

Abstract
Cure rates for patients with acute myeloid leukemia (AML) remain low despite ever-increasing dose intensity of cytotoxic therapy. In an effort to identify novel approaches to AML therapy, we recently reported a new method of chemical screening based on the modulation of a gene expression signature of interest. We applied this approach to the discovery of AML-differentiation-promoting compounds. Among the compounds inducing neutrophilic differentiation was DAPH1 (4,5-dianilinophthalimide), previously reported to inhibit epidermal growth factor receptor (EGFR) kinase activity. Here we report that the Food and Drug Administration (FDA)-approved EGFR inhibitor gefitinib similarly promotes the differentiation of AML cell lines and primary patient-derived AML blasts in vitro. Gefitinib induced differentiation based on morphologic assessment, nitro-blue tetrazolium reduction, cell-surface markers, genome-wide patterns of gene expression, and inhibition of proliferation at clinically achievable doses. Importantly, EGFR expression was not detected in AML cells, indicating that gefitinib functions through a previously unrecognized EGFR-independent mechanism. These studies indicate that clinical trials testing the efficacy of gefitinib in patients with AML are warranted. (Blood. 2005;106: 2841-2848)
Introduction
Despite an improved understanding of the pathogenesis of acute myeloblastic leukemia (AML), long-term survival remains poor. While the current approach to AML therapy is based on cytotoxic agents, increasing evidence points to the potential of therapy aimed at overcoming a block in differentiation that is characteristic of AML.1,2 This approach is particularly striking in acute promyelocytic leukemia (APL), where treatment of patients with all-trans retinoic acid (ATRA) results in the induction of myeloid differentiation of the leukemic blasts, and long-term survival has improved dramatically.3-5 Other AML subtypes similarly have defects in differentiation caused in part by mutations in differentiation-promoting transcription factors (eg, CCAAT/enhancer-binding protein alpha [C/EBPalpha], PU.1, globin transcription factor-1 [GATA-1]).6-9 The potential of differentiation therapy in non-APL AML has yet to be realized, however, in large measure because chemical inducers capable of triggering differentiation have yet to be described.
The challenges in identifying AML differentiation-inducing agents are 2-fold. First, in many cases the mechanism by which differentiation is abrogated is unknown, thereby precluding a traditional biochemical screen for compounds that activate the differentiation program. Second, even for those cases in which differentiation-blocking mutations are known, such mutations have been primarily in transcription factors, generally considered to be “undruggable.” We recently addressed these issues by developing a gene expression-based screening method (gene expression-based high-throughput screening [GE-HTS]), whereby a small molecule library was screened for compounds that induced the gene expression signature of myeloid differentiation.10 Among the chemicals confirmed to induce neutrophilic differentiation was DAPH1 (4,5-dianilinophthalimide). DAPH1 induced morphologic, biochemical, and functional changes indicative of myeloid maturation, consistent with its induction of a differentiation gene expression program.
DAPH1 was previously identified as an epidermal growth factor receptor (EGFR) kinase inhibitor but the compound has not been developed clinically, thus precluding its evaluation as differentiation therapy for patients with AML.11,12 In the present report we describe the preclinical efficacy of the Food and Drug Administration (FDA)-approved EGFR inhibitor gefitinib (Iressa). We find that gefitinib induces myeloid differentiation in AML cell lines and primary patient-derived AML blasts at concentrations that are achievable in humans. These results indicate that gefitinib warrants evaluation as potential differentiation therapy for patients with AML.
Patients, materials, and methods
Cells and gefitinib treatment
HL-60, Kasumi-1, and U937 cells were maintained in culture in RPMI 1640 with 10% fetal bovine serum and 1% penicillin-streptomycin at 37°C with 5% CO2. Primary patient AML blasts were collected from peripheral blood or bone marrow aspirate after obtaining patient informed consent under a Dana-Farber Cancer Institute Internal Review Board-approved protocol. They were isolated using Ficoll-Paque Plus (Amersham Biosciences, Uppsala, Sweden) separation and maintained in culture in RPMI 1640 with 10% fetal bovine serum and 1% penicillin-streptomycin at 37°C with 5% CO2. Cells were treated with gefitinib (Astra-Zeneca, London, United Kingdom; WuXi PharmaTech, Shanghai, China) resuspended in dimethyl sulfoxide (DMSO) at concentrations ranging from 10 μM down to 0.078 μM. DMSO was used as a vehicle control in differentiation and viability experiments. HL-60 cells were also treated with cetuximab (Bristol-Myers Squibb, Princeton, NJ) at 10 to 20 μg/mL and trastuzumab (Genentech, San Francisco, CA) at 0.1 to 1000 μg/mL.
