

Abstract
Shiga toxin 1 (Stx-1) and Stx-2 produced by enterohemorrhagic Escherichia coli cause the diarrhea-associated hemolytic uremic syndrome (HUS). This type of HUS is characterized by obstruction of the glomeruli and renal microvasculature by platelet-fibrin thrombi, acute renal failure, thrombocytopenia, microvascular hemolytic anemia, and plasma levels of von Willebrand factor (VWF)-cleaving protease (ADAMTS13) activity that are within a broad normal range. We investigated the mechanism of initial platelet accumulation on Stx-stimulated endothelial cells. Stx-1 or Stx-2 (1-10 nM) stimulated the rapid secretion of unusually large (UL) VWF multimeric strings from human umbilical vein endothelial cells (HUVECs) or human glomerular microvascular endothelial cells (GMVECs). Perfused normal human platelets immediately adhered to the secreted ULVWF multimeric strings. Nanomolar concentrations (1-10 nM) of the Shiga toxins were as effective in inducing the formation of ULVWF-platelet strings as millimolar concentrations (0.1-20 mM) of histamine. The rate of ULVWF-platelet string cleavage by plasma or recombinant ADAMTS13 was delayed by 3 to 10 minutes (or longer) in the presence of 10 nM Stx-1 or Stx-2 compared with 20 mM histamine. Stx-induced formation of ULVWF strings, and impairment of ULVWF-platelet string cleavage by ADAMTS13, may promote initial platelet adhesion above glomerular endothelial cells. These processes may contribute to the evolution of glomerular occlusion by platelet and fibrin thrombi in diarrhea-associated HUS.
Introduction
The hemolytic uremic syndrome (HUS) is a predominantly renal thrombotic microangiopathy associated with glomerular microvascular platelet adhesion/aggregation and fibrin polymer formation. The consequences are acute renal failure (sometimes accompanied by dysfunction of other organs), thrombocytopenia, and microangiopathic hemolytic anemia.1,2
HUS frequently occurs following an episode of hemorrhagic colitis/diarrhea. Among the offending microbes are Shigella dysenteriae serotype 1 or, more frequently, certain enterohemorrhagic serotypes of Escherichia coli (O157:H7).3-7 These bacteria produce the exotoxins Shiga toxin (Stx; made by S dysenteriae) or Stx-1 and Stx-2 (made by enterohemorrhagic E coli serotypes).6-10 The several Shiga toxins are composed of one A subunit (∼33 kDa) and 5 B (binding) subunits (∼7.7 kDa each; AB5).10
Toxin B subunits mediate Stx attachment to membrane globotriaosylceramide receptors (Gb3, or CD77).10,11 Stx and Stx-1 are nearly identical, whereas Stx-1 and Stx-2 are only 53% to 56% homologous.1,12 The E coli serotypes that produce Stx-2 may be more likely to cause HUS in humans.6-9
The injection of single bolus doses of 100 ng/kg of either Stx-1 or Stx-2 leads to HUS in baboons.13-15 Four divided 25 ng/kg doses of Stx-2, but not of the Stx-1, will also induce the syndrome in these primates.13
Gb3 is associated with cholesterol and ganglioside GM1 in mobile lipid membrane “rafts”16 and may be most effectively expressed on the surfaces of human renal endothelial, mesangial, and tubular cells. Similar Gb3 receptors are also present in the membranes of other human cells, including colonic and cerebral microvascular endothelial cells, monocytes, neutrophils, and platelets.10,11,17-22 GM1 molecules are receptors for cholera toxin, which is also composed of one A and 5 B subunits (AB5) that are structurally distinct from the Shiga toxins. (See “Materials and methods” for National Institutes of Health accession numbers for primary amino acid sequences.) Vibrio cholera toxin binding to GM1 receptors activates adenylate cyclase through a G-protein pathway.23-25
Infection of the terminal ileum and colon by enterohemorrhagic E coli 0157:H7 induces a “cytokine storm.” Microbial H7 flagellin stimulates colonic output of interleukin 8 (IL-8), other C-X-C chemokines, and granulocyte colony-stimulating factor (G-CSF), thereby inducing neutrophil production, attraction, and activation.1,26-28 Stx-1 and Stx-2 attach to monocytes/macrophages and renal cells and stimulate the release of tumor necrosis factor α (TNF-α), IL-1, and IL-6.15,29-32 These latter 3 cytokines up-regulate the membrane expression of Gb3 in renal, cerebral, and other microvascular endothelial cells by promoting the synthesis of enzymes responsible for the production of Gb3 molecules.10,33-35 Additional Stx molecules then attach to the increased number of Gb3 receptors in glomerular, and sometimes other, endothelial-cell membranes. It is likely that the relatively specific renal toxicities of Stx-1 and Stx-2 are the result of their transport via cells in the bloodstream to Gb3 receptors concentrated in the membranes of glomerular endothelial (and other renal) cells.
The precise explanation for the common observation of glomerular “hyaline” thrombus formation in HUS is not established. Thrombosis has been observed atop glomerular endothelial cells in renal biopsy and autopsy specimens.36-40 The endothelial cells in HUS may be swollen, usually remain attached to the underlying basement membrane, sometimes have a widened subendothelial space, and are not accompanied by vessel wall inflammation. Capillary lumina are often narrowed or completely obstructed.1,36-39
Fibrin is detectable in many to most of the occlusive glomerular thrombi found in postmortem renal sections of patients with HUS,1,39,41-44 or baboons injected with Stx.13 For example, in a retrospective autopsy study of 31 patients registered at the Johns Hopkins Hospital (Baltimore, MD) as HUS between 1960 and 1999,44 fibrin was frequently found. Staining for platelets using antibodies to “factor VIII” (presumably von Willebrand factor [VWF]) was, however, done only in a “minority of cases” and could have been negative if any platelets present had extruded their VWF-rich α-granules.
Whether fibrin formation is a primary or secondary phenomenon in HUS is unknown. Unlike the thrombocytopenia (often severe) of Stx-associated HUS, clotting studies are variable in these patients.1,39,45,46 Fibrinogen levels have been reported to be elevated in HUS, and fibrinogen survival was found to be normal in 2 infants studied 7 days after the onset of HUS symptoms. Heparin therapy in patients,39,45,47 or prophylaxis in a baboon model of Stx-HUS,48 has been ineffective in preventing development of the syndrome. The administration of hirudin to 3 greyhounds with Stx-induced HUS was, however, recently reported to be of possible benefit in 2 of the animals.49
Platelet adhesion, and subsequent aggregation, above Stx-stimulated renal glomerular microvascular endothelial cells may contribute to, or possibly even initiate, the formation of platelet-fibrin thrombi characteristic of the thrombotic microangiopathy in Stx-induced HUS.1,50 There are relatively few reports of detailed morphologic examination of renal biopsy specimens obtained during HUS episodes. Elegant examples include the electron microscopic studies of biopsies from 6 children with HUS by Courtecuisse et al37 and the detailed study by Habib et al51 (including electron microscopy) of biopsy samples from 28 patients with the syndrome. The works of Courtecuisse et al, in 1967,37 and Habib et al, in 1982,51 were published before the relationship between Stx and HUS was recognized. Nevertheless, these investigators observed that platelets, often numerous and in contact with endothelial cells, could partially or completely occlude the lumina of glomerular capillaries.
Thrombocytopenia, often progressive, accompanies the evolution of acute renal failure in Stx-induced HUS.1 It is, therefore, reasonable to suggest that platelet adhesion and aggregation contribute to the partial-complete obstruction of regions of the renal glomerular microcirculation in HUS.1 In one study of 4 infants with HUS examined 3 to 6 days after the appearance of symptoms, platelet lifespan was shortened in all patients.52 Thrombocytopenia, glomerular and peritubular capillary platelet microthrombi (observed by electron microscopy) and fibrin, and acute renal failure have also been observed in the baboon model of Stx-induced HUS.15,32
In one study of 4 patients who died of Stx-associated HUS,43 fibrin (but not VWF antigen) was detected in increased quantity in the glomerular thrombi of 3 patients. Both VWF antigen and fibrin were increased in the arterial and arteriolar thrombi from the fourth patient. The level of VWF expression in the glomerular capillary walls of these patients was not reported. In the baboons that developed HUS after injection of 4 divided doses of 25 ng/kg Stx-2, however, VWF antigenic expression was increased postmortem in glomerular and, especially, peritubular capillary walls.14 Furthermore, in the greyhound model of Stx-induced HUS, there was impressive accumulation of unusually large VWF (ULVWF) forms in the plasma of the 2 animals studied within 2 days of Stx injection.
Plasma VWF-cleaving metalloprotease (ADAMTS13) activity levels are usually ample in Stx-induced HUS compared with the absent or severely reduced levels in most patients with thrombotic thrombocytopenic purpura (TTP).1,43,53-56
Cultured human endothelial cells secrete ULVWF multimeric “strings” in response to high concentrations of histamine (0.025-20 mM). Platelets adhere quickly under flowing conditions to the ULVWF strings.57,58 The ULVWF strings are, however, normally cleaved rapidly by plasma ADAMTS13.57,58 Failure of ULVWF-platelet string cleavage as a result of absent or severely reduced plasma ADAMTS13 activity is characteristic of the congenital and acquired autoantibody-mediated types of TTP.53,57-61 In contrast to these types of TTP, plasma levels of ADAMTS13 in Stx-associated HUS are not usually absent or severely reduced.1,43,53
We have investigated the effects of Stx-1 and Stx-2 on ULVWF multimeric string secretion and on ADAMTS13-mediated string cleavage, using both human umbilical vein endothelial cells (HUVECs) and glomerular microvascular endothelial cells (GMVECs).
Materials and methods
Endothelial-cell cultures
HUVECs. Endothelial cells were obtained from human umbilical veins. Umbilical cords were washed with phosphate buffer (140 mM NaCl, 0.4 mM KCl, 1.3 mM NaH2PO4, 1.0 mM Na2HPO4, 0.2% glucose, pH 7.4) and then infused with a collagenase solution (0.02% in phosphate-buffered saline [PBS]; Invitrogen, Carlsbad, CA) for 30 minutes at room temperature. The cords were rinsed with 100 mL phosphate buffer, and eluates containing endothelial cells were centrifuged at 250g for 10 minutes. The cell pellets were resuspended in Medium 199 (Invitrogen) supplemented with 10% to 20% heat-inactivated fetal bovine serum (FBS), 3% penicillin-streptomycin-neomycin (PSN), and 0.2 mM l-glutamine. The endothelial cells were seeded on 35-mm culture dishes or 35-mm glass coverslips coated with 1% gelatin and grown until confluent (5-7 days). The HUVECs used in most experiments were primary or passage 1 cells (additional passages 2-4 were studied only by flow cytometry).
