

Abstract
Hemophilia A is a clinically important coagulation disorder caused by the lack or abnormality of plasma coagulation factor VIII (FVIII). Gene transfer of the FVIII cDNA to hepatocytes using lentiviral vectors is a potential therapeutic approach. We investigated the efficacy of feline immunodeficiency virus (FIV)–based vectors in targeting hepatocytes and correcting FVIII deficiency in a hemophilia A mouse model. Several viral envelope glycoproteins were screened for efficient FIV vector pseudotyping and hepatocyte transduction. The GP64 glycoprotein from baculovirus Autographa californica multinuclear polyhedrosis virus pseudo-typed FIV efficiently and showed excellent hepatocyte tropism. The GP64-pseudotyped vector was stable in the presence of human or mouse complement. Inclusion of a hybrid liver-specific promoter (murine albumin enhancer/human α1-antitrypsin promoter) further enhanced transgene expression in hepatocytes. We generated a GP64-pseudotyped FIV vector encoding the B domain–deleted human FVIII coding region driven by the liver-specific promoter, with 2 beneficial point mutations in the A1 domain. Intravenous vector administration conferred sustained FVIII expression in hemophilia A mice for several months without the generation of anti–human FVIII antibodies and resulted in partial phenotypic correction. These findings demonstrate the utility of GP64-pseudotyped FIV lentiviral vectors for targeting hepatocytes to correct disorders associated with deficiencies of secreted proteins.
Introduction
Hemophilia A is an X-linked disorder caused by a deficiency of plasma factor VIII (FVIII), affecting 1 in 5000 to 10 000 males1,2 and characterized by an abnormal bleeding tendency. In its severe form (less than 1% of the normal FVIII level) the disease may be fatal,2,3 and spontaneous hemorrhage into joints and muscles leading to permanent disability remains a significant clinical problem. Intravenous injections of FVIII concentrates purified from human plasma or produced by recombinant technology are effective in controlling bleeding episodes. However, prophylactic treatment of hemophilia A is problematic due to limited availability and high cost. Venous access and the risk of blood-borne transmissible diseases such as AIDS and hepatitis are also a concern. Moreover, the development of inhibitory antibodies can significantly reduce the efficacy of replacement therapy in about 20% of patients.4
Gene transfer provides an alternative therapeutic approach for long-term correction of FVIII deficiency. Transferring a functional FVIII gene to somatic cells, particularly hepatocytes, could provide sustained production of FVIII. Increasingly, prophylactic FVIII replacement therapy to maintain plasma levels above 1% is recommended in selected children and young adults by most hemophilia treatment centers in the United States and Europe.5,6 A stable level of FVIII in the bloodstream may prevent breakthrough bleeds and protect against spontaneous bleeding and chronic joint injury. Gene therapy is particularly suitable for hemophilia A,7,8 because as little as 5 ng/mL of plasma FVIII (normal levels about 200 ng/mL), equivalent to a production rate of 30 μg per 109 cells per day, is sufficient to convert severe hemophilia A to a mild form.7 Indeed, recent reports from many laboratories including ours have demonstrated the potential feasibility of retroviral gene therapy for hemophilia A in animal models.9,10 While an efficacious therapy has not yet been achieved in the clinic, a phase 1 dose escalation trial of intravenous infusion of retroviral vectors at low doses was safe, and expression persisted in some subjects for more than a year.11
Viral vectors currently used for in vivo FVIII gene transfer include Moloney leukemia virus (MLV),9,12,13 lentiviral vectors (HIV, feline immunodeficiency virus [FIV], and others),10,14-16 adeno-associated virus (AAV),17,18 and adenoviral vectors.19-22 Nonviral vector systems also demonstrate promise in gene delivery to hepatocytes.23-25 While each vector system has its advantages and drawbacks, lentiviral-based vectors are attractive because they transduce proliferating and nonproliferating cells at similar efficiencies and mediate stable integration, resulting in sustained expression.10,26 Furthermore, the packaging capacity of lentiviral vectors readily accommodates a promoter and the B domain–deleted FVIII cDNA.27
Cell tropism of lentiviral vectors is governed largely by interactions between the envelope glycoprotein and its host cell receptor. Retroviral vectors are most commonly pseudotyped with the vesicular stomatitis virus G (VSV-G) or amphotropic glycoproteins, neither of which directs efficient transduction of quiescent hepatocytes. Moreover, both envelope glycoproteins have limitations for potential clinical uses. The VSV-G is cytotoxic14 and may be inactivated by human serum,28 and the amphotropic envelope is fragile and does not tolerate most centrifugation concentration methods.29
Here we report development of a novel FIV-based vector for stable expression of human FVIII (hFVIII) in hepatocytes. Extensive screening of envelope glycoproteins from several virus families for their efficiency in directing liver gene transfer revealed that the GP64 glycoprotein from baculovirus Autographa californica multinuclear polyhedrosis virus (AcMNPV) was efficiently incorporated into FIV vector particles and conferred extensive hepatocyte gene transfer. FVIII production and secretion from the transduced hepatocytes was increased by incorporation of a liver-specific promoter and mutation of 2 amino acids in the FVIII A1 domain. Systemic administration of GP64-pseudotyped FIV vector encoding hFVIII produced sustained FVIII expression and clinically significant phenotypic correction in immunocompetent hemophilia A mice. These studies bring us closer to the clinical application of FIV for hemophilia A therapy.
Materials and methods
FIV vector constructs and modifications
FIV particles were generated by triple transfection of 293T cells with vector, envelope, and packaging plasmids, followed by collection of supernatants and particle concentration as described previously.30,31 The vector plasmid was based on the pVETL backbone containing the cytomegalovirus (CMV) promoter/enhancer in place of the 5′ U3, a truncated gag, and a multiple cloning site. Several FIV vector constructs were generated from this backbone for this study. The FIV vector encoding nuclear-targeted β-galactosidase (CMV-βgal) was generated by ligating the nuclear-targeted βgal cDNA, directed by the CMV promoter/enhancer, into the multiple cloning site of pVETL. The CMV promoter/enhancer was then replaced with the murine albumin enhancer/human α1-antitrypsin promoter (AlbE/AAT), as reported by Kramer and colleagues,32 to generate an FIV vector encoding nuclear-targeted βgal driven by a liver-specific promoter/enhancer (AlbE/AAT-βgal). A previously described FIV-RSV-hFVIII vector construct10 underwent 2 modifications to generate AlbE/AAT-ΔΔhFVIII. The RSV promoter was replaced with a liver-specific AlbE/hAAT promoter, and 2 amino acids in the FVIII A1 domain (L303E and F309S) were mutated using the QuikChange site-directed mutagenesis kit (Stratagene, La Jolla, CA).33 Vector DNA was purified and sequenced to confirm the presence of the mutations.
