

Abstract
The preclinical evaluation of investigational agents for Waldenström macroglobulinemia (WM) has been limited by the lack of in vivo models that enable the use of explanted patient cells. We describe here a novel in vivo model of human WM in severe combined immunodeficient (SCID) mice implanted with human fetal bone chips (SCID-hu mice) into which WM cells from patient bone marrow are engrafted directly into the human bone marrow (huBM) microenvironment. WM cells in SCID-hu mice produced human monoclonal paraprotein (immunoglobulin M [IgM] and/or κ or λ chain) detectable in mice sera. Immunohistochemical analysis of human bone retrieved from SCID-hu mice showed infiltration with CD20+, IgM+, and monotypic light chain+ lymphoplasmacytic cells. Mast cells were observed to be associated with the infiltrate in these sections. Treatment of SCID-hu mice bearing WM with rituximab induced tumor regression, associated with a decrease in serum paraprotein. This model, therefore, recapitulates the in vivo biology of WM and allows the study of novel investigational drugs targeting WM cells in the huBM milieu. (Blood. 2005;106:1341-1345)
Introduction
Waldenström macroglobulinemia (WM) is a distinct B-cell lympho-proliferative disorder characterized primarily by bone marrow (BM) infiltration with lymphoplasmacytic cells, along with demonstration of an immunoglobulin M (IgM) monoclonal gammopathy.1 This clinicopathologic condition is observed in the majority of cases defined pathologically as lymphoplasmacytic lymphoma in the Revised European-American Lymphoma and World Health Organization classification systems.2,3 Despite advances in therapy, WM remains an incurable disease, and most patients die of disease progression.4 In the absence of a preclinical model, evaluation of novel treatments for WM has been empiric and based on clinical trial data from related B-cell malignancies. Therefore, a need exists for an appropriate preclinical model for WM to validate new agents and to allow rapid bench-to-bedside translation.
To date, the only available animal model for the study of human WM is a subcutaneous tumor model developed by injecting a human WM cell line (WSU-WM) into the flank of immunodeficient mice.5 This cell line has been obtained from a patient with WM with advanced and therapy-resistant disease with cytogenetic abnormalities, reflecting the biologic behavior of an aggressive disease instead of a typical indolent WM.5,6 Therefore, the animal model based on subcutaneous xenograft of these cells does not recapitulate the in vivo biologic features of a typical WM nor does it reproduce the disease in a human bone marrow (huBM) milieu.
In the past, we and others have implanted human fetal bone chips into severe combined immunodeficient (SCID) mice (SCID-hu mice)7-15 and then directly engrafted tumor cells to allow in vivo growth of multiple myeloma (MM) cells.14,15 This model has advantages over other murine models16-18 because (1) tumor cells grow within the huBM microenvironment, (2) bone lesions develop, and (3) human paraprotein14,15 can be measured in mouse sera as an in vivo marker of tumor burden and response to therapy. This model, therefore, represents a biologically relevant in vivo experimental system that has provided important insight in the pathophysiology of MM and has been successfully used for preclinical evaluation of novel agents targeting tumor cells into huBM milieu.
Here, we characterize a novel in vivo SCID-hu model in which primary patient WM cells engraft in huBM in vivo and produce measurable levels of human IgM and/or κ or λ chain in mouse serum. This model recapitulates the in vivo biology of WM and is useful for preclinical evaluation of novel agents targeting WM cells in the BM milieu.
