

Abstract
The suppressive capacity of naturally occurring mouse CD4+CD25+ T cells on T-cell activation has been well documented. The present study is focused on the interaction of CD4+CD25+ T cells and B cells. By coculturing preactivated CD4+CD25+ T cells with B cells in the presence of polyclonal B-cell activators, we found that B-cell proliferation was significantly suppressed. The suppression of B-cell proliferation was due to increased cell death caused by the CD4+CD25+ T cells in a cell-contact–dependent manner. The induction of B-cell death is not mediated by Fas–Fas ligand pathway, but surprisingly, depends on the up-regulation of perforin and granzymes in the CD4+CD25+ T cells. Furthermore, activated CD4+CD25+ T cells preferentially killed antigen-presenting but not bystander B cells. Our results demonstrate that CD4+CD25+ T cells can act directly on B cells and suggest that the prevention of autoimmunity by CD4+CD25+ T cells can be explained, at least in part, by the direct regulation of B-cell function.
Introduction
CD4+CD25+ regulatory/suppressor T cells were originally identified by their capacity to primarily control immune responses to autoantigens,1-3 but recent studies have shown that they exert pleiotropic suppressive effects on immune responses to alloantigens,4 tumor antigens,5 and infectious agents.6,7 The functional phenotype of CD4+CD25+ T cells has been extensively characterized in vitro. CD4+CD25+ T cells do not proliferate when stimulated via the TCR but are able to proliferate when costimulated with IL-2.8,9 On activation via their TCR, they suppress the proliferation of both CD4+ T cells8 and CD8+ T cells10 by inhibiting the transcription of IL-2 mRNA.8 In vitro studies have demonstrated that the suppression is mediated exclusively by a cell-contact–dependent, cytokine-independent mechanism.4,9,10 However, suppression in some in vivo experimental models requires the cooperation of suppressive cytokines.11-13 The nature of the target cells for CD4+CD25+ T-cell–mediated suppression remains controversial. CD4+CD25+ T cells can suppress the activation of CD8+ T cells in the absence of professional antigenpresenting cells (APCs),10 but this does not exclude the possibility that they are also capable of acting on other cell types. One recent study has suggested that some (or all) of the suppressive effects of mouse CD4+CD25+ T cells on T-cell proliferation are secondary to CD4+CD25+ T-cell–mediated killing of the responder cells by a perforin-independent, granzyme-B–dependent pathway.14 In the day 3 thymectomy model of organ-specific autoimmune disease, CD4+CD25+ T cells suppressed the activation of autoantigen-specific T cells and inhibited the production of autoantibodies to the target organ.2,3 Similar results have been obtained in cell transfer studies using TCR transgenic T cells.15-18 However, it remains unclear whether these results are secondary to a direct effect of CD4+CD25+ T cells on B-cell function or whether the effects are mediated indirectly by an inhibition of T-helper function. One recent study has shown that activation of both human T regulatory 1 (Tr1)–like cells and CD4+CD25+ T cells by cross-linking CD3 and CD46 induces potent lytic activity against T cells, monocytes, and dendritic cells.19
In this study, we examined the effects of CD4+CD25+ T cells on B-cell function. We demonstrate that CD4+CD25+ T cells suppress B-cell proliferation in response to polyclonal B-cell activators by inducing death of the responding B cells. Most interestingly, B-cell death is not mediated by the Fas–Fas ligand (FasL) pathway, but instead is mediated by a granzyme-dependent, partially perforin-dependent pathway. The implications of these findings for the role of CD4+CD25+ T cells in the control of B-cell function in vivo are discussed.
Materials and methods
Mice
Female BALB/c and C57BL/6 mice were obtained from the National Cancer Institute (Frederick, MD). Mice expressing a transgenic TCR specific for influenza hemagglutinin peptide, HA110-119,20 were provided by Dr H. von Boehmer (Dana-Farber Cancer Institute, Boston, MA) and maintained at Taconic Farms (Germantown, NY) under an NIAID contract. OT-II mice were purchased from Taconic Farms. B6.MRL-Tnfrsf6 lpr, B6Smn.C3-Tnfsf gld, and perforin-deficient mice were purchased from Jackson Laboratories (Bar Harbor, ME).
Antibodies and flow cytometry
PE–anti–mouse CD25 (PC61), APC–rat anti–mouse CD4 (L3T4), FITC–anti–mouse B220, anti–mouse CD3 (2C11), FITC–anti–mouse H-2Dd, rat anti–mouse CD107a (LAMP-1), FITC–anti–mouse CD45.1, and annexin V–PE apoptosis detection kit were purchased from BD Pharmingen (San Diego, CA). Tricolor anti–mouse CD4 and PE–anti–human granzyme B (GB12) were from Caltag Laboratories (Burlingame, CA). The cross-reactivity of anti–human granzyme B for mouse protein has been confirmed previously.21 AffiniPure F (ab′)2 fragment goat anti–mouse IgM was purchased from Jackson ImmunoReasearch (West Grove, PA). LPS (Escherichia coli 026:B6) and 3,4-dichloroisocoumarin (DCI) were purchased from Sigma (St Louis, MO). HA110-119 peptide was synthesized by the Peptide Synthesis Laboratory (NIAID, NIH, Bethesda, MD). OVA323-339 was purchased from American Peptide (Sunnyvale, CA). Human IL-2 was obtained from the Preclinical Repository of the Biological Resources Branch of the National Cancer Institute. TNFR:Fc was a gift from Dr Peter Lipsky (National Institute of Arthritis and Musculoskeletal and Skin Diseases, NIH). rhFas:Fc and TRAIL-receptor 1-4:Fc were purchased from Alexis (San Diego, CA). Mouse CD19, CD8α (Ly-2), CD90, and anti-PE microbeads were purchased from Miltenyi Biotec (Auburn, CA). The hybridoma cell line producing monoclonal antibody (mAb) against CD40 (FGK 45) was a kind gift from Dr T. Rolink (Basel Institute for Immunology, Basel, Switzerland).
After staining, cells were subjected to 4-color flow cytometric analysis using a FACSCalibur flow cytometer (BD Biosciences) with CellQuest software (BD Biosciences) as described previously.10
Cell purification, activation, and culture
CD4+CD25+, CD4+CD25–, and CD8+ T cells were isolated by cell sorting on a FACStar Cell Sorter (Becton Dickinson, San Jose, CA) as previously described,10 resulting in a purity of 99.5%. Purified T cells were activated with plate-bound anti-CD3 (5 μg/mL) and IL-2 (100 U/mL) in complete RPMI 1640 media for 3 days and were expanded in complete media supplemented with IL-2 for an additional 2 to 4 days. APCs were prepared by depleting CD90+ cells from splenocytes on an auto-magnetic-activate cell sorter (autoMACS). APCs were irradiated with 3000 R. B cells were positively selected from CD90+-depleted splenocytes using CD19 microbeads. The purity of B cells ranged from 93% to 95%.