The envelope expression plasmid contains the CMV promoter, the simian virus 40 polyadenylation signal, and the envelope glycoprotein of interest. A series of viral glycoproteins were evaluated for their compatibility with FIV vector and their efficiency in directing gene transfer to the liver. These include env glycoproteins from (1) alphaviruses: Sindbis, Semliki Forest virus, and Ross River virus (RRV)34; (2) filoviruses (Marburg and Ebola virus)35; (3) the baculovirus AcMNPV GP64 36; (4) lymphocytic choriomeningitis virus (LCMV)37; (5) (VSV-G)29; and (6) the amphotropic envelope glycoprotein.38 To generate the AcMNPV baculovirus GP64 expression construct, the envelope glycoprotein cDNA from DH10B cells containing the “bacmid” with AcMNPV GP64 (Bac-to-Bac system; Invitrogen, Carlsbad, CA) was cloned. Using 300 ng of the bacmid as template, we performed polymerase chain reaction (PCR) using the following gene-specific primers: forward primer: 5′-ATGGTAAGCGCTATTG; reverse primer: 5′-TTAATATTGTCTATTACGG-3′. The PCR product was cloned into the TOPO Blunt vector (Invitrogen) and verified by sequencing. The verified insert was excised using the EcoRI sites and cloned into the pcDNA 4/V5-His vector (Invitrogen).
Transduction titers of the 250-fold concentrated viral preparations were determined by measurement of X-gal–positive cells in transduced HT1080 cells (for βgal-expressing FIV vectors) or by real-time PCR quantification of FIV proviral genome copy number in transduced HT1080 target cells for luciferase and FVIII vectors as described previously39 and expressed as transducing units (TU) per milliliter.
Mice
All mice used in this study were housed at the University of Iowa Animal Care Facilities and all animal procedures approved by the Animal Care and Use Review Committee at the University of Iowa. Hemophilia A mice containing a disruption of the murine FVIII gene in exon 16 40 were backcrossed with C57BL/6 mice for more than 6 generations10,41 and used in this study. Adult (4- to 8-week) hemizygous affected males and homozygous affected females were used for these studies.
In vivo gene-transfer protocols
To screen pseudotyped vectors for their efficiency in directing in vivo gene transfer to the liver, mice were injected via the tail vein with 2 doses of FIV-CMV-βgal pseudotyped with a selected envelope over 2 consecutive days. Polybrene at 40 μg/mL was included in all injections including buffer-injected control mice.9 One and 7 days after injection, we obtained blood samples from the retro-orbital plexus and assayed serum samples for the levels of glutamic oxaloacetic transaminase (SGOT) and glutamic pyruvic transaminase (SGPT), as described previously.34 Three weeks after injection, the mice were killed and perfused with cold phosphate-buffered saline (PBS) and 2% paraformaldehyde/PBS. Following the perfusion, samples of the remaining liver, spleen, kidney, lung, and heart were harvested for X-gal staining and βgal enzyme activity assays. To assess gene expression from the liver-specific AlbE/AAT promoter in vivo, mice received intravenously with the tail vein a single bolus of GP64-pseudotyped AlbE/AAT-βgal in a volume of 0.3 mL. The βgal expression was similarly measured 3 weeks after gene transfer.
To assess the efficacy of GP64-pseudotyped FIV vector in correcting FVIII deficiency, the hemophilia A mice were administered GP64/AlbE/AAT-ΔΔhFVIII (total delivered dose 5 × 108 TU, real-time PCR titer) over 2 consecutive days (1 injection per day). At the time points indicated in the legend of Figure 4A, blood samples were withdrawn from the retro-orbital plexus, sodium citrate was immediately added to a final concentration of 0.38% (wt/vol), and plasma samples prepared by centrifugation. The levels of hFVIII expression and the production of anti-hFVIII antibodies were measured by enzyme-linked immunosorbent assay (ELISA), described in “Measurement of plasma hFVIII expression levels and detection of anti-hFVIII antibodies.” To assess phenotypic correction of FVIII null mice, a tail clip assay was performed.10 Briefly, mice (about 30 weeks old) were restrained in a standard plastic mouse restraint, and a 1 cm length of tail (about 2 mm in diameter) was removed with a razor blade. Blood was allowed to drain into a 1.5 mL tube at room temperature for 45 minutes and blood loss determined by weight. If tail bleeding had not stopped after 45 minutes, thrombin was applied topically. Mice were observed for 4 hours and survival recorded. Untreated FVIII null mice and C57BL/6 mice served as controls.
Figure 4.

Persistent and therapeutic FVIII expression following systemic injection of GP64/AlbE/AAT-ΔΔhFVIII vector. About 5 × 108 TU (real-time PCR titer) GP64-pseudotyped FIV vector encoding hFVIII driven by the AlbE/AAT promoter was injected via the tail vein into hemophilia A mice over 2 consecutive days. (A) hVIII levels were measured by ELISA at the time points indicated (n = 5, mean ± SE). Results shown are representative of 2 independent experiments with 5 experimental mice per group. (B) Quantitation of blood loss over 45 minutes following tail vein cutting in wild-type mice (n = 4), FVIII–/– mice (n = 4), and FVIII–/– mice that received GP64/AlbE/AAT-ΔΔhFVIII (FIV/hFVIII, n = 10). Results shown are mean ± SE. FIV-treated animals showed significantly less blood loss (P < .03). (C) Functional hFVIII activity as assessed by Coamatic chromogenic assay. Results contrast wild-type mice, FVIII null mice, and FVIII null treated with GP64/AlbE/AAT-ΔΔhFVIII. Results shown are mean ± SE; *P < .05 when compared with untreated FVIII null mice; 4 to 5 mice per group.