Materials and methods
WM cells and reagents
Heparinized BM aspirates were obtained from patients with WM after they provided informed consent in accordance with the Declaration of Helsinki. Patients' data are provided in Table 1. BM cells were separated using Ficoll-Hypaque density gradient centrifugation. In some samples, to enrich for tumor cells, WM cells were sorted using CD19-immunomagnetic beads (Miltenyi Biotec, Auburn, CA) with purity of cells determined by flow cytometric analysis of CD20, κ or λ chain (Coulter Epics XL, Birmingham, United Kingdom) to be more than 85%. Unsorted, as well as CD19+-sorted, cells were either directly injected into mice or following incubation overnight at 37°C in a 5% CO2 atmosphere in RPMI-1640 medium (GIBCO, Grand Island, NY) supplemented with 20% fetal bovine serum (Hyclone, Logan, UT), l-glutamine, penicillin, and streptomycin (GIBCO). For in vivo treatments, the anti-CD20 monoclonal antibody rituximab (IDEC Pharmaceuticals, San Diego, CA, and Genentech, South San Francisco, CA) was administered at 25 mg/kg on alternate days for a total of 3 intraperitoneal injections.
Table 1.
Sample characteristics
| Patient | Isotype | Level of IgM in patient, mg/dL | Biopsy, % bone marrow infiltration | Prior therapy | Cells inoculated, × 106 | Weeks to detection of IgM | Level of IgM in mouse,*μg/mL | Level of light chain in mouse,*μg/mL |
|---|---|---|---|---|---|---|---|---|
| 1 | IgM κ | 1240 | 90 | RTX, THAL | 5 | 4 | 1.8 | Negative |
| 2 | IgM κ | 6130 | 60 | Prednisone | 5 | 16 | 10 | Negative |
| 3 | IgM κ | 4830 | 50 | Fl, RTX | 5 | 6 | 0.6 | NA |
| 4 | IgM κ | 1430 | 70 | THAL, DEX | 5 | 28 | 2.6 | 0.7 |
| 5 | IgM λ | 2590 | 60 | RTX | 10 | 4 | 1.5 | Negative |
| 6 | IgM κ | 2390 | 70-80 | None | 10 | 5 | 2.6 | NA |
| 7 | IgM, κ | 7630 | 90 | None | 2 | 13 | 0.4 | NA |
| 8 | IgM,κ | NA | NA | None | 2 | 20 | 0.2 | 0.4 |
| 9† | IgM κ | 4970 | 95 | CVP, RTX | 2 | 7 | 8.4 | 0.4 |
| 10† | IgM,κ | 2340 | 50 | None | 2 | 20 | 1.8 | NA |
RTX indicates rituximab; THAL, thalidomide; Fl, fludarabine; NA, not available; DEX, dexamethasone; CVP, cyclophosphamide, vincristine, and prednisone.
At time of first IgM detection.
In these cases, BM cells were CD19-immunomagnetically sorted before mice inoculation into human bone implant.
SCID-hu mouse model
Six- to 8-week-old male CB-17 SCID mice (Taconic, Germantown, NY) were housed and monitored in our Animal Research Facility. All experimental procedures and protocols had been approved by the Institutional Animal Care and Use Committee (Veteran's Administration [VA] Boston Healthcare System, Boston, MA). Procedures for SCID mouse implantation with human fetal long bone grafts (SCID-hu) have been previously described.7-14 Mice were surgically implanted with human bone chips of fetal femur or tibia from 19- to 23-week gestation human abortuses. Approximately 4 weeks following implantation, 2 to 10 × 106 whole BM mononuclear cells or 2 × 106 CD19-sorted cells, depending on number of cells available after Ficoll-Hypaque density gradient centrifugation procedures and/or following immunomagnetic CD19-cell sorting of BM aspirates, in 50 μL phosphate-buffered saline (PBS) were injected directly into human fetal bone implants within SCID-hu hosts. Increasing levels of circulating human paraprotein in mice sera were used to monitor tumor engraftment and growth of patient WM cells in SCID-hu mice. Mouse blood was collected from tail vein, and sera were serially tested for circulating IgM, IgG, and κ and λ chain by enzyme-linked immunosorbent assay (ELISA; Bethyl, Montgomery, TX), as previously described.19,20 This ELISA kit uses antibody specific for human immunoglobulins (Igs), which does not cross-react with murine Igs (1:100 dilution).
