

Abstract
Neuropilin-1 (Npn-1) is a receptor shared by class 3 semaphorins and heparin-binding forms of vascular endothelial growth factor (VEGF), protein families that regulate endothelial and neuronal-cell function. Ligand interaction with Npn-1 dictates the choice of signal transducer; plexins transduce semaphorin signals, and VEGF receptors transduce VEGF signals. It is not clear how class 3 semaphorins affect endothelial-cell function and how the shared receptor Npn-1 selects its ligand. We report that semaphorin3A (Sema3A) inhibits endothelial-cell lamellipodia formation, adhesion, survival, proliferation, and cord formation. VEGF165, but not VEGF121, could block all these effects of Sema3A. VEGF165 competed with Sema3A for binding to endothelial cells, effectively reduced cell-surface Npn-1, and promoted its internalization. Use of soluble forms of Npn-1 or VEGF receptor-1 to block VEGF165 binding to Npn-1 or to VEGF receptors provided evidence that surface Npn-1 and VEGF receptors are required for VEGF165-induced Npn-1 internalization. Sema3A also reduced cell-surface Npn-1 in endothelial cells and promoted its internalization, but required a higher concentration than VEGF165. These results demonstrate that preferential receptor binding and internalization by a ligand are mechanisms by which the common receptor Npn-1 can play an essential role in prioritizing conflicting signals.
Introduction
The vascular and nervous systems have a treelike branched structure originating from a center and reaching all tissues. Often vessels and nerves run parallel, suggesting that they are guided by common guidance mechanisms. Indeed, there is evidence that axonal guidance cues are also involved in blood-vessel formation. VEGF, an essential regulator of vascular development, has recently been shown to regulate neuronal development.1-4
Class 3 semaphorins, a family of 6 secreted glycoproteins that includes Sema3A-3F, function as axonal chemorepellents for growth cones during neuronal development.5-7 Their receptors are composed of a ligand-binding chain consisting of neuropilins and a signal-transducing chain consisting of plexins.8 Except for Sema3E, which directly binds to plexin-D1,9 Sema3A, 3B, 3C, and 3D need to bind to neuropilin-1 (Npn-1) or Npn-2 to signal.10-14 Class 3 semaphorins use distinct combinations of neuropilins and plexins as their receptor system13,15-17; Sema3A uses Npn-1–plexin-A1, -A2, or -A4.16,17 Recent studies in mice with targeted deletions of semaphorin signal components have revealed an essential role of semaphorin signaling in normal vascular development.9,15,18
The VEGF family of proteins is required for angiogenesis during development and after birth, and contributes to neuronal growth, immune regulation, and hematopoiesis.3,19-21 Npn-1 serves as a VEGF-A isotype-specific receptor.22 The VEGF-A gene is organized in 8 exons that generate different isoforms, VEGF121, VEGF145, VEGF165, VEGF189, and VEGF206, by alternative splicing. Exon 7, which codes for a Npn-1 binding site, is expressed in VEGF165, VEGF189, and VEGF206.23 Although VEGF receptors are required for VEGF165 signaling, Npn-1 acts as a coreceptor for VEGF165 that enhances VEGF165 binding to VEGF receptor-2 (VEGFR-2/KDR/Flk-1) and VEGF165 activity.24 VEGF-B isoforms (VEGF-B167 and VEGF-B186) and placenta growth factor-2 (PlGF-2) bind to Npn-1 and activate VEGFR-1 (Flt-1).25-27 The VEGF-like protein from orf virus NZ2 binds to Npn-1 and activates VEGFR-2.28
Due to its ability to bind various ligands, Npn-1 appears to serve as a “hub” receptor for different ligands. The multiple domain structure of Npn-1 extracellular domain may explain how Npn-1 interacts with various ligands. Npn-1 has a large extracellular domain of 860 amino acids, which consists of 3 subdomains, a1, a2 (CUB), b1, b2 (coagulation factor V/VIII), and c (MAM) domains. Sema3A binds to Npn-1 via a1a2b1 domains, whereas VEGF165 binds via b1b2 domains.29 Heparin and PlGF-2 also bind to the b1b2 domain.27 Consistent with Npn-1 playing multiple roles, Npn-1–deficient mice are embryonically lethal and display severe abnormalities in the nervous and cardiovascular systems.30,31 Endothelial-cell–specific Npn-1–null mice showed severe vascular defects.32 In vitro, Npn-1 plays a role in cell-to-cell adhesion.33
Npn-1 mediates distinct functions by choice of ligand and ligand-specific signal transducer; the semaphorin–Npn-1 complex uses plexin to signal, whereas the VEGF–Npn-1 complex uses VEGF receptors. In some cases, Npn-1 can mediate opposite signals. For example, VEGF165 promotes endothelial-cell adhesion to extracellular matrix and microvessel outgrowth, but Sema3A inhibits these functions.18,34 Sema3A evokes repulsive signals for growth cone guidance, whereas VEGF165 induces axonal outgrowth.6,35 It is not clear how Npn-1 prioritizes signals derived from different ligands, particularly when the signals are contrasting. To address this question, we have examined Npn-1 from a perspective of a receptor that is shared by multiple ligands. We show that Sema3A exerts a variety of activities on endothelial cells and that VEGF165 inhibits these activities. We also show that VEGF165 has a priority for use of Npn-1 over Sema3A due to its superior binding to cells and subsequent induction of Npn-1 internalization, which renders Npn-1 unavailable for Sema3A binding. This mechanism allows Npn-1 to select signals from its multiple ligands.
Materials and methods
Cytokines and reagents
Recombinant human VEGF165, VEGF121, VEGF-B167, VEGF-C, FGF-2, chimeric human Sema3A/Fc, soluble rat Npn-1/Fc (srNpn-1/Fc), human VEGFR-1/Fc, and human B7-1/Fc were from R&D Systems (Minneapolis, MN). Porcine heparin sodium salt and bovine fibronectin were from Sigma (St Louis, MO).
Cells and cell culture
Primary human umbilical vein endothelial cells (HUVECs) were prepared from umbilical cord and cultured as previously described.36 HUVECs were used between passages 2 and 5. The human stromal-cell line HS-5 (ATCC, Manassas, VA) was maintained in DMEM with 10% fetal bovine serum (FBS); the rat pheochromocytoma-cell line PC12 (ATCC) was maintained on fibronectin-coated dishes in DMEM with 10% FBS and 10% horse serum.
Endothelial-cell survival, adhesion, and proliferation assays
HUVECs were starved by overnight incubation in MEDIUM199 (Cellgro; Mediatech, Herndon, VA) with 1% FBS, and then cultured (7000 cells/well) in 96-well flat-bottom tissue culture plates (Costar, Corning, Corning, NY) in MEDIUM199 with 1% FBS and 2 μg/mL heparin. Cultures were supplemented with Sema3A alone or with VEGF165 or VEGF121. After 16 hours, Cell Counting Kit-8 (Dojindo, Kumamoto, Japan) was added (10 μL/well) and incubation continued for 2 hours, followed by measurement of absorbance at 450 nm. Subsequently, plates were patted dry, and adherent cells were fixed with 4% wt/vol paraformaldehyde (Electron Microscopy Sciences, Hatfield, PA) for 15 minutes and stained with 0.05% crystal violet in 20% ethanol. Adherent cells/field were counted at low-power magnification (× 40) by NIH image analysis. Data were obtained from triplicate wells. HUVECs (4000 cells/well) were cultured in 96-well tissue culture plates in MEDIUM199 with 10% FBS and 20 μg/mL heparin, with Sema3A, or Sema3A plus VEGF165 or VEGF121 for 3 days. Proliferation was measured by 3H thymidine uptake (0.6 μCi/well [0.022 MBq/well]; New England Nuclear, Boston, MA) during last 20 hours of culture. The results reflect mean cpm/culture.