Cells were cultured in RPMI 1640 medium supplemented with 10% heat-inactivated fetal calf serum (FCS), penicillin (100 U/mL), streptomycin (100 μg/mL), 2 mM l-glutamine, 10 mM HEPES, 0.1 mM nonessential amino acids, 1 mM sodium pyruvate (all from Biofluids, Rockville, MD), and 50 μM 2-ME (Sigma).
Proliferation assay
B cells (5 × 104) stimulated with 3 μg/mL LPS were cocultured with the indicated number of CD4+CD25+ or CD4+CD25– T cells in 96-well plates (0.2 mL) in the presence or absence of soluble anti-CD3 (2 μg/mL) for 72 hours. 3H-TdR was added during the last 6 hours of culture. All experiments were performed in triplicate. Transwell experiments were carried out in 24-well plates (0.4-μm pore size, Corning, Corning, NY). After 66 hours of culture, cells in the lower wells were harvested and transferred to 96-well plates. 3H-TdR incorporation was determined for the last 6 hours.
Apoptosis assay
Purified B cells were stimulated with LPS (5 μg/mL) in complete media for 20 hours. Viable B-cell blasts were isolated by Lympholyte-M gradient (CedarLane Labs, Hornby, ON, Canada) and cultured with preactivated CD4+CD25+ or CD4+CD25– T cells in the presence or absence of anti-CD3 (2 μg/mL) or HA peptide (16 μM) for indicated periods of time (8-18 hours). The effector-to-target (E/T) ratio was 5:1 or as specified. The cells were washed and stained with 7-aminoactinomycin (7-AAD), PE–anti–annexin V, and FITC–anti–mouse B220 in annexin V–binding buffer. The percentages of annexin V+ and 7-AAD+ cells were determined.
Real-time PCR
Total RNA was isolated with an RNeasy kit (Qiagen, Valencia, CA) from freshly purified or activated CD4+CD25+, CD4+CD25–, or CD8+ T cells. cDNA was synthesized using Superscript II reverse transcriptase with random primers (Invitrogen, Carlsbad, CA). Primers and FAM-labeled probes for granzyme B and perforin were purchased from Applied Biosystems (Foster City, CA). The primers for granzyme B are 5′-GGGAAGATGAAGATCCTCCTGC and 5′-TGATCTCCCCTGCCTTTGTC, and the probe for granzyme B is 5′-CTGCTGACCTTGTCTCTGGCCTCCA. The primers for perforin are 5′-CCCTAGGCCAGAGGCAAAC and 5′-AAAATTGGCTACCTTGGAGTGG, and the probe for perforin is 5′-TGCGCGCCTCCGTGGCT. The expression of granzyme B and perforin was normalized relative to respective 18S rRNA (Applied Biosystems). Unstimulated CD4+CD25– T cells were given an arbitrary value of 1.0 and the relative changes in gene expression in the samples were plotted relative to that value. All polymerase chain reaction (PCR) assays were performed in triplicate on an ABI Prism 7700 Sequence Detection System (Applied Biosystems).
Detection of granzyme B by intracellular staining
CD4+CD25+, CD4+CD25–, and CD8+ T cells were stained with anti-CD4 and anti-CD8, fixed with 4% paraformaldehyde at 37°C for 5 minutes, and permeabilized with 0.5% Triton. Cells were stained with PE–anti–mouse granzyme B or isotype control (mouse IgG1), and examined by fluorescence-activated cell sorting (FACS) analysis.
Cell stimulation and immunofluorescent staining for CD107a and granzyme B
Preactivated CD4+CD25+, CD4+CD25–, and CD8+ T cells (5 × 104) were restimulated with 1 μg/mL soluble anti-CD3 and irradiated T-depleted spleen cells (5 × 104) for the indicated periods of time with FITC-anti–mouse CD107a and GolgiStop (BD PharMingen) included in the culture during restimulation. The basal level of CD107a expression in each sample was determined prior to restimulation. After restimulation, the cells were stained with anti-CD107a, anti-CD4, and anti-CD8. The cells were then fixed, permeabilized, and stained with PE–anti–mouse granzyme B in the presence of 0.5% Triton-X at room temperature.
Results
Preactivated CD4+CD25+ T cells suppress B-cell proliferation in a cell-contact–dependent, but cytokine-independent, manner
To determine whether CD4+CD25+ T cells have an effect on B-cell proliferation, highly purified B cells were stimulated with LPS in the presence of CD4+CD25+ T cells (Figure 1A). Because CD4+CD25+ T cells must be activated via their TCR to exert their inhibitory effects on responder T cells, it was not surprising that inhibition of LPS-induced B-cell proliferation was not observed when freshly explanted CD4+CD25+ T cells were added in the absence of a TCR stimulus. However, addition of anti-CD3, soluble or plate-bound, to the cocultures of fresh CD4+CD25+ T cells with B cells also did not affect B-cell proliferation (Figure 1A and data not shown).
Figure 1.

CD4+CD25+ T cells suppress B-cell proliferation. (A) Preactivated, but not fresh, CD4+CD25+ T cells suppress LPS-primed B-cell proliferation in the presence of anti-CD3. B cells (5 × 104) from C57BL/6 mice were stimulated with LPS (3 μg/mL) and cocultured with the graded numbers of freshly explanted CD4+CD25+ T cells (▴, ▵), or preactivated CD4+CD25+ T cells (▪, □) in the presence (▪, ▴) or absence (▵, □) of soluble anti-CD3 (2 μg/mL). Note that preactivated CD4+CD25+ T cells cultured in the presence of LPS and anti-CD3, but in the absence of B cells, incorporated little if any radioactivity (○). *P < .01 between the absence and presence of anti-CD3 with preactivated CD4+CD25+ T cells. The statistical analysis was based on the Student t test. (B) The suppression of B-cell proliferation is specific to CD4+CD25+ T cells. B cells were stimulated with LPS (3 μg/mL) and cultured with irradiated activated CD4+CD25+ T cells (▪, □) or irradiated activated CD4+CD25– T cells (▴, ▵) in the presence (▴, ▪) or absence (▵, □) of soluble anti-CD3. *P < .01 between the CD4+CD25+ and CD4+CD25– cells in the presence of anti-CD3. (C) Activated CD4+CD25+ T cells suppress B-cell proliferation in response to restimulation with HA peptide. B cells stimulated with LPS were cultured with activated CD4+CD25+ T cells from HA-TCR transgenic mice in the absence (□) or presence (▪) of HA peptide (8 μM), or in the presence of anti-CD3 (▾). 3H-TdR incorporation was measured after the cells were pulsed for 6 hours during a total of 72 hours of culture. *P < .01 between the absence and presence of HA peptide with preactivated CD4+CD25+ cells. Results are expressed as the mean of triplicate cultures and are representative of at least 3 experiments.
We then tested CD4+CD25+ T cells that had been preactivated with plate-bound anti-CD3 for 3 days and further expanded in the presence of IL-2. These preactivated CD4+CD25+ T cells are capable of potently inhibiting T-cell proliferation in the absence of restimulation via their TCR. No suppression of B-cell proliferation was observed in the absence of TCR restimulation (Figure 1A), but potent suppression of B-cell proliferation was observed when the preactivated CD4+CD25+ T cells were restimulated with anti-CD3. Similar results were obtained when B cells were activated with anti-IgM F(ab′)2 or anti-CD40 (data not shown). Caramalho et al have reported that CD4+CD25+ T cells respond to LPS.22 However, this response was minimal and LPS did not induce significant proliferation of CD4+CD25+ T cells in our studies (Figure 1A).
