

Abstract
Fanconi anemia (FA) is a chromosomal instability disorder characterized by progressive bone marrow failure. Experimental evidence suggests that enhanced oxidant and myelosuppressive cytokine-mediated apoptosis of hematopoietic stem and progenitor cells contributes to the pathogenesis of marrow failure in FA. However, the molecular mechanisms responsible for the apoptotic phenotype in hematopoietic cells are incompletely understood. Recent data in Fancc-/- murine embryonic fibroblasts (MEFs) implicate increased oxidant-induced apoptotic signaling through the redox-dependent protein, apoptosis signal-regulating kinase 1 (Ask1). Here, we examined whether altered Ask1 signaling participated in the proapoptotic phenotype of primary Fancc-/- MEFs and hematopoietic progenitors treated with the myelosuppressive cytokine tumor necrosis factor-α (TNF-α). Our data indicate that TNF-α induces hyperactivation of Ask1 and the downstream effector p38 in Fancc-/- MEFs. In addition,Ask1 inactivation in Fancc-/- MEFs and hematopoietic progenitors restored survival to wild-type (WT) levels in the presence of TNF-α. Furthermore, targeting the Ask1 pathway by using either antioxidants or a p38 inhibitor protected Fancc-/- MEFs and c-kit+ cells from TNF-α-induced apoptosis. Collectively, these data argue that the predisposition of Fancc-/- hematopoietic progenitors to apoptosis is mediated in part through altered redox regulation and Ask1 hyperactivation.
Introduction
Fanconi anemia (FA) is a genetic disorder typified by bone marrow (BM) hypoplasia and increased cancer risk, with progression of marrow failure considered the major cause of death.1-5 Significant data support the idea that enhanced apoptosis of hematopoietic stem and progenitor cells is a major contributing factor in the pathogenesis of marrow failure in FA. In fact, FA hematopoietic progenitor cells are hypersensitive to multiple extracellular stimuli normally encountered in vivo, including oxidants and myelosuppressive cytokines.6-10 Interestingly, altered responsiveness to myelosuppressive cytokines, such as interferon-γ (IFN-γ) and tumor necrosis factor-α (TNF-α), also has a critical role in the pathogenesis of acquired aplastic anemia in which an immune-mediated increase in IFN-γ and TNF-α production results in apoptotic loss of hematopoietic stem and progenitor cells.11 In previous studies using murine and human progenitors deficient in the FA type C protein (FANCC), others6 and we7 have shown enhanced IFN-γ- and TNF-α-mediated apoptosis. In addition, recent studies showed a similar increase in sensitivity of human FANCA-deficient progenitors,12 substantiating a broader role for myelosuppressive cytokine hypersensitivity across FA complementation types. However, the molecular mechanisms underlying myelosuppressive cytokine hypersensitivity in FA progenitors remain incompletely understood.
Unlike most FA proteins (FANCA, FANCB, FANCD1/BRCA2, FANCD2, FANCE, FANCF, FANCG, and FANCL), FANCC is primarily localized in the cytoplasm.13,14 Additionally, protein-protein interaction studies support a cytoplasmic function for FANCC in regulating cytokine signaling pathways and intracellular redox metabolism.15-18 In this regard, recent studies from our laboratory showed that loss of Fancc in murine embryonic fibroblasts (MEFs) results in enhanced activity of a redox-dependent protein, apoptosis signal-regulating kinase 1 (Ask1), after oxidant stress.9 In addition, we demonstrated that increased oxidant-induced apoptosis in Fancc-/- MEFs is inhibited by blocking Ask1 activity, supporting the notion that Fancc-/- cells exhibit an altered redox state predisposing to Ask1-mediated apoptosis. These data are consistent with multiple previous studies suggesting that FA cells are in a pro-oxidant state (reviewed in Pagano and Youssoufian19).
Ask1 is a unique mitogen-activated protein kinase (MAPK) that is directly inhibited by binding to redox-sensitive proteins such as thioredoxin, glutathione S-transferase, and glutaredoxin, which sequester Ask1 in an inactive conformation.20-22 Oxidant stress is sensed by these redox active proteins through disulfide bond formation, resulting in dissociation from Ask1. Ask1 subsequently initiates an apoptotic program through its serine-threonine kinase activity.23 Interestingly, reactive oxygen species (ROS) generated after exposure to diverse stimuli, including growth factor withdrawal, H2O2, and TNF-α, activate Ask1 and induce apoptosis in a variety of cell types. However, most studies have been conducted in immortalized cell lines, and limited studies have been conducted in hematopoietic cells. To investigate the in vivo function of Ask1, Tobiume et al24 generated Ask1 knock-out (Ask1) mice and demonstrated that Ask1-/- MEFs are resistant to TNF-α and H2O2-induced apoptosis, confirming previous work in cell lines. Given these data together, with our previous observations demonstrating that oxidant hypersensitivity of Fancc-/- MEFs require Ask1, we questioned whether the molecular mechanism responsible for enhanced TNF-α-induced apoptosis of Fancc-/- MEFs and hematopoietic progenitors was mediated through Ask1.
Materials and methods
Mice
Fancc+/- mice in a C57Bl/6J genetic background were bred to generate Fancc-/- and wild-type (WT) mice (6-12 weeks of age) for hematopoietic progenitor assays and timed embryos for MEF cell lines, as previously described.9,25 To obtain mice with a genetic inactivation of both Fancc and Ask1, Fancc+/- mice were bred with Ask1+/- mice24 in a C57Bl/6J background. Multiple F1 double-heterozygous pairs (Fancc+/-; Ask1+/-) were bred to generate mice from the 4 F2 experimental genotypes: Fancc+/+; Ask1+/+ (WT), Fancc+/+; Ask1-/- (Ask1-/-), Fancc-/-; Ask1+/+ (Fancc-/-), and Fancc-/-; Ask1-/-. Fanca+/-) mice26 were generously provided by Dr Freerk Arwert (Vrije University, Amsterdam, The Netherlands), and these mice were backcrossed for 10 generations into a C57Bl/6J background. Syngeneic Fanca+/- mice were bred to generate Fanca-/- and WT mice for apoptosis studies. All studies were approved by the Indiana University Laboratory Animal Research Center.