Determination of β-galactosidase expression
For X-gal staining of various organs after intravenous vector injection, tissues were fixed in 2% paraformaldehyde/PBS overnight and then stained with X-gal overnight at 37°C. The overall expression of βgal was first examined by stereomicroscopy. The X-gal–stained tissues were then embedded in paraffin, 5 μm sections cut at 50 μm intervals, and counterstained with nuclear fast red or hematoxylin and eosin for quantification and histologic examination. To quantify the percentage of FIV-transduced cells in liver tissue, 6 to 7 representative fields from a minimum of 6 sections from each animal were examined under 20 × magnification, using an Olympus BX60 microscope, an Olympus DP70 digital camera, and UPlan FI 20×/0.5 NA objective (Olympus, Melville, NY); Scion acquisition software (Frederick, MD); and Adobe Photoshop (Adobe Systems, San Jose, CA) for image processing. Approximately 600 to 900 cells were counted from each section. The number of βgal-positive hepatocytes, endothelial cells, and Kupffer cells was identified using morphologic criteria.
The Galacto-light chemiluminescent reporter assay (Tropix, Bedford, MA) was used to measure tissue βgal activity as described previously.34 Tissues were homogenized and endogenous tissue βgal eliminated by heat inactivation in conjunction with protease inhibitors. Tissue extracts were then assayed for βgal activities and total protein concentrations. βGal levels were determined by interpolation off a standard curve established from 2-fold serial dilutions of purified βgal (Sigma, St Louis, MO) and expressed as nanograms of βgal per milligram of protein.
Stability of GP64-pseudotyped vectors in human or mouse serum
To examine the resistance of the GP64- or VSV-G–pseudotyped FIV vectors to inactivation by human serum, GP64- or VSV-G–pseudotyped FIV vectors were incubated with either 80% competent human sera or 80% heat-inactivated human sera at 37°C for 1 hour (gift from Dr Erik Edens, University of Iowa Complement Laboratory). The vectors were also similarly treated with mouse sera. After incubation, the vectors were titered on HT1080 cells. The results were means of 3 separate titering plates and were normalized to the heat-inactivated serum controls.
Measurement of plasma hFVIII expression levels and detection of anti-hFVIII antibodies
The levels of hFVIII expression in plasma samples were quantified by a sandwich ELISA as described previously.10 Briefly, 96-well immunoplates were coated with 2 murine monoclonal anti-hFVIII antibodies, a monoclonal antibody against hFVIII:c light chain (Boehringer Mannheim, Indianapolis, IN), and the ESH5 monoclonal antibody against hFVIII:c A1 heavy chain (American Diagnostica, Greenwich, CT). Horseradish peroxidase–conjugated ESH8 monoclonal antibody specific for hFVIII:c C2 domain (American Diagnostica) was used as the secondary antibody to detect bound FVIII antigen. Recombinant hFVIII (Baxter Healthcare, Glendale, CA) and pooled normal human plasma (George King Bio-medical, Overland Park, KS) were used as a standard with serial dilutions ranging from 0.3 to 79 ng/mL. The sensitivity of this assay was 0.3 ng/mL, and normal mouse plasma did not interfere with the assay. The production of immunoglogulin G (IgG) and IgM antibody immune responses against hFVIII in the recipient mice was measured by ELISA as described.10 Briefly, 96-well plates were coated overnight at 4°C with 0.8 μg/mL recombinant hFVIII in 0.05 M carbonate-bicarbonate (pH 9). After blocking with PBS/10% fetal bovine serum and washing, the plates were incubated overnight at 4°C with triplicate serial dilutions of test plasma samples and then probed with alkaline phosphatase–conjugated goat anti–mouse IgG and IgM (Southern Biotechnology Associates, Birmingham, AL). The plates were finally developed with P-nitrophenyl phosphate (Sigma), and the absorbance was read at 405 nm using an automated microtiter plate ELISA reader (Molecular Devices, Sunnyvale, CA). Monoclonal antibody against hFVIII:c light chain (Boehringer Mannheim) was used as a standard.
Determination of FVIII activity
FVIII activity in plasma was determined using the Coamatic Factor VIII Chromogenic Assay (DiaPharma, West Chester, OH), following the manufacturer's directions. Four FVIII null mice received GP64/AlbE/AAT-ΔΔhFVIII as described for Figure 4A. Plasma from 4 untreated FVIII null mice served as a negative control while plasma from 5 wild-type C57BL/6 served as a positive control. Assays were performed 4 and 8 weeks after gene transfer. Samples were read at 405 nm and analyzed (VersaMax microplate reader; Molecular Devices, Sunnyvale, CA). All standards and samples were performed in duplicate, and means were calculated.
Results
GP64 envelope glycoprotein directs efficient gene transfer to the liver
We reasoned that pseudotyping the vector with envelope glycoproteins that better target hepatocytes could augment FVIII expression. A number of heterologous env glycoproteins from enveloped viruses were screened to identify those compatible with FIV and showing enhanced hepatic tropism when contrasted with the VSV-G envelope. The envelope glycoproteins from Marburg virus, wild-type Ebola virus (Zaire strain), and Sindbis virus yielded FIV vector titers of less than 1 × 106 TU/mL after concentration and were not suitable for in vivo studies. The Semliki Forest virus envelope glycoprotein, LCMV WE54 envelope glycoprotein, VSV-G, amphotropic, a modified Ebola virus glycoprotein, and baculovirus GP64 were efficiently incorporated into FIV virions, generating concentrated vector titers of 1 × 108 TU/mL, and were thus selected for in vivo studies.