Lamellipodia spreading assay
Four-chamber glass slides (LAB-TEK; Nalge Nunc International, Rochester, NY) were coated with 10 μg/mL poly-l-lysine (Sigma) in PBS. HUVECs (40 000/chamber) were added to the chambers in MEDIUM199 with 1% FBS and 2 μg/mL heparin; Sema3A alone or with VEGF165 or VEGF121 was added to the cultures. After 30 minutes at 37°C, the medium was replaced with 4% wt/vol paraformaldehyde for 15-minute fixation. After removal of the plastic chambers, the glass slide was washed with PBS (3 ×) and mounted. Four nonoverlapping fields were counted at low-power magnification (× 40) to obtain an average ratio of the number of cells with lamellipodia spreading/the total number of adherent cells (40 to 90 cells/field). A cell was considered positive for spreading if it displayed lamellipodia in at least three fourths of the periphery.
Endothelial-cell retraction assay
Four-chamber glass slides were coated with 5 μg/mL fibronectin in PBS. HUVECs (15 000/chamber) were grown in HUVEC culture medium for 16 hours, washed with MEDIUM199 with 10% FBS and 2 μg/mL heparin, and incubated in the same medium with Sema3A (2 μg/mL or as indicated). After fixation with 4% wt/vol paraformaldehyde, slides were washed with PBS, and 4 nonoverlapping fields were observed to obtain an average retraction score (30 to 60 cells/field). Retraction scores were assigned blindly as described in the legend to Figure 2D.
Figure 2.
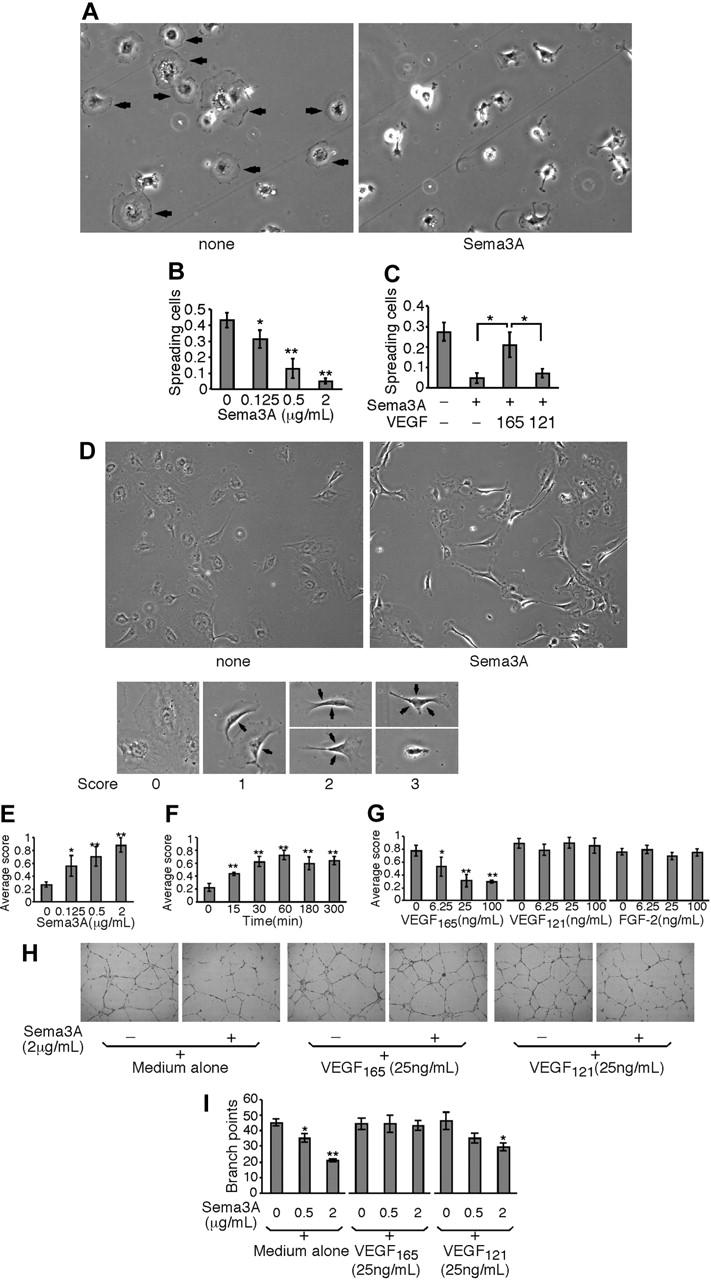
Opposing activities of Sema3A and VEGF165 on endothelial-cell lamellipodia spreading and retraction, and extracellular matrix–dependent cord formation. (A) Representative images depicting Sema3A-induced inhibition of lamellipodia spreading in endothelial cells. Endothelial cells were incubated for 30 minutes on poly-l-lysine–coated glass slides in medium alone or with Sema3A (2 μg/mL). Cells were visualized as described for Figure 1A, except for the use of a 10 ×/0.25 PhC objective lens. Original magnification, × 100. Arrows point to cells with lamellipodia. (B) Dose dependency of Sema3A-induced lamellipodia spreading inhibition. Results reflect the mean ratio (± SD triplicate wells) of cells with lamellipodia spreading/total number of attached cells. Representative of 3 experiments. (C) Effects of VEGF165 and VEGF121 on inhibition of lamellipodia spreading by Sema3A. Endothelial cells were incubated in medium alone, with Sema3A (0.5 μg/mL) alone, or with Sema3A (0.5 μg/mL) plus VEGF165 (25 ng/mL) or VEGF121 (25 ng/mL). The results reflect the mean (± SD of triplicate determinations). (D) Representative images depicting Sema3A-induced retraction of lamellipodia. After endothelial cells were allowed to attach on fibronectin-coated slides for 16 hours and nonadherent cells were removed, the adherent cells were further incubated (1 hour) in medium only or with Sema3A (2 μg/mL). Retraction scores: score 0 indicates cells have intact lamellipodia around; score 1, one side of the lamellipodia is retracted; score 2, 2 sides are retracted; and score 3, 3 sides are retracted or the cell has shrunk and is round. Arrows point to the retracted sides. Cells were visualized as described for Figure 1A, except for the use of a 10 ×/0.25 PhC objective lens. Original magnification: top panels, × 40; bottom panels, × 100 (bottom images enlarged with Photoshop [Adobe Systems, San Jose, CA]). (E) Concentration and (F) time dependency of Sema3A-induced lamellipodia retraction in endothelial cells. Adherent cells were incubated in medium alone or with Sema3A for 1 hour or for the indicated times. The results reflect the average retraction scores (± SD of triplicate determinations). (G) VEGF165 blocks lamellipodia retraction induced by Sema3A, but VEGF121 and FGF-2 do not. Adherent cells were incubated (1 hour) with Sema3A (2 μg/mL) alone or in the presence of VEGF165, VEGF121, or FGF-2. (H) Sema3A inhibits extracellular matrix–dependent cord formation by endothelial cells. Cells were incubated (16 hours) onto Matrigel-coated wells in medium alone, with Sema3A, VEGF165, VEGF121, or with Sema3A plus VEGF165 or VEGF121. Original magnification, × 40. (I) VEGF165 reconstitutes cord formation disrupted by Sema3A. Endothelial cells were cultured (16 hours) on Matrigel-coated wells in medium only, with Sema3A, VEGF165, with VEGF121, or with Sema3A plus VEGF165 or VEGF121. The results reflect the average number of branch points formed by intersecting endothelial cords (mean ± SD of triplicate wells). Representative experiment of 4 performed. *P < .05; **P < .01. For all micrographic images, photography, acquisition, and processing were performed as described for Figure 1A.
Cord-formation assay
The assay was performed as described.36 Cells were starved overnight in MEDIUM199 with 1% FBS. Wells (48-well plates) were coated with 150 μL Matrigel (BD Biosciences, San Jose, CA) at 4°C, and incubated for 30 minutes at 37°C. HUVECs (15 000 cells/well) were incubated with MEDIUM199 with 1% FBS and 2 μg/mL heparin for 18 hours onto the Matrigel-coated wells. Cells were photographed under phase-contrast microscopy at low-power magnification (× 40). Average numbers of branching points were counted from triplicate wells.