To determine if the suppression of B-cell proliferation was a unique property of activated CD4+CD25+ T cells, we cocultured LPS-primed B cells with irradiated, activated CD4+CD25+ or CD4+CD25– cells in the presence or absence of soluble anti-CD3. It was necessary to irradiate both populations of T cells in these studies because activated CD4+CD25– T cells will proliferate in response to restimulation with anti-CD3. Preactivated, irradiated CD4+CD25+ T cells, but not CD4+CD25– T cells, suppressed B-cell proliferation in a dose-dependent manner (Figure 1B). Similar results were also found when nonirradiated CD25+ and CD25– T cells were assayed for their ability to inhibit B-cell proliferation as measured by CFSE dilution (data not shown).
One possible explanation for the requirement for restimulation of the preactivated CD4+CD25+ T cells with anti-CD3 was that anti-CD3 mAb mediated redirected lysis of the activated B cells. To determine if restimulation of activated CD4+CD25+ T cells would result in inhibition of B-cell proliferation in the absence of Fc-mediated retargeting, CD4+CD25+ T cells were purified from mice that express a transgenic TCR specific for HA110-119 and were activated with anti-CD3 and IL-2 as described in Figure 1A. The preactivated CD4+CD25+ T cells were then cultured with LPS-stimulated B cells in the presence or absence of HA peptide. Suppression of B-cell proliferation was also observed when the TCR transgenic CD4+CD25+ T cells were restimulated with the peptide. The HA peptide was as effective as anti-CD3 in inducing the suppressive activity of the antigen-specific regulatory T cells (Figure 1C).
Suppression of B cells was not observed when the CD4+CD25+ T cells were separated from the B cells in a transwell (data not shown). Furthermore, addition of anti–IL-10, or anti–TGF-β, or both to the cocultures had no effect on suppression (data not shown).
Activated CD4+CD25+ T cells induce B-cell death
The decreased proliferation of B cells in the presence of activated CD4+CD25+ T cells could be caused by the induction of unresponsiveness in a manner similar to that seen in studies of the effects of CD4+CD25+ T cells on T-cell proliferation. However, the major effect of CD4+CD25+ T cells on T cells is inhibition of their autocrine production of IL-2. The fact that LPS-induced B-cell proliferation is not cytokine mediated, together with the requirement for restimulation of the activated T cells, raised the possibility that activated CD4+CD25+ T cells killed the LPS-stimulated B cells.
B cells preactivated with LPS (5 μg/mL) for 20 hours were cocultured with preactivated CD4+CD25+ T cells for 8 hours at an E/T ratio of 5:1. Under these conditions, we could observe significant lysis of the B cells in the presence of CD4+CD25+ T cells, whereas background cell death was minimal. Significant numbers of annexin V+, 7-AAD+ cells were seen only in cultures containing preactivated CD4+CD25+ T cells and anti-CD3 (Figure 2A). The induction of B-cell death was specific to CD4+CD25+ T cells because coculture with preactivated CD4+CD25– T cells, under the identical conditions, had no effect on the survival of B cells (Figure 2B). Fc-redirected lysis was not required because restimulation of CD4+CD25+ T cells from HA-TCR transgenic mice with peptide effectively induced B-cell death, whereas CD4+CD25– T cells from the same mice had little, if any, effect (Figure 2C). To determine whether activated CD4+CD25+ T cells selectively killed B cells, preactivated CD4+CD25+ T cells were cocultured with CD4+ T-cell blasts induced by stimulation with concanavalin A (conA). As a positive control, CD8+ T cells were preactivated and cultured with CD4+ T cells. ConA was added to retarget the effector cells to the CD4+ T-cell blasts during the 8-hour cytotoxicity assay. As expected, the activated CD8+ effector T cells readily lysed the CD4+ T-cell blasts in the presence of conA, but no lysis above background was seen when preactivated CD4+CD25+ T cells were used as effector cells (Figure 2D). Similar results were observed when anti-CD3 and T-cell–depleted splenocytes were used in place of conA to redirect lysis (data not shown). This result suggests that activated CD4+CD25+ T cells require recognition of an antigen on the target cell in addition to nonspecific activation of granule exocytosis by mitogenic stimulation.
Figure 2.
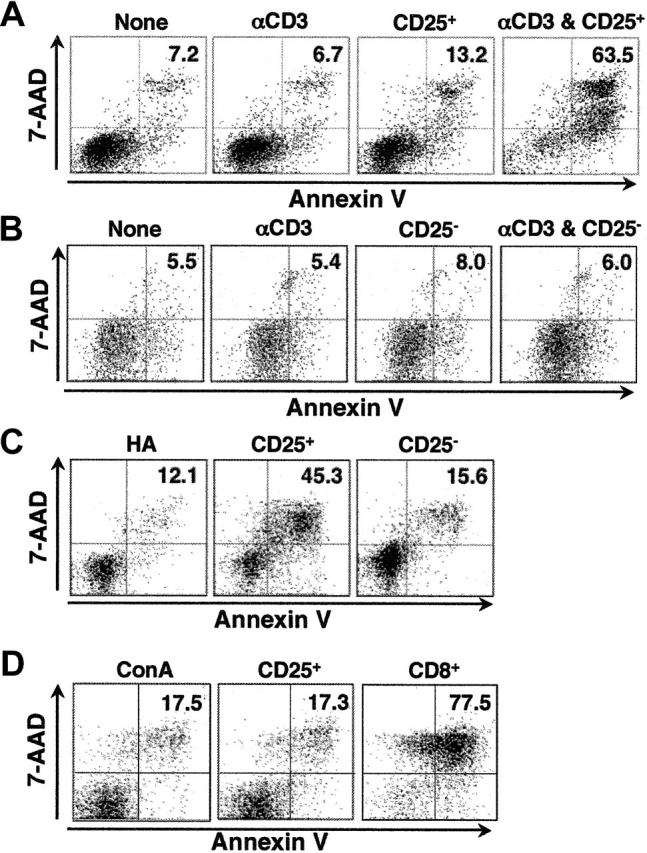
The induction of B-cell apoptosis is specific to activated CD4+CD25+ T cells. (A) Activated CD4+CD25+ T cells cause apoptosis of B cells on restimulation. CD4+CD25+ T cells preactivated for 6 days were cultured with B-cell blasts (preactivated with LPS for 20 hours) at a 5:1 ratio for 8 hours in the presence or absence of anti-CD3 (2 μg/mL). A low amount of LPS (0.5 μg/mL) was also added to the coculture. Annexin V expression and 7-AAD incorporation were evaluated on B220+ cells. (B) Activated CD4+CD25– T cells do not induce B-cell death. CD4+CD25– T cells were cultured with B-cell blasts as in panel A, and the cells were stained for B220, annexin V, and 7-AAD. (C) Activated CD4+CD25+T cells induce B-cell death in response to restimulation with HA peptide. CD4+CD25+ T cells from HA-TCR transgenic mice were cultured with B-cell blasts as in panel A except that HA peptide (16 μM) was used in place of anti-CD3. (D) Activated CD4+CD25+ T cells do not induce T-cell apoptosis. CD4+CD25– T cells were stimulated with irradiated T-depleted splenocytes and conA (3 μg/mL) for 20 hours, labeled with CFSE, and cocultured with activated CD4+CD25+ T cells or with CD8+ T cells that had been activated with plate-bound anti-CD3 and IL-2 for 3 days and expanded in IL-2 media for 3 days before use. ConA (3 μg/mL) was added to the cocultures to retarget the CD4+CD25+ T cells or the activated CD8+ T cells to the CD4+ blast target cells during the 8-hour coculture. Results are representative of at least 3 experiments.