GMVECs. GMVECs (passage 2) were purchased from the Applied Cell Biology Research Institute (Kirkland, WA). The cells were initially from decapsulated glomeruli isolated from normal human kidney cortical tissue and were more than 95% positive for cytoplasmic VWF and E-selectin by immunofluorescence. They were induced to proliferate according to the manufacturer's instructions in cell systems-certified (CS-C) complete medium containing PSN. Most experiments with GMVECs used cells with passage numbers 3 to 5 seeded onto gelatin-coated 35-mm culture dishes or 35-mm glass coverslips. Passage 6 GMVECs, as well as passage 4 cells, were studied using flow cytometry.
Gb3 (CD77) on HUVECs and GMVECs. HUVECs and GMVECs were detached with 5 mM EDTA (ethylenediaminetetraacetic acid), treated with 0.2% paraformaldehyde at 4°C and 0.2% Tween 20 at 37°C, blocked with 10% human serum/PBS, incubated with either IgM mouse anti-Gb3 (CD77)-fluorescein isothiocyanate (FITC) or irrelevant mouse IgM-FITC (as control), and analyzed by flow cytometry (Becton Dickinson FACScan, San Jose, CA). Table 1 shows that about 30% to 60% of HUVECs and GMVECs were positive for Gb3 (CD77). The flow cytometry data are also shown in Figure 1. Burkitt lymphoma cells (GA-10; American Type Culture Collection, Manassas, VA) were used as positive controls (data not shown).62 It is not possible to make direct quantitative comparisons between the expression of Gb3 on our cultured human endothelial cells in vitro and the expression of Gb3 on human endothelial cells in situ. Renal tissue has, however, previously been reported to contain relatively high concentrations of Gb3.63
Table 1.
Gb3 (CD77) receptors on cultured HUVECs and GMVECs
| Cell type and passage no. | Anti-CD77+ IgM-FITC, % | Anti-TNP+ IgM-FITC, % |
|---|---|---|
| HUVECs | ||
| P0 | 57.87 | 2.91 |
| P1 | 54.29 | 0.38 |
| P2 | 62.14 | 0.79 |
| P3 | 32.32 | 0.36 |
| P4 | 64.89 | 0.27 |
| GMVECs | ||
| P4 | 53.45 | 0.63 |
| P6 | 29.56 | 0.92 |
Two to 6 T-25 flasks of each cell type were combined for analysis.
TNP indicates trinitrophenol (irrelevant hapten); P, passage; P0, primary cells; P1, first passage, etc.
Figure 1.

Gb3 expression on HUVECs and GMVECs by flow cytometry. Labeling HUVECs (A) and GMVECs (D) with an isotype-matched irrelevant antibody (mouse IgM-FITC) determined the background FITC (FL1) fluorescence and is indicated by the horizontal line. HUVECs (B) and GMVECs (E) labeled with mouse IgM anti-Gb3 (CD77)-FITC expressing higher FITC fluorescence than background were considered positive. Burkitt lymphoma cells were used as positive controls (data not shown). The x-axis indicates increasing cell size and the y-axis indicates increasing FITC fluorescence. Overlay histograms of HUVECs (C) and GMVECs (F) show the background FITC fluorescence (cells labeled with irrelevant mouse IgM-FITC; first peak) and the increased FITC fluorescence due to Gb3 expression when cells were labeled with mouse IgM anti-Gb3 (CD77)-FITC (second peak). The x-axis indicates increasing FITC fluorescence and the y-axis indicates the events counted.
Stimulation of endothelial cells. To evaluate the capacity of HUVECs and GMVECs to secrete ULVWF multimeric strings, the HUVECS and GMVECs were stimulated for 10 minutes with 0.1 to 20 mM histamine (Sigma-Aldrich, St Louis, MO), 1 to 10 nM purified Stx-1,20,64 1 to 10 nM recombinant Stx-2 (Calbiochem, San Diego, CA), or 30 to 6000 nM cholera toxin (Calbiochem).
Purified Stx-1, suspended in PBS, migrated as 2 bands (30-kDa A subunit and 7.5-kDa B subunits) under reducing conditions. According to the manufacturer, recombinant Stx-2 migrated as 2 bands (30-kDa A subunit and 7.2-kDa B subunit). Purified Stx-1 and recombinant Stx-2 in concentrations as low as 10-13 M for 10 to 20 minutes stimulated HUVECs to secrete more than 25 ULVWF multimeric strings (3 experiments). To obtain consistently more than 100 ULVWF strings per 20 continuous fields, we used 10-9 to 10-8 M (1-10 nM) Stx-1 and Stx-2 in the design of subsequent experiments.
Purified Stx-1 may be contaminated by traces of lipopolysaccharide (LPS; undetectable by standard polyacrylamide gel electrophoresis), because this substance is difficult to exclude or remove. It is not known if recombinant Stx-2 also contains traces of LPS undetectable by gel electrophoresis. Consequently, in association with the experiments reported in “Results,” we also determined that the direct addition of 1 mg/mL LPS to HUVECs induced the release of only a modest number (11%-30%) of very short ULVWF strings. These results were in contrast to the extremely long ULVWF strings secreted by HUVECs and GMVECs in response to a wide range of Stx concentrations (or to millimolar concentrations of histamine). Additional evidence against any important contribution by LPS to our results on Stx-2-induced secretion of long ULVWF multimeric strings by HUVECs is described in the second paragraph of “Results.”
To stimulate the release of soluble VWF enriched in ULVWF forms, confluent HUVECs were washed with PBS and incubated with a serum-free M199 medium containing 10 μg/mL insulin, 5 μg/mL transferrin, 3% PSN, and 1% glutamine for 48 to 72 hours. The cultured cells were then treated with 0.1 mM histamine or 10 nM Stx-1 or Stx-2 for 10 minutes at 37°C. After incubation, the serum-free medium was collected and centrifuged at 150g for 10 minutes to remove cell debris. Samples of soluble VWF enriched in ULVWF were also obtained from the supernatant of proliferating GMVECs. In these samples, the VWF multimeric composition was evaluated by sodium dodecyl sulfate (SDS)-1% agarose gel electrophoresis and chemiluminescence using a polyclonal anti-human-VWF antibody and secondary anti-IgG-horseradish peroxidase (HRP) conjugate (Bethyl Laboratories, Montgomery, TX).
Endothelial-cell viability. Confluent primary HUVECs in gelatin-coated 24-well plates were exposed to Stx-1 or Stx-2 at concentrations of 0.1, 1, and 10 nM, 0.1 mM histamine, and complete Medium 199 (control) for 1 to 6 hours at 37°C. The capacity of 5 mg/mL 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide (MTT; Sigma-Aldrich) to enter HUVECs and undergo reduction to formazan was determined at 490 nm as a measurement of cell viability.65
Platelet and plasma preparations
Human blood was drawn from unmedicated healthy donors. Approval was obtained from the Baylor College of Medicine and Rice University Institutional Review Boards for these studies. Informed consent was provided according to the Declaration of Helsinki. To obtain washed platelets, blood was drawn into acid-citrate-dextrose anticoagulant (ACD; 85 mM sodium citrate, 111 mM glucose, and 71 mM citric acid, 10% vol/vol) containing 0.05 μg/mL prostaglandin I2. The blood was centrifuged at 150g for 15 minutes at 24°C to obtain platelet-rich plasma (PRP), which was then centrifuged at 900g for 10 minutes to separate platelet pellets. The platelet pellets were washed once with a CGS buffer (13 mM sodium citrate, 30 mM glucose, and 120 mM sodium chloride, pH 7.0) and resuspended in Ca++-, Mg++-free Tyrode buffer ([Tyrode], 138 mM sodium chloride, 5.5 mM glucose, 12 mM sodium bicarbonate, 2.9 mM potassium chloride, and 0.36 mM sodium phosphate dibasic, pH 7.4). Washed platelets were resuspended in Tyrode buffer equivalent to original PRP volume.
Formalin-fixed platelets were prepared from outdated blood bank normal donor platelets as previously described.66
For studies using PRP as the source of ADAMTS13, blood was drawn into 0.38% sodium citrate and centrifuged at 150g for 15 minutes at 24°C to obtain PRP.
Fluorescent microscopy studies under flow
ULVWF strings secreted from endothelial cells were examined by fluorescent microscopy (Nikon Diaphot TE300, Garden City, NY). HUVECs and GMVECs were grown to confluence on 35-mm round glass coverslips coated with 1% gelatin. The cells were stimulated with either 0.1 mM histamine, 1 nM Stx-1, or 1 nM Stx-2 for 3 minutes at 37°C. The cells were stained and washed directly in the flow chamber (GlycoTech, Rockville, MD) with Tyrode buffer at 0.2 mL/min (60 s-1 shear rate). ULVWF strings were stained with polyclonal rabbit anti-human-VWF IgG and secondary Alexa Fluor 488 F(ab′)2 anti-IgG (Molecular Probes, Eugene, OR) or anti-IgG-FITC (Vector Labs, Burlingame, CA). Cell nuclei were detected with either 1 nM DAPI (4′6-di-amidino-2-phenylindole di-lactate) or 20 μg/mL propidium iodide. For Gb3 (CD77) blocking studies, mouse monoclonal anti-human-CD77 was used at 50 ng/mL (BD PharMingen, San Diego, CA). Images were taken with a SensiCamQE CCD camera (Cooke, Romulus, MI) and IP Lab software version 3.9.4r4 (Scanalytics, Fairfax, VA) using a × 100 objective (× 1000 magnification), aperture 1.3, with appropriate fluorescent filters. In some fluorescent studies the HUVECs were fixed at 0.1 mL/min with 1% paraformaldehyde in PBS and permeabilized with 0.1% Triton-X.
Parallel-plate perfusion assay of ULVWF-platelet string formation and cleavage
The formation and cleavage of ULVWF-platelet strings secreted from stimulated endothelial cells was studied under flow in a parallel-plate flow chamber system (GlycoTech) and observed by phase-contrast microscopy using × 20/0.45 aperture and × 40/0.6 aperture objectives (× 200 and × 400 magnification). Endothelial cells were grown on 35-mm culture dishes that became the bottom of each flow chamber. A syringe pump connected to the outlet port pulled washed platelets or PRP through the chamber at 0.2 mL/min to generate a shear rate of approximately 60 s-1 (wall shear stress, ∼1 dyn/cm2).
The assembled parallel-plate flow chamber was mounted onto an inverted-stage microscope (Nikon, Eclipse TE300) equipped with a high-speed digital camera (Photometrics, Model Quantix, Tucson, AZ). Images were acquired using MetaMorph software version 5.0r7 (Universal Images, West Chester, PA). The ULVWF-platelet strings were counted in 20 continuous view-fields at × 400.