Apoptosis assays
MEFs were maintained as previously described,9 and c-kit+ cells were sorted by fluorescence cytometry using a BD FACStar cytometer (Becton Dickinson, San Jose, CA) as previously described.7 All studies using MEFs were conducted in at least 2 to 3 different cell lines per genotype, and only MEFs that were less than passage 5 were used. WT and Fancc-/- cells were pretreated with 20 μM selenomethionine (SeMet; Sigma, St Louis, MO) overnight, 4 mM N-acetylcysteine (NAC; Sigma) for 1 hour, 10 μM SB203580 overnight, 10 μM PD098059 overnight, or 10 μM SB202474 (Calbiochem, San Diego, CA) overnight before culturing with TNF-α (Peprotech, Rocky Hill, NJ) for 24 hours. Apoptosis was then examined by the terminal deoxynucleotidyl transferase-mediated digoxigenin-dUTP nick-end labeling (TUNEL) assay exactly according to the manufacturer's recommendations (Roche, Indianapolis, IN).7,27 At least 100 cells were scored per condition per experiment.
Retroviral constructs and transduction
GP + E86 packaging cell lines were previously developed for the control retrovirus encoding enhanced green fluorescence protein (EFGP) only (MIEG3) and the retrovirus containing the Ask1 dominant-negative cDNA and EGFP (MIEG3-Ask1-K709M).9 In some experiments, a transient transfection system was used to obtain high-titer retroviral supernatants. For these studies, the MigR1 retrovirus28 was used (generously provided by Dr Warren Pear, University of Pennsylvania, Philadelphia, PA). The Ask1-K709M cDNA was subcloned into MigR1 using standard techniques. Retroviral supernatants were collected, and early-passage (P0-P1) MEFs were transduced in the presence of polybrene, as previously described.9,25 Transduced MEFs were treated with 50 ng/mL TNF-α, and apoptosis was examined by TUNEL, as described, using a TMR-red fluorochrome (Roche). The percentage of apoptotic cells was calculated by dividing EGFP+TMR (tetramethyl-rhodamine)-red+ cells by total EGFP+ cells for each condition. At least 100 cells were scored per condition per experiment.
Ask1 in vitro kinase assay
Ask1 in vitro kinase assays were conducted as previously described.9 Briefly, WT and Fancc-/- MEFs were untreated or were treated with 50 ng/mL TNF-α for 5 minutes, washed twice with cold phosphate-buffered saline (PBS) containing 1 mM sodium orthovanadate, and lysed in nonionic lysis buffer. Protein concentrations were determined using the bicinchoninic acid assay (BCA) (Pierce Chemical, Rockford, IL). Ask1 immunoprecipitations were conducted using protein A Sepharose beads (Amersham, Piscataway, NJ) and anti-total-Ask1 antibody (Cell Signaling, Beverly, MA). Immunobeads were subjected to an in vitro kinase reaction using either myelin basic protein (MBP; Sigma) or MKK4 (Upstate Biotechnologies, Lake Placid, NY) as substrate for Ask1. After kinase reactions were terminated by the addition of sample buffer, protein samples were separated on a 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) gel (Invitrogen, Grand Island, NY), transferred to a nitrocellulose membrane, and subjected to autoradiography. Densitometric analysis of individual bands was conducted using National Institutes of Health (NIH) Image software (NIH, Bethesda, MD) to quantitate arbitrary density units. Mean arbitrary densitometry units were calculated from the 5 experiments conducted to demonstrate statistical significance. Equivalent protein loading was demonstrated by Ask1 immunoblotting.
p38 Western blotting
MEFs either were untreated or were treated with 50 ng/mL TNF-α for 45 minutes, washed with cold PBS containing 1 mM sodium orthovanadate, and lysed in nonionic lysis buffer. Whole cell extracts were separated on a 12% SDS-PAGE gel and transferred onto a nitrocellulose membrane. For immunodetection of active p38, a phospho-p38 (Thr180/Tyr182)-specific antibody was used at a 1:500 dilution (Cell Signaling), and the secondary antibody, anti-rabbit horseradish peroxidase (HRP) was used at a 1:3000 dilution before visualization by chemiluminescence. Densitometric analysis of individual bands was conducted using NIH Image software to quantitate arbitrary density units. Mean arbitrary densitometry units were calculated from the 5 experiments conducted to demonstrate statistical significance. To document equal protein loading, the membranes were stripped and reprobed with a p38 MAPK antibody that recognizes total p38 (Cell Signaling).
Hematopoietic progenitor assays
WT, Ask1-/-, Fancc-/-, and Fancc-/-; Ask1-/- low-density mononuclear cells were prepared as previously described7,29 and were resuspended in Iscove modified Dulbecco medium (IMDM; Gibco BRL, Gaithersburg, MD) supplemented with 20% fetal calf serum (FCS; BioWhittaker, Walkersville, MD). BM cells were plated in methylcellulose colony assays (3 × 104 cells/mL) in the presence of increasing TNF-α concentrations, as described previously.7 All conditions were conducted in triplicate. For inhibitor studies, either 10 μM SB203580 or vehicle control was added to progenitor cultures. Total colonies, including granulocyte macrophage erythroid-megakaryocyte colony-forming unit (CFU-GEMM), erythroid blast-forming unit (BFU-E), and granulocyte-macrophage colony-forming unit (CFU-GM), were scored 7 days after plating. Data from progenitor assays are presented as percentage of control colony formation, as previously described.7,25 Percentage of control colony formation was calculated by dividing colonies scored in each treatment by total colonies in the control untreated condition and multiplying by 100. Baseline colony formations of WT, Ask1-/-, Fancc-/-, and Fancc-/-; Ask1-/- were similar among genotypes. Further, the addition of SB203580 did not change baseline colony formation of WT or Fancc-/- progenitors.
Statistical analyses
For all data shown, an unpaired Student t test was conducted to evaluate for differences among treatment groups, and P ≤ .05 was considered significant. Data shown are mean ± SD.
Results
Enhanced TNF-α-induced apoptosis of Fancc-/- MEFs is mediated through the redox-regulated protein Ask1
TNF-α-induced apoptotic signaling is complex and includes ROS-dependent and -independent mechanisms. Recently, we demonstrated that Fancc-/- MEFs and progenitors exhibit increased H2O2-induced apoptosis.9 Thus, we questioned whether the hypersensitivity of Fancc-/- MEFs to TNF-α was caused by enhanced apoptotic signaling through the ROS-dependent Ask1 pathway. To examine whether enhanced TNF-α-mediated apoptosis of Fancc-/- MEFs occurs through an ROS-dependent mechanism, WT and Fancc-/- MEFs were pretreated with either 20 μM SeMet or 4 mM NAC before culturing with 50 ng/mL TNF-α for 24 hours. Apoptosis was then examined using a TUNEL assay, as previously described.9 Consistent with previous data, Fancc-/- MEFs exhibit increased TNF-α-mediated apoptosis compared with WT (Figure 1A). Furthermore, both antioxidants reduced TNF-α-mediated apoptosis of Fancc-/- MEFs to WT levels, suggesting that enhanced TNF-α-induced apoptosis in Fancc-/- MEFs is ROS dependent. Control WT and Fancc-/- MEF cultures containing each antioxidant alone did not change baseline apoptosis in either genotype (data not shown).