Histopathologic analysis
Excised tissues included human fetal bone grafts and mouse femur, spleen, liver, lung, kidney, and lymph nodes were fixed in Bouin or formalin solution, processed by standard methods, embedded in paraffin, sectioned at 5 μm, and stained with hematoxylin and eosin (H&E) for histopathologic examination, as previously described.14 For immunohistochemical evaluation, tissues were analyzed using antihuman reagents for expression of CD20 (murine monoclonal L26; DAKO, Carpinteria, CA), IgM (rabbit polyclonal; Biomeda, Foster City, CA), IgG (murine monoclonal A57H; DAKO), IgA (murine monoclonal 6E2C1; DAKO), κ immunoglobulin light chain (murine monoclonal R10-21-F3; DAKO), λ immunoglobulin light chain (murine monoclonal N10/2; DAKO), and anti-human mast-cell tryptase (murine monoclonal AA1; DAKO) and detected using the Envision Plus detection system (DAKO). Slides were counterstained with Harris hematoxylin and examined by standard light microscopy. Samples were analyzed using an Olympus BX41 microscope equipped with UPlan FL 40×/0.75 and 20×/0.50 objective lenses (Olympus, Melville, NY). Pictures were taken using Olympus QColor3 and analyzed using QCapture 2.60 software (QImaging, Burnaby, BC, Canada). Adobe Photoshop 6.0 was used to process images.
Results
Lymphoplasmacytic tumor cells were obtained from BM aspirates of patients with WM. We first evaluated the ability of primary patient WM cells to engraft in SCID mice. A total of 10 mice were injected subcutaneously with unsorted 5 × 106 BM cells/mouse from 5 different patients (2 mice/patient). Following 30 weeks of observation, we did not detect tumor growth or measurable levels of human paraproteins in mice sera (data not shown). We therefore next evaluated the ability of patient WM cells to engraft into human fetal bone implants in SCID-hu mice. WM cells from 10 different patients were used, and the characteristics of these patients are presented in Table 1. Unsorted 2 to 10 × 106 BM cells (n = 8 cases) or CD19+-sorted cells (n = 2 cases) per mouse were injected in human fetal bone implants in SCID-hu mice. Starting 2 weeks after cell inoculation, mice sera were serially collected and analyzed for appearance and level of human paraproteins. A total of 19 SCID-hu mice were used for this study. Eighteen of them were injected with cells from 9 patients (2 mice/patient sample). We observed patient WM-cell engraftment in 13 of 19 SCID-hu mice (69%), as demonstrated by appearance of human IgM and/or κ or λ chain in mice serum. As shown in Table 1, animals had detectable circulating IgM 4 to 28 weeks after cell injection. The kinetics of appearance and increase of human paraprotein was variable among patients. Both unsorted, as well as CD19+-sorted cells showed engraftment. Figure 1 shows the kinetics of human paraproteins (patient 1, 5, and 9 in Table 1) in 3 representative SCID-hu mice injected with 2 to 10 × 106/cells from 3 different patients with WM. In approximately 40% of cases, we detected an initial increase in human IgG in mice serum followed by subsequent decline to an undetectable level (Figure 1B-C), suggesting a progressive expansion of paraprotein-secreting monoclonal tumor cells and disappearance of nonclonal plasma cells.
Figure 1.

Engraftment of primary patient WM cells in SCID-hu mice. Blood was collected from mouse tail vein at different time points following inoculation of tumor cells directly in the human fetal bone chips, and sera were serially tested for circulating human IgM, IgG, and κ and λ chain by ELISA. The results demonstrate the appearance and increase in paraprotein, human IgM and/or light chain, in 3 representative SCID-hu mice, respectively, injected with patient IgMκ, IgMλ, and IgMκ WM cells. Panels A and B refer to mice injected with 5 and 10 × 106 BM cells, respectively, and panel C refers to a mouse injected with 2 × 106 CD19+-sorted cells.