For screening, FIV vector preparations encoding the Escherichia coli βgal reporter were injected via the tail vein into hemophilia A mice and βgal expression in the liver measured 3 weeks after injection. Despite high vector titers, Semliki Forest virus, LCMV WE54, VSV-G, amphotropic, and the modified Ebola pseudotypes directed poor liver transduction (data not shown). In contrast, GP64-pseudotyped FIV showed remarkable liver transduction compared with prior work using VSV-G and RRV envelopes.10,34
The AcMNPV GP64 FIV titers ranged from 1 × 108 to 10 × 108 TU/mL, comparable to those obtained with the VSV-G envelope.30,39 To quantify the relative tissue tropism of the GP64 envelope, we injected GP64-pseudotyped FIV into mice and assayed βgal activity in liver, spleen, heart, lung, and kidney 3 weeks later. The data in Figure 1A demonstrate that the predominant tissue transduced following systemic administration of GP64/CMV-βgal vector was the liver (about 33 ng/mg protein), with much lower levels of βgal activity observed in heart and spleen. A survey of X-gal–stained tissues (Figure 1B) revealed impressive staining in liver, with only occasional βgal–expressing cells in heart and spleen.
Figure 1.
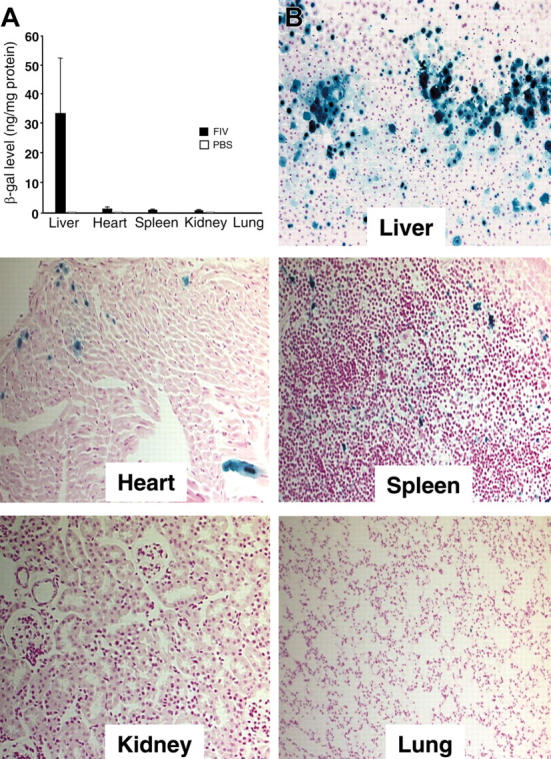
Tissue transduction. (A) Tissue distribution of βgal expression 3 weeks following delivery of 2.4 × 108 TU GP64/CMV-βgal. Control animals received injection of PBS (7 β-gal; 3 PBS). Error bars indicate mean ± SEM. (B) Localization of βgal expression in tissues (magnification, 20×). One-month-old mice were injected via tail vein over 2 consecutive days with 2.4 × 108 TU GP64/CMV-βgal. Three weeks after injection, the mice were killed and organs (liver, heart, spleen, kidney, and lung) harvested and X-gal stained.
Three weeks following gene transfer with GP64-pseudotyped FIV with the βgal marker gene, transgene expression remained robust. Figure 2A-C shows livers from representative control and vector-injected animals. Quantitation indicated that 15.2% of the liver cells were βgal positive (Table 1), a significant improvement over our previously reported results with RRV-pseudotyped FIV (6%) or with VSV-G–pseudotyped FIV (3%) (Table 1, with data in part from Kang et al34). Most of the transgene-expressing cells had morphologic characteristics of hepatocytes (98%), but some non-hepatocytes morphologically consistent with Kupffer cells were targeted. In some cases we observed neighboring cells expressing βgal, suggesting there may have been clonal expansion of a single transduced cell (Figure 2D).
Figure 2.
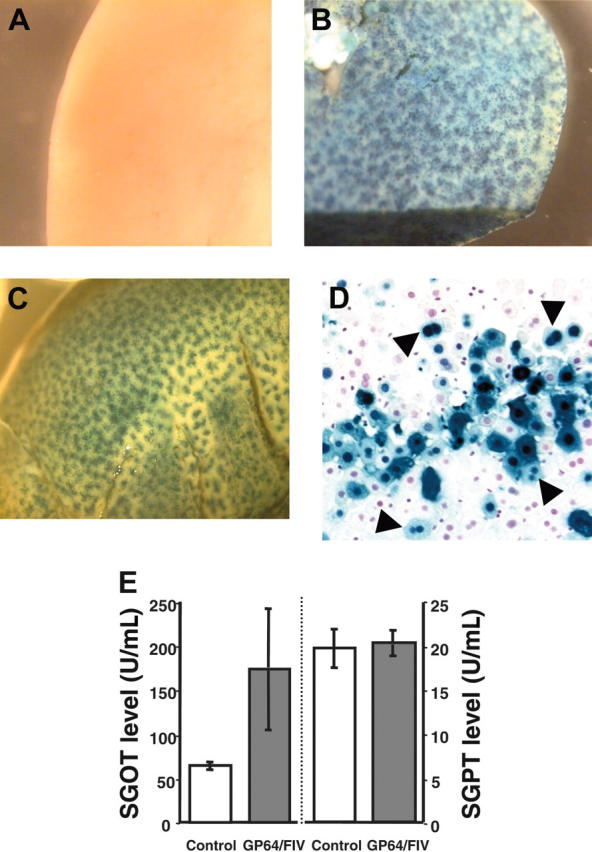
Liver staining. One-month-old mice received either control buffer (A) or GP64/CMV-βgal vector (B-C). Three weeks after injection, livers were X-gal stained and examined under stereomicroscopy (2 representative mice from 3 separate sets of experiments from 15 experimental and 8 control animals are shown). (D) Liver tissue section. Most of the nuclear-targeted β-gal–positive cells were hepatocytes. In some cases 2 neighboring cells had blue nuclei (arrowheads), suggesting clonal expansion. (E) Plasma SGOT (left) and SGPT (right) levels at 24 hours after injection with PBS or FIV vector (5 experimental and 3 control mice, mean ± SE).
Table 1.
Liver transduction with FIV constructs
|
Transduced cell type, %
|
|||
|---|---|---|---|
| Vector | % Transduction* | Hepatocytes | Nonhepatocytes |
| GP64/CMV-βgal; n = 8 | 15.2 (1.4) | 98 | 2 |
| GP64/AlbE/AAT-βgal; n = 3 | 23.4 (2.6) | 99.4 | 0.6 |
| RRV/FIV-RSV-cyto-β-gal; best34 | 6 | 75 | 25 |
| VSV-G/FIV-RSV-cyto-β-gal; best34 | 3 | 75 | 25 |
All mice received similar doses of vector (1 × 108 to 3.5 × 108 TU).