Figure 1.

TNF-α hypersensitivity of Fancc-/- MEFs is Ask1 dependent. (A) Antioxidants and MEF apoptosis assays. WT and Fancc-/- MEFs were grown in normal conditions, pretreated with 20 μM SeMet, or pretreated with 4 mM NAC before 50 ng/mL TNF-α treatment. Apoptosis was analyzed 24 hours after TNF-α treatment using TUNEL assay. For each experiment, at least 100 cells were evaluated (Hoechst+) to determine the percentage of apoptotic cells (TUNEL+) in each condition (n = 3; *P ≤ .002). (B) Ask1 in vitro kinase assays. Ask1 kinase activity was evaluated in WT and Fancc-/- MEFs after treatment with 50 ng/mL TNF-α. Ask1 immunoprecipitations were subjected to an in vitro kinase reaction, as described. Autoradiography of Ask1 kinase assays, densitometry analyses, and Western blots for total Ask1 are shown. Data in the left panel are representative of 5 independent experiments. The graph on the right depicts the mean arbitrary density units from the 5 experiments (*P < .001). (C) Dominant-negative Ask1 studies. WT and Fancc-/- MEFs were transduced with a retrovirus encoding a catalytically inactive, dominant-negative Ask1 (Ask1-K709M) or vector control. Transduced MEFs were treated with 50 ng/mL TNF-α for 24 hours before evaluating apoptosis by the TUNEL assay (TMR-red+). The percentage of apoptotic cells was calculated by dividing EGFP+TMR-red+ cells by total EGFP+ cells for each condition. At least 100 cells were scored per condition per experiment (n = 3; *P < .01) compared with vector control + TNF-α. Data shown represent the mean of multiple experiments, and error bars represent the SD.
We next examined whether aberrant Ask1 activation was involved in TNF-α hypersensitivity of Fancc-/- MEFs. Ask1 was immunoprecipitated from WT and Fancc-/- MEFs that were untreated or were treated with TNF-α and then used for Ask1 in vitro kinase assays. Consistently, Fancc-/- MEFs treated with TNF-α displayed an approximately 2-fold increase in Ask1 activity compared with WT (Figure 1B). To determine whether the mechanism for enhanced TNF-α-induced apoptosis of Fancc-/- MEFs was Ask1 dependent, WT and Fancc-/- MEFs were transduced with a retrovirus that coexpresses a catalytically inactive, dominant-negative Ask1 cDNA (Ask1-K709M)23 and EGFP or a retroviral vector control. Transduced MEFs were untreated or were treated with TNF-α and evaluated for apoptosis. These data revealed that Fancc-/- MEFs expressing Ask1-K709M exhibited significantly less apoptosis after TNF-α treatment compared with Fancc-/- MEFs transduced with vector control. Moreover, the levels of apoptosis in Fancc-/- MEFs expressing the dominant-negative Ask1 cDNA were comparable with those of WT controls (Figure 1C). Collectively, these data show that the predisposition of Fancc-/- MEFs to TNF-α-induced apoptosis is dependent on ROS generation and Ask1 activation.
Inhibition of p38 reduces TNF-α-induced apoptosis of Fancc-/- MEFs to WT levels
After ROS stress, Ask1 initiates an apoptotic cascade by phosphorylating MAPK, which in turn activates stress-activated protein kinases such as p38.30 Verma et al31 previously demonstrated a central role for p38 in regulating myelosuppressive cytokine growth inhibition of clonogenic hematopoietic progenitors. Thus, we questioned whether p38 was a downstream effector involved in Fancc-/- MEF hypersensitivity to TNF-α. WT and Fancc-/- MEFs treated with TNF-α were evaluated for p38 activation by Western blot analysis using an antibody specific for phosphorylated or active p38. These studies revealed a marked elevation of p38 activity in Fancc-/- MEFs treated with TNF-α compared with WT controls (Figure 2A). To examine whether the enhanced apoptosis of Fancc-/- MEFs treated with TNF-α was p38 dependent, we pretreated WT and Fancc-/- MEFs with a p38 inhibitor, SB203580, before TNF-α exposure. These data showed that p38 inhibition in Fancc-/- MEFs reduced TNF-α-induced apoptosis to levels near those of WT cells (WT + TNF-α compared with Fancc-/- + SB203580 + TNF-α; P < .07; Figure 2B). To ensure that this observation was specific to p38 inhibition, we conducted control studies with PD098059, an MEK inhibitor, and SB202474, a structural analog to SB203580 that does not inhibit p38 activity. These data showed that neither the PD098059 nor the SB202474 compound significantly inhibited TNF-α-mediated apoptosis in Fancc-/- MEFs (Figure 2C), suggesting that the effect detected with SB203580 is specific to p38 inhibition (Figure 2B). Collectively, these data showed that the Ask1/p38 pathway is aberrantly activated in Fancc-/- MEFs exposed to TNF-α. Moreover, these data suggest that enhanced TNF-α-induced apoptosis of Fancc-/- MEFs requires Ask1 and p38 activation.
Figure 2.

TNF-α hypersensitivity of Fancc-/- MEFs is p38 dependent. (A) p38 activity. WT and Fancc-/- MEFs were treated with 50 ng/mL TNF-α before assaying p38 activity by Western blot analysis using a phospho-specific p38 antibody. Western blots for phosphorylated p38 and total p38 are shown, as is densitometry analysis of phosphorylated p38. Data in the left panel are representative of 5 independent experiments. The graph on the right depicts the mean arbitrary density units from the 5 experiments (*P < .001). (B) p38 inhibitor and MEF apoptosis assays. WT and Fancc-/- MEFs were grown in normal conditions or were pretreated with the p38 inhibitor SB203580 before 50 ng/mL TNF-α treatment. Apoptosis was analyzed by TUNEL as previously described (n = 6; *P ≤ .001). (C) Control inhibitors and MEF apoptosis assays. WT and Fancc-/- MEFs were grown in normal conditions or pretreated with the SB203580 structural analog SB202474 or the MEK inhibitor PD098059 before 50 ng/mL TNF-α exposure for 24 hours. Apoptosis was analyzed by TUNEL, as previously described (n = 3; *P ≤ .003), compared with Fancc-/- control. Data shown represent the mean of multiple experiments, and error bars represent the SD.