To confirm that the serum paraprotein increase was due to huBM engraftment by patient WM cells, selected mice were killed, and the pathology of decalcified sections of huBM implants were analyzed. By H&E staining (Figure 2), the huBM implants examined showed significant intramedullary fibrosis and patchy infiltration with small lymphocytes, lymphoplasmacytoid cells, and plasma cells. We further evaluated the immunophenotype of these cells by immunohistochemical techniques. Figure 3 shows a representative case of a huBM injected with patient WM cells which expressed IgMκ. We observed cell-surface expression of CD20 (Figure 3A), cytoplasmic IgM (Figure 3B), and κ light chain (Figure 3C), but not IgG (Figure 3D) or λ light chain (Figure 3E) in the infiltrating lymphoplasmacytic cells, confirming the monotypic B-cell nature of these cells. We also found the presence of mast cells in these sections, as determined by immunohistochemical analysis for mast-cell tryptase (Figure 4), suggesting a potential role of these cells in WM pathogenesis. Interestingly, histologic evidence of residual fetal hematopoiesis was not observed. These overall findings indicate that the WM-cell growth is supported by the human BM microenvironment. To confirm the specific homing of patient WM cells into huBM, we further evaluated a variety of other tissues from injected mice. In mice that were injected with unsorted WM cells, we were unable to detect tissue infiltration by patient WM cells in murine BM, spleen, liver, lung, kidney, and lymph nodes (data not shown), suggesting a selective homing of primary tumor cells into huBM milieu. Interestingly, histopathologic examination of a mouse injected with CD19-sorted WM cells revealed tumor-cell infiltration in the murine spleen, liver, and bone marrow (data not shown), in addition to the huBM, suggesting a potential systemic spread of disease under these conditions.
Figure 2.
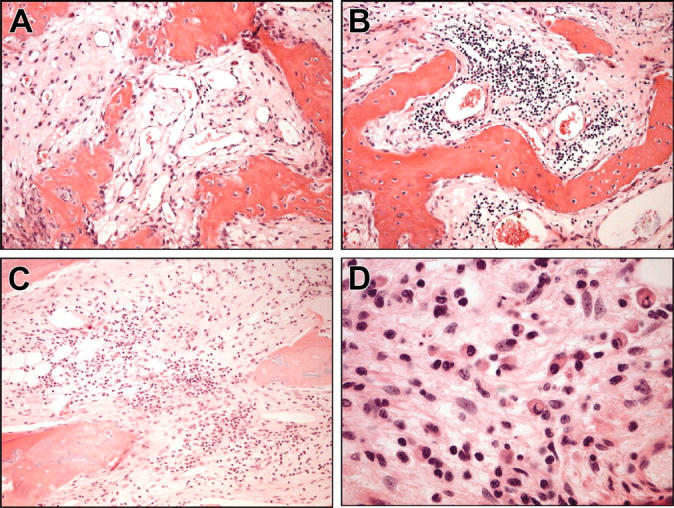
Histologic analysis of human fetal BM sections engrafted with WM cells. Analysis was performed on decalcified fetal bone chips implanted in SCID-hu mice and directly injected with patient unsorted WM cells. Bone chips were retrieved from mice after detection of rising levels of human paraprotein in murine serum and then stained with H&E. Four representative sections show infiltration with small lymphocytes, lymphoplasmacytoid cells, and plasma cells in the context of intramedullary fibrosis and scant residual fetal hematopoiesis. Original magnifications are (A) × 100, (B) × 100, (C) × 100, and (D) × 400.
Figure 3.

Immunohistochemical analysis of human fetal bone sections engrafted with WM cells. A fetal bone engrafted with IgMκ WM cells was retrieved from murine host after detection of rising levels of human paraprotein in mouse serum and examined. (A) Staining with anti-CD20 monoclonal antibody (mAb; original magnification, × 200), (B) anti-IgM polyclonal Ab (original magnification, × 200), and (C) anti-κ mAb (original magnification, × 200), demonstrates predominantly cytoplasmic reactivity within the plasmacytic component of the tumor only, (D) anti-IgG mAb (original magnification, × 400), and (E) anti-λ mAb (original magnification, × 200).