Best indicates highest expression levels achieved.
Mean (± SE).
Previous studies demonstrated that some vector envelopes, such as VSV-G, are associated with hepatotoxicity.14,16,34 To evaluate the acute effects of the vector on liver function, we measured SGOT and SGPT levels in mice 24 hours after injection with the GP64 pseudotype. As shown in Figure 2E, the GP64/CMV-βgal caused a modest elevation in SGOT but no significant change in SGPT compared with PBS-treated control animals. The SGOT levels returned to normal range when measured at day 7 after injection (data not shown). This contrasts with the 2- to 3-fold increases in these enzymes we noted in a previous study with VSV-G–pseudotyped FIV.34
GP64-pseudotyped FIV vectors are stable in human and mouse plasma
Enveloped viruses are subject to complement-mediated inactivation in plasma, a principle that applies also to gene transfer vectors, as previously reported for VSV-G–pseudotyped retroviruses28 and wild-type baculovirus vectors.42 To examine the sensitivity of the GP64- or VSV-G–pseudotyped FIV vectors to inactivation by human serum, the FIV vectors were incubated with either 80% competent human or mouse sera or 80% heat-inactivated human or mouse sera at 37°C for 1 hour. Following incubation, vectors were titered on HT1080 cells by limiting dilution. As shown in Figure 3, the VSV-G vector was inactivated in the presence of human serum, consistent with previous reports.28 In contrast the GP64 vector was stable in the presence of human or mouse serum.
Figure 3.

Stability of GP64-pseudotyped vector in human and mouse sera. GP64- or VSV-G–pseudotyped FIV vectors were incubated with either 80% competent human sera or 80% heat-inactivated human sera at 37°C for 1 hour. The vectors were similarly treated with mouse sera. After incubation, the vectors were titered on HT1080 cells. The results represent means of 3 separate titering plates and were normalized to the heat-inactivated serum controls. Data shown are representative of 2 replicate experiments. Dashed line indicates 100% of control titer.
A liver-specific promoter confers enhanced, long-term transgene expression in vivo
Initial experiments with GP64 FIV were performed using vector constructs driven by the CMV promoter (Figures 1, 2). Of note, the predominant cell type targeted was the hepatocyte (Table 1). We also noted that approximately 2% of transduced cells were Kupffer cells. We hypothesized that a liver-specific promoter would direct exclusive hepatocyte expression and improve persistence. To test this, we generated a hybrid promoter composed of the mouse albumin enhancer and the human AAT32 driving nuclear-targeted βgal and performed additional studies. Pilot experiments verified that the GP64/AlbE/AAT-βgal vector was efficiently expressed in human HepG2 cells, with little expression in 293 cells (data not shown). As shown in Table 1, the percentage of transgene-expressing cells in the liver was significantly increased in GP64/AlbE/AAT-βgal–versus GP64/CMV-βgal–treated animals (mean, 23.4%; range, 20.2% to 25.8%). Also, Kupffer cell transduction was remarkably reduced.
Gene transfer with GP64-pseudotyped FIV expressing hFVIII confers long-term correction of hemophilia A in a mouse model
We next prepared an expression construct containing B domain–deleted hFVIII cDNA with 2 point mutations (L303E and F309S) to enhance protein secretion33 and driven by the AlbE/AAT promoter (GP64/AlbE/AAT-ΔΔhFVIII). Approximately 5 × 108 TU (real-time PCR titer) of GP64/AlbE/AAT-ΔΔhFVIII was delivered via tail vein to immunocompetent hemophilia A mice over 2 consecutive days. Plasma levels of hF VIII levels were measured by ELISA at the times indicated (Figure 4A). Mice transduced with the GP64 FIV vector expressed therapeutically relevant levels of hFVIII throughout the almost 8-month duration of the study. Following an initial peak of expression of about 20 ng/mL, hFVIII protein levels stabilized around 5 to 10 ng/mL. In addition, there were no significant differences in hFVIII protein levels between male and female mice. Of interest, no antibodies to hFVIII were detected when assayed 2 weeks or 4 and 6 months following gene transfer. The lack of antibody responses may be strain related. Therefore, we treated wild-type BALB/c mice with the same gene FIV-FVIII transfer protocol used in Figure 4 and assayed for anti-hFVIII antibodies 2 and 4 weeks after injection. In contrast to the findings in FVIII null mice on a C57BL/6 background, BALB/c mice did develop some degree of antibodies to hFVIII (mean values of about 200 pg/mL at 2 weeks and about 500 pg/mL at 4 weeks after gene transfer).
To confirm that biologically relevant levels of FVIII were expressed, a tail-cutting experiment was performed. In comparison with untreated FVIII–/– mice, the GP64/AlbE/AAT-ΔΔhFVIII–treated animals showed significantly less blood loss over a 45-minute period, demonstrating persistent partial correction of the clotting disorder (Figure 4B). To further assess the functional significance of hFVIII expression, Coamatic chromogenic FVIII activity assays were performed. As shown in Figure 4C, mice treated with GP64/AlbE/AAT-ΔΔhFVIII demonstrated approximately 5% of wild-type FVIII activity 4 and 8 weeks after gene transfer compared with no detectable activity in untreated littermates.
Discussion
Several vector systems, including retroviruses and lentiviruses, are under investigation for the treatment of the hemophilias.9-16 The present studies demonstrate that systemic delivery of FIV vector expressing B domain–deleted hFVIII confers sustained, therapeutically relevant, and functional FVIII expression in a hemophilia A mouse model. Plasma FVIII levels stabilized after about 6 months at near 10 ng/mL, twice the levels necessary for a therapeutic effect. Vector modifications including the GP64 pseudotype and use of a liver-specific promoter facilitated a hepatocyte transduction rate of about 20% and contributed to these results. Based on minimal elevation of SGOT, transduction of the liver with GP64-pseudotyped FIV did not appear to be hepatotoxic, and the pseudotyped virions were stable in the presence of human and mouse complement. The findings are encouraging and indicate the potential utility of this vector for stable, long-term expression of FVIII in vivo.