Recombinant human ADAMTS13
A full-length cDNA of human ADAMTS13 was cloned into the mammalian expression vector pSectag2 (Invitrogen). This vector carries a multi-His tag and a Myc tag at the 3′ end of the insert for detection and purification, and a hygromycin-resistant marker. The ADAMTS13 cDNA was then transfected into Chinese hamster ovary (CHO) cells using a lipid carrier (Lipofectamine; Invitrogen). The transfected cells were grown in Dulbecco modified Eagle medium/F-12 with 10% FBS (Invitrogen) and increasing concentrations of hygromycin (final concentration 400 μg/mL). The cells were subjected to 2 runs of single-cell cloning using a flow cytometer/cell sorter set for high expression. Collecting culture medium and probing for ADAMTS13 by Western blot (monoclonal mouse anti-tetra-histidine IgG, Qiagen, Valencia, CA; goat anti-mouse IgG-HRP, Promega, Madison, WI) verified expression and release of recombinant human (rh) ADAMTS13. Stable cell lines were passed into multiple T-250 flasks for production and growth (90%-95% confluence). The medium was removed, and the cells were washed with PBS and incubated with the serum-free medium CD CHO-A with 1% glutamine (Invitrogen) for 24 hours. At the end of the incubation period, the medium was collected and centrifuged at 1500g to remove cell debris. The rhADAMTS13 was concentrated 10-fold by dialysis in 15-kDa molecular mass cutoff tubes surrounded by solid polyethylene glycol chips (35 000 molecular weight; Sigma-Aldrich). The rhADAMTS13 protein concentration was determined using a BCA protein kit (Pierce Chemicals, Rockford, IL) and stored at -80°C until use.
ADAMTS13 cleavage of ULVWF
Flowing conditions. The disappearance of ULVWF multimeric strings secreted by stimulated HUVECs or GMVECs was visualized under phase-contrast microscopy using adherent platelets or anti-VWF beads under flow. The disappearance of ULVWF-platelet strings (or, in some control experiments, strings of ULVWF with attached anti-VWF beads) represents ULVWF cleavage by ADAMTS13, as demonstrated by us in a series of reports over the past 3 years.57,58,67,68 Specific ADAMTS13-mediated cleavage of the ULVWF-platelet (or ULVWF-bead) strings was confirmed repeatedly throughout this study using EDTA in control runs. EDTA blocked the disappearance of ULVWF-platelet (or ULVWF-bead) strings during the perfusion of ADAMTS13 activity.
ADAMTS13 was present either in perfused normal PRP, or as rhADAMTS13 added to perfused washed platelets (or anti-VWF beads) in buffer. The points of endothelial-cell anchorage of the ULVWF-platelet (or ULVWF-bead) strings, as well as the entire length of the ULVWF-platelet (or ULVWF-bead) strings floating above the endothelial cells were observed continuously during the experiments reported. The floating ends of the ULVWF-platelet (or ULVWF-bead) strings were not seen to double back and attach to endothelial cells in response to the Stxs, histamine, or any other agent tested. Furthermore, ULVWF-platelet (or ULVWF-bead) strings were anchored to the endothelial cells at only one site, regardless of whether the cell stimulant was Stx-1, Stx-2, or histamine.
The endothelial cells were stimulated for 10 minutes with histamine, Stx-1, Stx-2, or, in some control experiments, cholera toxin. In one set of experiments, histamine stimulation of ULVWF multimeric string secretion by HUVECs was followed by perfusion over the stimulated cells of PRP containing about 100% ADAMTS13 activity. The perfused PRP had been mixed with either buffer or an equal volume of Stx-1 or Stx-2. The quantification of ULVWF strings in 20 fields or repeated photographing of the same field began 2 minutes after the beginning of perfusion with PRP, and so 2 minutes in these experiments is the actual “0” point. The rate of cleavage of ULVWF-platelet or ULVWF-anti-VWF/bead strings was quantified by string counting and photographed at 1- to 2-minute intervals following the addition of ADAMTS13 activity in the normal PRP that had been mixed with either buffer, Stx-1 or Stx-2.
In another set of experiments using rhADAMTS13, HUVECs or GMVECs were stimulated for 10 minutes with either histamine, Stx-1, or Stx-2. The stimulated cells were then perfused with washed normal platelets in Ca2+/Mg2+-free Tyrode buffer throughout the total period of each run. No additional histamine, Stx-1, or Stx-2 was added to the perfusate of washed platelets in buffer. During the initial 2 minutes after the addition of the washed platelets in buffer to the previously stimulated HUVECs or GMVECs, the platelets adhered to the ULVWF strings. During the next 2 minutes, the ULVWF strings were quantified in 20 fields or photographed in a single field. Then, rhADAMTS13 was added to the washed platelets in buffer, and the perfusion was continued for another 10 to 15 minutes with string counting in 20 fields or photographing of the same field every 1 to 2 minutes.
Static conditions. The rhADAMTS13 activity was measured under static conditions using HUVEC supernatant containing VWF enriched in ULVWF as the substrate. This method was modified from Furlan et al.53,59,60 Stx-1, Stx-2, or buffer was mixed with rhADAMTS13. A final concentration of 1 mM Pefabloc was added to inhibit serine protease activity (Roche Labs, Indianapolis, IN). The samples were diluted (1:5) with low ion strength Tris (tris(hydroxymethyl)aminomethane)-saline also containing 1 mM Pefabloc, and activated for 5 minutes with 1 mM BaCl2. The activated samples were then mixed at a ratio of 2:1 with supernatant containing VWF enriched in ULVWF. The mixture was dialyzed in 1.5 M urea for 24 hours at 37°C, and the reaction was stopped by the addition of 0.02 M EDTA. The samples were subjected to 1% agarose gel electrophoresis and then transferred to a polyvinylidene difluoride membrane. For display of VWF multimers, the membrane was blocked in 5% nonfat dry milk in Tris-buffered saline/1% Tween for 1 hour at room temperature, incubated with polyclonal goat anti-human-VWF antibody (Bethyl) for 2 hours, and then incubated for 2 hours with HRP-labeled rabbit anti-goat IgG secondary antibody (Bethyl). VWF multimers were displayed using enhanced chemiluminescence detection reagents (PerkinElmer, Fremont, CA).
Amino acid sequence comparisons
The primary amino acid sequences of Stx-1 subunit A (accession no. CAA85370), Stx-2 subunit A (CAA85780), Stx-1 subunit B (CAA85371), Stx-2 subunit B (CAA85781), VWF (P04275), ADAMTS13 (CAC83682), and IL-6 (NP_000591) were aligned using the BLAST2P program at the National Center for Biotechnology Information (NCBI; http://www.ncbi.nlm.nih.gov/). Cholera toxin subunits A (AAM22586) and B (CAA53976) were compared with subunits A and B of Stx-1 and Stx-2 using the same program.
Statistical analysis
All experimental data were presented as mean plus or minus average deviation of data points from their mean.
Results
ULVWF string secretion by HUVECs and GMVECs
Unstimulated HUVECs and GMVECs (Figures 2, 3) were packed with VWF+ granules (Figures 2A and 3C). Both endothelial-cell types secrete long ULVWF multimeric strings within 10 minutes after stimulation under flowing conditions by either millimolar concentrations (0.1-20 mM) of the known endothelial-cell VWF secretagogue, histamine,57,58 (Figure 2B) or nanomolar concentrations (1-10 nM) of Stx-1 or Stx-2 (Figures 2C-E and 3D).
Figure 2.
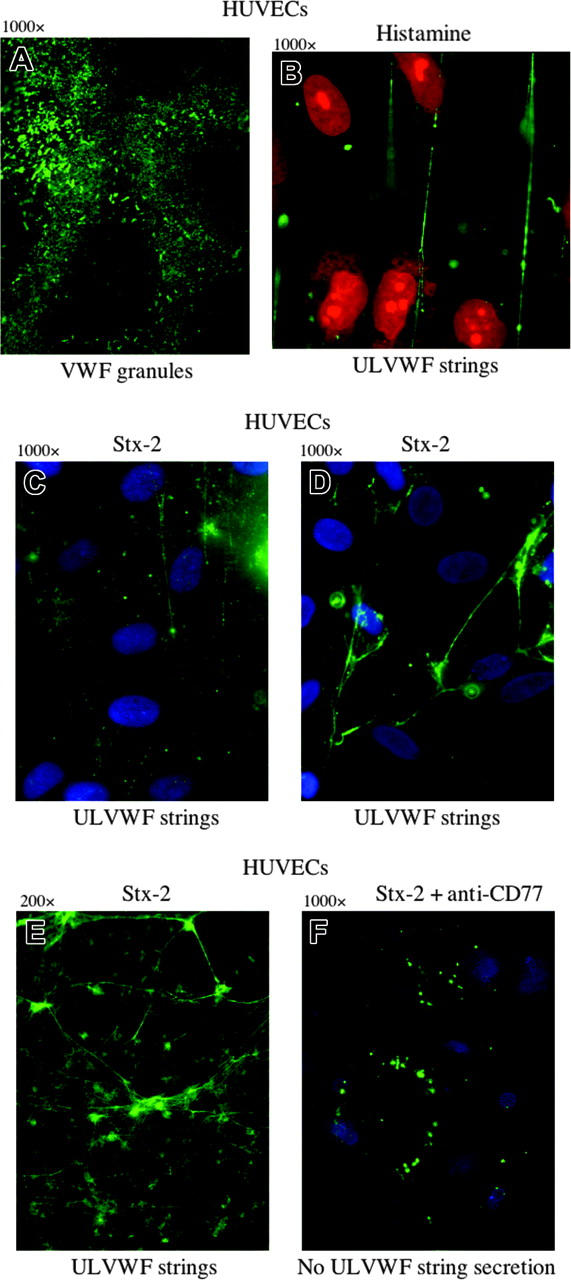
ULVWF strings are secreted from HUVECs stimulated by histamine or by the binding of Stx-2 to Gb3. Green VWF granules (A,F) or strings (B-E) were visualized by fluorescence microscopy using rabbit anti-human VWF IgG and goat anti-rabbit IgG-FITC or IgG-Alexa Fluor 488. (A) Unstimulated HUVECs permeabilized by Triton-X and fixed with paraformaldehyde contain cytoplasmic VWF-containing granules. (B) HUVECs stimulated under flowing conditions with the known VWF secretagogue, histamine (0.1 mM), secrete long ULVWF strings. HUVEC nuclei were stained by propidium iodide (red). In panels A and B, the cells were fixed with 1% paraformaldehyde in PBS and made permeable with Triton-X (in panel B after cell stimulation). The cells were unfixed in panels C-F. HUVECs stimulated by Stx-2 (1 nM) secrete ULVWF strings (C), which often become entangled (D-E). HUVEC nuclei in panels C, D, and are stained blue with DAPI. Original magnification × 1000 for all panels, except panel E, which is × 200. (F) Preincubation of HUVECs with monoclonal mouse anti-human Gb3 (CD77; 5 ng/mL) blocks Stx-2 stimulated ULVWF multimeric string secretion under flowing conditions. Only intracellular VWF granules in clumps near the cell surface are demonstrated in the unfixed cells with the VWF stimulation blocked. HUVECs in all experiments were either in primary culture or passage 1. Each of these photos is representative of 3 to 20 experiments.