Fluorescence-activated cell sorter (FACS) analysis
Endothelial cells were detached with 5 mM EDTA in PBS, washed with binding buffer (MEDIUM199, 1% FBS, 10 mM HEPES, 2 μg/mL heparin), and incubated at the indicated temperatures with VEGF165, VEGF121, or FGF-2. Cell-surface staining was performed with FITC-conjugated anti–human CD31 mAb (WM59; BD Pharmingen), PE-conjugated ant–ihuman VEGFR-2 mAb (89106; R&D Systems), or PE-conjugated anti–human BDCA-4 (Npn-1) mAb (AD5-17F6; Miltenyi Biotec, Auburn, CA) at 4°C for 20 minutes. For intracellular staining, cells were permeabilized with BD Cytofix/Cytoperm kit (BD Biosciences). Binding of Sema3A to cells was measured after incubation with 2 μg/mL Sema3A/Fc in binding buffer at 4°C for 60 minutes. After washing, Sema3A/Fc bound to cells was detected with FITC-conjugated F(ab′)2 goat anti–human IgG Fc (Jackson ImmunoResearch Laboratories, West Grove, PA). As a control, we used human B7-1/Fc. Data were collected using a FACScalibur cytofluorometer (Becton Dickinson, San Jose, CA) and analyzed using CELLQuest software (Becton Dickinson).
Enzyme-linked immunosorbent assay (ELISA)–based binding assays
Flat-bottom microtiter plates (96 well, Immulon 4HBX; Thermo Labsystems, Franklin, MA) were coated overnight with srNpn-1/Fc chimeric protein (2 μg/mL in PBS). After blocking with Superblock (PIERCE, Rockford, IL) for 30 minutes, VEGF165 (25 ng/mL), Sema3A/Fc (2 μg/mL), VEGF165 (25 ng/mL) plus srNpn-1 (0.05-3.2 μg/mL), or VEGF165 (25 ng/mL) plus sVEGFR-1 (0.05-3.2 μg/mL) in PBS 0.1% Tween20, 1% BSA were added with heparin at various concentrations. After incubation at 25°C for 1 hour, bound VEGF165 or Sema3A/Fc was detected with rabbit anti-VEGF antibody (A-20; Santa Cruz Biotechnology, Santa Cruz, CA) or rabbit anti-Sema3A antibody (H-300; Santa Cruz Biotechnology) at 1 μg/mL, followed by HRP-conjugated anti–rabbit IgG antibody (Bio-Rad, Hercules, CA) at 1:5000 in PBS 0.1% Tween20 1% BSA. Reactions were visualized with tetramethoxybenzene peroxidase substrate (Kirkegaard & Perry Laboratories, Gaithersburg, MD), followed by addition of 1 M H2SO4, and read at 450 nm.
Laser confocal microscopic analysis
Endothelial cells on glass chamber slides coated with 5 μg/mL fibronectin were incubated (MEDIUM199 with 10% FBS and 2 μg/mL heparin) with 25 ng/mL VEGF165 or 2 μg/mL Sema3A at 37°C for the indicated times. The medium was replaced with 4% wt/vol paraformaldehyde for fixation, cells were washed with PBS, and cell membranes were permeabilized with 0.1% Triton X100 in PBS. Cells were then stained with mouse anti–human BDCA-4 (Npn-1) mAb (AD5-17F6; Miltenyi Biotec) in 1% BSA and 3% goat serum in PBS at 4°C for 16 hours. Cells were washed with PBS and incubated with Alexa 488–conjugated goat anti–mouse IgG antibody (Molecular Probes, Eugene, OR) in PBS with 1% BSA and 3% goat serum at 25°C for 1 hour. Slides were mounted with FluorSave Reagent (Calbiochem, San Diego, CA). Images were visualized using an LSM 510 confocal microscope (Carl Zeiss, Thornwood, NY), and reflect the merging of fluorescent slice and DIC (differential interference contrast) images.
Statistical analysis
Results are expressed as means plus or minus SD. Student t test was applied to evaluate group differences; a P value of less than .05 was considered significant.
Results
Sema3A inhibits endothelial-cell attachment, survival, and proliferation: blocking by VEGF165
We examined the effects of Sema3A on endothelial-cell attachment, survival, and growth, and tested whether VEGF165, a VEGF-A isoform that binds Npn-1,24 modifies these activities of Sema3A. Previously, Sema3A was reported to inhibit endothelial-cell adhesion to extracellular matrix.18 As shown in Figure 1A, we found that Sema3A also inhibits endothelial-cell adhesion to tissue culture wells, in the absence of protein coating. This inhibition was blocked by VEGF165, but not by VEGF121, a VEGF-A isoform that lacks the Npn-1 binding domain.24 Since previous studies concluded that attachment is critical to endothelial-cell survival,37 we examined whether reduced endothelial-cell attachment correlated with decreased survival. In the same cultures, we measured first cell survival using a colorimetric assay, and after removal of nonadherent cells, we measured cell attachment (NIH image analysis). We found that Sema3A dose-dependently reduces endothelial-cell viability after 16-hour incubation in culture medium with 1% FBS (Figure 1B line graph). VEGF165 (25 ng/mL) alone increased endothelial-cell viability and protected the cells from death occurring in the presence of Sema3A. VEGF121 (25 ng/mL) alone also increased endothelial-cell viability comparably with VEGF165 but, unlike VEGF165, failed to protect the cells from death induced by Sema3A. In these cultures, endothelial-cell survival closely correlated with endothelial-cell attachment (Figure 1B bar graph), suggesting that the nonadherent cells are dead.
Figure 1.

Inhibition of endothelial-cell adhesion, survival, and proliferation by Sema3A and reversal by VEGF165. (A) Representative images depicting Sema3A inhibition of endothelial-cell attachment to tissue culture wells. HUVECs were incubated (16 hours) in medium (MEDIUM199, 1% FBS) only, with Sema3A, VEGF165, VEGF121, with Sema3A plus VEGF165 or VEGF121. Cells were visualized under an Olympus IX51 phase-contrast microscope equipped with a 4 ×/0.13 PhL objective lens and a 10 × eyepiece (Olympus Optical, Melville, NY), and were photographed with a Retiga 1300 digital camera (Qimaging, Burnaby, BC, Canada). Images obtained via QCapture software (Qimaging) were imported into Adobe Photoshop 6.0 software (Adobe Systems, San Jose, CA) for processing. Original magnification, × 40. (B) Correlation between endothelial-cell attachment and viability. Endothelial cells were incubated (16 hours) in medium alone, with Sema3A, VEGF165, VEGF121, or with Sema3A plus VEGF165 or VEGF121. Cell viability was measured by absorbance at 450 nm after addition of Cell Counting Kit-8. The adherent cells were counted by NIH image analysis after removal of nonadherent cells. The results reflect the mean (± SD) of triplicate cultures (representative experiment of 3 performed). (C) Sema3A inhibits endothelial-cell proliferation. Cells were cultured for 3 days in medium alone, Sema3A, VEGF165, VEGF121, or with Sema3A plus VEGF165 or VEGF121. 3H thymidine uptake was measured during the final 20 hours of incubation. The results reflect the mean cpm of triplicate cultures (representative experiment of 3 performed).
We also examined the effects of Sema3A on endothelial-cell proliferation after 3-day culture. As expected (Figure 1C), VEGF165 and VEGF121 dose-dependently promoted the proliferation of endothelial cells (open circles). Sema3A (1 μg/mL, Figure 1C closed triangles) reduced by 90% and 87% cell proliferation induced by 12.5 and 25 ng/mL VEGF121, respectively. By contrast, under the same conditions, Sema3A reduced by only 29% and 13% endothelial-cell proliferation induced by 12.5 and 25 ng/mL VEGF165, respectively (Figure 1C). These results demonstrate that Sema3A inhibits endothelial-cell attachment, survival, and proliferation, and that VEGF165, but not VEGF121, can counter these functions of Sema3A.