The induction of B-cell death is not mediated by the Fas/FasL pathway
To explore the mechanisms involved in CD4+CD25+ T-cell–mediated B-cell death, we first examined possible roles of death receptors: members of the tumor necrosis factor receptor (TNF-R) family containing an intracellular death domain (eg, Fas, TNF-R1, and TRAIL receptors). The Fas/FasL pathway has been described as an important regulator of T- and B-lymphocyte population size and function in vivo23-25 and it has been established as one of the main lytic mechanisms to maintain lymphocyte homeostasis.26,27 Inclusion of Fas:Fc (20 μg/mL) in the cocultures did not abrogate the suppression of B-cell proliferation (Figure 3A). Furthermore, the proliferative response of Fas-deficient B cells from MRL/lpr mice was suppressed to the same extent as that of wild-type (WT) B cells (Figure 3B). We also examined the capacity of FasL-deficient CD4+CD25+ T cells from gld/gld mice to suppress B-cell proliferation. Because both MRL/lpr and gld/gld mice develop autoimmune diseases,28-30 we used mice at a young age (about 4-6 weeks old) and confirmed that CD4+CD25+ T cells from gld/gld mice were not autonomously activated (data not shown). FasL-deficient CD4+CD25+ T cells suppressed the proliferation of both WT and Fas-deficient B cells to the same extent as WT CD4+CD25+ T cells (Figure 3C-D). All these results indicate that B-cell death mediated by preactivated CD4+CD25+ T cells was independent of the Fas/FasL pathway. We also evaluated whether the TNF/TNFR pathway and the TRAIL/TRAILR pathways were involved in CD4+CD25+ T-cell–mediated B-cell death. The addition of human Fas:Fc, TRAILR(1-4):Fc or human TNFRII-Fc (Enbrel) did not prevent the induction of B-cell death (Figure 3E and data not shown).
Figure 3.
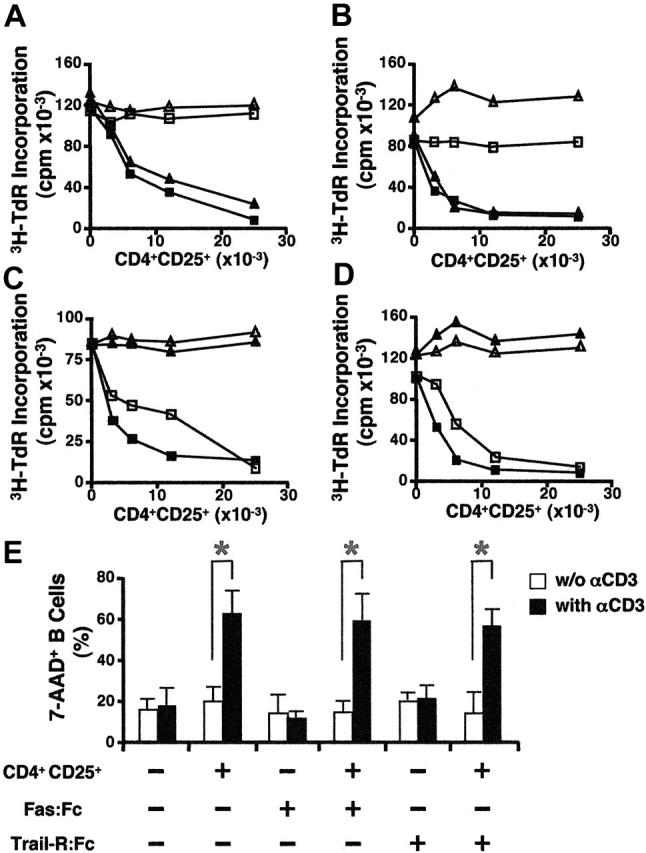
Suppression of B-cell proliferation by activated CD4+CD25+ T cells is not mediated by death receptors. (A) Fas:Fc has no effect on CD4+CD25+ T-cell–mediated suppression of B cells. LPS-primed B cells (5 × 104) were cultured with activated CD4+CD25+ T cells in the presence (▴, ▪) or absence (▵, □) of anti-CD3 (2 μg/mL) and in the presence (▴, ▵) or absence of Fas:Fc (20 μg/mL; ▪, ▪) for 72 hours and the proliferation of B cells was measured as in Figure 1A. (B) Suppression of the proliferation of Fas-deficient B cells by CD4+CD25+ T cells. Fas-deficient B cells (▪, □) from MRL/lpr or WT (▴, ▵) B cells (5 × 104) were stimulated with LPS and cultured with activated CD4+CD25+ T cells in the presence (▴, ▪) or absence (▵, □) of anti-CD3. (C-D) FasL-deficient CD4+CD25+ T cells suppressed the proliferation of both WT and Fas-deficient B cells. LPS-primed WT (C) or Fas-deficient (D) B cells (5 × 104) were cultured with activated CD4+CD25+ T cells from WT (▪, ▴) or FasL-deficient (gld/gld) mice (□, ▵) in the presence (□, ▪) or absence (▴, ▵) of anti-CD3. (E) The induction of B-cell death is not dependent on Fas/FasL and TRIALl/TRAILR pathways. Preactivated CD4+CD25+ T cells were cultured with LPS-primed B cells at a 5:1 ratio for 8 hours in the presence (▪) or absence (□) of anti-CD3. Where indicated, Fas:Fc (20 μg/mL), or TRAILR(1-4):Fc (a mixture of 10 μg/mL R1:Fc, 0.5 μg/mL R2:Fc, 50 ng/mL R3:Fc, and 10 μg/mL R4:Fc) was included in the coculture. B-cell apoptosis was analyzed as described in Figure 2A. Results are expressed as means ± SD (n = 3). *P < .01.