Figure 3.
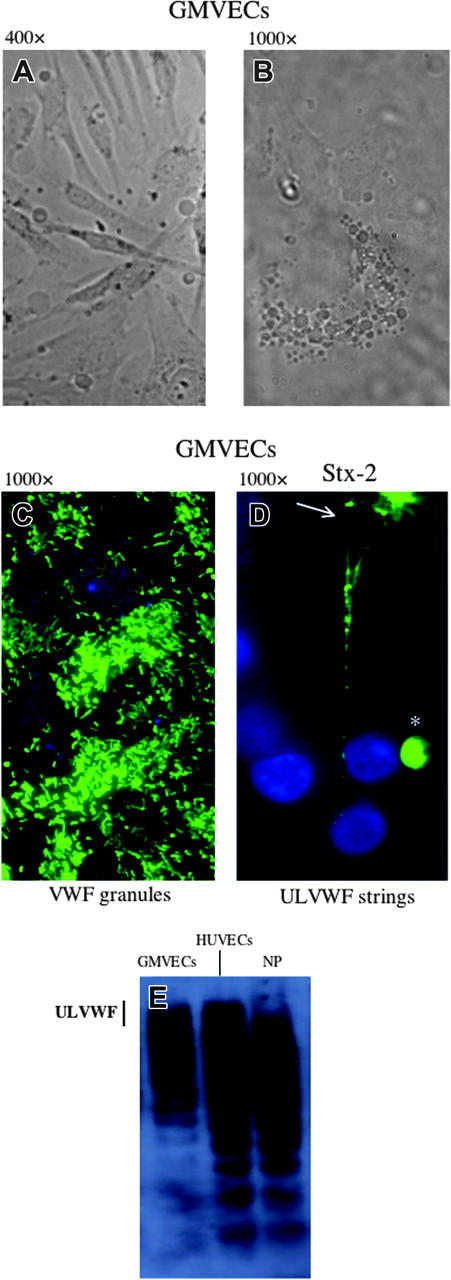
GMVECs. The cells were passage 4, grown on glass coverslips. Bright field images at different magnifications show elongated cells (A; original magnification × 400) and the rounded central portion of a single cell (B; original magnification × 1000). (C-D) GMVECs were stained with rabbit anti-VWF IgG and goat anti-rabbit-IgG-Alexa 488 (green), as well as the nuclear stain, DAPI (blue). The unstimulated GMVECs contain cytoplasmic VWF-containing granules (C). The granules are secreted as long ULVWF multimeric strings (D) in response to stimulation of the GMVECs for 3 minutes with Stx-2 (1 nM). The ULVWF strings frequently become entangled, as at the top of the frame (arrow). In some Stx-1- or Stx-2-stimulated GMVECs at passage number 4 (or higher), circular conglomerates of surface VWF (*) that do not unfurl into strings in response to stimulation are also seen. (E) VWF multimers enriched in ULVWF are also released slowly into the culture medium in soluble form by unstimulated GMVECs over the course of 24 hours. The VWF multimers were separated by 1% agarose/SDS gel electrophoresis, and then detected by membrane transfer, a polyclonal rabbit anti-VWF IgG, goat anti-rabbit IgG-HRP and chemiluminescence. VWF released from unstimulated HUVECs over 24 hours, or present in normal human platelet-poor plasma (NP), are shown for comparison. The vertical line indicates the gel location of ULVWF forms. These photos are representative of 3 experiments.
A monoclonal antibody to Gb3 receptors (CD77) added before Stx stimulation completely blocked the secretion of ULVWF strings (Figure 2F), demonstrating that the process requires Stx-Gb3 binding. This result also indicates that there is little or no contribution to Stx-2-stimulated ULVWF string secretion by any contaminating LPS in recombinant Stx-2 under the conditions of these experiments.
The morphology of the GMVECs is shown in Figure 3A-B. In all experiments reported in “Results” a low flow rate was used because of the fragility of the GMVECs (passages 3-5), that is, their propensity to peel from coverslips under higher flow conditions. Experiments were all completed within 30 minutes. After 1 hour of exposure to 10 nM of either Stx-1 or Stx-2, more than 90% of the HUVECs remained viable, and there was no detectable alteration in GMVEC number or morphology (eg, no cell peeling or shrinkage [data not shown]). After 6 hours of exposure to 10 nM of either Stx-1 or Stx-2, 75% of HUVECs remained viable (data not shown).
Platelet adherence to ULVWF strings above Stx-stimulated HUVECs and GMVECs
Flowing normal platelets, formalinized platelets (with intact glycoprotein Ibα [GPIbα], but not αIIb-β3, receptors), or anti-VWF beads57 adhered as singlets and small clumps along the length of individual ULVWF strings secreted by HUVECs and GMVECs in response to 20 mM histamine or 1 to 10 nM Stx-1 or Stx-2 (Figure 4).
Figure 4.

Platelets attach to ULVWF strings secreted from HUVECs or GMVECs stimulated by histamine in millimolar or Stxs in nanomolar concentrations. Washed normal human platelets in Ca2+/Mg2+-free Tyrode buffer were perfused over HUVECs (A-B) or GMVECs (C-D) and attached to the ULVWF strings formed as a result of cell stimulation by 20 mM histamine (A), or 10 nM of either Stx-1 (B-C) or Stx-2 (D). White arrows point to ULVWF strings with adherent platelets. Photographs of ULVWF-platelet strings were taken under flowing conditions 2 minutes after HUVEC stimulation. Each of these photos is representative of 3 to 20 experiments.
The ULVWF-platelet strings were quantified and photographed in real time using phase-contrast microscopy. Nanomolar concentrations (1-10 nM) of Stx-1 or Stx-2 induced the formation of as many ULVWF strings in 10 minutes as were induced by a millimolar concentration (20 mM) of histamine (Figure 5A). In separate experiments, small quantities of VWF enriched in ULVWF in soluble form could be detected within minutes in the supernatants of stimulated HUVECs (Figure 5B) or over the course of 24 to 48 hours in the supernatants of unstimulated HUVECs or GMVECs (Figure 3E).
Figure 5.

HUVECs produce similar quantities of ULVWF strings following stimulation by histamine in millimolar or Stxs in nanomolar concentrations. (A) HUVECs were incubated with histamine, buffer, or 10 nM Stx-1 or Stx-2 prior to perfusion with washed normal human platelets in Ca+2/Mg+2-free Tyrode buffer. The number of VWF-platelet strings formed in 20 microscope fields at × 400 magnification was quantified after 2 minutes of flow (n = 10-20). (B) In separate experiments, the multimeric patterns of VWF secreted into culture media were compared following HUVEC stimulation for 5 minutes with histamine (His, 20 mM) or Stx-2 or Stx-1 (10 nM). Separation and detection of VWF multimers were as described in Figure 3. The VWF multimeric pattern in normal plasma (NP) is shown for comparison. The vertical line indicates the location of ULVWF forms (n = 10-15).
Cholera toxin is an AB5 toxin analogous to Stx-1 and Stx-2 and binds to GM1 receptors in the membrane lipid “rafts” that also contain the Shiga toxin Gb3 receptors. Under identical flowing conditions, cholera toxin, in concentrations 30 to 6000-fold greater than Stx-1 or Stx-2, induced the output of only 10% to 20% as many ULVWF-strings from HUVECs and GMVECs as were induced by 10 nM Stx-1 or Stx-2 (data not shown).
Stx-mediated inhibition of ULVWF-platelet string cleavage by ADAMTS13
ADAMTS13 activity in normal citrated PRP was evaluated under flow by quantifying the persistence of ULVWF-platelet strings produced above HUVECs stimulated by 20 mM histamine. Cleavage of the histamine-induced ULVWF-platelet strings by ADAMTS13 was compared using normal PRP samples that had been mixed with either buffer or bovine serum albumin (BSA; as controls), Stx-1 or Stx-2, or EDTA, which is a known ADAMTS13 inhibitor53,57-61 (Figure 6A). After 2 minutes of flow (the first time point examined), ADAMTS13 cleavage of the ULVWF-platelet strings was impaired in PRP mixed with Stx-1, Stx-2, or EDTA (Figure 6A-B). In contrast to the irreversible inhibition of ADAMTS13 activity by EDTA, inhibition of ADAMTS13 by Stx-1 or Stx-2 became less pronounced as flow continued for an additional 2 (Figure 6B) to 6 minutes (not shown).
Figure 6.

Stxs inhibit ADAMTS13 activity in PRP. (A) HUVECs were stimulated with histamine (20 mM), followed by perfusion of normal platelet-rich-plasma (Nl PRP; containing approximately 100% ADAMTS13 activity). The PRP was mixed with an equal volume of either Tyrode buffer, BSA (1 mg/mL), or Stx-1 or Stx-2 (10 nM), or EDTA (1 mM). The number of ULVWF-platelet strings that remained uncleaved by plasma ADAMTS13 after 2 minutes of perfusion was quantified in 20 fields (original magnification × 200; n = 4-18). (B) Histamine-stimulated HUVECs were perfused with either PRP alone or PRP mixed with an equal volume of buffer, Stx-1, or Stx-2. The number of ULVWF-platelet strings that remained uncleaved by the ADAMTS13 in normal PRP was greater in the presence of Stx-1 or Stx-2 (10 nM) than in the presence of buffer after 2, 3, and 4 minutes of perfusion (n = 4-7).
The rhADAMTS13 cleaved ULVWF-platelet strings secreted by HUVECs stimulated by 20 mM histamine in a reaction that was dose dependent (Figure 7A) and inhibited by EDTA (not shown). The ULVWF-cleaving protease activity of rhADAMTS13 in dilutions of 1:5 to 1:10 was similar to the activity in the citrated PRP obtained from 20 healthy donors.
Figure 7.