Loss of Ask1 or inhibition of p38 protects Fancc-/- c-kit+ cells from TNF-α-induced apoptosis and corrects Fancc-/- hematopoietic progenitor hypersensitivity to TNF-α
Our data in MEFs elucidate a mechanistic role for aberrant redox signaling in the predisposition of Fancc-/- cells toward TNF-α-induced apoptosis. These observations in the murine system are encouraging and suggest a previously unrecognized molecular mechanism in the pathogenesis of bone marrow failure in FA. However, whether the proapoptotic phenotype of primary Fancc-/- hematopoietic progenitors exposed to TNF-α is mediated through an Ask1/p38 pathway remains unknown. This point was critical to address because a function for Ask1 in hematopoietic progenitors has not been reported and has provided the basis for our next set of studies. Initially, studies were conducted to verify that TNF-α-induced apoptosis in a phenotypically defined cell population enriched for hematopoietic progenitors (c-kit+ cells) was dependent on ROS production. For these studies, WT and Fancc-/- c-kit+ cells were purified from primary BM cells using fluorescence cytometry. Sorted c-kit+ cells were untreated or were pretreated with SeMet before culturing with 10 ng/mL TNF-α for 24 hours and were evaluated for apoptosis as previously described.7 Similar to Fancc-/- MEFs (Figure 1A), SeMet pretreatment of Fancc-/- c-kit+ cells reduced TNF-α-induced apoptosis to WT levels (Figure 3A), suggesting that enhanced TNF-α-mediated apoptosis in Fancc-/- hematopoietic progenitors is dependent on ROS generation. In addition, these data suggest that the TNF-α hypersensitivity of Fancc-/- c-kit+ cells may be attributed to alterations in redox-regulated apoptotic signaling, supporting further evaluation of Ask1 function in Fancc-/- progenitors.
Figure 3.

TNF-α hypersensitivity of Fancc-/- hematopoietic progenitors is dependent on Ask1 and p38. (A) TNF-α-induced apoptosis in c-kit+ cells. WT and Fancc-/- c-kit+ cells were pretreated with SeMet or were cultured in normal conditions before 24-hour exposure to 10 ng/mL TNF-α. Cytospins of cells from each experimental group were made before apoptosis was analyzed using a TUNEL assay (n = 3; *P ≤ .006). (B) TNF-α sensitivity of hematopoietic progenitors. WT, Fancc-/-, and Fancc-/-; Ask1-/- low-density BM cells were cultured in colony assays with increasing concentrations of TNF-α (n = 4 mice/genotype; *P ≤ .05). (C) TNF-α sensitivity of hematopoietic progenitors. WT and Ask1-/- low-density BM cells were cultured in colony assays with increasing concentrations of TNF-α (n = 5 mice/genotype). (D) p38 inhibitor and progenitor assays. WT and Fancc-/- low-density BM cells were cultured in colony assays with SB203580 or vehicle control and increasing concentrations of TNF-α (n = 6 mice/genotype; *P ≤ .001). All data points shown represent the mean of multiple experiments, and error bars represent SD.
We used a genetic approach to determine whether enhanced TNF-α sensitivity of Fancc-/- clonogenic progenitors requires Ask1. For these studies, Fancc+/- mice in a C57BL/6J strain were bred with Ask1+/- mice in the same genetic background to generate F1 Fancc+/-; Ask1+/- breeders. Multiple F1 breeders were intercrossed to obtain the F2 experimental groups Fancc+/+; Ask1+/+ (WT), Fancc+/+; Ask1-/- (Ask1-/-), Fancc-/-; Ask1+/+ (Fancc-/-), and Fancc-/-; Ask1-/-. BM cells from mice in each experimental group were isolated, and hematopoietic progenitor assays were conducted. As in our previous studies,7 Fancc-/- hematopoietic progenitor colony formation was significantly inhibited by increasing TNF-α concentrations compared with WT controls. However, genetic inactivation of Ask1 in Fancc-/- progenitors (Fancc-/-; Ask1-/-) completely restored TNF-α sensitivity to WT levels (Figure 3B). Importantly, control experiments showed that Ask1-/- and WT progenitors had equivalent responsiveness to TNF-α (Figure 3C). Collectively, these data demonstrate that the molecular mechanism responsible for enhanced TNF-α-induced apoptosis of Fancc-/- MEFs and progenitors is dependent on Ask1.
Because the stress-activated protein kinase p38 is a downstream effector of Ask123 and because of the importance of p38 in mediating TNF-α growth inhibition of human hematopoietic progenitors,31 we next examined whether p38 was required for TNF-α hypersensitivity of Fancc-/- hematopoietic progenitors. To answer this question, we conducted progenitor assays in the presence of the p38 inhibitor SB203580 or vehicle control with increasing TNF-α doses. Similar to our data using Fancc-/- MEFs (Figure 2B), TNF-α sensitivity of Fancc-/- progenitors was restored to WT levels in the presence of SB203580 (Figure 3D), suggesting that TNF-α hypersensitivity of Fancc-/- progenitors is mediated through p38. In addition, SB203580 reduced TNF-α-induced apoptosis of Fancc-/- c-kit+ cells to WT levels (data not shown). Collectively, these data suggest that the predisposition of Fancc-/- MEFs and hematopoietic progenitors to TNF-α-mediated apoptosis is dependent on the redox-regulated protein Ask1 and the downstream effector p38.
SeMet and SB203580 ameliorate the apoptotic predisposition of Fanca-/- c-kit+ cells treated with TNF-α
Recent studies in human FANCA-deficient hematopoietic progenitors demonstrate a dramatic reduction in colony formation after combined treatment with both IFN-γ and TNF-α compared with progenitors from a healthy donor.12 Given our data showing that SeMet and SB203580 protect Fancc-/- c-kit+ cells from TNF-α-induced apoptosis, we tested whether a similar survival enhancement could be demonstrated in Fanca-/- c-kit+ cells. Importantly, Fanca-/- c-kit+ cells treated with TNF-α exhibited an increase in apoptosis compared with WT c-kit+ cells that were similarly treated (Figure 4A-B). Moreover, both SeMet and SB203580 protected Fanca-/- c-kit+ cells from TNF-α-induced apoptosis. Collectively, these data in the murine system implicate a role for myelosuppressive cytokine hypersensitivity in FA-A and FA-C complementation types and suggest that antioxidants and p38 inhibitors may enhance the survival of hematopoietic progenitor cells from multiple FA complementation types.
Figure 4.