Role of perforin and granzymes in regulatory T-cell–induced B-cell death
Because B-cell death caused by CD4+CD25+ T cells did not appear to be mediated by a ligand-receptor pathway, we investigated whether factors released from regulatory T cells might be involved. We first examined the expression of granzyme B in both naive and activated T cells by quantitative PCR (Figure 4A). Granzyme B mRNA could not be detected in naive CD4+CD25+, CD4+CD25–, and CD8+ T cells, but was up-regulated following TCR activation of all 3 cell types with varying kinetics. The induction of granzyme B on CD8+ T cells was faster, reached a peak level 3 days after activation, and then declined. In contrast, the up-regulation of granzyme B in both CD4+CD25+ and CD4+CD25– T cells was slower, but continuous to day 7. The level of granzyme B mRNA was much higher in the CD4+CD25+ T cells than in the CD4+CD25– T cells. Similar results were obtained when granzyme B protein expression was measured by intracellular staining. CD8+ cells up-regulated granzyme B expression rapidly and maintained expression throughout the culture period (Figure 4B bottom rows). Granzyme B was induced with slower kinetics in CD4+CD25+ and CD4+CD25– T cells. Both the percentage of cells that expressed granzyme B and the level of protein expression as determined by mean fluorescence intensity (MFI) were always higher in the activated CD4+CD25+ T cells than in the activated CD4+CD25– T cells (Figure 4B top and middle rows).
Figure 4.

The induction and release of granzyme B in CD4+CD25+ T cells. (A) Induction of granzyme B mRNA after activation. CD4+CD25– (⋄), CD4+CD25+ (♦), and CD8+ (▴) T cells that were either freshly isolated or activated for indicated periods were collected, total RNA isolated, and reverse transcribed. Granzyme B mRNA was then analyzed by quantitative PCR. (B) Induction of granzyme B protein after activation. CD4+CD25–, CD4+CD25+, and CD8+ T cells were isolated by cell sorting and activated as indicated. Granzyme B expression was determined by intracellular staining. (C) Concomitant cell-surface expression of CD107a and release of granzyme B on restimulation. The 5-day preactivated CD4+CD25–, CD4+CD25+, and CD8+ T cells were restimulated with soluble anti-CD3 (1 μg/mL) and irradiated T-depleted splenocytes for the indicated periods. The cells were stained for cell-surface CD4, CD8, and CD107a, fixed, permeabilized, and stained for intracellular granzyme B. Representative results from at least 3 independent experiments are shown.
CD107a has been used as a functional marker for the cytolytic activity of both CD8+ cells31 and NK cells.32 CD107a is expressed only on the membrane of lytic granules in resting cells and is up-regulated after activation.33,34 The appearance of CD107a on cell membranes has been shown to be associated with the release of lytic granule proteins such as perforin and granzymes, and thus is used as an indicator of degranulation of effector T or NK cells.31,32 We characterized the kinetics of CD107 up-regulation on preactivated T cells, along with granzyme B expression over different restimulation periods. Before restimulation with soluble anti-CD3 and irradiated APCs, CD8+ T cells had low expression of CD107a but a very high level of granzyme B. On restimulation, CD8+ T cells continuously lost intracellular granzyme B with increased CD107a expression on the cell surface (Figure 4C bottom row), indicating the release of granzyme B from granules in CD8+ T cells. Granzyme B and CD107a in CD4+CD25+ T cells showed a virtually identical pattern of change in response to restimulation, suggesting that CD4+CD25+ T cells degranulated and as a result released significant amount of granzyme B (Figure 4C middle row). In contrast, activated CD4+CD25– T cells had much lower expression of granzyme B, but a slightly higher basal level of CD107a before restimulation. Only minimal changes in both CD107a and granzyme B were noted after restimulation (Figure 4C top rows). Taken together, these results suggest that CD4+CD25+ T cells release granzyme B as one of their effector molecules in a manner very similar to that of activated CD8+ T cells.
We also examined perforin expression in CD4+CD25–, CD4+CD25+, and CD8+ T cells by quantitative PCR. Unlike CD8+ cells, freshly isolated CD4+CD25+ and CD4+CD25– T cells did not express perforin. CD8+ T cells up-regulated perforin expression after activation, with similar kinetics to the induction of granzyme B (Figure 5A). CD4+CD25+ T cells up-regulated perforin mRNA expression slowly on stimulation. Perforin mRNA was not detectable in CD4+CD25– T cells at any time point after activation (Figure 5A). Because perforin permealization and granule exocytosis are largely Ca2+-dependent processes, we next examined if the presence of Ca2+ is required for the induction of B-cell death. EGTA was added to chelate calcium ions during the 8-hour coculture. EGTA completely protected B cells from lysis by the CD4+CD25+ T cells and did not exert any toxic effects on the B cells or CD4+CD25+ T cells, suggesting that release of granules or perforin might play a role (Figure 5B and data not shown).
Figure 5.

Perforin-deficient CD4+CD25+ T cells are partially deficient in their ability to kill B cells. (A) Induction of perforin expression in CD4+CD25–, CD4+CD25+, and CD8+ T cells after activation. Sorted resting or activated WT CD4+CD25– (⋄), CD4+CD25+ (♦), CD8+ (▴), and perforin-deficient CD4+CD25+ T cells (○) were collected and analyzed for perforin mRNA expression by quantitative PCR as in Figure 4A. (B) The induction of B-cell death is Ca2+ dependent. Activated CD4+CD25+ T cells were cultured with B-cell blasts from the same mouse at a 5:1 ratio for 8 hours in the presence or absence of anti-CD3. Where indicated, EGTA (3 mM) was included in the culture. The cells were stained for B220, annexin V, and 7-AAD to evaluate B-cell death. Results are expressed as means ± SD (n = 3). (C) Both perforin and granzyme B are required for CD4+CD25+ T-cell–mediated B-cell apoptosis. Preactivated WT or perforin-deficient CD4+CD25+ T cells were incubated with B-cell blasts for 8 hours under the conditions specified. Where indicated, CD4+CD25+ T cells were treated with DCI (30 μM) for 30 minutes and carefully washed before use in the coculture. (D) The suppression of T-cell proliferation is perforin independent. CD4+CD25– responder T cells were stimulated with soluble anti-CD3 and irradiated APCs and cultured with either activated WT (▪) or perforin-deficient (□) CD4+CD25+ T cells. The proliferation of responder T cells was determined as described in Figure 1A. Results are representative of at least 3 experiments.
To investigate these possibilities, we compared the ability of WT and perforin-deficient CD4+CD25+ T cells to lyse LPS-activated B cells in the 8-hour apoptosis assay. Surprisingly, CD4+CD25+ T cells from perforin-deficient mice were still capable of inducing apoptosis of LPS-activated B-cell blasts, but their cytolytic activity was compromised when compared with CD4+CD25+ T cells from WT mice. In several experiments of this type, CD4+CD25+ T cells from the perforin-deficient mice were approximately one third less potent effectors than WT CD4+CD25+ T cells (Figure 5C). The Fas/FasL pathway did not play a role in the suppression of B-cell proliferation mediated by perforin-deficient CD4+CD25+ T cells because the addition of Fas:Fc or TRAILR:Fc to the cocultures had no effect on the lytic activity of the CD4+CD25+ T cells from the perforin-deficient mice (data not shown). Because perforin deficiency only partially abrogated CD4+CD25+ T-cell–triggered B-cell death, we directly examined the role of granzyme B in this process. DCI is a serine proteinase inhibitor that efficiently inhibits granzyme B but not granzyme A,35 and has been used as a specific granzyme B inhibitor in assays with intact cytotoxic T lymphocytes (CTLs).36 DCI pretreatment of WT CD4+CD25+ T cells partially prevented B-cell death (Figure 5C). The lytic activity of DCI-pretreated perforin-deficient CD4+CD25+ T cells was almost completely abrogated (Figure 5C). In addition, CD4+CD25+ T cells from WT and perforin-deficient mice were equivalent in their capacity to suppress T-cell proliferation (Figure 5D). Collectively, these experiments demonstrate that the lytic activity of CD4+CD25+ T cells from perforin-deficient mice is mediated by a granule exocytosis pathway involving granzyme B.