Sema3A inhibits lamellipodia spreading and cord formation in endothelial cells: blocking by VEGF165
We examined in detail cell-attachment inhibition by Sema3A, and tested whether Sema3A inhibits the initial step of endothelial-cell spreading or rather promotes cell retraction and detachment once spreading has occurred. Endothelial cells usually spread their lamellipodia for firm adherence to poly-l-lysine–coated glass surfaces within 30 minutes (Figure 2A left panel). Lamellipodia spreading by endothelial cells is not observed on uncoated glass surfaces (not shown). In the presence of Sema3A (2 μg/mL), endothelial cells attached to the poly-l-lysine–coated surfaces but displayed virtually no spreading of lamellipodia (Figure 2A right panel). This effect was dependent upon Sema3A concentration (Figure 2B). VEGF165 (25 ng/mL) significantly reduced this effect of Sema3A, whereas VEGF121 (25 ng/mL) was ineffective (Figure 2C).
Next, we examined whether Sema3A affects endothelial cells that have already spread their lamellipodia. In neuronal cells, Sema3A induces axonal growth cone collapse, a process whereby filopodia become shorter and fewer, and lamellipodia shrink back from the sides of the growth cone.38 After 16-hour incubation on fibronectin-coated slides, most endothelial cells display spreading of lamellipodia (Figure 2D lower left panel). At low-power magnification, lamellipodia are not clearly visible on endothelial cells, but their presence is reflected by somewhat undefined cell boundaries (Figure 2D upper left). After incubation with Sema3A for 1 hour, the endothelial cells displayed retracted lamellipodia to a various degree (Figure 2D lower), which was reflected by more sharp cell boundaries (Figure 2D upper right). Similar Sema3A-induced lamellipodia retraction was noted in endothelial cells cultured on poly-l-lysine–coated glass surfaces (data not shown). To quantify lamellipodia retraction, we established a scoring system (Figure 2D lower panels). By blindly scoring at least 120 cells from 4 fields, we obtained an average retraction score. As shown in Figure 2E and 2F, respectively, Sema3A dose-dependently and time-dependently promoted lamellipodia retraction in endothelial cells. This effect reached a plateau after 60-minute incubation. VEGF165, but not VEGF121, dose-dependently inhibited Sema3A-induced retraction; almost complete inhibition was noted at 25 ng/mL (Figure 2G). VEGF165 had no or minimal effect on cell morphology when added alone (not shown). FGF-2, an endothelial-cell mitogenic factor reported to bind to Npn-1,39 minimally affected retraction induced by Sema3A (Figure 2G). These results provide evidence that in endothelial cells Sema3A inhibits lamellipodia spreading and promotes lamellipodia retraction.
Endothelial cells can form cordlike structures when incubated onto Matrigel, a crude extract of extracellular matrix proteins.40 We examined the effects of Sema3A on Matrigel-dependent endothelial-cell cord formation. As shown in Figure 2H, Sema3A inhibited formation of the characteristic reticular network. VEGF165 reversed this effect of Sema3A, whereas VEGF121 did not. Comparison of branching point numbers indicated that Sema3A significantly reduces cord formation by endothelial cells and that VEGF165, but not VEGF121, reverses this effect (Figure 2I).
Analysis of VEGF165 competition for Sema3A binding to Npn-1
To explore the mechanisms by which VEGF165 counters the biologic effects of Sema3A on endothelial cells, we examined whether VEGF165 reduces the binding of Sema3A to Npn-1, which serves as a common receptor for VEGF165 and Sema3A. Since it is known that heparin enhances VEGF165 binding to Npn-1,23 we first examined the effect of heparin on Sema3A binding to Npn-1. Using an ELISA-based assay in which Npn-1 is immobilized onto the well, we found that heparin enhances significantly the binding of Sema3A to Npn-1 as well as the binding of VEGF165 to Npn-1 (Figure 3A).
Figure 3.

Analysis of VEGF165 and Sema3A binding to Npn-1. (A) Heparin enhances VEGF165 and Sema3A binding to Npn-1. VEGF165 (25 ng/mL, ○) or Sema3A (2 μg/mL, □) was added to Npn-1–coated wells with heparin (0-50 000 ng/mL). Bound VEGF165 or Sema3A was measured by ELISA. The results reflect the means ± SD of 3 experiments. (B) Analysis of Sema3A binding to Npn-1 in the presence of VEGF165. Sema3A (2 μg/mL) was added to Npn-1–coated wells with VEGF165 (0-1600 ng/mL) with (○) or without (□) heparin (2 μg/mL). The results reflect the means ± SD of 3 experiments, and are expressed as the mean percent binding of Sema3A in the presence of VEGF165 compared with no VEGF165.
Based on these results, we examined whether VEGF165 interferes with the binding of Sema3A to Npn-1, with or without heparin (2 μg/mL). As shown in Figure 3B, VEGF165 dose-dependently inhibited the binding of Sema3A to Npn-1. However, even at the highest concentration tested (1600 ng/mL), reduction by VEGF165 was less than 50% with or without heparin. In the biologic assays above (Figures 1, 2), 25 ng/mL VEGF165 inhibited almost completely the activities of Sema3A (1-2 μg/mL), whereas this concentration of VEGF165 did not significantly inhibit the binding of Sema3A (2 μg/mL) to purified Npn-1. These results suggested that VEGF165 inhibits Sema3A activity in endothelial cells by mechanisms other than simple binding competition for Npn-1.
VEGF165 inhibits the binding of Sema3A to target cells
In the presence of VEGF165, Npn-1 forms a complex with VEGFR-2, which enhances the binding of VEGF165 to VEGFR-2.22 Also, Npn-1 forms a stable complex with plexin-A1, and this complex has a higher affinity for Sema3A than does Npn-1 alone.41 Since endothelial cells express plexin-A1 and VEGFR-2 in addition to Npn-1,18 we tested whether VEGF165 can compete for Sema3A binding to the surface of endothelial cells. FACS analysis showed that Sema3A/Fc bound to endothelial cells dose dependently, whereas a control B7-1/Fc did not (Figure 4A). When added to cells together with VEGF165 at 4°C for 1 hour, Sema3A/Fc (2 μg/mL) binding to cells was reduced by VEGF165 dose dependently (Figure 4B top). VEGF165 (12.5 ng/mL) reduced Sema3A/Fc binding to cells by approximately 50%. VEGF121, VEGF-B167, VEGF-C, and FGF-2 (all at 100 ng/mL) failed to reduce Sema3A/Fc binding to endothelial cells (Figure 4B).
Figure 4.

VEGF165 specifically inhibits Sema3A binding to endothelial, stromal, and neuronal cells. (A) Concentration dependency of Sema3A/Fc (○) binding to endothelial cells. Control B7-1/Fc (16 μg/mL, □). (B) Endothelial cells were incubated (4°C, 1 hour) with Sema3A/Fc (2 μg/mL) plus VEGF165 (0-100 ng/mL), VEGF121 (100 ng/mL), VEGF-B167 (100 ng/mL), VEGF-C (100 ng/mL), or FGF-2 (100 ng/mL). (C) Endothelial cells were preincubated (25°C, 1 hour) with or without VEGF165 (12.5 ng/mL), VEGF121 (100 ng/mL), or FGF-2 (100 ng/mL). Sema3A/Fc (2 μg/mL) was added and cells were further incubated at 4°C for 60 minutes. (D) Human stromal HS-5 and (E) rat pheochromocytoma PC12 cells were preincubated (25°C, 1 hour) with VEGF165 (0-250 ng/mL) or VEGF121 (250 ng/mL). Sema3A/Fc (2 μg/mL) was added and cells were further incubated at 4°C for 60 minutes. Sema3A/Fc binding was detected by anti-Fc mAb. Shaded graphs reflect control staining.
We examined Sema3A (2 μg/mL) binding to endothelial cells (1 hour at 4°C) after the cells were preincubated (25°C for 1 hour) with VEGF165 (12.5 ng/mL). VEGF165 was not removed. Under these conditions, VEGF165 at 12.5 ng/mL reduced Sema3A/Fc (2 μg/mL) binding to endothelial cells by approximately 80% (Figure 4C top). These results indicated that preincubation of cells with VEGF165 at 25°C is more effective at reducing Sema3A binding than simultaneous addition of VEGF165 and Sema3A at 4°C. Preincubation of cells with VEGF121 or FGF-2 did not affect the subsequent binding of Sema3A to cells (Figure 4C middle and bottom). These results provided evidence that VEGF165, but not VEGF121, VEGF-B167, VEGF-C, or FGF-2, can compete for the binding of Sema3A to endothelial cells. The observation that preincubation enhanced the ability of VEGF165 to inhibit Sema3A binding to endothelial cells suggested a contribution by mechanisms other than cell binding competition.