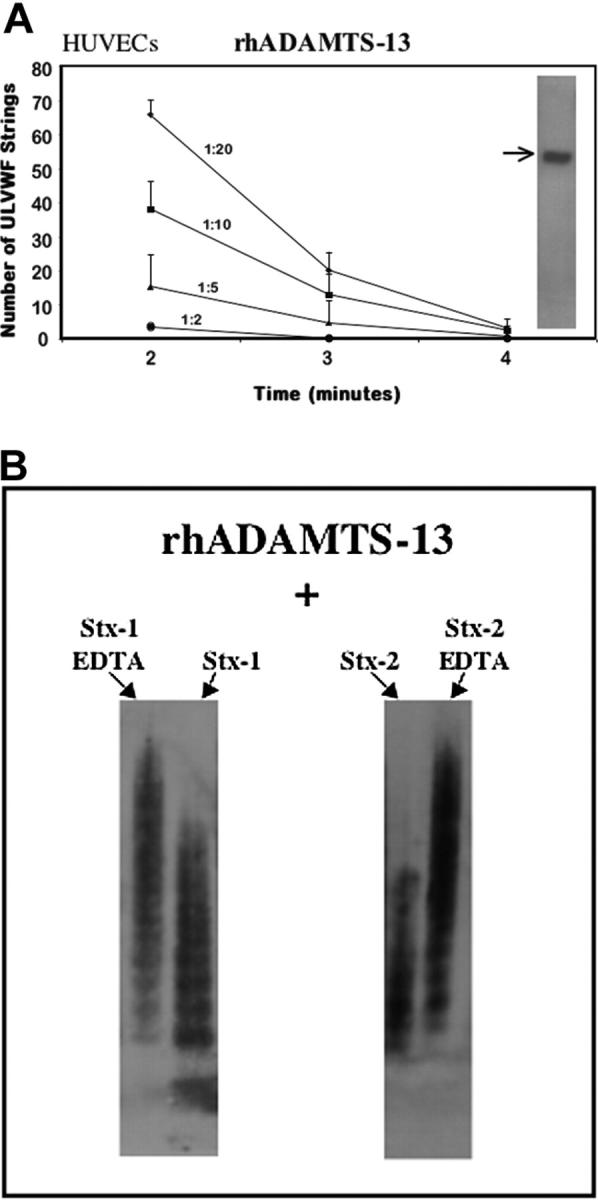
rhADAMTS13. (A) Cleavage of ULVWF-platelet strings by dilutions of rhADAMTS13. HUVECs were stimulated with histamine (20 mM) for 10 minutes and exposed to normal human platelets under flow. The ULVWF-platelet strings that formed were then cleaved over the course of 4 minutes by different dilutions of added rhADAMTS13 (n = 2-6 at each dilution). (Inset) Arrow shows rhADAMTS13 band in the gel lane at approximately 160 Mr, detected by anti-His tag antibody. (B) Neither Stx-1 nor Stx-2 inhibits the rhADAMTS13 cleavage of large or unusually large VWF forms under static nonphysiologic conditions. Inhibition of cleavage was observed under these conditions only if the known ADAMTS13 inhibitor, EDTA (1 mM), was also present. VWF enriched in ULVWF, derived from cultured HUVEC supernatant, was incubated with rhADAMTS13 for 24 hours in the presence of BaCl2 and urea, and 10 nM of either Stx-1 or Stx-2; VWF multimers were displayed as described in Figure 2(n = 3).
In addition, rhADAMTS13 cleaved soluble VWF enriched in ULVWF forms (from endothelial-cell supernatant) under static conditions. These conditions include prolonged incubation (24 hours), low ionic strength, and the presence of barium and urea. Under these nonphysiologic conditions, neither Stx-1 nor Stx-2 (Figure 7B) inhibited the capacity of rhADAMTS13 to cleave soluble large and unusually large VWF forms (which, therefore, disappeared from the gel lanes) unless EDTA was also added to the mixture.
In contrast, there was delayed cleavage by rhADAMTS13 (1:10) of ULVWF-platelet strings formed in response to stimulation of HUVECs or GMVECs by 10 nM Stx-1 or Stx-2, compared with endothelial-cell stimulation by 20 mM histamine (Table 2; Figure 8). ULVWF-platelet string counting data in 20 contiguous fields over time, as well as examples of single-field photographs of ULVWF-platelet strings during these time periods, are shown for confluent HUVECs in Table 2 and Figure 8. For GMVECs, single-field photographs of ULVWF-platelet strings during similar time periods are shown. GMVECs in culture grow as “islands” composed of many cells, and the cell islands may not be completely confluent. It is more difficult to count quickly 20 fields of ULVWF-platelet strings secreted from noncontiguous stimulated cells.
Table 2.
Cleavage of ULVWF-platelet strings by rhADAMTS-13
| Before rhADAMTS-13 | 2 min after rhADAMTS-13 | 3 min after rhADAMTS-13 | 4 min after rhADAMTS-13 | |
|---|---|---|---|---|
| Histamine | 119 ± 17.1 | 25 ± 12 | 3 ± 3 | 1 ± 1 |
| Stx-1 | 112 ± 28 | 34 ± 9 | 21 ± 7 | 12 ± 6 |
| Stx-2 | 122 ± 43 | 38 ± 15 | 24 ± 11 | 11 ± 4 |
The number of ULVWF-platelet strings secreted by HUVECs stimulated with 20 mM histamine or 10 nM Stx-1 or Stx-2 for 10 minutes prior to the experiment was quantified 2 minutes after beginning the perfusion of washed platelets in buffer over the previously stimulated cells. (No additional histamine or Stx was added to the washed platelet perfusate.) After counting the ULVWF strings, rhADAMTS13 was added to the washed platelets in buffer, and the perfusion was continued for another 10 to 15 minutes with string counting in 20 fields or photographing of the same field every 1 to 2 minutes.
Figure 8.

Stx-1 and Stx-2 inhibit ULVWF-platelet string cleavage by rhADAMTS13. Washed normal human platelets in Ca+2/Mg+2-free Tyrode buffer were perfused continuously over HUVECs (A-B) or GMVECs (C-D) that had been stimulated for 10 minutes with histamine (20 mM; A,C), Stx-1 (10 nM; B) or Stx-2 (10 nM; D). ULVWF-platelet strings formed during the initial 2 minutes were photographed in a single field in the next 2 minutes. Washed platelets in buffer containing rhADAMTS13 (1:10 dilution; no additional histamine or Stx) were then perfused over the endothelial cells, and the ULVWF-platelet strings were photographed repeatedly in the same microscopic field (original magnification × 400) at the time intervals shown. A vertical line indicates the addition of rhADAMTS13. Arrows indicate ULVWF-platelet strings that were cleaved by rhADAMTS13 in about 2 minutes during the perfusion of endothelial cells that had been stimulated previously by 20 mM histamine (A,C), but persisted for more than 3 to 5 minutes (> 10 minutes in many experiments) during the perfusion of endothelial cells that had been stimulated previously by 10 nM Stx-1 (B) or Stx-2 (D). Each of these photos is representative of 3 to 20 experiments.
The data shown in Table 2 and Figure 8 suggest that either Stx-1 or Stx-2 impairs the cleavage of ULVWF-platelet strings by interfering with normal human (plasma or recombinant) ADAMTS13 binding to, or cleavage of, ULVWF-platelet strings formed above stimulated endothelial cells. The inhibitory effect of the Stxs on ULVWF-platelet string cleavage by ADAMTS13 is more impressive using normal PRP than using normal platelets in buffer containing rhADAMTS13 (compare Figure 6 and Table 2). One possible explanation for this difference is that other proteins in plasma (eg, VWF) bind to plasma ADAMTS13 and delay or prevent some of the plasma ADAMTS13 molecules from gaining access to the ULVWF-platelet strings as readily as in the platelets-buffer-rhADAMTS13 system.69
Stx, which stimulates ULVWF string output via binding to Gb3, may interfere with ADAMTS13 cleavage by attaching to ULVWF strings as they are secreted from Stx-stimulated endothelial cells. This possibility is more likely than Stx attachment to rhADAMTS13 for the following reason. Stx-stimulated endothelial cells were perfused with washed platelets in buffer (containing no additional Stx) for a total of 4 minutes between the end of the Stx stimulation period and the beginning of perfusion with washed platelets in buffer and rhADAMTS13. It would be reasonable to presume that little, if any, free Stx would be available to interact with added rhADAMTS13 under these experimental conditions.
Further explanation of the mechanism of this inhibition is not presently available. The answer is not obvious from sequence comparisons of the proteins involved. That is, there are no detectable homologies among the primary amino acid sequences of Stx-1, Stx-2, the domains of ADAMTS13, or the VWF monomeric subunit that would suggest competitive interactions among peptide portions of these molecules (NCBI; http://www.ncbi.nlm.nih.gov/).
Discussion
This study demonstrates that nanomolar concentrations of Stx-1 or Stx-2 stimulate the secretion of ULVWF multimeric strings from both HUVECs and GMVECs. The Stx-stimulation of glomerular endothelial cells may contribute to the elevated levels of VWF found in the plasma of most patients with diarrhea-associated HUS.70-74 In the baboon model of Stx-induced HUS, however, it was concluded that LPS was predominantly responsible for the elevated plasma levels of VWF antigen in the small number of animals studied.14
ULVWF strings remain attached to the endothelial cells via P-selectin molecules secreted concurrently with ULVWF from Weibel-Palade bodies.75 Normal platelets, formalinized platelets (with functional GPIbα-IX receptors), beads coated with anti-VWF or, as previously shown,57 CHO cells expressing GPIbα-IX complexes adhere to the ULVWF strings as the platelets, beads, or CHO cells flow through the “thicket” of secreted ULVWF strings. Attachment of platelets to the ULVWF strings is, therefore, mediated by ULVWF-GPIbα-IX binding. Extensive platelet attachment to ULVWF strings secreted by Stx-stimulated glomerular endothelial cells in vivo may explain the thrombocytopenia and acute thrombotic obstruction of the renal microvasculature in patients with HUS. Thrombocytopenia and intrarenal thrombosis are even more likely under the higher flow/shear conditions of the glomerular microcirculation. At higher in vitro levels of shear stress (20-40 dynes/cm2), activation of platelets attached to the ULVWF strings occurs and results in the formation of platelet aggregates.76
Exposure of endothelial cells in culture to Stx-1 or Stx-2 for more than 1 hour results in progressive cell injury and death. In contrast, exposure to nanomolar concentrations of these toxins for minutes (as described in this study) causes rapid and profuse secretion of ULVWF multimeric strings from HUVECs and GMVECs. The 1- to 10-nM concentrations of Stx-1 and Stx-2 used in most of our experiments are more than 1000-fold greater than the levels expected in the blood of baboons after the injection of 25 to 100 ng/kg13,14,32 or greyhounds after the injection of 30 to 50 ng/kg.49 The lower doses of Stx given to the baboons and dogs, however, would be expected to recirculate repeatedly bound to blood cells and in plasma. In contrast, our larger doses of Stx-1 or Stx-2 were added to cultured endothelial cells as a one-time in vitro exposure. In addition, the endothelial cells in our in vitro experiments were primary or passage 1 HUVECs and passage 3 to 5 GMVECs, and likely had some metabolic and structural compromise associated with their isolation and maintenance in tissue culture, compared to endothelial cells in vivo in the baboon and greyhound models of HUS. The secreted ULVWF strings induce the immediate adhesion of flowing platelets. The cleavage and elimination of the ULVWF-platelet strings by plasma ADAMTS13 (∼100% activity) or by rhADAMTS13 (adjusted to ∼100% activity) is delayed by several minutes in the presence of either Stx-1 or Stx-2. This vigorous ULVWF string secretion, ULVWF-platelet string formation, and the associated delay in ULVWF-platelet string cleavage may combine to initiate platelet adhesion above Stx-stimulated glomerular endothelial cells. These events may exceed the counteractive ULVWF-platelet string cleaving capacity of circulating ADAMTS13, even though the plasma levels of ADAMTS13 are usually normal (or only modestly reduced), and promote thrombotic microangiopathy, acute renal failure, and HUS.