Activated CD4+CD25+ T cells preferentially kill B cells presenting antigen
One important question raised by these experiments is whether activated CD4+CD25+ T cells are capable of killing only B cells that present a ligand to the TCR (anti-CD3 via the Fc receptor or peptide-major histocompatibility complex class II complexes) or whether any activated B cell can be killed in a bystander fashion. Activated CD4+CD25+ T cells were prepared from OT-II mice that recognize OVA323-339 in association with I-Ab and were tested for their capacity to kill activated, nonpulsed B cells and activated, antigen-pulsed B cells that were present in the same culture. The 2 B-cell populations (pulsed versus nonpulsed) were distinguished by their expression of the congenic marker CD45. Interestingly, CD4+CD25+ T cells from OT-II mice preferentially killed OVA323-339 prepulsed B cells (Figure 6A). Similarly, B cells from C57BL/6, but not from BALB/c mice, were killed by OT-II CD4+CD25+ T cells when B cells were present in equal numbers and stimulated in the continuous presence of OVA peptide in the same culture. Both populations were equally susceptible to lysis in the presence of anti-CD3 (Figure 6B).
Figure 6.

CD4+CD25+ T cells preferentially kill antigen-presenting B cells. (A) CD45.1 and CD45.2 B cell blasts were prepulsed with OVA (100 μM) for 2 hours or left untreated, respectively, then were equally mixed and cocultured with activated OT-II CD4+CD25+ T cells at an E/T ratio of 5:1 for 8 hours. The culture was then stained with FITC-conjugated anti–mouse CD45.1, PE-conjugated anti–mouse B220, and 7-AAD. The gate was set on B220+ cells. Results are expressed as means ± SD (n = 4). P indicates cells that are pulsed with ova peptide; N, cells that are not pulsed with peptide; *P < .01 between pulsed and nonpulsed CD45.1 B cells; **P < .01 between nonpulsed and pulsed CD45.2 B cells. (B) B cells from C57BL/6 or BALB/c mice were equally mixed and cocultured with OVA (10 μM) or anti-CD3 (2μg/mL) in the presence or absence of activated OT-II CD4+CD25+ T cells. The cells were then stained with FITC-anti–mouse H-2Dd, PE–anti–mouse B220, and 7-AAD and gated on B220+ cells. Results are expressed as means ± SD (n = 4).
Discussion
The regulatory functions of CD4+CD25+ T cells on CD4+CD25– T cells have been extensively studied, and multiple mechanisms have been suggested1-5 as mediating their suppressive functions. Bystry et al previously demonstrated that CD4+CD25+ T cells inhibited LPS-mediated B-cell proliferation.37 Lim et al also recently demonstrated that regulatory T cells can suppress B-cell–dependent immunoglobulin production and class switch recombination in the absence of T-helper cells.38 However, the mechanism for the suppression remains unclear in these studies. We examined the direct effect of CD4+CD25+ T cells on B cells and found that, when restimulated with either anti-CD3 or antigen, preactivated CD4+CD25+ T cells mediated apoptosis of activated B cells by a cytotoxic granule-mediated mechanism. There are clear differences between the mechanisms of T-cell and B-cell suppression by CD4+CD25+ T cells. First, freshly isolated CD4+CD25+ T cells had no effect on B cells, but inhibited T-cell proliferation. The slow induction of the granule cytotoxicity pathway during culture of the CD4+CD25+ T cells might explain their inability to kill B cells when freshly isolated. Secondly, preactivated CD4+CD25+ T cells required restimulation via their TCR to kill B cells, whereas they do not require TCR restimulation to suppress T cells.9
These results differ from the recent report by Gondek et al14 that suggested that suppression of T-cell activation by CD4+CD25+ T cells was mediated by a perforin-independent, granzyme-B–dependent mechanism. We have previously demonstrated that suppression of T-cell activation results in inhibition of the transcription of IL-2 mRNA in the responding CD4+CD25– T cells. Inhibition of IL-2 mRNA production can be observed as early as 24 hours in the cocultures at a time when we have not observed a difference in the number of viable cells recovered from cultures in the presence or absence of CD4+CD25+ T cells.8 Furthermore, we have recently reported that CD4+CD25+ T cells are capable of suppressing IL-2 mRNA transcription even in cultures containing high concentrations of exogenous IL-2.39 In these studies, very high levels of cell viability were maintained and significant proliferation of both the suppressors and responders was observed.8,39 We did observe, as did Gondek et al,14 a marked decrease in the number of viable T cells recovered after 72 hours of coculture. We have interpreted this decrease in cell recovery as secondary to the G1-S cell-cycle arrest of the responding CD4+CD25– T cells.9 A G1-S arrest in cell cycle is frequently followed by death of the responding cells.40 Finally, even activated T cells did not appear to be susceptible to lysis by preactivated, restimulated CD4+CD25+ T cells. Taken together, we do not believe that an active killing mechanism is responsible for the suppression of T-cell activation by CD4+CD25+ T cells in vitro.
There are both similarities and differences between the killing of B cells by activated CD4+CD25+ T cells and killing by conventional activated CD8+ CTLs. Granzyme B and perforin expression were both up-regulated in CD4+CD25+ T cells during activation with slightly slower kinetics than those observed with CD8+ T cells stimulated under identical conditions. The levels of induced perforin and granzyme B expression were significantly higher in activated CD4+CD25+ T cells than in activated CD4+CD25– T cells at all time points. As has been reported with CD8+ CTLs, stimulation of the activated CD4+CD25+ T cells resulted in release of granules from the cytoplasm with expression of cell-surface CD107a. However, we think that the loss of granzymes and the expression of degranulation marker are not necessarily concomitant events. Thus, it is possible that some cells remain CD107a negative for some time after degranulation. In addition, we observed that CD8+ T cells still maintain granzyme B stores to some extent following degranulation. Our experiments indicated that after activation, CD8+ T cells express granzyme B more rapidly and possessed high levels of granzyme. Thus, we believe that at both transcriptional and translational levels, granzyme B can be more efficiently produced and continuously replenished after the restimulation of CD8+ T cells. However, a surprising aspect of our results as well as those of Gondek et al14 is the relative perforin-independent mechanism of granule-mediated cell death. The Fas/FasL pathway did not contribute to the B-cell death mediated by CD4+CD25+ T cells from WT or perforin-deficient animals. It is unlikely that granzymes were delivered via the cation-independent mannose-6-P receptor (CI-MPR) because CI-MPR–dependent delivery of granzymes requires internalized perforin to provide a signal to release granzymes from the endolysosomal compartments to cleave their substrates.41,42 It has been demonstrated recently that granzyme B acts extracellularly to induce smooth muscle cell apoptosis.43 Thus, it is possible that granzyme B released from preactivated CD4+CD25+ T cells may remain in the conjugation site and inflict cytolytic damage to B cells. Alternatively, our results raise the possibility that cytotoxic granule-mediated death by activated CD4+CD25+ T cells might be facilitated by a distinct pore-forming protein that is uniquely expressed by activated CD4+CD25+ T cells.