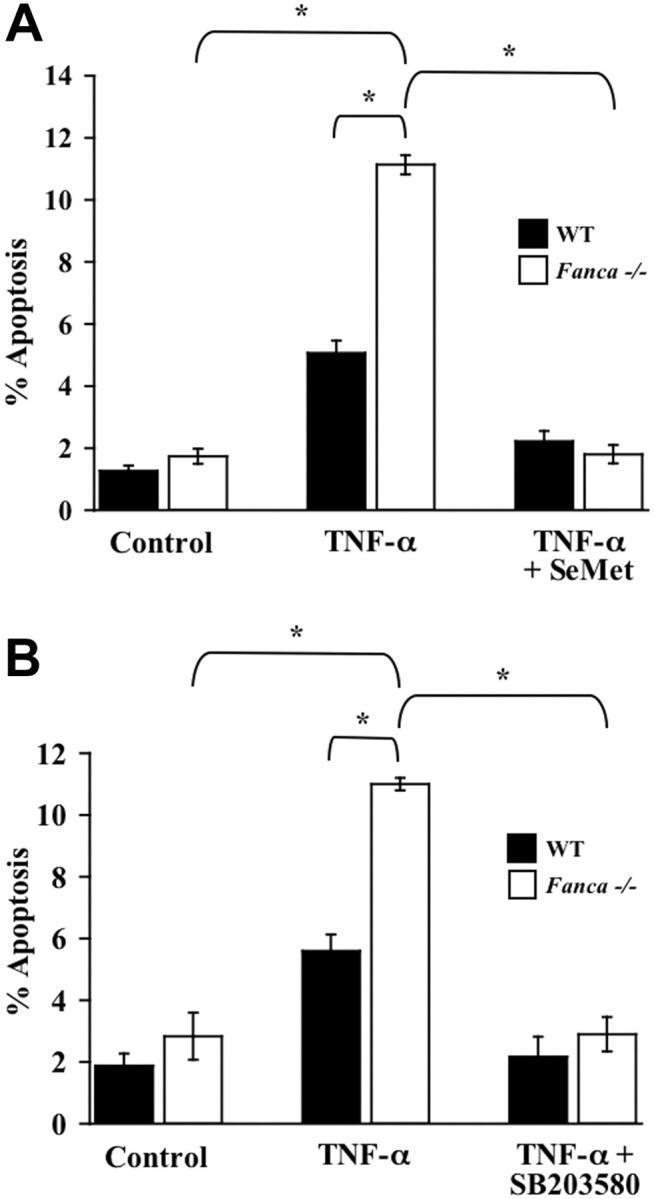
Enhanced TNF-α-mediated apoptosis of Fanca-/- c-kit+ cells is blocked by antioxidants and p38 inhibition. (A) Antioxidant and TNF-α-induced apoptosis of c-kit+ cells. WT and Fanca-/- c-kit+ cells were pretreated with SeMet or were cultured in normal conditions before 24-hour exposure to 10 ng/mL TNF-α. Cytospins of cells from each experimental group were made before apoptosis was analyzed using a TUNEL assay (n = 3; *P ≤ .001). (B) p38 inhibitor and TNF-α-induced apoptosis of c-kit+ cells. WT and Fanca-/- c-kit+ cells were pretreated with SB203580 or were cultured in normal conditions before 24-hour treatment with 10 ng/mL TNF-α. Apoptosis was analyzed as described (n = 3; *P ≤ .003). Data shown represent the mean of multiple experiments, and error bars represent the SD.
Discussion
Myelosuppressive cytokines have a critical role in the pathogenesis of acquired aplastic anemia, in which the increased production of IFN-γ and TNF-α results in apoptotic loss of hematopoietic stem and progenitor cells.11 In FA marrow failure, significant evidence in the Fancc murine model supports myelosuppressive cytokines and oxidants as endogenous stimuli involved in the proapoptotic phenotype of FA hematopoietic stem and progenitor cells. For instance, murine and human Fancc-/- progenitors are markedly hypersensitive to IFN-γ.6,32 Additionally, we previously showed that Fancc-/- progenitors exhibit enhanced apoptosis after stimulation with low, physiologically relevant in vitro doses of multiple inhibitory cytokines, including IFN-γ, TNF-α, MIP-1α,7 and TGF-β (L.S.H., unpublished data, February 2001), and in vivo administration of IFN-γ.33 As for oxidant sensitivity, Fancc-/- progenitors exposed to oxidants either in vitro or in vivo exhibit reduced colony formation, consistent with enhanced oxidant-mediated apoptosis.8,9 Moreover, recent studies using a murine model of ataxia-telangiectasia mutated (ATM), another chromosomal instability disorder, demonstrated increased ROS in ATM-/- hematopoietic stem cells, which led to marrow failure.34 Collectively, these data establish inhibitory cytokines and oxidants as critical endogenous regulators of hematopoiesis and suggest a pathogenic role in marrow failure.
In the studies reported here, we questioned whether altered redox signaling in Fancc-/- cells could be a potential mechanistic link between oxidant and TNF-α-induced apoptosis. TNF-α was selected for detailed study given its critical role in maintaining hematopoietic stem cell homeostasis35-37 and the literature indicating that TNF-α induces ROS-dependent apoptotic signaling.20,38 Because TNF-α also activates ROS-independent apoptotic programs, it was first necessary to determine whether the proapoptotic phenotype of Fancc-/- cells after TNF-α treatment was ROS dependent. By pretreating cells with antioxidants before TNF-α stimulation, our data demonstrate that enhanced TNF-α-mediated apoptosis in Fancc-/- MEFs and a hematopoietic population enriched for progenitor cells (c-kit+ cells) were dependent on ROS production. Given our recent data showing that enhanced H2O2-induced apoptosis in Fancc-/- MEFs is mediated through Ask1,9 we hypothesized that increased Ask1 activation in Fancc-/- MEFs was also responsible for the observed TNF-α hypersensitivity. As predicted, Fancc-/- MEFs treated with TNF-α exhibited Ask1 hyperactivation. Furthermore, studies using a dominant-negative Ask1 construct revealed that TNF-α-induced apoptosis in Fancc-/- MEFs is Ask1 dependent. Collectively, these observations support the idea that Fancc-/- cells exhibit an altered intracellular redox environment, predisposing to stimuli that activate Ask1 through ROS generation, such as oxidants and TNF-α.