The days of delay between the initial entrance of Stx into the bloodstream and the development of overt HUS in patients may be related to the cumulative processes of ULVWF-platelet adhesion and subsequent aggregation atop Stx-stimulated renal glomerular endothelial cells. The plasma ADAMTS13 activity in Stx-associated HUS is not usually reduced. Our in vitro observations suggest that Stx-induced HUS may be associated with (1) increased, persistent ULVWF-platelet string formation atop Stx-stimulated glomerular endothelial cells and (2) modestly decreased rates of cleavage of ULVWF-platelet strings by plasma ADAMTS13 in the presence of Stx-1 or Stx-2.
The combination of these 2 factors (ie, points 1 and 2) may account for a continuous increase, over the course of days, in the number of glomeruli that become partially or completely obstructed by platelet (and/or platelet-fibrin) thrombi. When platelet production can no longer compensate for the putative intrarenal platelet deposition, then patients would be expected to develop thrombocytopenia of an extent that reflects the production/deposition disparity. When the function of a sufficient number of glomeruli becomes compromised by platelet, platelet-fibrin or, eventually, fibrin thrombotic occlusion, then patients would be expected to manifest signs of acute renal failure.
These effects of Stx-1 and Stx-2 are likely to be potentiated by the “cytokine storm” that accompanies enterohemorrhagic E coli infection. We have shown previously67 that TNF-α, IL-8, and IL-6 (in complex with the soluble IL-6 receptor) all stimulate endothelial-cell secretion of ULVWF multimeric strings accompanied by the rapid adhesion of flowing platelets. Of these 3 cytokines, IL-6 has effects similar to Stx-1 and Stx-2 in that it both stimulates ULVWF multimeric string secretion (and platelet adherence) and impairs the rate of ULVWF-platelet string cleavage by ADAMTS13.67 The effects in vitro of the 2 Stxs and 3 cytokines on endothelial-cell ULVWF secretion and ULVWF-platelet string cleavage by ADAMTS13 are summarized in Table 3.
Table 3.
Summary of in vitro effects by Shiga toxins and cytokines associated with HUS on cultured endothelial-cell ULVWF string secretion and cleavage by ADAMTS-13
| ULVWF secretion | Inhibition of ADAMTS-13 | |
|---|---|---|
| TNF-α | Yes | No |
| IL-8 | Yes | No |
| IL-6 | Yes* | Yes |
| Stx-1 | Yes | Yes |
| Stx-2 | Yes | Yes |
The concentrations of substances used for the in vitro experimental results summarized in this table are greater than the concentrations expected immediately after injection in vivo in the blood of baboons13,32 and greyhounds49 that develop HUS.
IL-6 stimulation of ULVWF string secretion requires the presence of soluble IL-6 receptor67
Both cholera toxin and the Stxs are AB5 toxins that bind to separate specific receptors in the lipid rafts of plasma membranes (Gb3 for the Stxs and GM1 for cholera toxin). In vitro, cholera toxin stimulated only about one tenth to one fifth as much ULVWF string secretion by endothelial cells as did Stx-1 and Stx-2 at 1000-fold lower concentrations. It is not known if any toxin other than Stx-1 and Stx-2 (at nanomolar-micromolar concentrations) is also capable of stimulating ULVWF string secretion by endothelial cells, or delaying ADAMTS13 cleavage of ULVWF-platelet strings. Only the Stxs may cause these effects in vivo, and predominantly in the glomerular microcirculation, because they are unique in being transported on monocytes and platelets through the bloodstream to congregate on glomerular endothelial Gb3 receptors.
The mechanism of this inhibitory effect by Stx-1, Stx-2, and IL-6 is not yet known. None of the 3 inhibits the ADAMTS13 cleavage of soluble VWF enriched in ULVWF forms during prolonged incubation with BaCl2 and urea under static conditions. The Stxs cause a delay in ULVWF-platelet string cleavage by ADAMTS13 that is not detected when the interaction between soluble ULVWF and ADAMTS13 occurs over the course of many hours, in the presence of Ba2+, and under denaturing conditions.
Nishio et al77 recently reported that the VWF A1 domain exerts a negative influence on VWF cleavage by ADAMTS13. These investigators found that the binding of the VWF A1 domain to platelet GPIbα renders the adjacent VWF A2 domain more susceptible to proteolysis by ADAMTS13. Our evaluation of the rate of ADAMTS13 cleavage is based on the timed disappearance of strings of platelets adherent to ULVWF multimers. Stx-1 and Stx-2 impair ULVWF-platelet string cleavage by ADAMTS13 but not ADAMTS13 cleavage of soluble ULVWF in the presence of chemical additives. One possible explanation for these results is that some structural alteration in the VWF A2 domain, caused by platelet attachment to the VWF A1 domain, is required before Stxs can interfere with ADAMTS13 cleavage of ULVWF-platelet strings. It is not yet known if Stx-1 and Stx-2 (and, perhaps, IL-6) delay ULVWF-platelet string cleavage by competitive inhibition of ADAMTS13, by affecting VWF A1-GPIbα interactions in a manner that maintains some of the negative A1 modulation on A2 cleavage, or by some other mechanism currently unknown.
Previous in vitro experiments under flowing conditions using dermal microvascular endothelial cells (and HUVECs) by Morigi et al33 demonstrated that Stx-1 induced the increased cell-surface exposure of αIIb-β3, αv-β3, P-selectin, and platelet endothelial-cell adhesion molecule 1. These investigators also found that VWF was of critical importance in platelet thrombus formation above the perfused endothelial cells. P-selectin contains a transmembrane domain that enables it to anchor secreted ULVWF multimeric strings to endothelial-cell membranes. The arginine-glycine-aspartate (RGD) region at amino acid residues 1744-1746 of mature VWF monomeric subunits binds to αIIb-β3 and αv-β3 integrins and may allow secreted ULVWF multimeric strings to fold over onto the surface of some Stx-stimulated endothelial cells under flowing conditions and induce platelet adhesion.
Our in vitro results are compatible with in vivo observations in a primate model of Stx-induced HUS. Pysher et al14 found in baboons that the single intravenous injection of 100 ng/kg Stx-1 caused increased VWF expression in glomerular and peritubular capillaries and increased circulating levels of TNF-α, IL-8, and IL-6. The intravenous injection of 4 doses of 25 ng/kg Stx-2 into the animals was associated with increased IL-6 levels.13 The outcome in the baboons given injections of either Stx-1 or Stx-2 was glomerular thrombotic microangiopathy and HUS.13,14 Our in vitro results are also compatible with the findings by Raife et al49 that injection of 30 to 50 ng/kg into greyhounds induced the rapid appearance of ULVWF forms in dog plasma and symptoms of HUS.
Our in vitro experiments could not distinguish important differences between the actions of directly added Stx-1 and Stx-2 at 1 to 10 nM. Both stimulate endothelial-cell ULVWF secretion and delay ADAMTS13 cleavage. Furthermore, either intravenous Stx-1 or Stx-2 is capable of inducing HUS in baboons and greyhounds.13,14,32,49 These findings together suggest that the reported greater propensity for HUS in individuals exposed to Stx-2, as opposed to Stx-1, may be explained by events at the portal of entry of the Stxs. For example, perhaps there is increased efficiency of transcolonic transport of Stx-2 into the circulation of humans.
The signals transduced rapidly by Stx-1 or Stx-2 attachment to Gb3 to stimulate ULVWF multimeric string secretion are unknown. Candidates include nuclear factor κB (NF-κB), activator protein 1 (AP-1),78 and several protein kinases.79 Ishii et al78 reported that the generation of NF-κB and AP-1 activates the tissue factor promoter region of chromosome 1 and increases cell exposure of tissue factor. Up-regulation of tissue factor on cultured human proximal tubular epithelial cells has also been reported recently.41 Binding and activation of factor VII would then be expected to lead to the thrombin generation and fibrin polymer formation that contribute to the intrarenal thrombi in HUS.1,2,41 The thrombin generation (and fibrin formation) may be potentiated by Stx disruption of the interaction between thrombin and thrombomodulin and consequent interference with the protein C/S coagulation control pathway.80,81
As the thrombotic lesions progress, Stx-stimulated ULVWF string secretion, along with ADAMTS13 inhibition, may continue to induce platelets to accumulation in the HUS lesions.
In summary, we have described profuse HUVEC and GMVEC secretion of ULVWF multimeric strings, rapid adherence of flowing platelets to the ULVWF strings, and delayed ADAMTS13 cleavage of the ULVWF-platelet strings in the presence of Stx-1 or Stx-2. The effects described may initiate ULVWF-mediated platelet adhesion and thrombus formation above Stx-stimulated glomerular endothelial cells in diarrhea-associated HUS, even with plasma ADAMTS13 levels within the normal range. These in vitro findings may help to explain the elevated plasma VWF, thrombocytopenia, and acute renal failure in these patients.
Prepublished online as Blood First Edition Paper, August 30, 2005; DOI 10.1182/blood-2005-05-2111.
Supported by grants from the National Institutes of Health (P50-HL65967 and HL71895), the American Heart Association (J.-f.D., Established Investigator), the American Heart Association-Texas Affiliate, and the Mary R. Gibson Foundation. L.H.N. and N.A.T. contributed equally to this study.
An Inside Blood analysis of this article appears at the front of this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 U.S.C. section 1734.
