

Abstract
The chromosomal translocation t(12; 22)(p13;q11) in human myeloid leukemia generates an MN1-TEL (meningioma 1-translocation-ETS-leukemia) fusion oncoprotein. This protein consists of N-terminal MN1 sequences, a transcriptional coactivator fused to C-terminal TEL sequences, an ETS (E26 transformation-specific) transcription factor. Enforced expression of MN1-TEL in multipotent hematopoietic progenitors in knock-in mice perturbed growth and differentiation of myeloid as well as lymphoid cells. Depending on obligatory secondary mutations, these mice developed T-cell lympholeukemia. Here we addressed the role of MN1-TEL in myeloid leukemogenesis using the same mouse model. Expression of MN1-TEL enhanced the growth of myeloid progenitors in an interleukin 3/stem cell factor (IL-3/SCF)–dependent manner in vitro whereas 10% of MN1-TEL–expressing mice developed altered myelopoiesis with severe anemia after long latency. Coexpression of MN1-TEL and IL-3, but not SCF, rapidly caused a fatal myeloproliferative disease rather than acute myeloid leukemia (AML). Because MN1-TEL+ AML patient cells overexpress HOXA9 (homeobox A9), we tested the effect of coexpression of MN1-TEL and HOXA9 in mice and found that 90% of MN1-TEL+/HOXA9+ mice developed AML much more rapidly than control HOXA9+ mice. Thus, the leukemogenic effect of MN1-TEL in our knock-in mice is pleiotropic, and the type of secondary mutation determines disease outcome.
Introduction
The MN1-TEL fusion oncoprotein is the product of the t(12;22)(p13; q11) in human myeloid leukemia, consisting of N-terminal MN1 sequences, a transcriptional coactivator fused to C-terminal TEL sequences, an ETS transcription factor.1 The TEL/ETV6 gene on human chromosome 12p13 is frequently rearranged in leukemia2 and its protein product harbors a highly conserved ETS DNA binding domain at its C-terminus,3 which is retained in the fusion protein. The TEL gene was originally identified by cloning the breakpoint of t(5;12)(q33;p13) in leukemia, and to date more than 30 different rearrangements involving TEL have been reported.4,5 Gene targeting studies in mice have shown that TEL is essential for normal hematopoiesis.6 On the other hand, the MN1 gene on human chromosome 22q11 was originally cloned from the breakpoint of t(4;22)(p16;q11) in meningioma7 and its product is a ubiquitously expressed nuclear protein. Its N-terminus, which is retained in the fusion, functions as a transcriptional coactivator through recruitment of retinoic acid receptor interacting protein (RAC3) and E1A binding protein (p300).8 We previously reported that MN1-TEL transformed NIH3T3 fibroblasts, an activity dependent on a functional ETS DNA binding domain and N-terminal sequences of MN1.9 Thus, dysregulated expression of TEL target genes might mediate cellular transformation. However, the mechanism causing disease remains to be determined.
In the accompanying article (Kawagoe and Grosveld,10 beginning on page 4278), we have identified MN1-TEL as a bona fide hematopoietic oncogene in vivo by generating a mouse model of MN1-TEL leukemia. After long latency, 30% of the MN1-TEL knock-in (KI) mice, which express the MN1-TEL transgene in multipotent hematopoietic progenitors, developed T-lymphoid tumors, whereas 10% of these mice developed severe anemia with altered myelopoiesis but not myeloid leukemia. We also found that MN1-TEL prompted the proliferation of normal myeloid as well as lymphoid progenitors and partially blocked their differentiation in vivo. Thus, the growth stimulatory effects of MN1-TEL are not restricted to the myeloid lineage, although the fusion protein has been exclusively found in human myeloid disease such as chronic myeloid leukemia (CML), acute myeloid leukemia (AML), and myelodysplastic syndrome (MDS).1 In addition, T-cell lymphomagenesis was accelerated by N-ethyl-N-nitrosourea–induced mutations, indicating that MN1-TEL required secondary mutations to induce leukemia. Based on these data, we hypothesized that MN1-TEL, when expressed in the multipotent progenitors, affects both myeloid and lymphoid lineages, and the disease phenotype might be determined by the type of secondary cooperative mutation. To prove this hypothesis, we have analyzed the primary effect of MN1-TEL on myeloid hematopoiesis in our KI mouse and identified cofactors of MN1-TEL that bias disease outcome toward a myeloid phenotype.
Materials and methods
Hematopoietic progenitor assays
In vitro hematopoietic differentiation of embryonic stem (ES) cells was performed using ES cult (StemCell Technologies, Vancouver, BC, Canada) following the manufacturer's directions. ES cells expressing MN1-TEL and control cells (MN1-TELKI/wild type [WT] ES cells not treated with Cre and WT ES cells) were cultured in methylcellulose-based media (MC-1) with stem cell factor (SCF) to generate embryoid bodies (EBs). Number, size, and morphology of EBs from Cre-treated ES cells were identical to those from controls (data not shown). After 15 or 18 days, cells obtained from dissociated EBs were plated in methylcellulose cultures (MC-2) with SCF, interleukin 3 (IL-3), and IL-6. For secondary colony-forming assays, cells were harvested from the MC-2 and replated in MC-3. Colonies were scored after 10 days of culture.
Analysis of cell proliferation
MN1-TEL+ cells were grown in liquid culture in Iscoves modified Dulbecco medium (IMDM) supplemented with 20% fetal calf serum (FCS), SCF (50 ng/mL), and/or IL-3 (20 ng/mL). Numbers of viable cells were determined by trypan blue dye exclusion at 2, 4, and 6 days after the initiation of cultures.
Western blot
Western blots were performed using anti-TEL C-terminal,9 anti-FLAG (enterokinase cleavage site M2; Sigma, St Louis, MO), and anti-Gapdh (anti–glyceraldehyde phosphate dehydrogenase; c-11; Santa Cruz Biotechnology, Santa Cruz, CA) antibodies.
FCM analysis
Expression of surface markers and green fluorescent protein/yellow fluorescent protein (GFP/YFP) expression was determined by flow cytometric (FCM) analysis (FACS Caliber or LSRII; BD Immunocytometry Systems, Franklin Lakes, NJ) as described.11
Histologic analysis
Tissues were stained by hematoxylin-eosin or immunohistochemical methods as described.11 Cytospin slides were stained with May-Giemsa. Images of tissue sections and cytospins were obtained using a BX-50 microscope (equipped with UplanFl 40×/0.75 NA or 100×/1.30 NA objectives; Olympus, Tokyo, Japan) with a SPOT camera and SPOT Advanced imaging software (Diagnostic Instruments, Sterling Heights, MI). Original magnification for tissue sections or cytospins was 400× or 1000× respectively. Images of colonies were obtained using a Gel Doc EQ System (Bio-Rad, Hercules, CA).
Clonality analysis
The clonality of MN1-TEL+ tumor cells was analyzed by Southern blot with a probe for Il3 or HOXA9 (entire coding sequence) on BglII-digested genomic DNA.