While the potential use of the AcMNPV baculovirus for gene transfer to mammalian cells, including hepatocytes, was reported previously,43 the large baculovirus genome, its restricted cell tropism, and complement inactivation in serum are limitations.42,44 Recently, the potential for pseudotyping retroviral vectors with the AcMNPV GP64 envelope and transducing cells in vitro was recognized.36 Here we extended this observation, demonstrating the compatibility of the GP64 envelope with the FIV lentivirus and the remarkable tropism this pseudotype exhibits for hepatocytes. The efficiency of hepatocyte gene transfer was improved over that observed previously with the RRV or VSV-G envelopes using the same vector.10,34 Importantly, GP64-pseudotyped FIV is stable in the presence of human or murine complement and, of production relevance, GP64 can withstand concentration by centrifugation and purification by ion exchange chromatography (B.L.D. and P.B.M., unpublished data, September 2004). We speculate that the expression of the GP64 env in mammalian packaging cells eliminates insect glycosylation patterns that lead to complement-mediated destruction of the wild-type baculovirus following in vivo delivery.42 In related studies we found that AcMNPV GP64-pseudotyped FIV also transduces respiratory epithelia and cells of the CNS in vivo (P.B.M., B.L.D., unpublished data, January 2004). The cellular receptor for GP64 is currently unknown. Interestingly, AcMNPV GP64 shares homology with the envelopes of the orthomyxovirus influenza D viruses.45 Studies with AcMNPV in insect cells indicate that infection is ammonium chloride sensitive, consistent with entry in a low pH endosomal compartment.46 We found that blockers of endosomal acidification inhibited GP64-pseudotyped FIV transduction of human A549 cells (P.B.M., unpublished data, January 2004).
The inclusion of a liver-specific promoter in our studies conferred both hepatocyte-specific and long-term expression. Compared with our previous findings with FIV and the RSV promoter,34 the use of a tissue-specific promoter restricted expression almost entirely to hepatocytes. Surprisingly, none of the FVIII null animals on a C57BL/6 background developed antibodies against hFVIII. This result may reflect the limited expression of hFVIII in cells with antigen-presenting capability, as a consequence of using a liver-specific promoter.47,48 In agreement with others,9,14,15 we previously noted that VSV-G–pseudotyped FIV with hFVIII driven by the ubiquitously expressed RSV promoter was associated with development of hFVIII antibodies in the same mouse model.10 It is also possible that GP64 may fortuitously be unable to direct transduction of potent antigen-presenting cells, such as dendritic cells, in vivo. In support of this possibility, Schauber and colleagues recently reported poor transduction of murine dendritic cells with a GP64-pseudotyped HIV vector.49 Furthermore, the FVIII null mice used in the present study are congenic C57BL/6, and this strain may be less likely to generate antibodies against FVIII.50,51 Our finding that BALB/c mice receiving the GP64/AlbE/AAT-ΔΔhFVIII vector generated anti-hFVIII antibodies supports this notion. Direct comparison of VSV-G and GP64 pseudotypes with identical expression cassettes in this congenic strain could help elucidate the immunologic basis for the lack of an anti-FVIII antibody response. A potential advantage of GP64-pseudotyped vectors for human gene transfer applications is that preexisting immunity is highly unlikely. An alternative strategy to enhance gene expression, not pursued in the present study, involves the induction of tolerance to the therapeutic protein.52,53
In addition to the envelope modification and inclusion of a tissue specific promoter, the B domain–deleted hFVIII cDNA was altered to include 2 point mutations in the A1 domain previously demonstrated to improve protein secretion without interfering with function, presumably by disrupting binding with immunoglobulin-binding protein (BiP).33 Additional modifications of the FVIII cDNA may further improve the levels of secreted protein.54
This study demonstrates the potential utility of GP64/FIV pseudotype in the treatment of hemophilia A. Vector modifications including a liver-specific promoter conferred hepatocyte-specific, long-term expression. Ongoing manipulations of this vector system, including dosing optimization and vector purification, will facilitate future experiments in larger animal models.
Acknowledgments
We thank David Sanders for providing the Sindbis and Semliki Forest virus envelope constructs and Linda Bulinga for generating the AcMNPV GP64 construct. We thank Christine Wohlford-Lenane for technical assistance. We thank Erik Edens for providing human and mouse serum with reference laboratory–validated complement activity.
Prepublished online as Blood First Edition Paper, May 10, 2005; DOI 10.1182/blood-2004-11-4358.
Supported by the National Institutes of Health (NIH) (HL079023 and NS34568), the National Hemophilia Foundation (Y.K.), the Hemophilia Association of New York (Y.K.), a pilot and feasibility grant from the Center for Gene Therapy for Cystic Fibrosis (Y.K.), and the Gene Transfer Vector Core and Cell Morphology Core partially supported by the Cystic Fibrosis Foundation, National Heart, Lung, and Blood Institute (NHLBI) (PPG HL51670), and the Center for Gene Therapy for Cystic Fibrosis (NIH P30 DK54759).
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 U.S.C. section 1734.
References
- 1.Sadler J. Hemophilia A, Hemophilia B and von Willebrand's disease. In: Stamatoyannopoulos G, Nienhuis AW, Leder P, Majerus PW, eds. Molecular Basis of Blood Diseases. Philadelphia, PA: Saunders; 1987: 575-630.
- 2.Hoyer LW. Hemophilia A. N Engl J Med. 1994; 330: 38-47. [DOI] [PubMed] [Google Scholar]
- 3.Levine P. Clinical manifestations and therapy of hemophilia A and B. In: Colman R, Hirsh J, Marder V, Saizman E, eds. Hemostasis and Thrombosis. 2nd ed. Philadelphia, PA: JB Lippincott; 1987: 97-111.