Figure 4.

Immunohistochemical analysis for mast-cell tryptase in human fetal BM sections after engraftment of unsorted WM cells. A fetal bone engrafted with IgMκ WM cells was retrieved from murine host after detection of rising levels of human paraprotein in mouse serum and then examined for the presence of mast cells as assessed by immunostaining for mast-cell tryptase. Original magnification, × 400.
To evaluate the potential utility of this model to investigate the efficacy of therapeutic agents, we used rituximab, which has demonstrated antitumor activity against WM.21-25 SCID-hu mice bearing patient WM cells were treated with rituximab (25 mg/kg, intraperitoneally) on every other day for a total of 3 injections, and serum human paraproteins were measured to monitor WM growth. As seen in Figure 5A, treatment with rituximab induced a dramatic tumor regression, as demonstrated by decrease in IgM to undetectable serum levels. We detected high IgG levels following rituximab administration and its progressive slow decline because of the clearance of the circulating chimeric mAb from serum (Figure 5B). These results indicate that this SCID-hu model not only allows for the study of WM biology, but it is also a useful system for the preclinical evaluation of novel agents potentially active against WM.
Figure 5.

Evaluation of response to rituximab in the SCID-hu WM model. Kinetics of change in human IgM paraprotein in murine serum from a representative SCID-hu mouse. (A) Serum samples were collected from mice bearing WM cells before and after rituximab therapy (25 mg/kg) every other day for a total of 3 injections. (B) Kinetics of change in IgG levels in mouse serum treated with rituximab; the IgG increase is related to the injection of rituximab (chimeric IgG). Arrows indicate the first day of treatment.
Discussion
In this study, we demonstrate that primary patient WM cells engraft in huBM microenvironment implanted in immunocompromised mice. To our knowledge, this is the first report demonstrating the potential of primary patient WM cells to engraft and grow in SCID-hu mice recapitulating the biologic features of human disease.
Murine models of human cancer are extremely useful in studying the pathobiology of cancer and for preclinical evaluation of experimental therapies. Unfortunately, most of these murine models are obtained by xenografting tumor cell lines instead of primary patient tumor cells. These cell lines are generally established in vitro using cells from metastatic lesions or effusions and are maintained through numerous in vitro passages that allow accumulation of genetic abnormalities, conferring a progressive dedifferentiated and aggressive phenotype not similar to the original disease. Furthermore, in these models, human tumor xenografts engraft and proliferate in a murine microenvironment, with growth characteristics not similar to primary clinical presentation (eg, cutaneous, or other tissues, versus bone marrow).
To date, the only available animal model of WM uses WSU-WM cell line xenografted in a murine subcutaneous microenvironment. This cell line has been obtained from a pleural effusion of a patient with IgMκ WM with advanced and therapy-resistant disease and has been characterized as aggressive WM rather than more typical indolent disease.5,6 Cytogenetically this cell line has been characterized by an 8;14 chromosomal translocation and c-myc rearrangement, not characteristic of typical WM, but usually associated with non-Hodgkin lymphomas.26 In addition, the WSU-WM cell line shows a switch from the original κ to λ chain, further supporting the evidence of accumulation of genetic abnormalities that confer aggressive biologic behavior to WSU-WM cells. These cells grow in the murine microenvironment and, unlike primary WM, do not require a human BM microenvironment. Therefore, this model does not recapitulate the in vivo biologic features of a typical WM nor does it reproduce the disease in a huBM milieu.
In contrast, our model has a number of characteristics that mimics the human WM: (1) the model supports growth of primary WM cells from patient BM, (2) the cell growth is dependent on human BM microenvironment, (3) the progression of disease is determined accurately by measuring human IgM and/or light chain in murine serum similar to the patient setting, and (4) we have confirmed efficacy of anti-CD20 mAb in this model as a measure of reproducibility of the model for preclinical investigations. Therefore, this model, which recapitulates the pathologic features of human disease, will help us understand important aspects of the disease biology, for example, the role of BM stromal cells and mast cells in WM growth and survival and the presence of WM stem cells. This model will also allow preclinical investigation of novel agents targeting WM cells as well as its microenvironment prior to their evaluation in patients.