Because enhanced apoptosis of hematopoietic progenitors contributes to the development of marrow failure in FA, it was imperative to evaluate whether the loss of Ask1 function in Fancc-/- progenitors would also protect from TNF-α hypersensitivity. Interestingly, genetic inactivation of Ask1 in Fancc-/- progenitors completely abrogated TNF-α hypersensitivity and restored progenitor growth to WT and Ask1-/- progenitor levels. Additionally, the loss of Ask1 in Fancc-/- progenitors also corrected H2O2 hypersensitivity to WT and Ask1-/- levels (data not shown). These data are the first demonstration that Ask1 has a role in regulating hematopoietic progenitor homeostasis. In fact, few studies have investigated the role of Ask1 in primary hematopoietic cells. In Ask1-/- thymocytes treated with Fas ligand, sustained activation of c-Jun N-terminal kinase (JNK) and p38 was reduced, but the proportion of apoptotic cells was similar to that of WT thymocytes, indicating an Ask1-independent mechanism for Fas-mediated apoptosis.24 In acute myeloid leukemia blasts, Schepers et al39 showed that p21waf1/cip1 binds Ask1, inhibiting p21waf1/cip1 nuclear activity and downstream stress-activated kinases, though Ask1 activity was not assessed. We found that WT and Ask1-/- hematopoietic progenitors have similar TNF-α and H2O2 (Figure 3C; L.S.H., unpublished data, March 2004) dose-response curves, which alone might suggest that Ask1 has no role in regulating ROS-induced apoptosis in hematopoietic progenitors. However, Fancc-/-; Ask1-/- progenitors are clearly protected against the same oxidant stimuli compared with Fancc-/- progenitors. Therefore, Ask1 may not be critical for steady state hematopoiesis, but it becomes important under conditions of hematopoietic stress, which would include Fancc hematopoiesis.
To further examine alterations in Ask1-mediated signaling in Fancc-/- cells, we investigated the downstream effector, p38. Fancc-/- MEFs treated with TNF-α exhibited p38 hyperactivation. In addition, studies using the p38 inhibitor SB203580 revealed that TNF-α-induced apoptosis in Fancc-/- MEFs and c-kit+ cells is p38 dependent, which is consistent with enhanced Fancc-/- hematopoietic progenitor formation in the presence of TNF-α and the p38 inhibitor. Interestingly, Verma et al31 demonstrated that human CD34+ cells (enriched for hematopoietic stem and progenitor cells) treated with either IFN-γ or TNF-α exhibit increased p38 activity. In addition, the inhibition of human progenitor growth by these myelosuppressive cytokines and TGF-β is abrogated by p38 inhibition.31,40 Of clinical relevance, progenitors from patients with acquired aplastic anemia exhibit enhanced colony formation in the presence of a p38 inhibitor. Thus, it has been suggested that p38 may serve as a common downstream effector for multiple myelosuppressive cytokines and could be targeted to treat marrow failure syndromes.31,41 In fact, clinical trials testing the safety and efficacy of p38 inhibitors are under way.42-44
It is unclear whether myelosuppressive cytokine signals converge on p38 through distinct signaling pathways or whether the point of intersection is at a common upstream molecule such as Ask1. Our data support a role for Ask1 in mediating increased TNF-α-induced apoptosis in Fancc-/- MEFs and progenitors; however, it remains unknown whether a similar Ask1-dependent mechanism is involved in the hypersensitivity of Fancc-/- cells to other myelosuppressive cytokines. Fancc-/- MEFs and human FA-C lymphoblast cells treated with IFN-γ alone or IFN-γ and TNF-α together produce increased double-stranded RNA-dependent kinase (PKR)-mediated apoptosis through disrupted HSP70 and PKR binding to FANCC.18,45,46 In addition, Pearl-Yafe et al47 demonstrated that IFN-γ-induced apoptosis of FA-C lymphoblast cells was partially blocked by p38 inhibition. Interestingly, p38 is also a downstream effector of PKR.48,49 Thus, it is conceivable that the hypersensitivity of Fancc-/- progenitors to multiple myelosuppressive cytokines is not mediated through the same upstream molecules (ie, Ask1 and PKR), but these apoptotic pathways converge on p38. Support for this idea is provided by structure-function studies showing that distinct functional domains of FANCC mediate H2O2 and IFN-γ hypersensitivity.9,50 Future studies to clarify the contribution of the distinct functions of the FANCC protein in sustaining normal hematopoietic stem cell function and in protecting hematopoietic stem cells from malignant transformation will be required so that the pathogenesis of marrow failure and leukemia in FA can be understood and novel treatments can be developed.
Optimal design of therapeutic agents for the treatment of patients with FA will require broad applicability across multiple FA complementation types. Our data demonstrating that Fanca-/- c-kit+ cells undergo enhanced TNF-α-mediated apoptosis, together with the observation that human FANCA-efficient progenitors cultured in the presence of IFN-γ and TNF-α have a profound reduction in colony formation,12 suggest that targeting myelosuppressive cytokine signaling pathways may be a reasonable strategy. Our studies showing that the antioxidant SeMet and the p38 inhibitor SB203580 protected Fanca-/- c-kit+ cells from TNF-α-induced apoptosis in a manner similar to that for Fancc-/- c-kit+ cells support such an approach.
In summary, we report that Fancc-/- MEFs exhibit TNF-α-induced hyperactivation of Ask1 and the downstream effector, p38. In addition, TNF-α-induced apoptosis in Fancc-/- MEFs and progenitors is corrected to WT levels by the disruption of Ask1 function. Furthermore, we show that antioxidants and a p38 inhibitor enhance the survival of Fancc-/- and Fanca-/- cells treated with TNF-α. We propose that by defining the molecular pathways responsible for oxidant and myelosuppressive cytokine hypersensitivity of FA hematopoietic cells, the potential may exist to develop therapeutic agents to improve the survival of FA hematopoietic stem and progenitor cells for the treatment or prevention of marrow failure.
Acknowledgments
We thank Drs Manuel Buchwald (Hospital for Sick Children, University of Toronto) and Freerk Arwert (Vrije University) for providing the Fancc+/- and Fanca+/- mice, respectively. We thank Dr John Critser (University of Missouri-Columbia) for rederiving Fanca mice. We thank Drs Clapp, Ingram, and Yoder (Indiana University) for thoughtful critique of the manuscript, and Arliene Britt for excellent administrative support.
Prepublished online as Blood First Edition Paper, August 18, 2005; DOI 10.1182/blood-2005-05-2096.
Supported by United States Public Health Service grants K08 HLDK04071 (L.S.H.), R01 HL077175 (L.S.H.), P01 HL53586 (L.S.H.), R01 HL075816 (R.K.), P30 DK49218, and P30 CA82709 and by the Fanconi Anemia Research Fund (L.S.H.) and the Riley Children's Foundation (L.S.H., R.K.).
K.B.-V. and A.W. performed research and analyzed data; M.R.S. designed and performed research and analyzed data; K.A.W.M. performed research; R.K. and H.I. contributed vital new reagents; L.S.H. analyzed data and wrote the paper.
K.B.-V. and M.R.S. contributed equally to this work.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 U.S.C. section 1734.
References
- 1.Alter BP, Young NS. The bone marrow failure syndromes. In: Nathan D, Oski F, eds. Hematology of Infancy and Childhood. Vol. 1. 5th ed. Philadelphia, PA: WB Saunders; 1998: 237-249.