Our results also differ from the studies by Grossman et al19 on the induction of cytotoxic T cells by stimulation of either human CD4+CD25– or CD4+CD25+ T cells with a combination of anti-CD3 and anti-CD46. The induction of cytotoxic activity in this model appeared to require costimulation with anti-CD46 because very little activity was induced with the combination of anti-CD3 and anti-CD28. Anti-CD3/CD46–activated CD4+CD25+ T cells appeared to selectively express granzyme A, whereas anti-CD3/CD46–activated CD4+CD25– T cells selectively expressed granzyme B. Although it was concluded in these studies that killing was mediated by perforin-dependent mechanisms based on the calcium dependence and concanamycin A dependence of the CTL activity, the presence of perforin was not directly assayed. Most importantly, both the activated CD4+CD25– and the activated CD4+CD25+ T cells recognized a broad range of targets including activated CD4+ and CD8+ T cells, CD14+ monocytes, and immature/mature dendritic cells, and did not require reactivation via their TCR to exhibit lytic activity. The effect of the activated regulatory cells on B cells was not tested. We did not evaluate the expression of granzyme A in the activated CD4+CD25+ T cells in our studies, but the presence of granzyme B was readily detectable at the protein and mRNA levels. In addition, the pretreatment of regulatory cells with DCI, a serine proteinase inhibitor, that preferentially inhibits granzyme B, significantly blocked the induction of B-cell apoptosis by the activated CD4+CD25+ T cells. The differences between these studies may be secondary to the stimulatory conditions used or to differences between mouse and human regulatory T cells.
The results of our studies certainly suggest that one should re-evaluate the concept of “cytotoxic/suppressor cell” because it appears that, at least in vitro, CD4+CD25+ T cells can mediate both of these functions. The major question to be addressed in the future is whether CD4+CD25+ T cells develop into CTLs in vivo. One of the most interesting findings in our study was that activated CD4+CD25+ T cells selectively killed antigen-presenting B cells and not bystander B cells in the cocultures. This result raises the possibility that one site of action of regulatory T cells in mediating suppression in vivo is the APC. Because only highly activated, TCR-stimulated, CD4+CD25+ T cells can kill B cells, there appears to be greater regulation of this aspect of their function compared to their capacity to suppress T-cell activation by acting directly on responding T cells. Such a control mechanism may facilitate a rapid response of B cells to pathogen-derived thymic-independent antigens and thus avoid suppression by CD4+CD25+ T cells during the early phases of an immune response. After activation and expansion in vivo following multiple rounds of antigen stimulation, B-cell apoptosis induced by CD4+CD25+ T cells may result in a decrease not only in autoantibody production but, more importantly, in a decrease in the antigen-presenting function of activated B cells. If activated CD4+CD25+ T cells are also lytic for activated dendritic cells, lysis of APCs by activated regulatory T cells may be an important mechanism whereby they produce lasting suppression of T-cell activation in vivo.
Acknowledgments
We thank Drs Pierce Henkart and Itzhak Mendel for helpful discussions and Ms Sarah Tanksley for cell sorting.
Prepublished online as Blood First Edition Paper, January 17, 2006; DOI 10.1182/blood-2005-11-4502.
An Inside Blood analysis of this article appears at the front of this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 U.S.C. section 1734.
References
- 1.Mason D, Powrie F. Control of immune pathology by regulatory T cells. Curr Opin Immunol. 1998; 10: 649-655. [DOI] [PubMed] [Google Scholar]
- 2.Shevach EM. Regulatory T cells in autoimmunity. Annu Rev Immunol. 2000;18: 423-429. [DOI] [PubMed] [Google Scholar]
- 3.Sakaguchi S. Regulatory T cells: key controllers of immunologic self-tolerance. Cell. 2000;101: 455-458. [DOI] [PubMed] [Google Scholar]
- 4.Ng WF, Duggan PJ, Ponchel F, et al. Human CD4+CD25+ cells: a naturally occurring population of regulatory T cells. Blood. 2001;98: 2736-2744. [DOI] [PubMed] [Google Scholar]
- 5.Shimizu J, Yamazaki S, Sakaguchi S. Induction of tumor immunity by removing CD25+ T cells: a common basis between tumor immunity and autoimmunity. J Immunol. 1999;163: 5211-5218. [PubMed] [Google Scholar]
- 6.Hori S, Carvalho TL, Demengeot J. CD25+ regulatory T cells suppress CD4+ T cell-mediated pulmonary hyperinflammation driven by Pneumocystis carinii in immunodeficient mice. Eur J Immunol. 2002;32: 1282-1291. [DOI] [PubMed] [Google Scholar]
- 7.Belkaid Y, Piccirillo CA, Mendez S, Shevach EM, Sacks DL. CD4+CD25+ regulatory T cells control Leishmania major persistence and immunity. Nature. 2002;420: 502-507. [DOI] [PubMed] [Google Scholar]
- 8.Thornton AM, Shevach EM. CD4+CD25+ immunoregulatory T cells suppress polyclonal T cell activation in vitro by inhibiting interleukin 2 production. J Exp Med. 1998;188: 287-296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Thornton AM, Shevach EM. Suppressor effector function of CD4+CD25+ immunoregulatory T cells is antigen nonspecific. J Immunol. 2000; 164: 183-190. [DOI] [PubMed] [Google Scholar]
- 10.Piccirillo CA, Shevach EM. Cutting edge: control of CD8+ T cell activation by CD4+CD25+ immunoregulatory cells. J Immunol. 2001;167: 1137-1140. [DOI] [PubMed] [Google Scholar]
- 11.Asseman C, Mauze S, Leach MW, Coffman RL, Powrie F. An essential role for interleukin 10 in the function of regulatory T cells that inhibit intestinal inflammation. J Exp Med. 1999;190: 995-1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Powrie F, Carlino J, Leach MW, Mauze S, Coffman RL. A critical role for transforming growth factor-beta but not interleukin 4 in the suppression of T helper type 1-mediated colitis by CD45RBlowCD4+ T cells. J Exp Med. 1996;183: 2669-2674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Seddon B, Mason D. Regulatory T cells in the control of autoimmunity: the essential role of transforming growth factor beta and interleukin 4 in the prevention of autoimmune thyroiditis in rats by peripheral CD4+CD45RC– cells and CD4+CD8– thymocytes. J Exp Med. 1999;189: 279-288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gondek DC, Lu LF, Quezada SA, Sakaguchi S, Noelle RJ. Cutting edge: contact-mediated suppression by CD4+CD25+ regulatory cells involves a granzyme B-dependent, perforin-independent mechanism. J Immunol. 2005;174: 1783-1786. [DOI] [PubMed] [Google Scholar]