Acknowledgments
We thank Janice Williams, Donna Skinner, and Vuong Nguyen for technical assistance in histopathologic examinations.
Prepublished online as Blood First Edition Paper, May 10, 2005; DOI 10.1182/blood-2004-11-4477.
Supported by Multiple Myeloma Research Foundation Awards (N.C.M. and K.C.A.); Department of Veteran's Affairs merit review award, and Leukemia and Lymphoma Society Scholar in Translational Research Award (N.C.M.); the National Institute of Health (NIH) grants P50-100707 and PO1-78378 (K.C.A., N.C.M.) and RO1-50947 (K.C.A.); the Doris Duke Distinguished Clinical Research Scientist Award (K.C.A.); and an NIH Career Development Award grant K23-CA087977 (S.P.T.).
An Inside Blood analysis of this article appears at the front of the issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 U.S.C. section 1734.
References
- 1.Owen RG, Treon SP, Al-Katib A, et al. Clinicopathological definition of Waldenstrom's macroglobulinemia: consensus panel recommendations from the Second International Workshop on Waldenstrom's Macroglobulinemia. Semin Oncol. 2003;30: 110-115. [DOI] [PubMed] [Google Scholar]
- 2.Harris NL, Jaffe ES, Stein H, et al. A revised European-American classification of lymphoid neoplasms: a proposal from the International Lymphoma Study Group. Blood. 1994;84: 1361-1392. [PubMed] [Google Scholar]
- 3.Harris NL, Jaffe ES, Diebold J, et al. World Health Organization classification of neoplastic diseases of the hematopoietic and lymphoid tissues: report of the Clinical Advisory Committee meeting-Airlie House, Virginia, November 1997. J Clin Oncol. 1999;17: 3835-3849. [DOI] [PubMed] [Google Scholar]
- 4.Ghobrial IM, Gertz MA, Fonseca R. Waldenstrom macroglobulinaemia. Lancet Oncol. 2003;4: 679-685. [DOI] [PubMed] [Google Scholar]
- 5.Al-Katib A, Mohammad R, Hamdan M, Mohamed AN, Dan M, Smith MR. Propagation of Waldenstrom's macroglobulinemia cells in vitro and in severe combined immune deficient mice: utility as a preclinical drug screening model. Blood. 1993;81: 3034-3042. [PubMed] [Google Scholar]
- 6.Al-Katib AM, Mensah-Osman E, Aboukameel A, Mohammad R. The Wayne State University Waldenstrom's Macroglobulinemia preclinical model for Waldenstrom's macroglobulinemia. Semin Oncol. 2003;30: 313-317. [DOI] [PubMed] [Google Scholar]
- 7.McCune JM, Namikawa R, Kaneshima H, Shultz LD, Lieberman M, Weissman IL. The SCID-hu mouse: murine model for the analysis of human hematolymphoid differentiation and function. Science. 1988;241: 1632-1639. [DOI] [PubMed] [Google Scholar]
- 8.Namikawa R, Ueda R, Kyoizumi S. Growth of human myeloid leukemias in the human marrow environment of SCID-hu mice. Blood. 1993;82: 2526-2536. [PubMed] [Google Scholar]
- 9.Kyoizumi S, Baum CM, Kaneshima H, McCune JM, Yee EJ, Namikawa R. Implantation and maintenance of functional human bone marrow in SCID-hu mice. Blood. 1992;79: 1704-1711. [PubMed] [Google Scholar]
- 10.Kyoizumi S, Murray LJ, Namikawa R. Preclinical analysis of cytokine therapy in the SCID-hu mouse. Blood. 1993;81: 1479-1488. [PubMed] [Google Scholar]