References
- 1.Moake JL. Thrombotic microangiopathies. N Engl J Med. 2002;347: 589-600. [DOI] [PubMed] [Google Scholar]
- 2.Tarr PI, Gordon CA, Chandler WL. Shiga-toxin-producing Escherichia coli and haemolytic uraemic syndrome. Lancet. 2005;365: 1073-1086. [DOI] [PubMed] [Google Scholar]
- 3.Cleary TG. Cytotoxin producing Escherichia coli and the hemolytic uremic syndrome. Pediatr Clin North Am. 1988;35: 485-501. [DOI] [PubMed] [Google Scholar]
- 4.Karmali MA, Petric M, Lim C, Fleming PC, Arbus GS, Lior H. The association between idiopathic hemolytic uremic syndrome and infection by verotoxin-producing Escherichia coli. J Infect Dis. 1985;151: 775-782. [DOI] [PubMed] [Google Scholar]
- 5.Banatvala N, Griffin PM, Greene KD, et al. The United States National Progressive Hemolytic Uremic Syndrome Study: microbiologic, serologic, clinical, and epidemiologic findings. J Infect Dis. 2001;183: 1063-1070. [DOI] [PubMed] [Google Scholar]
- 6.Brett KN, Hornitzky MA, Bettelheim KA, Walker MJ, Djordjevic SP. Bovine non-O157 Shiga toxin 2-containing Escherichia coli isolates commonly possess stx2-EDL933 and/or stx2vhb subtypes. J Clin Microbiol. 2003;41: 2716-2722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Misselwitz J, Karch H, Bielazewska M, et al. Cluster of hemolytic-uremic syndrome caused by Shiga toxin-producing Escherichia coli O26:H11. Pediatr Infect Dis J. 2003;22: 349-354. [DOI] [PubMed] [Google Scholar]
- 8.Matussek A, Lauber J, Bergau A, et al. Molecular and functional analysis of Shiga toxin-induced response patterns in human vascular endothelial cells. Blood. 2003;102: 1323-1332. [DOI] [PubMed] [Google Scholar]
- 9.Eklund M, Leino K, Siitonen A. Clinical Escherichia coli strains carrying stx genes: stx variants and stx-positive virulence profiles. J Clin Microbiol. 2002;40: 4585-4593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Obrig TG. Pathogenesis of Shiga toxin (verotoxin)-induced endothelial-cell injury. In: Kaplan BS, Trompeter RS, Moake JL, eds. Hemolytic-Uremic Syndrome and Thrombotic Thrombocytopenic Purpura. New York, NY: Marcel Dekker; 1992: 405-419.
- 11.Lindberg AA, Brown JE, Stromberg N, et al. Identification of the carbohydrate receptor for Shiga toxin produced by Shigella dysenteriae type I. J Biol Chem. 1987;262: 1779-1785. [PubMed] [Google Scholar]
- 12.Fraser ME, Fujinaga M, Cherney MM, et al. Structure of shiga toxin type 2 (Stx2) from Escherichia coli O157:H7. J Biol Chem. 2004;279: 27511-27517. [DOI] [PubMed] [Google Scholar]
- 13.Siegler RL, Obrig TG, Pysher TJ, Tesh VL, Denkers ND, Taylor FB. Response to Shiga toxin 1 and 2 in a baboon model of hemolytic uremic syndrome. Pediatr Nephrol. 2003;18: 92-96. [DOI] [PubMed] [Google Scholar]
- 14.Pysher TJ, Siegler RL, Tesh VL, Taylor FB Jr. von Willebrand Factor expression in a Shiga toxin-mediated primate model of hemolytic uremic syndrome. Pediatr Dev Pathol. 2002;5: 472-479. [DOI] [PubMed] [Google Scholar]
- 15.Siegler RL, Pysher TJ, Lou R, Tesh VL, Taylor FB. Response to Shiga toxin-1, with and without lipopolsaccharide, in a primate model of hemolytic uremic syndrome. Am J Nephrol. 2001;21: 420-425. [DOI] [PubMed] [Google Scholar]
- 16.Kovbasnjuk O, Edidin M, Donowitz M. Role of lipid rafts in Shiga toxin 1 interaction with the apical surface of Caco-2 cells. J Cell Sci. 2001;114: 4025-4031. [DOI] [PubMed] [Google Scholar]
- 17.Karpman D, Papadopoulou D, Nilsson K, Sjogren A-C, Mikaelsson C, Lethagen S. Platelet activation by Shiga toxin and circulatory factors as a pathogenetic mechanism in the hemolytic uremic syndrome. Blood. 2001;97: 3100-3108. [DOI] [PubMed] [Google Scholar]
- 18.Kiyokawa N, Taguchi T, Mori T, et al. Induction of apoptosis in normal human renal tubular epithelial cells by Escherichia coli Shiga toxins 1 and 2. J Infect Dis. 1998;178: 178-184. [DOI] [PubMed] [Google Scholar]
- 19.Karmali MA. Infection by Shiga toxin-producing Escherichia coli: an overview. Mol Biotechnol. 2004;26: 117-122. [DOI] [PubMed] [Google Scholar]
- 20.Simon M, Cleary TG, Hernandez JD, Abboud HE. Shiga toxin 1 elicits diverse biologic responses in mesangial cells. Kidney Int. 1998;54: 1117-1127. [DOI] [PubMed] [Google Scholar]
- 21.Uchida H, Kiyokawa N, Horie H, Fujimoto J, Takeda T. The detection of shiga toxin in the kidney of a patient with hemolytic uremic syndrome. Pediatr Res. 1999;45: 133-137. [DOI] [PubMed] [Google Scholar]
- 22.Cooling LL, Walker KE, Gille T, Koerner TA. Shiga toxin binds human platelets via globotriaosylceramide (Pk antigen) and a novel platelet glycosphingolipid. Infect Immun. 1998;66: 4355-4366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Aman AT, Fraser S, Merritt EA, et al. A mutant cholera toxin B subunit that binds GM1-ganglioside but lacks immunomodulatory or toxic activity. Proc Natl Acad Sci U S A. 2001;98: 8536-8541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.De Haan L, Hirst TR. Cholera toxin: a paradigm for multi-functional engagement of cellular mechanisms [review]. Mol Membr Biol. 2004;21: 77-92. [DOI] [PubMed] [Google Scholar]
- 25.Holmgren J, Lonnroth I, Svennerholm L. Tissue receptor for cholera exotoxin: postulated structure from studies with GM1 ganglioside and related glycolipids. Infect Immun. 1973;8: 208-214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Proulx F, Toledano B, Phan V, Clermont MJ, Mariscalco MM, Seidman EG. Circulating granulocyte colony-stimulating factor, C-X-C, and C-C chemokines in children with Escherichia coli O157:H7 associated hemolytic uremic syndrome. Pediatr Res. 2002;52: 928-934. [DOI] [PubMed] [Google Scholar]
- 27.Thorpe CM, Hurley BP, Lincicome LL, Jacewitz MS, Keusch MS, Acheson DW. Shiga toxins stimulate secretion of interleukin-8 from intestinal epithelial cells. Infect Immunol. 1999;67: 5985-5993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Thorpe CM, Smith WE, Hurley BP, Acheson DW. Shiga toxins induce, superinduce, and stabilize a variety of C-X-C chemokine mRNAs in intestinal epithelial cells, resulting in increased chemokine expression. Infect Immunol. 2001;69: 6140-6147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Eisenhauer PB, Chaturvedi P, Fine RE, et al. Tumor necrosis factor alpha increases human cerebral endothelial cell Gb3 and sensitivity to Shiga toxin. Infect Immunol. 2001;69: 1889-1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Foster GH, Tesh VL. Shiga toxin 1-induced activation of c-Jun NH(2)-terminal kinase and in the human monocytic cell line THP-1: possible involvement in the production of TNF-alpha. Leukoc Biol. 2002;71: 107-114. [PubMed] [Google Scholar]
- 31.Hughes AK, Stricklett PK, Kohan DE. Shiga toxin-1 regulation of cytokine production by human glomerular epithelial cells. Nephron. 2001;88: 14-23. [DOI] [PubMed] [Google Scholar]
- 32.Siegler RL, Pysher TJ, Tesh VL, Taylor FB. Response to single and divided doses of Shiga toxin-1 in a primate model of hemolytic uremic syndrome. J Am Soc Nephrol. 2001;12: 1458-1467. [DOI] [PubMed] [Google Scholar]
- 33.Morigi M, Galbusera M, Binda E, et al. Verotoxin-1 induced up-regulation of adhesive molecules renders microvascular endothelial cells thrombogenic at high shear stress. Blood. 2001;98: 1828-1835. [DOI] [PubMed] [Google Scholar]
- 34.Ray PE, Liu XH. Pathogenesis of Shiga toxin-induced hemolytic uremic syndrome. Pediatr Nephrol. 2001;16: 823-839. [DOI] [PubMed] [Google Scholar]
- 35.Stricklett PK, Hughes AK, Ergonul Z, Kohan DE. Molecular basis for up-regulation by inflammatory cytokines of Shiga toxin 1 cytotoxicity and globotriaosylceramide expression. J Infect Dis. 2002;186: 976-982. [DOI] [PubMed] [Google Scholar]
- 36.Habib R. Pathology of the hemolytic uremic syndrome. In: Kaplan B, Trompeter R, Moake J, eds. Hemolytic-Uremic Syndrome and Thrombotic Thrombocytopenic Purpura. New York, NY: Marcel Dekker; 1992: 315-353.