- 15.Olivares-Villagomez D, Wang Y, Lafaille JJ. Regulatory CD4+ T cells expressing endogenous T cell receptor chains protect myelin basic protein-specific transgenic mice from spontaneous autoimmune encephalomyelitis. J Exp Med. 1998;188: 1883-1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McHugh RS, Shevach EM, Margulies DH, Natarajan K. A T cell receptor transgenic model of severe spontaneous organ-specific autoimmunity. Eur J Immunol. 2001;31: 2094-2103. [DOI] [PubMed] [Google Scholar]
- 17.Curotto de Lafaille MA, Muriglan S, Sunshine MJ, et al. Hyper immunoglobulin E response in mice with monoclonal populations of B and T lymphocytes. J Exp Med. 2001;194: 1349-1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Seo SJ, Fields ML, Buckler JL, et al. The impact of T helper and T regulatory cells on the regulation of anti-double-stranded DNA B cells. Immunity. 2002;16: 535-546. [DOI] [PubMed] [Google Scholar]
- 19.Grossman WJ, Verbsky JW, Barchet W, et al. Human T regulatory cells can use the perforin pathway to cause autologous target cell death. Immunity. 2004;21: 589-601. [DOI] [PubMed] [Google Scholar]
- 20.Kirberg J, Baron A, Jakob S, et al. Thymic selection of CD8+ single positive cells with a class II major histocompatibility complex-restricted receptor. J Exp Med. 1994;180: 25-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wherry EJ, Teichgraber V, Becker TC, et al. Lineage relationship and protective immunity of memory CD8 T cell subsets. Nat Immunol. 2003; 4: 225-234. [DOI] [PubMed] [Google Scholar]
- 22.Caramalho I, Lopes-Carvalho T, Ostler D, et al. Regulatory T cells selectively express toll-like receptors and are activated by lipopolysaccharide. J Exp Med. 2003;197: 403-411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kagi D, Ledermann B, Burki K, Zinkernagel RM, Hengartner H. Molecular mechanisms of lympho-cyte-mediated cytotoxicity and their role in immunological protection and pathogenesis in vivo. Annu Rev Immunol. 1996;14: 207-232. [DOI] [PubMed] [Google Scholar]
- 24.Russell JH, Ley TJ. Lymphocyte-mediated cytotoxicity. Annu Rev Immunol. 2002;20: 323-370. [DOI] [PubMed] [Google Scholar]
- 25.Nagata S. Apoptosis by death factor. Cell. 1997; 88: 355-365. [DOI] [PubMed] [Google Scholar]
- 26.Berke G. The binding and lysis of target cells by cytotoxic T lymphocytes: molecular and cellular aspects. Annu Rev Immunol. 1994;12: 735-773. [DOI] [PubMed] [Google Scholar]
- 27.Cohen PL, Eisenberg RA. Lpr and gld: single gene models of systemic autoimmunity and lymphoproliferative disease. Annu Rev Immunol. 1991;9: 243-269. [DOI] [PubMed] [Google Scholar]
- 28.Alderson MR, Tough TW, Davis-Smith T, et al. Fas ligand mediates activation-induced cell death in human T lymphocytes. J Exp Med. 1995;181: 71-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lynch DH, Watson ML, Alderson MR, et al. The mouse Fas-ligand gene is mutated in gld mice and is part of a TNF family gene cluster. Immunity. 1994;1: 131-136. [DOI] [PubMed] [Google Scholar]
- 30.Takahashi T, Tanaka M, Brannan CI, et al. Generalized lymphoproliferative disease in mice, caused by a point mutation in the fas ligand. Cell. 1994;76: 969-976. [DOI] [PubMed] [Google Scholar]
- 31.Betts MR, Brenchley JM, Price DA, et al. Sensitive and viable identification of antigen-specific CD8+ T cells by a flow cytometric assay for degranulation. J Immunol Methods. 2003;281: 65-78. [DOI] [PubMed] [Google Scholar]
- 32.Alter G, Malenfant JM, Altfeld M. CD107a as a functional marker for the identification of natural killer cell activity. J Immunol Methods. 2004;294: 15-22. [DOI] [PubMed] [Google Scholar]
- 33.Kannan K, Stewart RM, Bounds W, et al. Lysosome-associated membrane proteins h-LAMP1 (CD107a) and h-LAMP2 (CD107b) are activation-dependent cell surface glycoproteins in human peripheral blood mononuclear cells which mediate cell adhesion to vascular endothelium. Cell Immunol. 1996;171: 10-19. [DOI] [PubMed] [Google Scholar]
- 34.Bossi G, Griffiths GM. Degranulation plays an essential part in regulating cell surface expression of Fas ligand in T cells and natural killer cells. Nat Med. 1999;5: 90-96. [DOI] [PubMed] [Google Scholar]
- 35.Odake S, Kam CM, Narasimhan L, et al. Human and murine cytotoxic T lymphocyte serine proteases: subsite mapping with peptide thioester substrates and inhibition of enzyme activity and cytolysis by isocoumarins. Biochemistry. 1991;30: 2217-2227. [DOI] [PubMed] [Google Scholar]
- 36.Song Q, Burrows SR, Smith G, et al. Interleukin-1β-converting enzyme-like protease cleaves DNA-dependent protein kinase in cytotoxic T cell killing. J Exp Med. 1996;184: 619-626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bystry RS, Aluvihare V, Welch KA, Kallikourdis M, Betz AG. B cells and professional APCs recruit regulatory T cells via CCL4. Nat Immunol. 2001; 2: 1126-1132. [DOI] [PubMed] [Google Scholar]
- 38.Lim HW, Hillsamer P, Banham AH, Kim CH. Cutting edge: direct suppression of B cells by CD4+ CD25+ regulatory T cells. J Immunol. 2005;175: 4180-4183. [DOI] [PubMed] [Google Scholar]
- 39.Thornton AM, Donovan EE, Piccirillo CA, Shevach EM. Cutting edge: IL-2 is critically required for the in vitro activation of CD4+CD25+ T cell suppressor function. J Immunol. 2004;172: 6519-6523. [DOI] [PubMed] [Google Scholar]
- 40.Ashwell JD, Cunningham RE, Noguchi PD, Hernandez D. Cell growth cycle block of T cell hybridomas upon activation with antigen. J Exp Med. 1987;165: 173-194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Motyka B, Korbutt G, Pinkoski MJ, et al. Mannose 6-phosphate/insulin-like growth factor II receptor is a death receptor for granzyme B during cytotoxic T cell-induced apoptosis. Cell. 2000;103: 491-500. [DOI] [PubMed] [Google Scholar]
- 42.Pinkoski MJ, Hobman M, Heibein JA, et al. Entry and trafficking of granzyme B in target cells during granzyme B-perforin-mediated apoptosis. Blood. 1998;92: 1044-1054. [PubMed] [Google Scholar]
- 43.Choy JC, Hung VH, Hunter AL, et al. Granzyme B induces smooth muscle cell apoptosis in the absence of perforin involvement of extracellular matrix degradation. Arterioscler Thromb Vasc Biol. 2004;24: 2245-2250. [DOI] [PubMed] [Google Scholar]