Npn-1 is expressed on various cell types.42,43 We examined whether VEGF165 can inhibit the binding of Sema3A to human bone marrow stromal HS-5 and rat pheochromocytoma PC12 cells. Preincubation with VEGF165, but not VEGF121, dose-dependently inhibited the binding of Sema3A to stromal HS-5 (Figure 4D) and rat pheochromocytoma PC12 cells (Figure 4E). These results indicate that VEGF165 can inhibit the binding of Sema3A to cells of various lineages.
VEGF165 reduces endothelial-cell–surface Npn-1 and induces its internalization
To clarify the mechanisms by which VEGF165 reduces the binding of Sema3A to target cells, we examined endothelial-cell–surface expression of Npn-1. Using FACS analysis, we found that 1-hour preincubation at 25°C with VEGF165, but not VEGF121, dose-dependently reduced surface Npn-1 (Figure 5A left). At the concentration of 12.5 ng/mL (0.3 nM), VEGF165 reduced by approximately 50% Npn-1 levels detected on control cells. In parallel experiments, both VEGF165 and VEGF121 reduced surface VEGFR-2 (Figure 5A right) but did not affect surface CD31 in these cells (not shown). VEGF165 also specifically reduced surface Npn-1 in HS-5 cells (not shown; the effect on rat PC12 cells could not be tested because the antibody fails to recognize rat Npn-1).
Figure 5.

VEGF165 reduces levels of cell-surface Npn-1 and induces its internalization. (A) (Left) Npn-1 detected on endothelial cells incubated (25°C, 1 hour) with VEGF165 (0-100 ng/mL) or VEGF121 (0 or 100 ng/mL). (Right) VEGFR-2 detected on endothelial cells incubated with VEGF165 (0 or 25 ng/mL) or VEGF121 (0 or 25 ng/mL). Shaded graphs reflect control staining. (B) Npn-1 detected on endothelial cells stimulated (25°C, 1 hour) with VEGF165 (250 ng/mL) plus heparin (0-400 ng/mL). Unstimulated cells: dotted line. (C) Temperature dependency of VEGF165-induced surface Npn-1 reduction. Endothelial cells were incubated with VEGF165 (25 ng/mL) and heparin (2 μg/mL) at 4°C, 25°C, or 37°C for 5, 15, 30, or 60 minutes. Results reflect mean percent signal intensities with stimulation compared with no stimulation. (D) Npn-1 detected on endothelial cells cultured (37°C, 30 hours) with 25 ng/mL VEGF165, VEGF121, or FGF-2. (E) Npn-1 detected on the endothelial-cell–surface (left) and after cell permeabilization (right). Cells were incubated (25°C, 1 hour) with VEGF165 (0, 25, or 250 ng/mL). (F) Npn-1 is internalized after stimulation with VEGF165. Endothelial cells grown on fibronectin-coated glass slides were incubated (37°C; 0, 5, 30, or 60 minutes) with VEGF165 (25 ng/mL). Npn-1 was visualized under an LSM510 confocal microscope equipped with a Plan-Neofluar 40 ×/1.3 objective lens (Carl Zeiss, Thornwood, NY). Images reflect the merging of fluorescent slice and differential interference contrast (DIC) images. Images were imported into Adobe Photoshop 6.0 (Adobe Systems) for processing. Scale bar, 20 μm.
Since heparin is required for maximal binding of VEGF165 to Npn-1, we examined the effect of heparin on VEGF165 (250 ng/mL)–induced reduction of Npn-1 on endothelial cells. We found that heparin enhances VEGF165-induced reduction of surface Npn-1, with maximal Npn-1 reduction achieved at a heparin concentration of 80 ng/mL (Figure 5B). We also examined the effect of temperature on VEGF165-induced reduction of surface Npn-1. At 4°C, levels of surface Npn-1 reached a plateau after 15 minutes, whereas Npn-1 continued to decrease at 25°C and 37°C (Figure 5C). The reduction of Npn-1 was somewhat more rapid and linear at 37°C than seen at 25°C. When endothelial cells were incubated with VEGF165 (25 ng/mL) at 37°C for 30 hours, levels of surface Npn-1 were approximately 75% lower than those measured on endothelial cells cultured under the same conditions with 25 ng/mL VEGF121 or FGF-2 (Figure 5D). This time- and temperature-dependent pattern of surface Npn-1 decrease induced by VEGF165 suggested either that Npn-1 undergoes structural change, which renders it undetectable under these conditions, or that levels of surface Npn-1 decrease.
We examined whether internalization contributes to the reduction of surface Npn-1 by VEGF165. Therefore, we compared the effects of VEGF165 on cell-surface and total cell-associated levels of Npn-1. By FACS analysis, VEGF165 (25 and 250 ng/mL, 25°C 1 hour) substantially reduced the levels of surface Npn-1 but only slightly reduced the levels of Npn-1 detected after cell permeabilization, which include cell-surface and intracellular Npn-1 (Figure 5E), suggesting that internalization contributed to the reduction of surface Npn-1 levels by VEGF165. Using confocal microscopy, we traced Npn-1 distribution in endothelial cells after VEGF165 stimulation (Figure 5F). After 5-minute incubation with VEGF165 (25 ng/mL, 37°C), Npn-1 was detectable more intensely at the endothelial-cell rim, especially on lamellipodia, whereas after 30 minutes Npn-1 was detectable more intensely in the cytoplasm at the perinuclear region with a dotlike distribution. After 60 minutes, Npn-1 staining was detected almost exclusively at the perinuclear region. VEGF121 and FGF-2 did not induce such changes (not shown). Based on these results, we conclude that VEGF165 reduces cell-surface Npn-1, at least in part, by promoting its internalization.
Characterization of requirements for VEGF-induced reduction of surface Npn-1
The experiments shown in Figures 1, 2, 3, 4, 5 indicated that VEGF165 inhibits Sema3A function in endothelial cells (Figures 1, 2) by competing with Sema3A for binding to cell-surface receptors (Figure 4) and inducing Npn-1 internalization (Figure 5). Since VEGF165 is ineffective at directly competing with Sema3A for binding to Npn-1 (Figure 3B), we examined the potential contribution of VEGF receptors present on endothelial cells. First, we compared a soluble rat Npn-1 (srNpn-1) and a soluble VEGFR-1 (sVEGFR-1) for their ability to compete with VEGF165 binding to immobilized Npn-1. srNpn-1, which is expected to serve as a direct competitor for VEGF165, inhibited binding of VEGF165 to immobilized Npn-1 (Figure 6A left). By contrast, sVEGFR-1, which interferes with VEGF binding to both VEGFR-1 and VEGFR-2,44 minimally affected the binding of VEGF165 to immobilized Npn-1, even at the highest VEGFR-1 concentration (3.2 μg/mL) tested (Figure 6A right). Thus, sVEGFR-1 does not directly compete with VEGF165 binding to Npn-1.
Figure 6.