- 4.Fakharzadeh SS, Kazazian HH Jr. Correlation between factor VIII genotype and inhibitor development in hemophilia A. Semin Thromb Hemost. 2000;26: 167-171. [DOI] [PubMed] [Google Scholar]
- 5.Plug I, Van Der Bom JG, Peters M, et al. Thirty years of hemophilia treatment in the Netherlands, 1972-2001. Blood. 2004;104: 3494-3500. [DOI] [PubMed] [Google Scholar]
- 6.Petrini P, Chambost H, Nemes L. Towards the goal of prophylaxis: experience and treatment strategies from Sweden, France and Hungary. Haemophilia. 2004;10(suppl 4): 94-96. [DOI] [PubMed] [Google Scholar]
- 7.Kaufman RJ. Advances toward gene therapy for hemophilia at the millennium. Hum Gene Ther. 1999;10: 2091-2107. [DOI] [PubMed] [Google Scholar]
- 8.Chuah MK, Collen D, VandenDriessche T. Gene therapy for hemophilia. J Gene Med. 2001;3: 3-20. [DOI] [PubMed] [Google Scholar]
- 9.VandenDriessche T, Vanslembrouck V, Goovaerts I, et al. Long-term expression of human coagulation factor VIII and correction of hemophilia A after in vivo retroviral gene transfer in factor VIII-deficient mice. Proc Natl Acad Sci U S A. 1999;96: 10379-10384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stein CS, Kang Y, Sauter SL, et al. In vivo treatment of hemophilia A and mucopolysaccharidosis type VII using nonprimate lentiviral vectors. Mol Ther. 2001;3: 850-856. [DOI] [PubMed] [Google Scholar]
- 11.Powell JS, Ragni MV, White GC II, et al. Phase 1 trial of FVIII gene transfer for severe hemophilia A using a retroviral construct administered by peripheral intravenous infusion. Blood. 2003;102: 2038-2045. [DOI] [PubMed] [Google Scholar]
- 12.Xu L, Gao C, Sands MS, et al. Neonatal or hepatocyte growth factor-potentiated adult gene therapy with a retroviral vector results in therapeutic levels of canine factor IX for hemophilia B. Blood. 2003;101: 3924-3932. [DOI] [PubMed] [Google Scholar]
- 13.Greengard JS, Jolly DJ. Animal testing of retroviral-mediated gene therapy for factor VIII deficiency. Thromb Haemost. 1999;82: 555-561. [PubMed] [Google Scholar]
- 14.Park F, Ohashi K, Kay MA. Therapeutic levels of human factor VIII and IX using HIV-1-based lentiviral vectors in mouse liver. Blood. 2000;96: 1173-1176. [PubMed] [Google Scholar]
- 15.Kootstra NA, Matsumura R, Verma IM. Efficient production of human FVIII in hemophilic mice using lentiviral vectors. Mol Ther. 2003;7: 623-631. [DOI] [PubMed] [Google Scholar]
- 16.Park F. Correction of bleeding diathesis without liver toxicity using arenaviral-pseudotyped HIV-1-based vectors in hemophilia A mice. Hum Gene Ther. 2003;14: 1489-1494. [DOI] [PubMed] [Google Scholar]
- 17.Sarkar R, Tetreault R, Gao G, et al. Total correction of hemophilia A mice with canine FVIII using an AAV 8 serotype. Blood. 2004;103: 1253-1260. [DOI] [PubMed] [Google Scholar]
- 18.Chao H, Mao L, Bruce AT, Walsh CE. Sustained expression of human factor VIII in mice using a parvovirus-based vector. Blood. 2000;95: 1594-1599. [PubMed] [Google Scholar]
- 19.Connelly S, Andrews JL, Gallo AM, et al. Sustained phenotypic correction of murine hemophilia A by in vivo gene therapy. Blood. 1998;9: 3273-3281. [PubMed] [Google Scholar]
- 20.Andrews JL, Kadan MJ, Gorziglia MI, Kaleko M, Connelly S. Generation and characterization of E1/E2a/E3/E4-deficient adenoviral vectors encoding human factor VIII. Mol Ther. 2001;3: 329-336. [DOI] [PubMed] [Google Scholar]
- 21.Gallo-Penn AM, Shirley PS, Andrews JL, et al. Systemic delivery of an adenoviral vector encoding canine factor VIII results in short-term phenotypic correction, inhibitor development, and biphasic liver toxicity in hemophilia A dogs. Blood. 2001;97: 107-113. [DOI] [PubMed] [Google Scholar]
- 22.Lipshutz GS, Sarkar R, Flebbe-Rehwaldt L, Kazazian H, Gaensler KM. Short-term correction of factor VIII deficiency in a murine model of hemophilia A after delivery of adenovirus murine factor VIII in utero. Proc Natl Acad Sci U S A. 1999;96: 13324-13329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yant SR, Meuse L, Chiu W, Ivics Z, Izsvak Z, Kay MA. Somatic integration and long-term transgene expression in normal and haemophilic mice using a DNA transposon system. Nat Genet. 2000;25: 35-41. [DOI] [PubMed] [Google Scholar]
- 24.Olivares EC, Hollis RP, Chalberg TW, Meuse L, Kay MA, Calos MP. Site-specific genomic integration produces therapeutic factor IX levels in mice. Nat Biotechnol. 2002;20: 1124-1128. [DOI] [PubMed] [Google Scholar]
- 25.Miao CH, Ye X, Thompson AR. High-level factor VIII gene expression in vivo achieved by nonviral liver-specific gene therapy vectors. Hum Gene Ther. 2003;14: 1297-1305. [DOI] [PubMed] [Google Scholar]
- 26.Naldini L, Blomer U, Gallay P, et al. In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector. Science. 1996;272: 263-267. [DOI] [PubMed] [Google Scholar]
- 27.Kumar M, Keller B, Makalou N, Sutton RE. Systematic determination of the packaging limit of lentiviral vectors. Hum Gene Ther. 2001;12: 1893-1905. [DOI] [PubMed] [Google Scholar]
- 28.DePolo NJ, Reed JD, Sheridan PL, et al. VSV-G pseudotyped lentiviral vector particles produced in human cells are inactivated by human serum. Mol Ther. 2000;2: 218-222. [DOI] [PubMed] [Google Scholar]