- 2.Liu J. Fanconi's anemia. In: Young NS, ed. Bone Marrow Failure Syndromes. Philadelphia, PA: WB Saunders; 2000: 47-68.
- 3.Joenje H, Patel KJ. The emerging genetic and molecular basis of Fanconi anaemia. Nat Rev Genet. 2001;2: 446-457. [DOI] [PubMed] [Google Scholar]
- 4.D'Andrea AD, Grompe M. The Fanconi anaemia/BRCA pathway. Nat Rev Cancer. 2003;3: 23-34. [DOI] [PubMed] [Google Scholar]
- 5.Venkitaraman AR. Tracing the network connecting BRCA and Fanconi anaemia proteins. Nat Rev Cancer. 2004;4: 266-276. [DOI] [PubMed] [Google Scholar]
- 6.Rathbun RK, Faulkner GR, Ostroski MH, et al. Inactivation of the Fanconi anemia group C gene augments interferon-gamma-induced apoptotic responses in hematopoietic cells. Blood. 1997;90: 974-985. [PubMed] [Google Scholar]
- 7.Haneline LS, Broxmeyer HE, Cooper S, et al. Multiple inhibitory cytokines induce deregulated progenitor growth and apoptosis in hematopoietic cells from Fac-/-mice. Blood. 1998;91: 4092-4098. [PubMed] [Google Scholar]
- 8.Hadjur S, Ung K, Wadsworth L, et al. Defective hematopoiesis and hepatic steatosis in mice with combined deficiencies of the genes encoding Fancc and Cu/Zn superoxide dismutase. Blood. 2001;98: 1003-1011. [DOI] [PubMed] [Google Scholar]
- 9.Saadatzadeh MR, Bijangi-Vishehsaraei K, Hong P, Bergmann H, Haneline LS. Oxidant hypersensitivity of Fanconi anemia type C-deficient cells is dependent on a redox-regulated apoptotic pathway. J Biol Chem. 2004;279: 16805-16812. [DOI] [PubMed] [Google Scholar]
- 10.Hadjur S, Jirik FR. Increased sensitivity of Fancc-deficient hematopoietic cells to nitric oxide and evidence that this species mediates growth inhibition by cytokines. Blood. 2003;101: 3877-3884. [DOI] [PubMed] [Google Scholar]
- 11.Young NS, Maciejewski J. The pathophysiology of acquired aplastic anemia. N Engl J Med. 1997;336: 1365-1372. [DOI] [PubMed] [Google Scholar]
- 12.Zhang X, Li J, Sejas DP, Rathbun KR, Bagby GC, Pang Q. The Fanconi anemia proteins functionally interact with the protein kinase regulated by RNA (PKR). J Biol Chem. 2004;279: 43910-43919. [DOI] [PubMed] [Google Scholar]
- 13.Youssoufian H. Localization of Fanconi anemia C protein to the cytoplasm of mammalian cells. Proc Natl Acad Sci U S A. 1994;91: 7975-7979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Heinrich MC, Silvey KV, Stone S, et al. Posttranscriptional cell cycle-dependent regulation of human FANCC expression. Blood. 2000;95: 3970-3977. [PubMed] [Google Scholar]
- 15.Cumming RC, Lightfoot J, Beard K, Youssoufian H, O'Brien PJ, Buchwald M. Fanconi anemia group C protein prevents apoptosis in hematopoietic cells through redox regulation of GSTP1. Nat Med. 2001;7: 814-820. [DOI] [PubMed] [Google Scholar]
- 16.Kruyt FA, Hoshino T, Liu JM, Joseph P, Jaiswal AK, Youssoufian H. Abnormal microsomal detoxification implicated in Fanconi anemia group C by interaction of the FAC protein with NADPH cytochrome p450 reductase. Blood. 1998;92: 3050-3056. [PubMed] [Google Scholar]
- 17.Pang Q, Fagerlie S, Christianson TA, et al. The Fanconi anemia protein FANCC binds to and facilitates the activation of STAT1 by gamma interferon and hematopoietic growth factors. Mol Cell Biol. 2000;20: 4724-4735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pang Q, Keeble W, Christianson TA, Faulkner GR, Bagby GC. FANCC interacts with Hsp70 to protect hematopoietic cells from IFN-gamma/TNF-alpha-mediated cytotoxicity. EMBO J. 2001;20: 4478-4489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pagano G, Youssoufian H. Fanconi anaemia proteins: major roles in cell protection against oxidative damage. Bioessays. 2003;25: 589-595. [DOI] [PubMed] [Google Scholar]
- 20.Saitoh M, Nishitoh H, Fujii M, et al. Mammalian thioredoxin is a direct inhibitor of apoptosis signal-regulating kinase (ASK) 1. EMBO J. 1998;17: 2596-2606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Song JJ, Rhee JG, Suntharalingam M, Walsh SA, Spitz DR, Lee YJ. Role of glutaredoxin in metabolic oxidative stress: glutaredoxin as a sensor of oxidative stress mediated by H2O2. J Biol Chem. 2002;277: 46566-46575. [DOI] [PubMed] [Google Scholar]
- 22.Cho SG, Lee YH, Park HS, et al. Glutathione S-transferase μ modulates the stress-activated signals by suppressing apoptosis signal-regulating kinase 1. J Biol Chem. 2001;276: 12749-12755. [DOI] [PubMed] [Google Scholar]
- 23.Ichijo H, Nishida E, Irie K, et al. Induction of apoptosis by ASK1, a mammalian MAPKKK that activates SAPK/JNK and p38 signaling pathways. Science. 1997;275: 90-94. [DOI] [PubMed] [Google Scholar]
- 24.Tobiume K, Matsuzawa A, Takahashi T, et al. ASK1 is required for sustained activations of JNK/p38 MAP kinases and apoptosis. EMBO Rep. 2001;2: 222-228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Haneline LS, Li X, Ciccone SL, et al. Retroviral-mediated expression of recombinant Fancc enhances the repopulating ability of Fancc-/-hematopoietic stem cells and decreases the risk of clonal evolution. Blood. 2003;101: 1299-1307. [DOI] [PubMed] [Google Scholar]
- 26.Cheng NC, van De Vrugt HJ, van Der Valk MA, et al. Mice with a targeted disruption of the Fanconi anemia homolog Fanca. Hum Mol Genet. 2000;9: 1805-1811. [DOI] [PubMed] [Google Scholar]
- 27.Freie B, Li X, Ciccone SL, et al. Fanconi anemia type C and p53 cooperate in apoptosis and tumorigenesis. Blood. 2003;102: 4146-4152. [DOI] [PubMed] [Google Scholar]
- 28.Pear WS, Miller JP, Xu L, et al. Efficient and rapid induction of a chronic myelogenous leukemia-like myeloproliferative disease in mice receiving P210 bcr/abl-transduced bone marrow. Blood. 1998;92: 3780-3792. [PubMed] [Google Scholar]