Viability assays
Viability experiments were performed in 96-well format in replicates of 4 using the Promega Cell-Titer Glo (Madison, WI) adenosine triphosphate (ATP)-based assay per the manufacturer's instructions. Cells were evaluated at 0, 3 days, and 6 days with gefitinib in a 2-fold dilution from 10 μM down to 0.078 μM versus DMSO control-treated cells. The concentration at which cell viability was reduced to 50% of DMSO-treated controls (EC50) was determined. Values for EC50 were calculated by interpolating a polynomial fit to the measured viability data. Curve fitting was performed in MATLAB (Mathworks, Natick, MA) using the least-squares curve-fitting function (polyfit). Model order for the polynomial was set to n = 3 except in the cases where the estimated EC50 point falls outside the range of the measured values where it was set to n = 1 (one of the healthy controls). The EC50 value was found by interpolating the curve for the value of 50 with the MATLAB one-dimensional interpolation function (interp1).
Differentiation assays
Differentiation induction with gefitinib was confirmed by morphology, nitro-blue tetrazolium (NBT) reduction, cell surface marker expression, and whole-genome changes in expression. For morphologic assessment, cytospin preparations of treated AML blasts stained with May-Grünwald Giemsa were evaluated with light microscopy. For NBT reduction assays, experiments were performed in triplicate. Gefitinib-treated cells were compared with DMSO-treated controls after 5 days of treatment. Cells were incubated at 37°C for 1 hour in a mixture containing total medium, 0.1% NBT (Sigma, St Louis, MO), and 1 μg/mL TPA (12-O-tetradecanoylphorbol-13-acetate; Sigma). The percentage of blue cells was counted by light microscopy for at least 200 cells per sample. Gefitinib-treated cells were compared with DMSO-treated cells with a one-tailed t test analysis assuming 2 samples with unequal variance.13 Analysis for myeloid maturation with cell-surface markers was performed by fluorescence-activated cell sorting (FACS) with fluorescein isothiocyanate (FITC)-labeled antibodies for CD11b and CD14 (Becton Dickinson, San Jose, CA). Live cells were gated based upon forward and side scatter patterns. Fluorescence was analyzed by FACS with a Becton Dickinson FACScan and CELLQuest analytic software (Becton Dickinson).
Expression profiling
HL-60 and Kasumi-1 cells were treated in replicate of 3 with 10 μM gefitinib or DMSO vehicle control for 6 hours and 24 hours. Primary patient APL cells (patient 1) were treated with 5 μM gefitinib or DMSO in duplicate or triplicate for 3 days. Primary patient M5-AML cells (patient 2) were treated in duplicate with 5 μM gefitinib or DMSO in duplicate for 6 hours. Primary patient M4-AML cells (patient 7) were treated in triplicate with 2.5 μM gefitinib or DMSO for 6 hours, 24 hours, and 3 days and with 5 μM gefitinib for 6 hours and 24 hours. RNA was extracted with Trizol (Invitrogen, Carlsbad, CA) as per the manufacturer's guidelines, and 10 μg was used to create target for hybridization to Affymetrix U133A DNA microarrays (Affymetrix, Santa Clara, CA) as previously described.14 GeneChip MAS5 Software (Affymetrix) was used for preprocessing of the raw data, and all scans within an experiment were scaled to the array with the median overall microarray intensity, as previously described.10 Raw microarray data are available in Stegmaier et al.15