- 11.Akkina RK, Rosenblatt JD, Campbell AG, Chen IS, Zack JA. Modeling human lymphoid precursor cell gene therapy in the SCID-hu mouse. Blood. 1994;84: 1393-1398. [PubMed] [Google Scholar]
- 12.Chen BP, Galy A, Kyoizumi S, et al. Engraftment of human hematopoietic precursor cells with secondary transfer potential in SCID-hu mice. Blood. 1994;84: 2497-2505. [PubMed] [Google Scholar]
- 13.Sandhu JS, Clark BR, Boynton EL, et al. Human hematopoiesis in SCID mice implanted with human adult cancellous bone. Blood. 1996;88: 1973-1982. [PubMed] [Google Scholar]
- 14.Urashima M, Chen BP, Chen S, et al. The development of a model for the homing of multiple myeloma cells to human bone marrow. Blood. 1997;90: 754-765. [PubMed] [Google Scholar]
- 15.Yaccoby S, Barlogie B, Epstein J. Primary myeloma cells growing in SCID-hu mice: a model for studying the biology and treatment of myeloma and its manifestations. Blood. 1998;92: 2908-2913. [PubMed] [Google Scholar]
- 16.Anderson KC. Targeted therapy for multiple myeloma. Semin Hematol. 2001;38: 286-294. [DOI] [PubMed] [Google Scholar]
- 17.Hideshima T, Anderson KC. Molecular mechanisms of novel therapeutic approaches for multiple myeloma. Nat Rev Cancer. 2002;2: 927-937. [DOI] [PubMed] [Google Scholar]
- 18.Anderson KC. Moving disease biology from the lab to the clinic. Cancer. 2003;97: 796-801. [DOI] [PubMed] [Google Scholar]
- 19.Tassone P, Gozzini A, Goldmacher V, et al. In vitro and in vivo activity of the maytansinoid immunoconjugate huN901-N2′-deacetyl-N2′-(3-mercapto-1-oxopropyl)-maytansine against CD56+ multiple myeloma cells. Cancer Res. 2004;64: 4629-4636. [DOI] [PubMed] [Google Scholar]
- 20.Tassone P, Goldmacher VS, Neri P, et al. Cytotoxic activity of the maytansinoid immunoconjugate B-B4-DM1 against CD138+ multiple myeloma cells. Blood. 2004;104: 3688-3696. [DOI] [PubMed] [Google Scholar]
- 21.Byrd JC, White CA, Link B, et al. Rituximab therapy in Waldenstrom's macroglobulinemia: preliminary evidence of clinical activity. Ann Oncol. 1999;10: 1525-1527. [DOI] [PubMed] [Google Scholar]
- 22.Foran JM, Rohatiner AZ, Cunningham D, et al. European phase II study of rituximab (chimeric anti-CD20 monoclonal antibody) for patients with newly diagnosed mantle-cell lymphoma and previously treated mantle-cell lymphoma, immunocytoma, and small B-cell lymphocytic lymphoma. J Clin Oncol. 2000;18: 317-324. [DOI] [PubMed] [Google Scholar]
- 23.Treon SP, Anderson KC. The use of rituximab in the treatment of malignant and nonmalignant plasma cell disorders. Semin Oncol. 2000;27: 79-85. [PubMed] [Google Scholar]
- 24.Treon SP, Agus DB, Link B, et al. CD20-directed antibody-mediated immunotherapy induces responses and facilitates hematologic recovery in patients with Waldenstrom's macroglobulinemia. J Immunother. 2001;24: 272-279. [PubMed] [Google Scholar]
- 25.Dimopoulos MA, Zervas C, Zomas A, et al. Treatment of Waldenstrom's macroglobulinemia with rituximab. J Clin Oncol. 2002;20: 2327-2333. [DOI] [PubMed] [Google Scholar]
- 26.Schop RF, Fonseca R. Genetics and cytogenetics of Waldenstrom's macroglobulinemia. Semin Oncol. 2003;30: 142-145. [DOI] [PubMed] [Google Scholar]