- 29.Haneline LS, Gobbett TA, Ramani R, et al. Loss of FancC function results in decreased hematopoietic stem cell repopulating ability. Blood. 1999;94: 1-8. [PubMed] [Google Scholar]
- 30.Matsuzawa A, Nishitoh H, Tobiume K, Takeda K, Ichijo H. Physiological roles of ASK1-mediated signal transduction in oxidative stress- and endoplasmic reticulum stress-induced apoptosis: advanced findings from ASK1 knockout mice. Antioxid Redox Signal. 2002;4: 415-425. [DOI] [PubMed] [Google Scholar]
- 31.Verma A, Deb DK, Sassano A, et al. Cutting edge: activation of the p38 mitogen-activated protein kinase signaling pathway mediates cytokine-induced hemopoietic suppression in aplastic anemia. J Immunol. 2002;168: 5984-5988. [DOI] [PubMed] [Google Scholar]
- 32.Whitney MA, Royle G, Low MJ, et al. Germ cell defects and hematopoietic hypersensitivity to γ-interferon in mice with a targeted disruption of the Fanconi anemia C gene. Blood. 1996;88: 49-58. [PubMed] [Google Scholar]
- 33.Li X, Yang Y, Yuan J, et al. Continuous in vivo infusion of interferon-gamma (IFN-γ) preferentially reduces myeloid progenitor numbers and enhances engraftment of syngeneic wild-type cells in Fancc-/- mice. Blood. 2004;104: 1204-1209. [DOI] [PubMed] [Google Scholar]
- 34.Ito K, Hirao A, Arai F, et al. Regulation of oxidative stress by ATM is required for self-renewal of haematopoietic stem cells. Nature. 2004;431: 997-1002. [DOI] [PubMed] [Google Scholar]
- 35.Dybedal I, Bryder D, Fossum A, Rusten LS, Jacobsen SE. Tumor necrosis factor (TNF)-mediated activation of the p55 TNF receptor negatively regulates maintenance of cycling reconstituting human hematopoietic stem cells. Blood. 2001;98: 1782-1791. [DOI] [PubMed] [Google Scholar]
- 36.Bryder D, Ramsfjell V, Dybedal I, et al. Self-renewal of multipotent long-term repopulating hematopoietic stem cells is negatively regulated by Fas and tumor necrosis factor receptor activation. J Exp Med. 2001;194: 941-952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rebel VI, Hartnett S, Hill GR, Lazo-Kallanian SB, Ferrara JL, Sieff CA. Essential role for the p55 tumor necrosis factor receptor in regulating hematopoiesis at a stem cell level. J Exp Med. 1999;190: 1493-1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jones PL, Ping D, Boss JM. Tumor necrosis factor α and interleukin-1β regulate the murine manganese superoxide dismutase gene through a complex intronic enhancer involving C/EBP-beta and NF-κB. Mol Cell Biol. 1997;17: 6970-6981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schepers H, Geugien M, Eggen BJ, Vellenga E. Constitutive cytoplasmic localization of p21(Waf1/Cip1) affects the apoptotic process in monocytic leukaemia. Leukemia. 2003;17: 2113-2121. [DOI] [PubMed] [Google Scholar]
- 40.Verma A, Deb DK, Sassano A, et al. Activation of the p38 mitogen-activated protein kinase mediates the suppressive effects of type I interferons and transforming growth factor-β on normal hematopoiesis. J Biol Chem. 2002;277: 7726-7735. [DOI] [PubMed] [Google Scholar]
- 41.Platanias LC. The p38 mitogen-activated protein kinase pathway and its role in interferon signaling. Pharmacol Ther. 2003;98: 129-142. [DOI] [PubMed] [Google Scholar]
- 42.Parasrampuria DA, de Boer P, Desai-Krieger D, Chow AT, Jones CR. Single-dose pharmacokinetics and pharmacodynamics of RWJ 67657, a specific p38 mitogen-activated protein kinase inhibitor: a first-in-human study. J Clin Pharmacol. 2003;43: 406-413. [DOI] [PubMed] [Google Scholar]
- 43.Branger J, van den Blink B, Weijer S, et al. Inhibition of coagulation, fibrinolysis, and endothelial cell activation by a p38 mitogen-activated protein kinase inhibitor during human endotoxemia. Blood. 2003;101: 4446-4448. [DOI] [PubMed] [Google Scholar]
- 44.Branger J, van den Blink B, Weijer S, et al. Anti-inflammatory effects of a p38 mitogen-activated protein kinase inhibitor during human endotoxemia. J Immunol. 2002;168: 4070-4077. [DOI] [PubMed] [Google Scholar]
- 45.Pang Q, Christianson TA, Keeble W, Koretsky T, Bagby GC. The anti-apoptotic function of Hsp70 in the interferon-inducible double-stranded RNA-dependent protein kinase-mediated death signaling pathway requires the Fanconi anemia protein, FANCC. J Biol Chem. 2002;277: 49638-49643. [DOI] [PubMed] [Google Scholar]
- 46.Pang Q, Keeble W, Diaz J, et al. Role of double-stranded RNA-dependent protein kinase in mediating hypersensitivity of Fanconi anemia complementation group C cells to interferon-γ, tumor necrosis factor-α, and double-stranded RNA. Blood. 2001;97: 1644-1652. [DOI] [PubMed] [Google Scholar]
- 47.Pearl-Yafe M, Halperin D, Scheuerman O, Fabian I. The p38 pathway partially mediates caspase-3 activation induced by reactive oxygen species in Fanconi anemia C cells. Biochem Pharmacol. 2004;67: 539-546. [DOI] [PubMed] [Google Scholar]
- 48.Goh KC, deVeer MJ, Williams BR. The protein kinase PKR is required for p38 MAPK activation and the innate immune response to bacterial endotoxin. EMBO J. 2000;19: 4292-4297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Patel CV, Handy I, Goldsmith T, Patel RC. PACT, a stress-modulated cellular activator of interferon-induced, double-stranded RNA activated protein kinase, PKR. J Biol Chem. 2000;275: 37993-37998. [DOI] [PubMed] [Google Scholar]
- 50.Pang Q, Christianson TA, Keeble W, et al. The Fanconi anemia complementation group C gene product: structural evidence of multifunctionality. Blood. 2001;98: 1392-1401. [DOI] [PubMed] [Google Scholar]