sNpn-1 and sVEGFR-1 counteract VEGF165 effects on Npn-1 expression and Sema3A function. (A) Binding of VEGF165 (25 ng/mL) to Npn-1 (2 μg/mL)–coated wells with srNpn-1 (0-3.2 μg/mL) or sVEGFR-1 (0-3.2 μg/mL). Error bars depict ± SD of triplicate determinations. (B) Npn-1 on endothelial cells incubated (25°C, 1 hour) with or without srNpn-1 (5 μg/mL) (top left); VEGF165 (25 ng/mL) plus srNpn-1 (0-800 ng/mL) (bottom left); VEGF165 (25 ng/mL) plus control B7-1/Fc (0 or 800 ng/mL) (top right); and VEGF165 (25 ng/mL) plus sVEGFR-1 (0-800 ng/mL) (bottom right). Dotted line: Npn-1 without stimulation of VEGF165. (C) Npn-1 detected by confocal microscopy (scale bar, 20 μm) in cells incubated (37°C; 30 minutes) in medium only (left panel), with VEGF165 (25 ng/mL), with VEGF165 plus srNpn-1 (3.2 μg/mL), or with VEGF165 plus sVEGFR-1 (3.2 μg/mL) (right panel). Images were acquired and processed as described for Figure 5F. (D) Lamellipodia retraction in endothelial cells incubated (37°C, 1 hour) with Sema3A (2 μg/mL), Sema3A (2 μg/mL) plus VEGF165 (25 ng/mL), with or without sVEGFR-1 (0.8 or 3.2 μg/mL). Error bars depict ± SD of triplicate determinations. *P < .05; **P < .01.
We then compared the effects of srNpn-1 and sVEGFR-1 on VEGF165-induced reduction of cell-surface Npn-1. To detect human endothelial-cell–surface Npn-1, we used the anti–human Npn-1 mAb AD5-17F6. This mAb specifically recognizes human Npn-1 and does not cross-react with rat Npn-1 because excess amounts of srNpn-1 (5 μg/mL) did not significantly alter recognition of human Npn-1 by this antibody (Figure 6B upper left). The species specificity of this antibody to human Npn-1 contrasts with the lack of species specificity of human VEGF165 and Sema3A, which can both bind to srNpn-1 (Figure 3A). Taking advantage of this anti–human Npn-1 mAb for detection of surface Npn-1 on human endothelial cells, we found that srNpn-1 dose-dependently blocked VEGF165-induced reduction of cell-surface Npn-1 (Figure 6B lower left). These results indicated that a direct interaction of VEGF165 with membrane-bound Npn-1 is essential for VEGF165 to reduce cell-surface Npn-1. By confocal microscopy, we found that srNpn-1 reduced VEGF165-induced internalization of Npn-1, indicating that binding to cell-surface Npn-1 is also essential for VEGF165 to promote Npn-1 internalization (Figure 6C).
To examine whether interaction with VEGFR-1 and/or VEGFR-2 is necessary for VEGF165 to reduce cell-surface Npn-1 and promote Npn-1 internalization, we used sVEGFR-1, which does not compete for VEGF165 binding to Npn-1 (Figure 6A) but neutralizes VEGF bioactivity by competing for VEGF binding to VEGFR-1 and VEGFR-2.44 Endothelial cells were incubated with VEGF165 (25 ng/mL) plus sVEGFR-1 (0-800 ng/mL). Dose dependently, sVEGFR-1 blocked VEGF165-induced reduction of cell-surface Npn-1 (Figure 6B lower right), whereas a control protein (B7-1/Fc) had no effect (Figure 6B upper right). In addition, by confocal microscopy, we found that sVEGFR-1 reduced VEGF165-induced internalization of Npn-1 (Figure 6C). These results indicated that VEGF165 must interact with endothelial-cell–surface VEGFR-1 and/or VEGFR-2 to effectively reduce cell-surface Npn-1 and promote its internalization, perhaps by bridging VEGFR-1 and/or VEGFR-2 with Npn-1.
Finally, we investigated the effect of sVEGFR-1 on functional assays in which Sema3A promotes endothelial-cell retraction and VEGF165 reduces this effect. Dose dependently, sVEGFR-1 reduced VEGF165 inhibition of the Sema3A-induced retraction of endothelial cells (Figure 6D). This result is consistent with the results shown in Figure 6B-C that sVEGFR-1 reduces Npn-1 internalization induced by VEGF165, thereby enhancing surface Npn-1 available for binding to Sema3A.
Sema3A induces Npn-1 internalization
It was reported that Sema3A could induce Npn-1 endocytosis in the COS7-cell line, which coexpress Npn-1 and the L1 cell-adhesion molecule.45 We examined whether Sema3A can also induce Npn-1 internalization in primary endothelial cells. We found that Sema3A dose-dependently reduced endothelial-cell–surface Npn-1 (Figure 7A), and that approximately 1 μg/mL (4 nM) Sema3A was required to achieve a 50% reduction in Npn-1 levels compared with control. This indicates that approximately 13-fold more Sema3A is required to achieve levels of surface Npn-1 reduction comparable with those induced by VEGF165 under the same experimental conditions (Figure 5A). As expected, srNpn-1 blocked reduction of surface Npn-1 induced by Sema3A, whereas sVEGFR-1 was ineffective (data not shown). By using confocal microscopy, we confirmed that Sema3A could induce Npn-1 internalization (Figure 7B). After 5-minute stimulation with Sema3A, Npn-1 accumulated mainly in the cell-to-cell junctional regions, and after 30 minutes Npn-1 accumulated in the cytoplasmic region similar to the pattern noted after stimulation with VEGF165. These results demonstrate that Sema3A can induce Npn-1 internalization in endothelial cells.
Figure 7.

Sema3A down-regulates surface Npn-1 and promotes Npn-1 internalization. (A) Surface Npn-1 detected on endothelial cells incubated (25°C, 1 hour) with Sema3A (0-4 μg/mL). Shaded graphs reflect control staining. (B) Npn-1 detected by confocal microscopy in endothelial cells stimulated with Sema3A (2 μg/mL) at 37°C for 0, 5, 30, or 60 minutes. Scale bar, 20 μm. Images were acquired and processed as described for Figure 5F.
Discussion
We show that Sema3A inhibits endothelial-cell lamellipodia formation, attachment, survival, and growth, and that VEGF165 counters these activities of Sema3A. We further show that this functional competition between Sema3A and VEGF165 is attributable to preferential binding of VEGF165 to target cells and internalization of the shared receptor Npn-1. This potency difference at promoting Npn-1 internalization can dictate a priority for Npn-1 use by VEGF165 over Sema3A, effectively blocking Sema3A function. Thus, our results demonstrate that control of surface levels of the common receptor is a critical mechanism for functional prioritization among ligands that share a receptor. Sema3A could also reduce surface levels of Npn-1 by promoting its internalization but required a higher concentration than VEGF165 at reducing Npn-1 surface levels. The conclusion that Sema3A is less potent than VEGF165 may not apply to the various endothelial environments in vivo. Relative effective concentrations of endogenous Sema3A and VEGF165 in vivo are unknown and may depend on untested variables, such as levels of plexin-A and VEGF receptors in various cells, the contribution of posttranscriptional modifications, and protein degradation.
We established a requirement for linkage between cell-surface Npn-1 and VEGFR-1 or VEGFR-2 in VEGF165-induced Npn-1 internalization. This conclusion comes from the observation that soluble VEGFR-1 extracellular domain, which does not interfere with the binding of VEGF165 to Npn-1 (Figure 6A) but competes for VEGF165 binding to VEGFR-1 or VEGFR-2,44 blocked VEGF165-induced down-regulation of surface Npn-1 (Figure 6B). Consistent with this conclusion, VEGF121, which does not bind Npn-1 but binds to VEGFRs, did not affect Npn-1 levels. VEGFRs are subject to endocytosis and subsequent ubiquitination by the E3 ubiquitin ligase/multiadaptor protein Cbl.46,47 It will be important to establish whether Npn-1 is also degraded. Of interest, we observed PEST sequences for protein degradation in the region of amino acids 823 to 840 of human Npn-1.48
In addition to VEGF165, PlGF-2 is known to inhibit Sema3A-induced growth cone collapse in dorsal root ganglion neurons.49 PlGF-2 has the potential to link Npn-1 to VEGF receptors, and thus may down-regulate Npn-1 by the same mechanism of VEGF165. FGF family members can also interact with Npn-1.39 However, our results show that FGF-2 did not change Npn-1 levels and did not inhibit endothelial-cell retraction induced by Sema3A. Thus, FGF-2 may not bridge Npn-1 to FGF receptor. Also, VEGF165 could inhibit other class 3 semaphorin family members that use the common receptor Npn-1. If so, our observations could explain why VEGF165, but not VEGF121, inhibited Sema3B-induced apoptosis in lung and breast cancers.50 Also, since VEGF165 can bind to Npn-251 in addition to Npn-1, it will be interesting to examine whether VEGF165 can induce internalization of Npn-2 and thereby inhibit Npn-2–mediated effects of class 3 semaphorins.