The Mantel test was used to assess whether gefitinib induced changes on a whole-genome level consistent with differentiation. A Mantel test is a nonparametric, randomization-based procedure that estimates the correlation between 2 distance matrices.16 We compared previously reported sets of primary AML versus normal mature neutrophils10 to sets of undifferentiated versus gefitinib-treated AML cells according to their level of expression to see if genes across the whole genome were being up-regulated and down-regulated similarly. Specifically, we compared the expression patterns (measured on Affymetrix's U133A microarray) observed in data sets composed of 9 primary AML versus 3 normal, mature neutrophil samples to those expression patterns observed in data sets composed of DMSO-treated samples versus gefitinib-treated samples. Next, we compared previously reported sets of primary AML versus normal mature monocytes to sets of gefitinib-treated AML cells according to level of expression to see if genes across the whole genome were being up-regulated and down-regulated similarly. Specifically, we compared the expression patterns observed in the data sets composed of 9 primary AML versus 3 normal, mature monocyte samples to those expression patterns observed in data sets composed of DMSO-treated samples versus gefitinib-treated samples (Table S1, available on the Blood website; see the Supplemental Table link at the top of the online article). For a given gene expression data set X and its corresponding class labels, the distance of each feature from the class labels was calculated using the signal-to-noise statistic. The signal-to-noise statistic is calculated as follows:
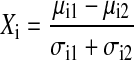 |
The Pearson correlation was calculated as follows:
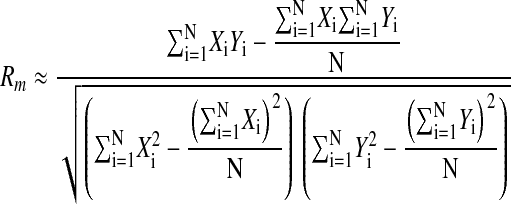 |
where Xi is the signal-to-noise statistic for feature i of sample set X and Yi is the signal-to-noise statistic for feature i of sample set Y. The Mantel correlation Rm was used as the reference value in the Mantel test. To calculate the significance level, the elements of one of the vectors were randomly permuted to produce a permuted vector X*. As before, the Mantel statistic Rm* was computed between X* and Y. The permutation-computation steps were repeated 2500 times and the resulting distribution was used to estimate the P value by examining the proportion of Rm* values that are greater than Rm.
Reverse transcription-polymerase chain reaction (RT-PCR)
Total RNA was isolated from HL-60 and Kasumi-1 cells using TRIZOL Reagent (Invitrogen). Universal Human Reference RNA (Stratagene, La Jolla, CA) was used as a positive control. cDNA was synthesized from 1 μg of total RNA from each sample using SuperScript III Reverse Transcriptase (Invitrogen) and oligo d(T)16 primers in a 20-μL reaction system. Two microliters of cDNA was amplified using HotStarTaq DNA Polymerase (Qiagen, Valencia, CA) in the DNA Engine (PTC-200) Peltier Thermal Cycler (MJ Research, Waltham, MA) in a 20-μL reaction system. PCR was performed at 94°C for 9 minutes followed by 40 cycles of denaturation at 94°C for 30 seconds, annealing at 60°C for 30 seconds, and extension at 72°C for 30 seconds. The primer pairs were as follows: glyceraldehyde-3-phosphate dehydrogenase (GAPD): 5′ AGCCACATCGCTCAGACAC 3′, 5′ CTCCATGGTGGTGAAGACG 3′; EGFR (erythroblastic leukemia viral oncogene homologue 2): 5′ CGGGACATAGTCAGCAGTGA 3′, 5′ ACTGGTTGTGGCAGCAGTC 3′; ERBB2: 5′ GTTTGAGTCCATGCCCAATC 3′,5′ GTAACTGCCCTCACCTCTCG 3′.