Vector constructs
FLAG-tagged HOXA9 (Invitrogen, Carlsbad, CA; IMAGE 2987818), IL-3 (RDB1506; Riken, Tokyo, Japan), or SCF (RDB1528; Riken) cDNAs were cloned into the EcoRI site of pSRα-IRES (internal ribosome entry site)–YFP (yellow fluorescent protein) retroviral vector (the EcoRI site is 5′ of the IRES sequence, generating pSRα-HOXA9-IRES-YFP, pSRα–IL-3–IRES–YFP, or pSRα-SCF-IRES-YFP, respectively).
Retroviral transduction
Ecotropic retroviral vectors were generated as previously described.12 Lineage depletion of bone marrow (BM) cells was performed using streptavidine-conjugated magnetic beads (Dynal Biotech ASA, Oslo, Norway) and biotin-labeled mouse monoclonal antibodies (BD PharMingen, San Jose, CA) to CD5, B220, Mac1, Gr1, and Ter119 (lin- cells). Lin- cells were cultured for 24 hours in IMDM with 20% FCS, 50 ng/mL murine SCF, 20 ng/mL murine IL-3, and 30 ng/mL human IL-6, and 1 × 106 cells were plated in a well coated with Retronectin (Takara-Bio, Tokyo, Japan) and incubated with 2 mL of virus supernatant in the presence of the same cytokines. Fresh virus supernatants were applied 5 times every 8 or 16 hours.
Transplantation
Retrovirally transduced BM cells (1 × 106) were injected intravenously into 9.5-Gy irradiated syngeneic recipient mice. Tumor cells (1 × 106) of mice were transplanted into 7.5-Gy irradiated syngeneic secondary recipients.
Microarray analysis
Microarray analysis was performed using the Affymetrix GeneChip (Mouse Genome 430 2.0 array; Santa Clara, CA) as described.13
Quantitative PCR
Quantitative reverse transcription–polymerase chain reaction (RT-PCR) was performed as previously described.12 For the normalization of each sample the expression level of the gene was divided by that of GAPDH for human samples or β-actin for mouse samples. The relative expression level was determined by comparing the normalized value (GAPDH or β-actin) to that in the reference sample included in the same reaction. Primers for human HOXA9 and GAPDH and mouse β-actin have been described previously.14,15 We used the following primers for mouse and human N-Myc, respectively: 5′-TGAGCGACTCAGATGATGAGGAT-3′ and 5′-TGGTTACCGCCTTGTTGTTAGA-3′; 5′-CACCCTGAGCGATTCAGATGA-3′ and 5′-CCTTGGTGTTGGAGGAGGAA-3′.
Statistical analysis
Survival analysis was done using the Kaplan-Meier method and log-rank test. Statistical comparison of 2 independent groups was done by Student t test. Statistical significance was assigned when the probability that there was no difference between 2 variables was less than .05.
Results
MN1-TEL enhances proliferation and repopulation of embryonic myeloid progenitors
We have shown that MN1-TEL enhanced the repopulating ability of myeloid progenitors using adult BM cells of our KI mice, as described in the accompanying paper.10 To further confirm this finding, we performed methylcellulose-based cultures with MN1-TELKI/WT ES cell–derived hematopoietic cells. MN1-TELKI/WT ES cells harbor the loxP-SV40 transcriptional stop/poly-adenylation (polyA) sequence-loxP cassette followed by the IRES–MN1-TEL–IRES–GFP (green fluorescent protein)–polyA transgene, which was inserted downstream of exon 4 of the Aml1 gene via homologous recombination. Expression of Aml1 from the KI allele was maintained by inserting the coding sequence of the human AML1 cDNA in-frame downstream of mouse exon 4. The stop/polyA cassette and the IRES–MN1-TEL–IRES–GFP transgene were inserted downstream of the AML1 cDNA sequence.10 Thus, the endogenous Aml1 regulatory sequences control expression of the transgene in this model. MN1-TELKI/WT ES cells were transiently transfected with a bicistronic murine stem cell virus (MSCV)–Cre-IRES-YFP plasmid and sorted for expression of YFP to remove the transcriptional stop cassette and induce expression of MN1-TEL and GFP. We identified subclones of a single parental ES cell, which had deleted the transcriptional stop cassette. The removal of the stop cassette was confirmed by Southern blot (data not shown). The ES cells were first cultured in semisolid media with SCF to generate hematopoietic cells in embryoid bodies (EBs). These EB–derived hematopoietic cells expressing MN1-TEL or not (MN1-TELKI/WT ES cells not treated with Cre and WT ES cells) were then cultured in methylcellulose-based media with SCF, IL-3, and IL-6 to compare their ability to generate myeloid colony-forming cells (CFCs). Cultures of Cre-treated cells produced 3-fold (day-15 EB culture) to 6-fold (day-18 EB culture) more myeloid colonies than controls, suggesting that MN1-TEL strongly stimulated this activity (Figure 1A). This is different from the data showing that the number of primary myeloid colonies generated by MN1-TEL–expressing adult mouse BM cells was almost equal to that of controls.10 (see accompanying article by Kawagoe and Grosveld,10 beginning on page 4278). This difference was possibly caused by the presence of SCF in the EB culture. After replating into secondary methylcellulose cultures, the Cre-treated cells produced 10-fold more and much larger myeloid colonies (Figure 1B-C) than control cells, consistent with results of the adult BM culture. Most of the Cre-treated CFCs were GFP+ whereas those of nontreated cells were GFP- (Figure 1D). This suggested that MN1-TEL stimulated the proliferation and repopulation ability of embryonic myeloid progenitors as well as adult progenitors in the presence of cytokines.
Figure 1.

MN1-TEL enhances cytokine-dependent growth of myeloid progenitors. (A) In vitro hematopoietic differentiation of ES cells. MN1-TELKI/WT ES cells treated with Cre or not or WT ES cells were cultured in methylcellulose-based media (MC-1) with stem cell factor (SCF) to generate embryoid bodies (EBs). After 15 or 18 days, cells from dissociated EBs were plated into MC-2 in the presence of SCF, interleukin 3 (IL-3), and IL-6. Numbers of myeloid colonies were scored after 10 days of culture. Mean ± standard error (SE) is shown (n = 3). Experiments repeated with 3 different ES cell clones yielded identical results. (B) Colony-forming efficiency of MC-2 ES-derived hematopoietic progenitors in MC-3. Colony-forming cells (CFCs) were harvested from the MC-2 and replated into MC-3 with the same cytokines (replating assay). Numbers of myeloid colonies were scored after 10 days. Mean ± SE is shown (n = 3). Experiments repeated in triplicate using different ES clones yielded identical results. (C) Colonies in MC-3. Representative pictures captured on day 10 of culture are shown. Images were obtained as previously described.12 (D) GFP expression in ES-derived CFCs. GFP expression was analyzed by flow cytometry (FCM) after 10 days of culture in MC-2. Solid line indicates KI-ES–derived cells; broken line, control WT-ES–derived cells. (E) Cytokine-dependent growth of MN1-TEL+ myeloid progenitors. MN1-TEL/Mx1-Cre BM cells treated with polyinosinic-polycytidylic acid (pI-pC) or not were plated in MC. After the MC-3, cells were harvested and grown in liquid cultures. Numbers of viable cells are plotted. • indicates IL-3 + SCF; ○, IL-3; ▴, SCF; and ▵, no cytokine. Mean ± SE is shown (n = 3). Experiments repeated in triplicate using different cells yielded identical results. (F) May-Giemsa (M-G) staining of MN1-TEL+ cells in culture. Image obtained as previously described.12 (G) Fluorescence-activated cell sorter (FACS) analysis of Sca-1/c-Kit expression of an MN1-TEL+ cell line.