- 29.Burns JC, Friedmann T, Driever W, Burrascano M, Yee J-K. Vesicular stomatitis virus G glycoprotein pseudotyped retroviral vectors: concentration to very high titer and efficient gene transfer into mammalian and nonmammlian cells. Proc Natl Acad Sci U S A. 1993;90: 8033-8037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Johnston JC, Gasmi M, Lim LE, et al. Minimum requirements for efficient transduction of dividing and nondividing cells by feline immunodeficiency virus vectors. J Virol. 1999;73: 4991-5000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stein CS, Davidson BL. Gene transfer to the brain using feline immunodeficiency virus-based lentivirus vectors. Methods Enzymol. 2002;346: 433-454. [DOI] [PubMed] [Google Scholar]
- 32.Kramer MG, Barajas M, Razquin N, et al. In vitro and in vivo comparative study of chimeric liver-specific promoters. Mol Ther. 2003;7: 375-385. [DOI] [PubMed] [Google Scholar]
- 33.Swaroop M, Moussalli M, Pipe SW, Kaufman RJ. Mutagenesis of a potential immunoglobulin-binding protein-binding site enhances secretion of coagulation factor VIII. J Biol Chem. 1997;272: 24121-24124. [DOI] [PubMed] [Google Scholar]
- 34.Kang Y, Stein CS, Heth JA, et al. In vivo gene transfer using a nonprimate lentiviral vector pseudotyped with Ross River Virus glycoprotein. J Virol. 2002;76: 9378-9388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sinn PL, Hickey MA, Staber PD, et al. Lentivirus vectors pseudotyped with filoviral envelope glyco-proteins transduce airway epithelia from the apical surface independently of folate receptor alpha. J Virol. 2003;77: 5902-5910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kumar M, Bradow BP, Zimmerberg J. Large-scale production of pseudotyped lentiviral vectors using baculovirus GP64. Hum Gene Ther. 2003;14: 67-77. [DOI] [PubMed] [Google Scholar]
- 37.Beyer WR, Westphal M, Ostertag W, von Laer D. Oncoretrovirus and lentivirus vectors pseudo-typed with lymphocytic choriomeningitis virus glycoprotein: generation, concentration, and broad host range. J Virol. 2002;76: 1488-1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang G, Williams G, Xia H, et al. Apical barriers to airway epithelial cell gene transfer with amphotropic retroviral vectors. Gene Ther. 2002;9: 922-931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang G, Slepushkin VA, Zabner J, et al. Feline immunodeficiency virus vectors persistently transduce nondividing airway epithelia and correct the cystic fibrosis defect. J Clin Invest. 1999;104: R49-R56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bi L, Lawler AM, Antonarakis SE, High KA, Gearhart JD, Kazazian HH Jr. Targeted disruption of the mouse factor VIII gene produces a model of haemophilia A. Nat Genet. 1995;10: 119-121. [DOI] [PubMed] [Google Scholar]
- 41.Qian J, Collins M, Sharpe AH, Hoyer LW. Prevention and treatment of factor VIII inhibitors in murine hemophilia A. Blood. 2000;95: 1324-1329. [PubMed] [Google Scholar]
- 42.Huser A, Rudolph M, Hofmann C. Incorporation of decay-accelerating factor into the baculovirus envelope generates complement-resistant gene transfer vectors. Nat Biotechnol. 2001;19: 451-455. [DOI] [PubMed] [Google Scholar]
- 43.Boyce FM, Bucher NL. Baculovirus-mediated gene transfer into mammalian cells. Proc Natl Acad Sci U S A. 1996;93: 2348-2352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tani H, Limn CK, Yap CC, et al. In vitro and in vivo gene delivery by recombinant baculoviruses. J Virol. 2003;77: 9799-9808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pearson MN, Rohrmann GF. Transfer, incorporation, and substitution of envelope fusion proteins among members of the Baculoviridae, Orthomyxoviridae, and Metaviridae (insect retrovirus) families. J Virol. 2002;76: 5301-5304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hefferon KL, Oomens AG, Monsma SA, Finnerty CM, Blissard GW. Host cell receptor binding by baculovirus GP64 and kinetics of virion entry. Virology. 1999;258: 455-468. [DOI] [PubMed] [Google Scholar]
- 47.Brown BD, Shi CX, Rawle FE, et al. Factors influencing therapeutic efficacy and the host immune response to helper-dependent adenoviral gene therapy in hemophilia A mice. J Thromb Haemost. 2004;2: 111-118. [DOI] [PubMed] [Google Scholar]
- 48.Follenzi A, Battaglia M, Lombardo A, Annoni A, Roncarolo MG, Naldini L. Targeting lentiviral vector expression to hepatocytes limits transgene-specific immune response and establishes long-term expression of human antihemophilic factor IX in mice. Blood. 2004;103: 3700-3709. [DOI] [PubMed] [Google Scholar]
- 49.Schauber CA, Tuerk MJ, Pacheco CD, Escarpe PA, Veres G. Lentiviral vectors pseudotyped with baculovirus gp64 efficiently transduce mouse cells in vivo and show tropism restriction against hematopoietic cell types in vitro. Gene Ther. 2004;11: 266-275. [DOI] [PubMed] [Google Scholar]
- 50.Fields PA, Armstrong E, Hagstrom JN, et al. Intravenous administration of an E1/E3-deleted adenoviral vector induces tolerance to factor IX in C57BL/6 mice. Gene Ther. 2001;8: 354-361. [DOI] [PubMed] [Google Scholar]
- 51.Rawle FE, Shi CX, Brown B, et al. Heterogeneity of the immune response to adenovirus-mediated factor VIII gene therapy in different inbred hemophilic mouse strains. J Gene Med. 2004;6: 1358-1368. [DOI] [PubMed] [Google Scholar]
- 52.Mingozzi F, Liu YL, Dobrzynski E, et al. Induction of immune tolerance to coagulation factor IX antigen by in vivo hepatic gene transfer. J Clin Invest. 2003;111: 1347-1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dobrzynski E, Mingozzi F, Liu YL, et al. Induction of antigen-specific CD4+ T-cell anergy and deletion by in vivo viral gene transfer. Blood. 2004;104: 969-977. [DOI] [PubMed] [Google Scholar]
- 54.Miao HZ, Sirachainan N, Palmer L, et al. Bioengineering of coagulation factor VIII for improved secretion. Blood. 2004;103: 3412-3419. [DOI] [PubMed] [Google Scholar]