Immunoprecipitation and Western blot analysis
A431 whole-cell lysate (20 μg; sc-2201; Santa Cruz Biotechnology, Santa Cruz, CA) was used as a positive control for EGFR detection. HL-60 and Kasumi-1 cells were collected by centrifugation at 482.5g for 5 minutes. Cells were lysed in RIPAbuffer (10 mM Tris-Cl [pH 7.6], 100 mM NaCl, 1 mM EDTA[ethylenediaminetetraacetic acid], 1% Triton X, 0.5% sodium deoxycholate, and 0.1% sodium dodecyl sulfate [SDS]) with protease inhibitor (Complete Mini EDTA-free protease inhibitor tablets; Roche Diagnostics, Mannheim, Germany) and phosphatase inhibitor (1 mM sodium vanadate) and incubated on ice for 30 minutes. The supernatant was retained and the protein concentration was determined using the BioRad Protein Assay reagent per the manufacturer's instructions (BioRad Laboratories, Hercules, CA). For total EGFR determination, 50 μg of whole-cell lysate was denatured by boiling in SDS sample buffer, separated on a 10% Tris-HCl precast Ready Gel (BioRad Laboratories), and transferred to Millipore Immobilon polyvinylidene difluoride (P-PVDF) membranes (Millipore, Billerica, MA). The membrane was blocked in nonfat dry milk (5% in TBST) for 1 hour at room temperature and blotted with anti-EGFR antibody (sc-03; Santa Cruz Biotechnology) overnight at 4°C. The membrane was washed 3 times in Tris-buffered saline Tween-20 (TBST) and incubated with anti-rabbit secondary horseradish peroxidase-linked antibody (Amersham Biosciences, Little Chalfont, United Kingdom) for 45 minutes at room temperature. It was then washed 3 times in TBST. Antibody binding was detected using Western Lightning Chemiluminescence Reagent Plus (PerkinElmer Life Sciences, Boston, MA) and exposed to Hyperfilm enhanced chemiluminescence (ECL; Amersham Biosciences) film. Membranes were stripped and reprobed using a pan-actin antibody (ACTN05; NeoMarkers, Fremont, CA) to assure consistent sample loading.
Immunoprecipitation was performed as follows. Cells were washed with ice-cold phosphate-buffered saline and then incubated with 0.5 mL of 1X ice-cold cell lysis buffer (Cell Signaling Technology, Beverly, MA) containing protease inhibitor (Complete Mini EDTA-free protease inhibitor tablets) for 30 minutes. Five hundred micrograms of each lysate (HL-60 and SKBR3) was incubated overnight at 4°C with anti-ERBB2 antibody (neu Ab-11; Neomarkers) or mouse immunoglobulin G (IgG) control antibody (Jackson Labs, West Grove, PA) and then incubated with Ultralink Protein G (Pierce, Rockford, IL) at room temperature for 2 hours. The immunoprecipitants were then washed 5 times with 1X cell lysis buffer (Cell Signaling Technology) with protease inhibitor. They were resuspended in SDS sample buffer, heated, and then separated by electrophoresis with a 5% Tris-HCl precast Ready Gel (BioRad Laboratories) and transferred to Millipore Immobilon P-PVDF membranes (Millipore). They were analyzed as described above with antiphosphotyrosine antibodies 4G10 (Upstate, Waltham, MA) and pY100 (Cell Signaling Technology) and anti-ERBB2 (neu Ab-17; Neomarkers).
Results
Gefitinib induces differentiation in AML cell lines
Using a new chemical genomic screening method, we previously screened a small molecule library for agents inducing myeloid maturation in the AML cell line HL-60. One of the chemicals confirmed to induce HL-60 differentiation based upon multiple phenotypic and functional assays of differentiation was DAPH1, a 4,5-dianilinophthalimide class member, initially developed as an EGFR inhibitor. We therefore hypothesized that inhibition of EGFR or a related kinase may be an important mechanism of myeloid differentiation and as such may have clinical implications for AML therapy.
Because DAPH1 is not an FDA-approved drug, we extended testing to the FDA-approved EGFR inhibitor gefitinib. HL-60 cells treated with 10 μM gefitinib for 4 days underwent striking changes consistent with differentiation with condensation and lobulation of the nucleus consistent with neutrophil maturation (Figure 1A-B), and the t(8;21)-containing AML cell line Kasumi-1 similarly demonstrated morphologic evidence of gefitinib-induced neutrophilic differentiation (Figure 1C-D). The monocytic AML cell line U937 underwent evidence of macrophage differentiation (Figure 1E-F) consistent with its proclivity toward monocyte/macrophage differentiation. Thus, it appears that gefitinib induces not only neutrophilic differentiation but also monocyte/macrophage differentiation depending on the cellular context.