Cytokine-dependent growth of MN1-TEL–expressing myeloid progenitors
MN1-TEL/Mx1-Cre mice were generated by crossing MN1-TEL KI mice, generated with germ line transmitting MN1-TELKI/WT ES cells, with Mx1-Cre transgenic mice.16 The injection of polyinosinic-polycytidylic acid (pI-pC) activates interferon-inducible Cre expression,16 which removes the transcriptional stop cassette and allows expression of MN1-TEL and GFP in these mice (see accompanying article by Kawagoe and Grosveld10). We isolated BM of pI-pC–induced MN1-TELKI/WT/Mx1-Cre+/WT (MN1-TEL/Mx1-Cre) mice and plated them in methylcellulose (MC) cultures (Kawagoe and Grosveld10). After 3 rounds of MC culture, colonies were collected and inoculated into liquid culture in the presence of IL-3 and SCF. We then examined their cytokine requirement in these liquid cultures. As shown in Figure 1E, MN1-TEL+ cells grew much faster than control cells but their proliferation was entirely dependent on IL-3 and SCF. The cells are GFP+ (data not shown) and readily produced MN1-TEL+ cell lines in the presence of both cytokines. Morphologically, some of these cells showed granulation, indicating their myeloid differentiation (Figure 1F). They were stem cell antigen-1–positive (Sca-1+)/c-Kit+ (Figure 1G) and negative for lineage markers including Mac1, Gr1, B220, CD3, and Ter119 (data not shown), suggesting that they were immature cells. This was consistent with data of experiments using wild-type BM cells transduced with an MN1-TEL retrovirus (Cintia Carella and G.C.G., unpublished data, May 2005). Transplantation of these MN1-TEL+ cell lines did not cause leukemia in mice during a 3-month observation period (data not shown). Taken together, MN1-TEL provides a growth and repopulation advantage to myeloid progenitors in the presence of cytokines but does not transform these cells. Indeed, the numbers and morphology of peripheral white blood cells (WBCs), red blood cells, and platelets were normal in healthy, randomly selected MN1-TEL–expressing mice at 1 or 8 months after pI-pC induction (data not shown).
MN1-TEL causes altered myeloid hematopoiesis in mice
As described in the accompanying paper (Kawagoe and Grosveld10), 30% of MN1-TEL+ mice developed T-lymphoid tumors de novo after an 8-month (mean) latency. In addition, 10% of MN1-TEL–expressing mice (28% of sick mice) developed severe anemia (mean ± SE of hemoglobin level, 48 ± 8.7 g/L [4.8 ± 0.87 g/dL]; hematocrit, 0.162 ± 0.0245 [16.2% ± 2.45%]; n = 3) without evidence of bleeding. We did not find invasion of myeloid cells into any of their organs. These animals all showed an increased percentage of GFP+ cells in the BM (Figure 2A; mean ± SE, 38.8% ± 7.1%; n = 3), which was considerably higher than that in activated healthy MN1-TEL/Mx1-Cre BM at 1 (13.9% ± 1.6%; n = 5) or 8 months (20.1% ± 0.3%; n = 3). Their peripheral WBC and platelet counts were normal (4.68 ± 1.76 × 109/L and 456 ± 80.5 × 109/L, respectively; n = 3) and their BM myeloid cells showed terminal differentiation (data not shown). The percentage of myeloblasts in their BM (n = 3) was higher than that in the BM of age-matched control uninduced MN1-TEL/Mx1-Cre mice (n = 5; 7.9% ± 1.3% vs 3.6% ± 0.27%, respectively), although the numbers did not reach abnormal levels (> 20%, Bethesda proposal [Kogan et al17]). Consistent with the data showing that MN1-TEL partially blocks granulocyte (Gr1+) differentiation (Kawagoe and Grosveld10), the BM of these mice contained increased numbers of Mac1+/Gr1- cells at the expense of the Mac1+/Gr1+ or Mac1-/Gr1+ populations (Figure 2B), suggesting that MN1-TEL expression perturbed normal myeloid differentiation, although the mice never showed signs of leukemia. The mean fluorescence intensity (MFI) of GFP of these Mac1+/Gr1- cells was 2-fold higher than that of the control Mac1+/Gr1- cells of uninduced mice (7.8 ± 1.2 vs 3.2 ± 0.6, respectively; n = 3), indicating that they expressed MN1-TEL/GFP. The percentage of GFP+ cells in the Mac1+/Gr1- cell population was 41.9% ± 3.8% (n = 3). As we discussed in the accompanying paper,10 it is possible that we underestimate the percentage of the MN1-TEL–expressing cells by using GFP as a marker, since GFP is translated from the last cistron of the tricistronic mRNA via a second IRES, which is usually translated less effectively than the upstream cistrons.18 Indeed, the GFP expression monitored by MFI in our Aml1–IRES–MN1-TEL–IRES–GFP mouse BM cells treated with pI-pC was 45% lower than that in BM of Aml1-IRES-GFP mice,19 although they share the same Aml1 promoter to drive the expression of the transgene (n = 6; 6.1 ± 0.5 vs 11.3 ± 0.6, respectively). Thus, the percentage of GFP+ cells within the Mac1+/Gr1- population in these mice is a minimal estimate of the number of MN1-TEL+ cells. To further characterize their BM cells, we evaluated the profile of Mac1/Gr1 in GFP+ and GFP- cells. To clearly discriminate MN1-TEL+ from MN1-TEL- cells, we gated the 10% GFP-brightest or -dimmest cells. Consistent with the result described in the accompanying paper (Kawagoe and Grosveld10), the percentage of GFP+/Mac1+/Gr1- cells in these mice was increased compared with that in age-matched control Aml1-IRES-GFP mice (46.1% ± 8.3% vs 16.6% ± 0.6%, respectively; n = 3), whereas the percentage of GFP+/Mac1+/Gr1+ cells was decreased (18.7% ± 2.5% vs 81.3% ± 1.2%, respectively; n = 3; Figure 2C). These GFP+/Mac1+/Gr1- cells showed high forward scatter (FSC) and side scatter (SSC), indicating that they are myeloid cells (Figure 2C). This is consistent with the data that 90% of the GFP+ cells in healthy MN1-TEL–expressing mice have a myeloid morphology (data not shown). It is known that a subpopulation of B lymphocytes is Mac1+/Gr1-,20 but expression of B220 in GFP+ cells of these mice was virtually absent (data not shown). Among the GFP- cells in these mice, we also found Mac1+/Gr1- cells (22.3% ± 7.0%; n = 3), which showed a different profile of FSC/SSC than GFP+/Mac1+/Gr1- cells (Figure 2C). The percentage of GFP-/Mac1+/Gr1- cells was higher than that in control age-matched Aml1-IRES-GFP mice (10.0% ± 1.4%; n = 3). In contrast to GFP+/Mac1+/Gr1- cells, these GFP-/Mac1+/Gr1- cells showed a low FSC/SSC profile (Figure 2C), suggesting that they were lymphocytes. Given their cell surface marker expression they are probably B lymphocytes. The reason for the