrombocytopenic purpura in patients under treatment with concentrates of anti-hemophilic factor (factor VIII). N Engl J Med. 1983;308: 439-442. [DOI] [PubMed] [Google Scholar]
- 6.Karpatkin S. Autoimmune (idiopathic) thrombocytopenic purpura. Lancet. 1997;349: 1531-1536. [DOI] [PubMed] [Google Scholar]
- 7.Karpatkin S, Nardi M, Hymes K. Sequestration of anti-platelet GPIIIa antibody in rheumatoid factor-immune complexes of human immunodeficiency virus 1 thrombocytopenic patients. Proc Natl Acad Sci U S A. 1995;92: 2263-2267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nardi M, Karpatkin S. Antiidiotype antibody against platelet anti-GPIIIa contributes to the regulation of thrombocytopenia in HIV-1-ITP patients. J Exp Med. 2000;191: 2093-2100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nardi M, Tomlinson S, Greco M, Karpatkin S. Complement-independent, peroxide induced antibody lysis of platelets in HIV-1-related immune thrombocytopenia. Cell. 2001;106: 551-561. [DOI] [PubMed] [Google Scholar]
- 10.Nardi MA, Liu LX, Karpatkin S. GPIIIa-(49-66) is a major pathophysiologically relevant antigenic determinant for anti-platelet GPIIIa of HIV-1-related immunologic thrombocytopenia. Proc Natl Acad Sci U S A. 1997;94: 7589-7594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nardi M, Feinmark S, Liang H, Li Z, Karpatkin S. Complement-independent Ab-induced peroxide lysis of platelets requires 12-lipoxygenase and a platelet NADPH oxidase pathway. J Clin Invest 2004;113: 973-980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Los Alamos National Library. http://www.hiv.lanl.gov/content/index. Accessed May 2004.
- 13.Damian R. Molecular mimickry: antigen sharing by parasite and host and its consequences. Am Nat 1964;98: 129-149. [Google Scholar]
- 14.Rose N, Mackay I. Molecular mimicry: a critical look at exemplary instances in human diseases. Cell Mol Life Sci. 2000;57: 542-551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Silvestris F, Williams RC Jr, Dammacco F. Autoreactivity in HIV-1 infection: the role of molecular mimicry. Clin Immunol Immunopathol. 1995;75: 197-205. [DOI] [PubMed] [Google Scholar]
- 16.Zandman-Goddard G, Shoenfeld Y. HIV and autoimmunity. Autoimmun Rev. 2002;1: 329-337. [DOI] [PubMed] [Google Scholar]
- 17.Schneider T, Harthus HP, Hildebrandt P, et al. Epitopes of the HIV-1-negative factor (nef) reactive with murine monoclonal antibodies and human HIV-1-positive sera. AIDS Res Hum Retroviruses. 1991;7: 37-44. [DOI] [PubMed] [Google Scholar]
- 18.Serres PF. Molecular mimicry between the trimeric ectodomain of the transmembrane protein of immunosuppressive lentiviruses (HIV-SIV-FIV) and interleukin 2. C R Acad Sci III 2000;323: 1019-1029. [DOI] [PubMed] [Google Scholar]
- 19.Trujillo JR, Rogers RA, Brain JD. Shared antigenic epitopes on the V3 loop of HIV-1 gp120 and proteins on activated human T cells. Virology. 1998;246: 53-62. [DOI] [PubMed] [Google Scholar]
- 20.Bettaieb A, Fromont P, Louache F, et al. Presence of cross-reactive antibody between human immunodeficiency virus (HIV) and platelet glycoproteins in HIV-related immune thrombocytopenic purpura. Blood. 1992;80: 162-169. [PubMed] [Google Scholar]
- 21.Chia WK, Blanchette V, Mody M, Wright JF, Freedman J. Characterization of HIV-1-specific antibodies and HIV-1-crossreactive antibodies to platelets in HIV-1-infected haemophiliac patients. Br J Haematol. 1998;103: 1014-1022. [DOI] [PubMed] [Google Scholar]
- 22.Wright JF, Blanchette VS, Wang H, et al. Characterization of platelet-reactive antibodies in children with varicella-associated acute immune thrombocytopenic purpura (ITP). Br J Haematol. 1996;95: 145-152. [DOI] [PubMed] [Google Scholar]
- 23.Bettaieb A, Oksenhendler E, Duedari N, Bierling P. Cross-reactive antibodies between HIV-gp120 and platelet gpIIIa (CD61) in HIV-related immune thrombocytopenic purpura. Clin Exp Immunol. 1996;103: 19-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kiepiela P, Leslie AJ, Honeyborne I, et al. Dominant influence of HLA-B in mediating the potential co-evolution of HIV and HLA. Nature. 2004; 432: 769-775. [DOI] [PubMed] [Google Scholar]