Npn-1 is known to serve as a cell-to-cell adhesion molecule,33 which facilitates contact between resting T cells and dendritic cells and promotes primary immune responses.52 Npn-1 is expressed in stromal cells, B lymphocytes, monocytes, and erythrocytes.42,53 Our observation that VEGF165 reduces surface levels of Npn-1 may explain some of the functions of VEGF on immune and hematopoietic cells.54
In essence, receptor components shared by multiple ligands play additional roles besides ligand binding. If the signal-transducing chain is shared, as in the case of gp130 that is shared by different members of the IL-6 family of cytokines, the common signaling chain serves as a convergence mechanism for function.55 As a result, distinct cytokines, which first bind to their specific receptor and then the ligand/ligand-specific receptor complexes engage the common signal transducer gp130, display overlapping functions. By contrast, if the ligand-binding chain is shared and the signaling chain differs, ligands with different and even contrasting activities converge on the same ligand-binding receptor. Npn-1 represents an example of a common receptor system, which binds multiple ligands as a hub receptor, but ligand/receptor complexes use specific signal transducers to transduce their respective signal. In this case, the common binding chain serves as a convergence of different ligands onto the same target cell, which provides a means for distinct ligands, even with opposing function, to share the same cell target. In such a receptor system, a mechanism for ligand prioritization appears essential to proper cell function. Here, we describe how a common receptor can serve as a cell membrane gatekeeper by selecting a specific ligand and undergoing internalization.
Acknowledgments
We thank Ms Yumi Narazaki for blindly scoring retraction assays; Ms Susan Garfield and Mr Stephen Wincovitch for technical assistance with confocal microscopy; Dr Lei Yao for isolation of HUVECs; and Drs Anna Virginia Gulino, Takayuki Nakayama, and Yoshiyasu Aoki for helpful suggestions.
Prepublished online as Blood First Edition Paper, January 19, 2006; DOI 10.1182/blood-2005-10-4113.
Supported by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 U.S.C. section 1734.
References
- 1.Serini G, Bussolino F. Common cues in vascular and axon guidance. Physiology (Bethesda). 2004;19: 348-354. [DOI] [PubMed] [Google Scholar]
- 2.Weinstein BM. Vessels and nerves: marching to the same tune. Cell. 2005;120: 299-302. [DOI] [PubMed] [Google Scholar]
- 3.Storkebaum E, Lambrechts D, Carmeliet P. VEGF: once regarded as a specific angiogenic factor, now implicated in neuroprotection. Bioessays. 2004;26: 943-954. [DOI] [PubMed] [Google Scholar]
- 4.Carmeliet P, Tessier-Lavigne M. Common mechanisms of nerve and blood vessel wiring. Nature. 2005;436: 193-200. [DOI] [PubMed] [Google Scholar]
- 5.Kolodkin AL, Matthes DJ, Goodman CS. The semaphorin genes encode a family of transmembrane and secreted growth cone guidance molecules. Cell. 1993;75: 1389-1399. [DOI] [PubMed] [Google Scholar]
- 6.Tessier-Lavigne M, Goodman CS. The molecular biology of axon guidance. Science. 1996;274: 1123-1133. [DOI] [PubMed] [Google Scholar]
- 7.Pasterkamp RJ, Kolodkin AL. Semaphorin junction: making tracks toward neural connectivity. Curr Opin Neurobiol. 2003;13: 79-89. [DOI] [PubMed] [Google Scholar]
- 8.Tamagnone L, Comoglio PM. Signalling by semaphorin receptors: cell guidance and beyond. Trends Cell Biol. 2000;10: 377-383. [DOI] [PubMed] [Google Scholar]
- 9.Gu C, Yoshida Y, Livet J, et al. Semaphorin 3E and plexin-D1 control vascular pattern independently of neuropilins. Science. 2005;307: 265-268. [DOI] [PubMed] [Google Scholar]
- 10.He Z, Tessier-Lavigne M. Neuropilin is a receptor for the axonal chemorepellent semaphorin III. Cell. 1997;90: 739-751. [DOI] [PubMed] [Google Scholar]
- 11.Kolodkin AL, Levengood DV, Rowe EG, Tai YT, Giger RJ, Ginty DD. Neuropilin is a semaphorin III receptor. Cell. 1997;90: 753-762. [DOI] [PubMed] [Google Scholar]
- 12.Feiner L, Koppel AM, Kobayashi H, Raper JA. Secreted chick semaphorins bind recombinant neuropilin with similar affinities but bind different subsets of neurons in situ. Neuron. 1997;19: 539-545. [DOI] [PubMed] [Google Scholar]
- 13.Takahashi T, Nakamura F, Jin Z, Kalb RG, Strittmatter SM. Semaphorins A and E act as antagonists of neuropilin-1 and agonists of neuropilin-2 receptors. Nat Neurosci. 1998;1: 487-493. [DOI] [PubMed] [Google Scholar]
- 14.Chen H, Chedotal A, He Z, Goodman CS, Tessier-Lavigne M. Neuropilin-2, a novel member of the neuropilin family, is a high affinity receptor for the semaphorins Sema E and Sema IV but not Sema III. Neuron. 1997;19: 547-559. [DOI] [PubMed] [Google Scholar]
- 15.Gitler AD, Lu MM, Epstein JA. PlexinD1 and semaphorin signaling are required in endothelial cells for cardiovascular development. Dev Cell. 2004;7: 107-116. [DOI] [PubMed] [Google Scholar]
- 16.Takahashi T, Strittmatter SM. Plexina1 autoinhibition by the plexin sema domain. Neuron. 2001; 29: 429-439. [DOI] [PubMed] [Google Scholar]
- 17.Yaron A, Huang PH, Cheng HJ, Tessier-Lavigne M. Differential requirement for plexin-A3 and -A4 in mediating responses of sensory and sympathetic neurons to distinct class 3 semaphorins. Neuron. 2005;45: 513-523. [DOI] [PubMed] [Google Scholar]
- 18.Serini G, Valdembri D, Zanivan S, et al. Class 3 semaphorins control vascular morphogenesis by inhibiting integrin function. Nature. 2003;424: 391-397. [DOI] [PubMed] [Google Scholar]
- 19.Gabrilovich DI, Chen HL, Girgis KR, et al. Production of vascular endothelial growth factor by human tumors inhibits the functional maturation of dendritic cells. Nat Med. 1996;2: 1096-1103. [DOI] [PubMed] [Google Scholar]
- 20.Ferrara N, Gerber HP, LeCouter J. The biology of VEGF and its receptors. Nat Med. 2003;9: 669-676. [DOI] [PubMed] [Google Scholar]
- 21.Rosenstein JM, Krum JM. New roles for VEGF in nervous tissue: beyond blood vessels. Exp Neurol. 2004;187: 246-253. [DOI] [PubMed] [Google Scholar]
- 22.Soker S, Takashima S, Miao HQ, Neufeld G, Klagsbrun M. Neuropilin-1 is expressed by endothelial and tumor cells as an isoform-specific receptor for vascular endothelial growth factor. Cell. 1998;92: 735-745. [DOI] [PubMed] [Google Scholar]
- 23.Soker S, Fidder H, Neufeld G, Klagsbrun M. Characterization of novel vascular endothelial growth factor (VEGF) receptors on tumor cells that bind VEGF165 via its exon 7-encoded domain. J Biol Chem. 1996;271: 5761-5767. [DOI] [PubMed] [Google Scholar]
- 24.Soker S, Miao HQ, Nomi M, Takashima S, Klagsbrun M. VEGF165 mediates formation of complexes containing VEGFR-2 and neuropilin-1 that enhance VEGF165-receptor binding. J Cell Biochem. 2002;85: 357-368. [DOI] [PubMed] [Google Scholar]