increase of this cell type in these mice is not clear. However, it is known that human MDS is frequently accompanied by alterations in B-cell subpopulations.21,22 We speculate that in our mice a similar mechanism affecting the development of B cell occurs. Based on these data, we conclude that GFP+/Mac1+/Gr1- myeloid cells were increased in these mice. To confirm the FCM data showing the increase of Mac1+/Gr1- myeloid cells, which are relatively immature,23 we also analyzed BM cells of these mice morphologically. The percentage of immature myeloid cells (myeloblast, promyelocyte, myelocyte, and meta-myelocyte) in their BM was increased (42.0% ± 1.4%; n = 3) compared with that in BM of control age-matched uninduced MN1-TEL/Mx1-Cre mice (25.4% ± 1.4%; n = 5). The ratio of immature to mature myeloid cells (band and segmented) was also higher in these mice than in controls (immature/mature, 1.27 ± 0.11 vs 0.60 ± 0.03, respectively). Although it is difficult to pinpoint at which stage myeloid differentiation is inhibited in these mice, the data suggest that there was a shift to a generally more immature phenotype. Spectral karyotyping (SKY) of BM cells of 2 of these mice after 1 to 3 weeks in culture showed that 7 (27%) of 26 and 12 (57%) of 21 metaphases contained nonclonal numeric and/or structural chromosome abnormalities (data not shown). Such an increase of abnormal cells was not detected by SKY in normal control BM cells cultured for 1 week. We believe that additional mutations caused by chromosomal abnormalities might have contributed to the amplification of MN1-TEL–expressing myeloid cells. Their peripheral blood data, anemia with normal WBC and platelet counts, satisfied one of the criteria for mouse MDS (Bethesda proposal [Kogan et al17); however, their BM cells did not show dysplasia, therefore they cannot be diagnosed to have MDS, although the disease showed similar features. However, we concluded that MN1-TEL–expressing (GFP+) cells, which consisted of 90% myeloid cells, were increased in these mice while their myeloid differentiation (Mac1/Gr1 expression) was altered. Thus, we consider this to be the result of accelerated proliferation of MN1-TEL–expressing myeloid cells.
Figure 2.

MN1-TEL–expressing mice develop altered myelopoiesis de novo. (A) Increased GFP+ cells in BM of MN1-TEL–expressing mice, which died of severe anemia without T-lymphoid tumor (anemic cases). Numbers indicated are mean ± SE percentage of GFP+ cells (n = 3). MT/Cre indicates control MN1-TEL/Mx1-Cre BM cells 1 month (1 mo.) or 8 months (8 mo.) after treatment with pI-pC. (B) Increased Mac1+Gr1- cells in MN1-TEL–expressing mice. The entire population of BM cells was analyzed by FCM. (Left) Age-matched control MN1-TEL/Mx1-Cre BM cells not induced with pI-pC. (Right) BM cells of MN1-TEL–expressing mice with increased number of GFP+ cells. Numbers indicated are mean ± SE percentage (n = 3). (C) Mac1+/Gr1- cells in GFP+ or GFP- BM cells of MN1-TEL–expressing mice with increased number of GFP+ cells. Gates were set to select the 10% GFP-brightest or -dimmest population in MN1-TEL–expressing mice to clearly discriminate MN1-TEL+ and MN1-TEL- cells. Numbers indicated are mean ± SE percentage (n = 3). (Top) Mac1/Gr1 expression in GFP+ or GFP- BM cells. (Bottom) FSC and SSC of Mac1+/Gr1- cells in GFP+ or GFP- BM cells.
IL-3–overexpressing MN1-TEL cells cause aggressive myeloproliferative disease in vivo
Autocrine (or paracrine) expression of IL-3 or abnormal SCF/c-Kit signaling has been described in human myeloid leukemia.24-26 Therefore, these cytokines were candidate cofactors for MN1-TEL–directed leukemogenesis. To examine if autocrine stimulation of MN1-TEL+ hematopoietic cells in vivo rendered these cells tumorigenic, we transduced BM cells of MN1-TEL/Mx1-Cre mice treated with pI-pC or not with pSRα–IL-3–IRES–YFP or control pSRα-IRES-YFP retrovirus and transplanted them into lethally irradiated syngeneic recipient mice (n = 10). The transduction efficiency was monitored by FCM analysis of YFP expression (Figure 3A) and was almost identical for MN1-TEL+ and MN1-TEL- cells (data not shown). Consistent with a previous report,27 recipients of BM overexpressing IL-3 alone all developed myeloid disease and died within 3 months after transplantation (Figure 3B). Controls that received transplants of MN1-TEL- BM or MN1-TEL+ BM transduced with pSRα-IRES-YFP all survived, whereas recipients of BM expressing MN1-TEL and IL-3 all died of fulminant disease within 2 weeks after transplantation (Figure 3B). Their peripheral WBC count was markedly increased (97.3 ± 28.0 × 109/L; n = 5) while they showed severe anemia (hemoglobin level 50 ± 8 g/L [5.0 ± 0.8 g/dL]; n = 5). BM of these mice consisted almost exclusively of GFP+/YFP+ myeloid cells (Figure 4A,C) with a differentiated morphology (Figure 4A right panel, arrow), which extensively infiltrated organs such as liver, spleen, and lung (Figure 4B arrows). The percentage of myeloblast in their BM was 7.1% ± 3.2% (n = 5), which failed to satisfy the criteria for AML (Bethesda proposal [Kogan et al17]). The disease was polyclonal as determined by Southern blot analysis with an IL3 probe (Figure 4D). We repeated the experiment with pSRα-SCF-IRES-YFP, but unlike IL-3, combined expression of MN1-TEL with SCF did not cause myeloid disease within 3 months after transplantation (data not shown).
Figure 3.

MN1-TEL causes aggressive myeloproliferative disease when coexpressed with IL-3 in vivo. (A) Retroviral transduction of MN1-TEL+ BM cells. BM cells were harvested from 6- to 8-week-old MN1-TEL/Mx1-Cre mice 2 weeks after the last injection with pI-pC (MN1-TEL+) or phosphate-buffered saline (PBS; MN1-TEL-). Sorted Lin- cells were transduced with pSRα–IL-3–IRES–YFP (IL-3) or pSRα-IRES-YFP (vector) retroviral vectors. Transduction efficiency was examined by FCM. A representative result of MN1-TEL+ cells transduced with IL-3 virus is shown. Uninfected WT BM cells were used as a negative control. Quadrants were determined using uninfected WT cells and GFP or YFP single-positive cells. (B) Survival analysis. Retrovirus transduced cells were transplanted into lethally irradiated syngeneic recipient mice (n = 10 in each group). Survival curves were generated according to the Kaplan-Meier method using StatView version 5.0 software (SAS Institute Inc, Cary, NC).
Figure 4.