Figure 1.

Morphologic and functional changes induced by gefitinib in AML cell lines. May-Grünwald Giemsa staining of HL-60 cells treated with (A) 0.02% DMSO and (B) 10 μM gefitinib for 4 days, Kasumi-1 cells treated with (C) 0.01% DMSO and (D) 5 μM gefitinib for 3 days, and U937 cells treated with (E) 0.02% DMSO and (F) 8 μM gefitinib for 3 days. Doses were chosen at which optimal differentiation occurs. Images were acquired with an Olympus BH-2 microscope (Melville, NY), × 100/1.25 magnification under oil, an Olympus Q-Color 5 digital camera, and Adobe Photoshop CS version 8.0 software (San Jose, CA). (G) HL-60 cells were treated in triplicate for 5 days with gefitinib in a 2-fold dose response series from 10 μM to 1.25 μM and the percentage of NBT-positive cells compared with DMSO-treated controls (Ctl) with a one-tailed t test analysis.
We next performed an NBT reduction assay to assess for functional evidence of myeloid maturation. One feature of myeloid maturation is the production of superoxide anion used by the mature myeloid cell to kill ingested microorganisms. Superoxide anion production can be measured by the reduction of NBT. After 5 days of treatment with gefitinib at 10 μM, more than 80% of the HL-60 cells were positive for NBT reduction (P < .001). At concentrations of gefitinib as low as 1.25 μM at 5 days, there was enhanced induction of NBT reduction compared with DMSO controls (P = .001; Figure 1G). Gefitinib treatment of U937 cells with 7.5 to 10 μM for 5 days increased NBT reduction compared with DMSO-treated controls from 1% positive to 13% (P = .04) and 34% (P = .03), respectively (data not shown).
We then evaluated for the cell-surface markers associated with myeloid maturation, CD14 and CD11b. HL-60 cells and Kasumi-1 cells showed a striking increase in expression of both CD14 and CD11b by flow cytometric analysis, whereas the U937 cells showed only a subtle increase in CD11b and CD14 (Figure 2). Although CD14 expression is generally associated with a more monocytic phenotype, it is also expressed on mature neutrophils and CD11b is expressed on neutrophils and monocytes.17 Thus, a neutrophil versus monocyte differentiation distinction cannot be made solely based on limited cell surface marker expression.
Figure 2.

Gefitinib induces maturation-associated myeloid cell surface markers. Fluorescence-activated cell sorting analysis was performed with FITC-labeled antibodies for CD14 and CD11b. AML cell lines were incubated for 5 days before analysis with DMSO vehicle or with gefitinib at a dose optimized for morphologic changes: HL-60 (10 μM), Kasumi-1 (5 μM), and U937 (7.5 μM). Viable cells were gated based on forward and side scatter patterns and fluorescence was determined. Shaded in gray are the DMSO-treated controls and outlined in black are the gefitinib-treated cells.
Gefitinib induces gene expression program of myeloid differentiation
To obtain a more global, nonbiased view of gefitinib effects on AML cells, we evaluated whether gefitinib induces genome-wide gene expression changes consistent with neutrophil or monocyte maturation. HL-60 and Kasumi-1 cells were treated in replicates of 3 with gefitinib at 10 μM or DMSO, and RNA was prepared at 6 and 24 hours for hybridization to Affymetrix U133A microarrays. In order to evaluate whether gefitinib induced a whole-genome program of differentiation, we applied the Mantel test, a statistical, global measurement of similarity. We compared the genes that distinguish primary patient-derived AML blasts from normal human neutrophils or monocytes to the genes that distinguish DMSO-treated HL-60 cells from gefitinib-treated HL-60 cells. As early as 6 hours, gefitinib treatment of HL-60 cells recapitulated the gene expression pattern of bona fide neutrophil maturation (P < .001), and the differentiation program was similarly evident following 24 hours of gefitinib treatment (P < .001; Figure 3). By 24 hours, gefitinib treatment of Kasumi-1 cells recapitulated the gene expression pattern of neutrophil differentiation (P = .006), whereas a gene expression pattern of monocyte differentiation was not significantly induced in either cell line (P > .1). This result indicates that gefitinib induced global changes in gene expression consistent with neutrophil differentiation in the HL-60 and Kasumi-1 cell lines. Detailed results are available in Stegmaier et al and in Table S1.15
Figure 3.