- 25.Makinen T, Olofsson B, Karpanen T, et al. Differential binding of vascular endothelial growth factor B splice and proteolytic isoforms to neuropilin-1. J Biol Chem. 1999;274: 21217-21222. [DOI] [PubMed] [Google Scholar]
- 26.Migdal M, Huppertz B, Tessler S, et al. Neuropilin-1 is a placenta growth factor-2 receptor. J Biol Chem. 1998;273: 22272-22278. [DOI] [PubMed] [Google Scholar]
- 27.Mamluk R, Gechtman Z, Kutcher ME, Gasiunas N, Gallagher J, Klagsbrun M. Neuropilin-1 binds vascular endothelial growth factor 165, placenta growth factor-2, and heparin via its b1b2 domain. J Biol Chem. 2002;277: 24818-24825. [DOI] [PubMed] [Google Scholar]
- 28.Wise LM, Veikkola T, Mercer AA, et al. Vascular endothelial growth factor (VEGF)-like protein from orf virus NZ2 binds to VEGFR2 and neuropilin-1. Proc Natl Acad Sci U S A. 1999;96: 3071-3076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gu C, Limberg BJ, Whitaker GB, et al. Characterization of neuropilin-1 structural features that confer binding to semaphorin 3A and vascular endothelial growth factor 165. J Biol Chem. 2002;277: 18069-18076. [DOI] [PubMed] [Google Scholar]
- 30.Kitsukawa T, Shimizu M, Sanbo M, et al. Neuropilin-semaphorin III/D-mediated chemorepulsive signals play a crucial role in peripheral nerve projection in mice. Neuron. 1997;19: 995-1005. [DOI] [PubMed] [Google Scholar]
- 31.Kawasaki T, Kitsukawa T, Bekku Y, et al. A requirement for neuropilin-1 in embryonic vessel formation. Development. 1999;126: 4895-4902. [DOI] [PubMed] [Google Scholar]
- 32.Gu C, Rodriguez ER, Reimert DV, et al. Neuropilin-1 conveys semaphorin and VEGF signaling during neural and cardiovascular development. Dev Cell. 2003;5: 45-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Takagi S, Kasuya Y, Shimizu M, et al. Expression of a cell adhesion molecule, neuropilin, in the developing chick nervous system. Dev Biol. 1995; 170: 207-222. [DOI] [PubMed] [Google Scholar]
- 34.Miao HQ, Soker S, Feiner L, Alonso JL, Raper JA, Klagsbrun M. Neuropilin-1 mediates collapsin-1/semaphorin III inhibition of endothelial cell motility: functional competition of collapsin-1 and vascular endothelial growth factor-165. J Cell Biol. 1999;146: 233-242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sondell M, Lundborg G, Kanje M. Vascular endothelial growth factor has neurotrophic activity and stimulates axonal outgrowth, enhancing cell survival and Schwann cell proliferation in the peripheral nervous system. J Neurosci. 1999;19: 5731-5740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Salvucci O, Yao L, Villalba S, Sajewicz A, Pittaluga S, Tosato G. Regulation of endothelial cell branching morphogenesis by endogenous chemokine stromal-derived factor-1. Blood. 2002;99: 2703-2711. [DOI] [PubMed] [Google Scholar]
- 37.Ruoslahti E, Vaheri A. Cell-to-cell contact and extracellular matrix. Curr Opin Cell Biol. 1997;9: 605-607. [DOI] [PubMed] [Google Scholar]
- 38.Fan J, Mansfield SG, Redmond T, Gordon-Weeks PR, Raper JA. The organization of F-actin and microtubules in growth cones exposed to a brain-derived collapsing factor. J Cell Biol. 1993;121: 867-878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.West DC, Rees CG, Duchesne L, et al. Interactions of multiple heparin binding growth factors with neuropilin-1 and potentiation of the activity of fibroblast growth factor-2. J Biol Chem. 2005;280: 13457-13464. [DOI] [PubMed] [Google Scholar]
- 40.Lubarsky B, Krasnow MA. Tube morphogenesis: making and shaping biological tubes. Cell. 2003; 112: 19-28. [DOI] [PubMed] [Google Scholar]
- 41.Takahashi T, Fournier A, Nakamura F, et al. Plexin-neuropilin-1 complexes form functional semaphorin-3A receptors. Cell. 1999;99: 59-69. [DOI] [PubMed] [Google Scholar]
- 42.Tordjman R, Ortega N, Coulombel L, Plouet J, Romeo PH, Lemarchandel V. Neuropilin-1 is expressed on bone marrow stromal cells: a novel interaction with hematopoietic cells? Blood. 1999; 94: 2301-2309. [PubMed] [Google Scholar]
- 43.Schwamborn JC, Fiore R, Bagnard D, Kappler J, Kaltschmidt C, Puschel AW. Semaphorin 3A stimulates neurite extension and regulates gene expression in PC12 cells. J Biol Chem. 2004;279: 30923-30926. [DOI] [PubMed] [Google Scholar]
- 44.Kendall RL, Thomas KA. Inhibition of vascular endothelial cell growth factor activity by an endogenously encoded soluble receptor. Proc Natl Acad Sci U S A. 1993;90: 10705-10709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Castellani V, Falk J, Rougon G. Semaphorin3A-induced receptor endocytosis during axon guidance responses is mediated by L1 CAM. Mol Cell Neurosci. 2004;26: 89-100. [DOI] [PubMed] [Google Scholar]
- 46.Duval M, Bedard-Goulet S, Delisle C, Gratton JP. Vascular endothelial growth factor-dependent down-regulation of Flk-1/KDR involves Cbl-mediated ubiquitination: consequences on nitric oxide production from endothelial cells. J Biol Chem. 2003;278: 20091-20097. [DOI] [PubMed] [Google Scholar]
- 47.Kobayashi S, Sawano A, Nojima Y, Shibuya M, Maru Y. The c-Cbl/CD2AP complex regulates VEGF-induced endocytosis and degradation of Flt-1 (VEGFR-1). Faseb J. 2004;18: 929-931. [DOI] [PubMed] [Google Scholar]
- 48.Rechsteiner M, Rogers SW. PEST sequences and regulation by proteolysis. Trends Biochem Sci. 1996;21: 267-271. [PubMed] [Google Scholar]
- 49.Cheng L, Jia H, Lohr M, et al. Anti-chemorepulsive effects of vascular endothelial growth factor and placental growth factor-2 in dorsal root ganglion neurons are mediated via neuropilin-1 and cyclooxygenase-derived prostanoid production. J Biol Chem. 2004;279: 30654-30661. [DOI] [PubMed] [Google Scholar]
- 50.Castro-Rivera E, Ran S, Thorpe P, Minna JD. Semaphorin 3B (SEMA3B) induces apoptosis in lung and breast cancer, whereas VEGF165 antagonizes this effect. Proc Natl Acad Sci U S A. 2004;101: 11432-11437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gluzman-Poltorak Z, Cohen T, Herzog Y, Neufeld G. Neuropilin-2 is a receptor for the vascular endothelial growth factor (VEGF) forms VEGF-145 and VEGF-165 [corrected]. J Biol Chem. 2000; 275: 18040-18045. [DOI] [PubMed] [Google Scholar]
- 52.Tordjman R, Lepelletier Y, Lemarchandel V, et al. A neuronal receptor, neuropilin-1, is essential for the initiation of the primary immune response. Nat Immunol. 2002;3: 477-482. [DOI] [PubMed] [Google Scholar]
- 53.Yamada Y, Oike Y, Ogawa H, et al. Neuropilin-1 on hematopoietic cells as a source of vascular development. Blood. 2003;101: 1801-1809. [DOI] [PubMed] [Google Scholar]
- 54.Gabrilovich D, Ishida T, Oyama T, et al. Vascular endothelial growth factor inhibits the development of dendritic cells and dramatically affects the differentiation of multiple hematopoietic lineages in vivo. Blood. 1998;92: 4150-4166. [PubMed] [Google Scholar]
- 55.Kishimoto T, Akira S, Narazaki M, Taga T. Interleukin-6 family of cytokines and gp130. Blood. 1995;86: 1243-1254. [PubMed] [Google Scholar]