Characteristics of the myeloproliferative disease induced by coexpression of MN1-TEL and IL-3. (A) Myeloproliferation in mice that received transplants of MN1-TEL+/IL-3 cells. (Left) Hematoxylin-Eosin (H-E) staining of BM; (middle) myeloperoxidase staining of BM; (right) M-G staining of BM cells. An arrow indicates a cell with differentiated morphology. (B) Aggressive organ infiltration by myeloid cells in mice that received transplants of MN1-TEL+/IL-3 bone marrow. H-E staining of liver (left), spleen (middle), and lung (right). Arrows indicate infiltrations of the myeloid cells. Images were obtained as previously described.12 (C) GFP and YFP expression in BM cells from mice that received transplants of MN1-TEL+/IL-3 cells. FCM analysis was performed as described in Figure 3A. (D) Clonality of MN1-TEL+/IL-3+ myeloproliferative disease (MPD). Genomic DNA of MPD BM cells was digested with BglII and hybridized with an Il3 probe. Control indicates untransduced BM; and G, germ line band.
MN1-TEL causes myeloid leukemia in combination with overexpression of HOXA9
Expression of MN1-TEL in multipotent progenitors led to development of T-lymphoid tumors and in some mice to expansion of MN1-TEL+ myeloid progenitors, whereas the fusion protein in humans is strictly associated with myeloid leukemia. Because MN1-TEL requires secondary mutations to cause full malignant transformation as shown in the accompanying paper (Kawagoe and Grosveld10), we hypothesized that mutations affecting myeloid differentiation might be important for development of MN1-TEL+ myeloid leukemia in our mice. One candidate gene was HOXA9, a member of the HOX transcription factors important for normal myeloid differentiation and proliferation.28 Elevated HOXA9 expression is frequently observed in human myeloid leukemia,29 and quantitative reverse transcription (RT)–PCR analysis of BM RNA of 2 MN1-TEL AML patients1 showed a 17- and 55-fold higher level of HOXA9 mRNA than in control BM, respectively (Figure 5A). To mimic the situation in humans we tested whether elevated HOXA9 expression cooperated with MN1-TEL in myeloid leukemogenesis. We transduced BM cells of MN1-TEL/Mx1-Cre mice treated with pI-pC or not with pSRα-HOXA9-IRES-YFP or control pSRα-IRES-YFP retrovirus and transplanted each into 10 lethally irradiated syngeneic recipients. The transduction efficiency of MN1-TEL+ and MN1-TEL- cells was identical (data not shown) as monitored by FCM (Figure 5B). Consistent with a previous report,30 HOXA9 alone induced leukemia 10 months after transplantation (Figure 5C), whereas 60% of mice that received transplants of MN1-TEL+/vector cells died of T-cell disease at 10 months after transplantation. In contrast, 9 of 10 mice that received transplants of MN1-TEL+/HOXA9+ cells died of AML within 3 to 6 months after transplantation, whereas the remaining animal died of a T-lymphoid tumor (Figure 5C). The peripheral WBC count was markedly increased (105.5 ± 35.4 × 109/L; n = 9) in the AML mice while they showed severe anemia (hemoglobin level 42 ± 5.3 g/L [4.2 ± 0.53 g/dL]; n = 9). The leukemic BM cells expressed HOXA9 and MN1-TEL (Figure 6A-B) and showed a typical morphology of myeloid blasts, featuring a large cleaved nucleus and scant cytoplasm (Figure 6C). The percentage of blasts in their total BM cells was 83.2% ± 3.8% (n = 5), which satisfied the criteria for AML (Bethesda proposal [Kogan et al17]). The cells only expressed the myeloid markers Mac1 and Gr1 (Figure 6D) and extensively invaded the spleen and liver (Figure 6E). Southern blot with a HOXA9 probe showed that the leukemias were oligoclonal to apparently monoclonal (Figure 6F). In addition, the disease was readily transplantable into syngeneic recipients (not shown). Taken together, these data suggested that enforced expression of HOXA9 cooperated with MN1-TEL in leukemogenesis and strongly skewed disease outcome toward a myeloid phenotype.
Figure 5.

HOXA9 and MN1-TEL cooperatively cause acute myeloid leukemia (AML) in mice. (A) Expression of HOXA9 in AML patient samples with MN1-TEL. A quantitative RT-PCR result is shown. NBM indicates control normal BM (n = 3); and Pt, patient samples (n = 2). (B) Retroviral transduction of MN1-TEL+ BM cells. Sorted MN1-TEL+ or MN1-TEL- Lin- BM cells were transduced with pSRα-HOXA9-IRES-YFP (HOXA9) or pSRα-IRES-YFP (vector) retrovirus. The FCM analysis of GFP/YFP expression in MN1-TEL+ BM cells transduced with HOXA9 virus is shown on the right. Control indicates uninfected WT BM cells. (C) Survival analysis. Retrovirally transduced BM cells (MN1-TEL+/HOXA9+, MN1-TEL+/Vector, MN1-TEL-/HOXA9+, MN1-TEL-/Vector) were transplanted into lethally irradiated syngeneic recipients (n = 10 in each group). Survival curves were generated by the Kaplan-Meier method. ○ indicates AML; and •, T-lymphoid leukemia.
Figure 6.
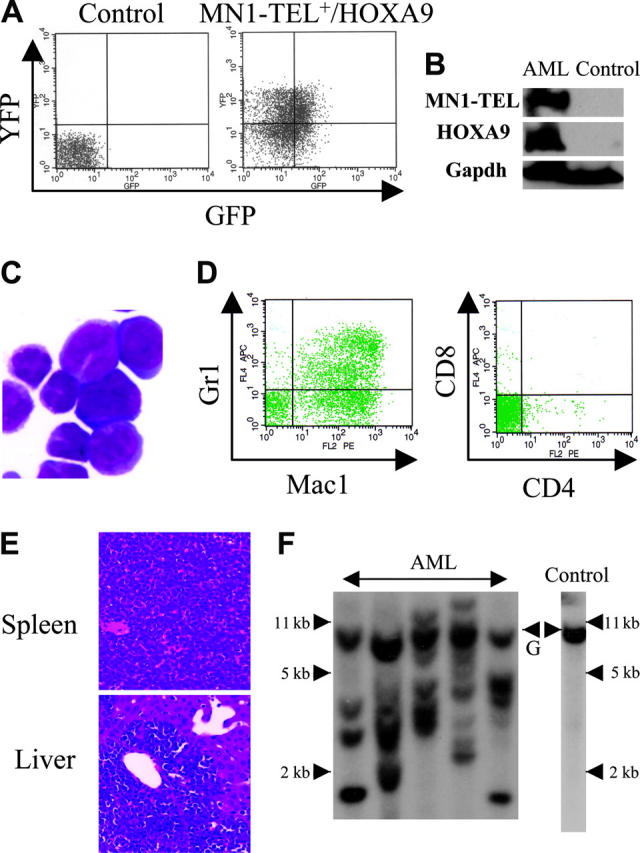
Characteristics of AML caused by MN1-TEL+/HOXA9+ BM cells. (A) GFP and YFP expression in AML cells. BM cells of mice that received leukemic MN1-TEL+/HOXA9 transplants were analyzed by FCM. (B) Expression of MN1-TEL and HOXA9 in AML cells. Western blot of BM cells was incubated with TEL C-terminal or FLAG (for HOXA9) antibody. Gapdh detection was used as a loading control. Control indicates untransduced normal BM. (C) M-G staining of AML cells. Images were obtained as previously described.12 (D) Surface marker analysis of AML cells by FCM. (E) Aggressive organ infiltration of AML cells. H-E staining of liver and spleen sections is shown. (F) Clonality of AML cells. Genomic DNA of AML cells was digested with BglII and hybridized with a HOXA9 probe. Control indicates untransduced BM; and G, germ line band.