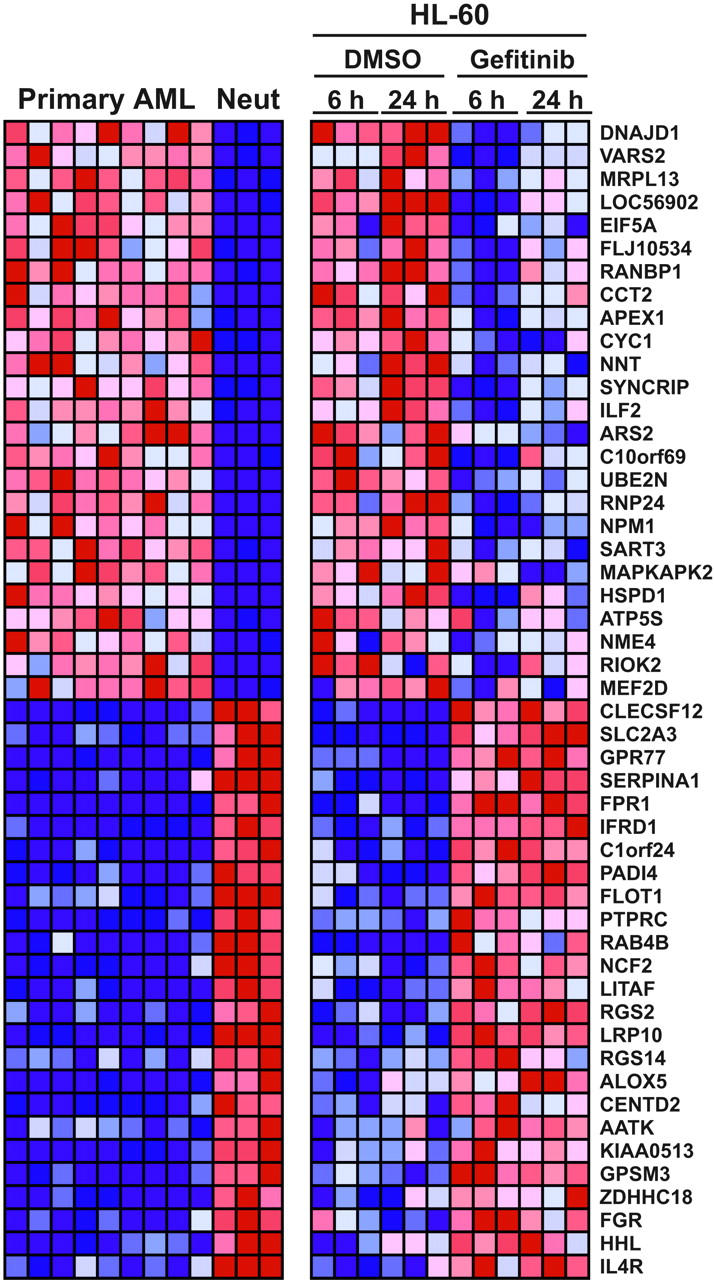
Gefitinib induces whole-genome expression modulation consistent with neutrophil maturation. Gene expression profiling was done in triplicate 6 hours and 24 hours after treatment with either 10 μM gefitinib or 0.02% DMSO in HL-60 cells. These patterns were compared with expression profiles distinguishing primary patient AML cells from normal human neutrophils. Using the signal-to-noise (SNR) metric, the genes distinguishing the 9 primary AML samples from the 3 normal neutrophil samples (Neut) were identified and then reordered according to their degree of regulation by gefitinib in HL-60 cells. The top 25 genes in each direction are shown. Expression levels greater than the mean are shown in red; those less than the mean are shown in blue.