- 37.Courtecuisse V, Habib R, Monnier C. Nonlethal hemolytic and uremic syndromes in children: an electron-microscope study of renal biopsies from six cases. Exp Mol Pathol. 1967;7: 327-347. [DOI] [PubMed] [Google Scholar]
- 38.Inward CD, Howie AJ, Fitzpatrick MM, Rafaat F, Milford DV, Taylor CM. Renal histopathology in fatal cases of diarrhoea-associated haemolytic uraemic syndrome. British Association for Paediatric Nephrology. Pediatr Nephrol. 1997;11: 556-559. [DOI] [PubMed] [Google Scholar]
- 39.Gianantonio CA, Vitacco M, Mendilaharzu F, Gallo GE, Sojo ET. The hemolytic-uremic syndrome. Nephron. 1973;11: 174-192. [DOI] [PubMed] [Google Scholar]
- 40.Argyle JC, Hogg RJ, Pysher TJ, Silva FG, Siegler RL. A clinicopathological study of 24 children with hemolytic uremic syndrome. A report of the Southwest Pediatric Nephrology Study Group. Pediatr Nephrol. 1990;4: 52-58. [DOI] [PubMed] [Google Scholar]
- 41.Nestoridi E, Kushak RI, Duguerre D, Grabowski EF, Ingelfinger JR. Up-regulation of tissue factor activity on human proximal tubular epithelial cells in response to Shiga toxin. Kidney Int. 2005;67: 2254-2266. [DOI] [PubMed] [Google Scholar]
- 42.Chandler WL, Jelacic S, Boster DR, et al. Prothrombotic coagulation abnormalities preceding the hemolytic-uremic syndrome. N Engl J Med. 2002;346: 23-32. [DOI] [PubMed] [Google Scholar]
- 43.Tsai H-M, Chandler WL, Sarode R, et al. von Willebrand factor and von Willebrand factor-cleaving metalloprotease activity in Escherichia coli 0157: H7-associated hemolytic uremic syndrome. Pediatr Res. 2001;49: 653-659. [DOI] [PubMed] [Google Scholar]
- 44.Hosler GA, Cusumano AM, Hutchins GM. Thrombotic thrombocytopenic purpura and hemolytic uremic syndrome are distinct pathologic entities: a review of 56 autopsy cases. Arch Pathol Lab Med. 2003;127: 834-839. [DOI] [PubMed] [Google Scholar]
- 45.Remuzzi G. HUS and TTP: variable expression of a single entity. Kidney Int. 1987;32: 292-308. [DOI] [PubMed] [Google Scholar]
- 46.Drummond KN. Hemolytic uremic syndrome—then and now. N Engl J Med. 1985;312: 116-118. [DOI] [PubMed] [Google Scholar]
- 47.Vitacco M, Sanchez Avalos J, Gianantonio CA. Heparin therapy in the hemolytic-uremic syndrome. J Pediatr. 1973;83: 271-275. [DOI] [PubMed] [Google Scholar]
- 48.Siegler RL, Pysher TJ, Tesh VL, Denkers ND, Taylor FB. Prophylactic heparinization is ineffective in a primate model of hemolytic uremic syndrome. Pediatr Nephrol. 2002;17: 1053-1058. [DOI] [PubMed] [Google Scholar]
- 49.Raife T, Friedman KD, Fenwick B. Lepirudin prevents lethal effects of Shiga toxin in a canine model. Thromb Haemost. 2004;92: 387-393. [DOI] [PubMed] [Google Scholar]
- 50.Moake JL. Haemolytic-uraemic syndrome: basic science. Lancet. 1994;343: 393-397. [DOI] [PubMed] [Google Scholar]
- 51.Habib R, Levy M, Gagnadoux M-F, Broyer M. Prognosis of the hemolytic uremic syndrome in children. In: Hamburger J, Crosnier J, Grunfeld J-P, eds. Advances in Nephrology. Vol. 2 Chicago, IL: Yearbook Medical; 1982: 99-128. [Google Scholar]
- 52.Katz J, Krawitz S, Sacks PV, et al. Platelet, erythrocyte, and fibrinogen kinetics in the hemolyticuremic syndrome of infancy. J Pediatr. 1973;83: 739-748. [DOI] [PubMed] [Google Scholar]
- 53.Furlan M, Robles R, Solenthaler M, Lammle B. Acquired deficiency of von Willebrand factor-cleaving protease in a patient with thrombotic thrombocytopenic purpura. Blood. 1998;91: 2839-2846. [PubMed] [Google Scholar]
- 54.Hunt BJ, Lammle B, Nevard CH, Haycock GB, Furlan M. von Willebrand factor-cleaving protease in childhood diarrhoea-associated haemolytic uraemic syndrome. Thromb Haemost. 2001;85: 975-978. [PubMed] [Google Scholar]
- 55.Gerritsen HE, Turecek PL, Schwarz HP, Lammle B, Furlan M. Assay of von Willebrand factor (vWF)-cleaving protease based on decreased collagen binding affinity of degraded vWF: a tool for the diagnosis of thrombotic thrombocytopenic purpura (TTP). Thromb Haemost. 1999;82: 1386-1389. [PubMed] [Google Scholar]
- 56.Veyradier A, Obert B, Houllier A, Meyer D, Girma JP. Specific von Willebrand factor-cleaving protease in thrombotic microangiopathies: a study of 111 cases. Blood. 2001;98: 1765-1762. [DOI] [PubMed] [Google Scholar]
- 57.Dong J-f, Moake JL, Nolasco L, et al. ADAMTS-13 rapidly cleaves newly secreted ultra-large von Willebrand factor multimers on the endothelial surface under flowing conditions. Blood. 2002;100: 4033-4039. [DOI] [PubMed] [Google Scholar]
- 58.Dong J-f, Moake JL, Bernardo A, et al. ADAMTS-13 metalloprotease interacts with the endothelial cell-derived ultra-large von Willebrand factor. J Biol Chem. 2003;278: 29633-29639. [DOI] [PubMed] [Google Scholar]
- 59.Furlan M, Robles R, Solenthaler M, Wassmer M, Sandoz P, Lammle B. Deficient activity of von Willegrand factor-cleaving protease in chronic relapsing thrombotic thrombocytopenic purpura. Blood. 1997;89: 3097-3103. [PubMed] [Google Scholar]
- 60.Furlan M, Robles R, Galbusera M, et al. von Willebrand factor-cleaving protease in thrombotic thrombocytopenic purpura and hemolytic-uremic syndrome. N Engl J Med. 1998;339: 1578-1584. [DOI] [PubMed] [Google Scholar]
- 61.Tsai HM, Lian EC-Y. Antibodies of von Willebrand factor cleaving protease in acute thrombotic thrombocytopenic purpura. N Engl J Med. 1998;339: 1585-1594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rapoport AP, Simons-Evelyn M, Chen T, et al. Flavopiridol induces apoptosis and caspase-3 activation of a newly characterized Burkitt's lymphoma cell line containing mutant p53 genes. Blood Cells Mol Dis. 2001;27: 610-624. [DOI] [PubMed] [Google Scholar]
- 63.Boyd B, Lingwood C. Verotoxin receptor glycolipid in human renal tissue. Nephron. 1989;51: 207-210. [DOI] [PubMed] [Google Scholar]
- 64.Prado D, Cleary TG, Pickering LK, et al. The relation between production of cytotoxin and clinical features in shigellosis. J Infect Dis. 1986;154: 149-155. [DOI] [PubMed] [Google Scholar]
- 65.Hansen MB, Nielsen SE, Berg K. Re-examination and further development of a precise and rapid dye method for measuring cell growth/cell kill. J Immunol Methods. 1989;119: 203-210. [DOI] [PubMed] [Google Scholar]
- 66.Moake JL, Olson JD, Troll JH, Weinger RS, Peterson DM, Cimo PL. Interaction of platelets, von Willebrand factor and ristocetin during platelet agglutination. J Lab Clin Med. 1980;96: 168-184. [PubMed] [Google Scholar]
- 67.Bernardo A, Ball C, Nolasco L, Moake JF, Dong JF. Effects of inflammatory cytokines on the release and cleavage of the endothelial cell-derived ultralarge von Willebrand factor multimers under flow. Blood. 2004;104: 100-106. [DOI] [PubMed] [Google Scholar]
- 68.Liu L, Choi H, Bernardo A, et al. Platelet-derived VWF-cleaving metalloprotease ADAMTS-13. J Thromb Haemost. Prepublished on September 13, 2005, as DOI 10.1111/j.1538-7836.2005.01561.x. [DOI] [PubMed]
- 69.Fujikawa K, Suzuki H, McMullen B, Chung D. Purification of von Willebrand factor-cleaving protease and its identification as a new member of the metalloproteinase family. Blood. 2001;98: 1662-1666. [DOI] [PubMed] [Google Scholar]
- 70.Sutor AH, Thomas KB, Prufer FH, Grohmann A, Brandis M, Zimmerhackl LB. Function of von Willebrand factor in children with diarrhea-associated hemolytic-uremic syndrome (D + HUS). Semin Thromb Hemost. 2001;27: 287-292. [DOI] [PubMed] [Google Scholar]
- 71.Moake JL, Byrnes JJ, Troll JH, et al. Abnormal VIII: von Willebrand factor patterns in the plasma of patients with the hemolytic-uremic syndrome. Blood. 1984;64: 592-598. [PubMed] [Google Scholar]
- 72.Moake JL, McPherson PD. Abnormalities of von Willebrand factor multimers in thrombotic thrombocytopenic purpura and hemolytic-uremic syndrome. Am J Med. 1989;87(3N): 9N-15N. [PubMed] [Google Scholar]
- 73.Galbusera M, Benigni A, Paris S, et al. Unrecognized pattern of von Willebrand factor abnormalities in hemolytic uremic syndrome and thrombotic thrombocytopenic purpura. J Am Soc Nephrol. 1999;10: 1234-1241. [DOI] [PubMed] [Google Scholar]
- 74.Gordjani N, Sutor AH. Coagulation changes associated with the hemolytic uremic syndrome. Semin Thromb Hemost. 1998;24: 577-582. [DOI] [PubMed] [Google Scholar]
- 75.Padilla A, Bernardo A, Nolasco L, Moake J, Lopez JA, Dong J-F. A potential role for P-selectin in anchoring ultra-large von Willebrand factor multimers onto the endothelial cell surface. Blood. 2002;100: 123a. Abstract 459. [Google Scholar]
- 76.Bernardo A, Ball C, Nolasco L, Choi H, Moake JL, Dong JF. Platelets adhered to endothelial cell-bound ultra-large von Willebrand factor strings support leukocyte tethering and rolling under high shear stress. J Thromb Haemost. 2005;3: 562-570. [DOI] [PubMed] [Google Scholar]
- 77.Nishio K, Anderson PJ, Zheng XL, Sadler JE. Binding of platelet glycoprotein Ibalpha to von Willebrand factor domain A1 stimulates the cleavage of the adjacent domain A2 by ADAMTS13. Proc Natl Acad Sci U S A. 2004;101: 10578-10583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ishii H, Takada K, Higuchi T, Sugiyama J. Verotoxin-1 induces tissue factor expression in human umbilical vein endothelial cells through activation of NF-kappaB/Rel and AP-1. Thromb Haemost. 2000;84: 712-721. [PubMed] [Google Scholar]
- 79.Takenouchi H, Kiyokawa N, Taguchi T, et al. Shiga toxin binding to globotriaosyl ceramide induces intracellular signals that mediate cytoskeleton remodeling in human renal carcinoma-derived cells. J Cell Sci. 2004;117: 3911-3922. [DOI] [PubMed] [Google Scholar]
- 80.Williams JM, Taylor CM. Decrease of thrombomodulin contributes to the procoagulant state of endothelium in haemolytic uraemic syndrome. Pediatr Nephrol. 2005;20: 243; author reply 244. [DOI] [PubMed] [Google Scholar]
- 81.Fernandez GC, Te Loo MW, van der Velden TJ, van der Heuvel LP, Palermo MS, Monnens LL. Decrease of thrombomodulin contributes to the procoagulant state of endothelium in hemolytic uremic syndrome. Pediatr Nephrol. 2003;18: 1066-1068. [DOI] [PubMed] [Google Scholar]