Elevated expression of N-Myc in mouse and human MN1-TEL+ AML
Coexpression of MN1-TEL and HOXA9 induced AML in mice but it still took 3 to 6 months for the AML to develop, suggesting that additional cooperative factors are required to complete the leukemogenic process. To identify candidates for such factors, we compared gene expression profiles of MN1-TEL+/HOXA9+ AML BM cells (n = 5) with that of control uninduced MN1-TEL/Mx1-Cre BM cells (n = 3) using Affymetrix microarray analysis. We found 144 genes significantly upregulated (> 2-fold; P < .05) in the tumors and 82 genes significantly downregulated (> 2-fold; P < .05). The lists of highly upregulated or downregulated genes (> 5-fold; P < .05) is shown in Tables S1 and S2 (available on the Blood website; see the Supplemental Tables link at the top of the online article). The most upregulated oncogene in the tumors was N-Myc, which is known to be upregulated in human AML.31 More precise expression levels were determined by quantitative RT-PCR in mouse MN1-TEL+/HOXA9+ AML BM and in the 2 human MN1-TEL AML patient samples showing upregulated HOXA9 expression. This confirmed that the expression levels of N-Myc were highly elevated in 4 of 5 mouse and 2 of 2 human MN1-TEL+/HOXA9+ AML samples compared with normal BM controls (Figure 7). Affymetrix analysis also revealed that N-Myc was not up-regulated in GFP+/MN1-TEL+ nonleukemic BM cells obtained from MN1-TEL/Mx1-Cre mice 1 month after pI-pC injection (data not shown). Therefore, it is unlikely that N-Myc is a direct target of MN1-TEL in BM cells. In addition, quantitative RT-PCR analysis also showed that MN1-TEL+HOXA9 did not directly up-regulate N-Myc in nonleukemic BM cells (data not shown). Also, N-Myc was not up-regulated in BM of MN1-TEL/IL-3 mice compared with the control normal BM, suggesting that the N-Myc up-regulation was specific to the MN1-TEL+/HOXA9+ AML (data not shown). Although N-Myc is not a direct target of these transcription factors, its up-regulation could contribute to AML pathogenesis.
Figure 7.

Elevated expression of N-Myc in human and mouse MN1-TEL+/HOXA9+ AML. A quantitative RT-PCR result is shown. (Left) Expression of N-Myc mRNA in mouse MN1-TEL+/HOXA9+ AML cells (BM); (right) expression of N-MYC mRNA in 2 human MN1-TEL AML patient samples. NBM indicates control normal BM (n = 3); and Pt, patient samples (n = 2).
To address the mechanisms of the N-Myc upregulation in these tumors, we performed fluorescence in situ hybridization (FISH) for the N-Myc gene in the AML cells of MN1-TEL+/HOXA9+ mice, which highly express N-Myc. However, we found no amplification of N-Myc (data not shown). Since the N-Myc locus is a frequent target of retrovirus integration/activation in mouse lymphomas,32 we examined if the HOXA9 retroviral vector integrated into this locus by FISH. We did not find any colocalization of signals of the N-Myc gene and the vector in these tumors. Thus, these previously known mechanisms of N-Myc up-regulation were not found in these tumors. Thus another as yet unknown mechanism may activate N-Myc expression in MN1-TEL+/HOXA9+ AML.
Discussion
By using a conditional KI mouse model, we addressed the role of MN1-TEL in the etiology of myeloid leukemia. MN1-TEL markedly enhanced the in vitro proliferation and repopulation of myeloid progenitors derived from both MN1-TEL KI mice and MN1-TEL–KI ES cell–derived hematopoietic cells, indicating that MN1-TEL affected both adult and embryonic myeloid hematopoiesis, albeit in a strictly IL-3– and SCF–dependent manner. In our mice the MN1-TEL transgene is driven by the endogenous Aml1 promoter. Aml1 is expressed in multipotent progenitors as well as in committed myeloid progenitors and thymocytes.19 This Aml1-directed expression of MN1-TEL not only affected myeloid cells in KI mice but also resulted in an increased fraction of thymocytes in S-phase in vivo and partially inhibited thymocyte differentiation, as described in the accompanying paper (Kawagoe and Grosveld10). Thus, the effect of MN1-TEL is not lineage specific. Although MN1-TEL was expressed in myeloid progenitors and accelerated their growth in vitro, none of the MN1-TEL–expressing mice developed myeloid leukemia de novo. Nonetheless in 10% of MN1-TEL–expressing mice, we observed an increase in MN1-TEL+ BM cells with altered myeloid differentiation. Possibly, these mice were in a preleukemic phase because their clinical features, anemia and altered myeloid differentiation, were similar to those observed in MDS, a preleukemic disease associated with MN1-TEL expression in humans.1 This suggested that MN1-TEL contributes to myeloid leukemogenesis through growth stimulation of myeloid progenitors and partially blocking their differentiation but is insufficient to cause myeloid leukemia. Similar to MN1-TEL+ T-lymphoid leukemia, MN1-TEL appeared to require cooperative mutations to cause myeloid leukemia.
Since MN1-TEL is found in human myeloid leukemia, we wished to identify cofactors that would promote myeloid leukemogenesis in our mice. Survival of MN1-TEL+ myeloid cell lines was strictly dependent on SCF and IL-3. The up-regulation of IL-3 has been described in CD34+/BCR-ABL+ CML cells and in AML cells that can efficiently engraft nonobese diabetic–severe combined immunodeficiency (NOD/SCID) mice, indicating that autocrine expression of IL-3 plays a role in the etiology of myeloid leukemia.24,25 It is also known that constitutive activation of SCF/c-Kit signaling is associated with AML.26 Therefore, these cytokines were candidate cofactors for MN1-TEL–directed leukemogenesis. Overexpression of IL-3 but not SCF in MN1-TEL+ BM cells caused an aggressive myeloproliferative disease in recipient mice. This disease resembled human CML or MDS in which MN1-TEL was found. The disease developed very rapidly and was polyclonal, strongly suggesting that the combination of IL-3 signaling, which provides survival signals,33 and MN1-TEL expression is sufficient to cause hyperproliferation of myeloid cells but insufficient to block differentiation and induce full leukemic transformation. This result suggested that aberrant expression of genes affecting myeloid differentiation might cooperate with MN1-TEL to cause myeloid leukemia. One such gene, HOXA9, which is frequently expressed at elevated levels in human myeloid leukemia,29 was overexpressed in both MN1-TEL+ patients we tested. Hoxa9 was cloned from a retroviral integration site in BXH2 mice suffering from AML,34 and its overexpression in hematopoietic cells perturbs normal myeloid differentiation.30,35 The most striking evidence that HOXA9 plays a role in leukemia is that this gene is fused to the NUP98 gene by chromosomal translocation in human AML with t(7;11)(p15;p15).36 Overexpression of HOXA9 in MN1-TEL+ BM cells caused AML in 90% of the recipient mice whereas 10% developed T-lymphoid leukemia. This finding suggested that MN1-TEL causes AML in mice if the interaction of different genetic alterations (MN1-TEL and overexpression of HOXA9) collectively blocks normal myeloid differentiation. Obviously, this combination accelerated myeloid leukemogenesis, which in 90% of the mice outpaced the “default” development of T lymphoma. Although MN1-TEL stimulates growth of both lymphoid and myeloid progenitors, disease outcome is also determined by the mode of fusion gene expression. We base this conclusion on the observation that mice that received transplants of BM expressing MN1-TEL from a retroviral vector all developed AML rather than T lymphoma (Cintia Carella and G.C.G., unpublished observation, May 2005). One difference with our KI mice is that the level of retroviral-driven MN1-TEL expression is much higher than in the Aml1 KI setting. This higher level of expression might cause additional enlargement of the myeloid progenitor compartment and thus bias disease outcome toward a myeloid direction. Given the relatively low level of expression of MN1-TEL in patient samples, this might also explain the necessity for upregulation of HOXA9 to ensure sufficient expansion of the myeloid progenitor pool. It is also known that overexpression of HOXA9 blocks differentiation of mouse hematopoietic cells in vitro in the presence of IL-3 or granulocyte-macrophage CSF (GM-CSF), suggesting cooperation between the HOXA9 and IL-3 pathways in leukemic transformation.37,38 Therefore, we speculate that both these pathways cooperate with the MN1-TEL pathway in our MN1-TEL+/HOXA9+ AML. However, we did not find up-regulation of IL3 in the Affymetrix analysis of the tumor cells. Thus, although we did not find any evidence for dysregulation of the IL-3 signaling pathway in MN1-TEL+/HOXA9+ AML, it might be an interesting issue to address in future studies. Despite the cooperation between MN1-TEL and HOXA9, the latency of the disease was still long, suggesting that yet additional changes are necessary to provoke MN1-TEL–mediated myeloid leukemogenesis. The elevated expression of N-Myc in both mouse and human MN1-TEL+/HOXA9+ AML suggests that it is one of the candidate genes for the third oncogenic step. Indeed, N-Myc up-regulation has been found in other types of primary human AML, whereas it is relatively rare in lymphoid leukemia.31,39 Thus, MN1-TEL is expressed in multipotent progenitors as well as in committed myeloid and lymphoid progenitors in our mice, which mainly affects their proliferation, whereas disease outcome is determined by the type of secondary mutations. This is similar to the E2A-PBX140,41 or MLL-ENL42 fusion oncogenes, which can cause either myeloid or lymphoid leukemia in humans and/or mice.
Supplementary Material
Acknowledgments
We thank Dr James Downing for providing Aml1-IRES-GFP KI mice; Dr Richard Ashmun, Dr Ann-Marie Hamilton Easton, Edward Wingfield, and Sam Lucas for FCM analysis and cell sorting; Dr Kelli Boyd for pathologic analysis; and John Ellis for technical assistance. We also thank the Hartwell Center for microarray analysis and the Cancer Center Core Cytogenetic Laboratory for SKY and FISH analysis. We thank Charlette Hill for secretarial assistance.
Prepublished online as Blood First Edition Paper, August 16, 2005; DOI 10.1182/blood-2005-04-1679.
Supported by National Cancer Institute (NCI) grant CA72999-07 and the Cancer Center (CORE) support grant CA217G and by the American Lebanese Syrian Associated Charities (ALSAC) of St Jude Children's Research Hospital.
H.K. designed research, performed research, analyzed data, and wrote the paper; G.C.G. designed research and wrote the paper.
The online version of the article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 U.S.C. section 1734.
References
- 1.Buijs A, Sherr S, van Baal S, et al. Translocation (12;22) (p13;q11) in myeloproliferative disorders results in fusion of the ETS-like TEL gene on 12p13 to the MN1 gene on 22q11. Oncogene. 1995;10: 1511-1519. [PubMed] [Google Scholar]
- 2.Rubnitz JE, Pui CH, Downing JR. The role of TEL fusion genes in pediatric leukemias. Leukemia. 1999;13: 6-13. [DOI] [PubMed] [Google Scholar]
- 3.Sharrocks AD, Brown AL, Ling Y, Yates PR. The ETS-domain transcription factor family. Int J Biochem Cell Biol. 1997;29: 1371-1387. [DOI] [PubMed] [Google Scholar]
- 4.Golub TR, Barker GF, Lovett M, Gilliland DG. Fusion of PDGF receptor beta to a novel ets-like gene, tel, in chronic myelomonocytic leukemia with t(5;12) chromosomal translocation. Cell. 1994;77: 307-316. [DOI] [PubMed] [Google Scholar]
- 5.Rowley JD. The role of chromosome translocations in leukemogenesis. Semin Hematol. 1999;36: 59-72. [PubMed] [Google Scholar]
- 6.Hock H, Meade E, Medeiros S, et al. Tel/Etv6 is an essential and selective regulator of adult hematopoietic stem cell survival. Genes Dev. 2004;18: 2336-2341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lekanne Deprez RH, Riegman PH, Groen NA, et al. Cloning and characterization of MN1, a gene from chromosome 22q11, which is disrupted by a balanced translocation in a meningioma. Oncogene. 1995;10: 1521-1528. [PubMed] [Google Scholar]
- 8.van Wely KH, Molijn AC, Buijs A, et al. The MN1 oncoprotein synergizes with coactivators RAC3 and p300 in RAR-RXR-mediated transcription. Oncogene. 2003;22: 699-709. [DOI] [PubMed] [Google Scholar]
- 9.Buijs A, van Rompaey L, Molijn AC, et al. The MN1-TEL fusion protein, encoded by the translocation (12;22)(p13;q11) in myeloid leukemia, is a transcription factor with transforming activity. Mol Cell Biol. 2000;20: 9281-9293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kawagoe H, Grosveld GC. MN1-TEL myeloid oncoprotein expressed in multipotent progenitors perturbs both myeloid and lymphoid growth, and causes T-lymphoid tumors in mice. Blood. 2005;106: 4278-4286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Higuchi M, O'Brien D, Kumaravelu P, Lenny N, Yeoh EJ, Downing JR. Expression of a conditional AML1-ETO oncogene bypasses embryonic lethality and establishes a murine model of human t(8;21) acute myeloid leukemia. Cancer Cell. 2002;1: 63-74. [DOI] [PubMed] [Google Scholar]
- 12.Kawagoe H, Potter M, Ellis J, Grosveld GC. TEL2, an ETS factor expressed in human leukemia, regulates monocytic differentiation of U937 cells and blocks the inhibitory effect of TEL1 on ras-induced cellular transformation. Cancer Res. 2004;64: 6091-6100. [DOI] [PubMed] [Google Scholar]
- 13.Lee Y, Miller HL, Jensen P, et al. A molecular fingerprint for medulloblastoma. Cancer Res. 2003;63: 5428-5437. [PubMed] [Google Scholar]
- 14.Drabkin HA, Parsy C, Ferguson K, et al. Quantitative HOX expression in chromosomally defined subsets of acute myelogenous leukemia. Leukemia. 2002;16: 186-195. [DOI] [PubMed] [Google Scholar]
- 15.Ortman CL, Dittmar KA, Witte PL, Le PT. Molecular characterization of the mouse involuted thymus: aberrations in expression of transcription regulators in thymocyte and epithelial compartments. Int Immunol. 2002;14: 813-822. [DOI] [PubMed] [Google Scholar]
- 16.Kuhn R, Schwenk F, Aguet M, Rajewsky K. Inducible gene targeting in mice. Science. 1995;269: 1427-1429. [DOI] [PubMed] [Google Scholar]
- 17.Kogan SC, Ward JM, Anver MR, et al. Bethesda proposals for classification of nonlymphoid hematopoietic neoplasms in mice. Blood. 2002;100: 238-245. [DOI] [PubMed] [Google Scholar]
- 18.Fux C, Langer D, Kelm JM, Weber W, Fussenegger M. New-generation multicistronic expression platform: pTRIDENT vectors containing size-optimized IRES elements enable homing endonuclease-based cistron swapping into lentiviral expression vectors. Biotechnol Bioeng. 2004;86: 174-187. [DOI] [PubMed] [Google Scholar]
- 19.Lorsbach RB, Moore J, Ang SO, Sun W, Lenny N, Downing JR. Role of RUNX1 in adult hematopoiesis: analysis of RUNX1-IRES-GFP knock-in mice reveals differential lineage expression. Blood. 2004;103: 2522-2529. [DOI] [PubMed] [Google Scholar]
- 20.Lagasse E, Weissman IL. Flow cytometric identification of murine neutrophils and monocytes. J Immunol Methods. 1996;197: 139-150. [DOI] [PubMed] [Google Scholar]
- 21.Hamblin T. Immunologic abnormalities in myelodysplastic syndromes. Hematol Oncol Clin North Am. 1992;6: 571-586. [PubMed] [Google Scholar]
- 22.Srivannaboon K, Conley ME, Coustan-Smith E, Wang WC. Hypogammaglobulinemia and reduced numbers of B-cells in children with myelodysplastic syndrome. J Pediatr Hematol Oncol. 2001;23: 122-125. [DOI] [PubMed] [Google Scholar]
- 23.Biermann H, Pietz B, Dreier R, Schmid KW, Sorg C, Sunderkotter C. Murine leukocytes with ring-shaped nuclei include granulocytes, monocytes, and their precursors. J Leukoc Biol. 1999;65: 217-231. [DOI] [PubMed] [Google Scholar]
- 24.Ailles LE, Gerhard B, Kawagoe H, Hogge DE. Growth characteristics of acute myelogenous leukemia progenitors that initiate malignant hematopoiesis in nonobese diabetic/severe combined immunodeficient mice. Blood. 1999;94: 1761-1772. [PubMed] [Google Scholar]
- 25.Jiang X, Lopez A, Holyoake T, Eaves A, Eaves C. Autocrine production and action of IL-3 and granulocyte colony-stimulating factor in chronic myeloid leukemia. Proc Natl Acad Sci U S A. 1999;96: 12804-12809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Beghini A, Ripamonti CB, Cairoli R, et al. KIT activating mutations: incidence in adult and pediatric acute myeloid leukemia, and identification of an internal tandem duplication. Haematologica. 2004;89: 920-925. [PubMed] [Google Scholar]
- 27.Perkins AC, Cory S. Conditional immortalization of mouse myelomonocytic, megakaryocytic and mast cell progenitors by the Hox-2.4 homeobox gene. EMBO J. 1993;12: 3835-3846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Buske C, Humphries RK. Homeobox genes in leukemogenesis. Int J Hematol. 2000;71: 301-308. [PubMed] [Google Scholar]
- 29.Golub TR, Slonim DK, Tamayo P, et al. Molecular classification of cancer: class discovery and class prediction by gene expression monitoring. Science. 1999;286: 531-537. [DOI] [PubMed] [Google Scholar]
- 30.Kroon E, Krosl J, Thorsteinsdottir U, Baban S, Buchberg AM, Sauvageau G. Hoxa9 transforms primary bone marrow cells through specific collaboration with Meis1a but not Pbx1b. EMBO J. 1998;17: 3714-3725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hirvonen H, Hukkanen V, Salmi TT, Pelliniemi TT, Alitalo R. L-myc and N-myc in hematopoietic malignancies. Leuk Lymphoma. 1993;11: 197-205. [DOI] [PubMed] [Google Scholar]
- 32.van Lohuizen M, Breuer M, Berns A. N-myc is frequently activated by proviral insertion in MuLV-induced T cell lymphomas. EMBO J. 1989;8: 133-136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Packham G, White EL, Eischen CM, et al. Selective regulation of Bcl-XL by a Jak kinase-dependent pathway is bypassed in murine hematopoietic malignancies. Genes Dev. 1998;12: 2475-2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nakamura T, Largaespada DA, Shaughnessy JD Jr, Jenkins NA, Copeland NG. Cooperative activation of Hoxa and Pbx1-related genes in murine myeloid leukaemias. Nat Genet. 1996;12: 149-153. [DOI] [PubMed] [Google Scholar]
- 35.Fujino T, Yamazaki Y, Largaespada DA, et al. Inhibition of myeloid differentiation by Hoxa9, Hoxb8, and Meis homeobox genes. Exp Hematol. 2001;29: 856-863. [DOI] [PubMed] [Google Scholar]
- 36.Borrow J, Shearman AM, Stanton VP Jr, et al. The t(7;11)(p15;p15) translocation in acute myeloid leukaemia fuses the genes for nucleoporin NUP98 and class I homeoprotein HOXA9. Nat Genet. 1996;12: 159-167. [DOI] [PubMed] [Google Scholar]
- 37.Calvo KR, Sykes DB, Pasillas M, Kamps MP. Hoxa9 immortalizes a granulocyte-macrophage colony-stimulating factor-dependent promyelocyte capable of biphenotypic differentiation to neutrophils or macrophages, independent of enforced meis expression. Mol Cell Biol. 2000;20: 3274-3285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schnabel CA, Jacobs Y, Cleary ML. HoxA9-mediated immortalization of myeloid progenitors requires functional interactions with TALE cofactors Pbx and Meis. Oncogene. 2000;19: 608-616. [DOI] [PubMed] [Google Scholar]
- 39.Hirvonen H, Hukkanen V, Salmi TT, et al. Expression of L-myc and N-myc proto-oncogenes in human leukemias and leukemia cell lines. Blood. 1991;78: 3012-3020. [PubMed] [Google Scholar]
- 40.Troussard X, Rimokh R, Valensi F, et al. Heterogeneity of t(1;19)(q23;p13) acute leukaemias: French Haematological Cytology Group. Br J Haematol. 1995;89: 516-526. [DOI] [PubMed] [Google Scholar]
- 41.Sykes DB, Kamps MP. E2a/Pbx1 induces the rapid proliferation of stem cell factor-dependent murine pro-T cells that cause acute T-lymphoid or myeloid leukemias in mice. Mol Cell Biol. 2004;24: 1256-1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rubnitz JE, Behm FG, Curcio-Brint AM, et al. Molecular analysis of t(11;19) breakpoints in childhood acute leukemias. Blood. 1996;87: 4804-4808. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.