Gefitinib-induced differentiation is EGFR independent
Gefitinib is well characterized as an inhibitor of in vitro and in vivo EGFR tyrosine kinase activity.18,19 In order to assess whether gefitinib is inducing AML differentiation via EGFR inhibition, we first evaluated whether EGFR transcript or protein is expressed. EGFR transcript was undetectable in HL-60 and Kasumi-1 cells (Figure 4A). Accordingly, no protein expression was detected by Western immunoblotting with anti-EGFR antibody, whereas EGFR-expressing A431 epidermoid carcinoma cells were strongly positive (Figure 4B). Additionally, stimulation of cells with 100 ng/mL EGF failed to induce expression of EGFR protein, and the monoclonal anti-EGFR antibody cetuximab failed to induce evidence of myeloid differentiation by morphology and NBT reduction (data not shown). These findings indicate that EGFR, the principle target of gefitinib, is not expressed in AML cells, consistent with prior reports, and therefore is unlikely to be the target of gefitinib in AML.20,21
Figure 4.

Gefitinib-induced differentiation is EGFR independent. (A) RT-PCR (40 cycles) was performed to evaluate for EGFR and ERBB2 transcript presence in HL-60 and Kasumi-1 cells. GAPD was amplified as a control to demonstrate intact cDNA. Negative water controls and a positive cDNA control (uRNA) synthesized from universal Stratagene RNA were also included. (B) Western immunoblotting was performed to evaluate for the presence of EGFR protein in HL-60 and Kasumi-1 whole-cell lysates (50 μg) with anti-EGFR antibody. An EGFR-expressing positive control, A431 epidermoid carcinoma cells, was included. (C) Immunoprecipitation (IP) was performed with anti-ERBB2 and a mouse IgG control antibody to evaluate for the presence of ERBB2 protein in HL-60 cells with and without 10 μM gefitinib stimulation. Total lysate from SKBR3, a breast cancer cell line known to overexpress ERBB2, was used as a positive control. Western immunoblotting was performed with ERBB2 or a cocktail of 4G10 and pY100 antiphosphotyrosine (PTyr) antibodies.
Gefitinib and DAPH1 have both been reported to inhibit the activity of the EGFR-related receptor tyrosine kinase ERBB2 (HER-2/neu).12,18,22 While ERBB2 transcript was detected (Figure 4A), ERBB2 protein was undetectable either by total lysate Western blotting or by anti-ERBB2 immunoprecipitation followed by antiphosphotyrosine or anti-ERBB2 Western blotting (Figure 4C), consistent with recent observations of lack of detectable ERBB2 protein in ERBB2 transcript-expressing AML cells.23 To further exclude ERBB2 as the target of gefitinib's differentiation-inducing activity, HL-60 cells were treated with the anti-ERBB2 monoclonal antibody trastuzumab, and this failed to induce any evidence of myeloid maturation (data not shown). These data, taken together, suggest that gefitinib induces myeloid differentiation via a non-EGFR, non-ERBB2 mechanism, thereby implicating a new mechanism of action for gefitinib.
Gefitinib inhibits viability and induces differentiation in primary AML blasts
The effect of small molecules in cell lines does not always parallel that in primary patient material. We therefore extended the evaluation of gefitinib-induced differentiation to primary patient AML blasts obtained from patients with de novo or relapsed AML. We first examined the effects of gefitinib on cellular viability in a gefitinib dose-response series. Six of 8 patients responded with an EC50 less than 5 μM, regardless of French-American-British (FAB) subtype (mean EC50 2.0 μM among responders; range, 1.0-8.1 μM; Figure 5; Table 1). In contrast, healthy donor peripheral blood mononuclear cells from 5 independent donors showed decreased viability in response to gefitinib only at high dose (mean EC50 = 9.15, SD = 2.6). Consistent with this lack of a gefitinib effect on normal hematopoietic cell viability, no hematologic toxicity has been observed in clinical trials evaluating gefitinib in patients with lung cancer and other solid tumors.24-26
Figure 5.

Gefitinib inhibits cell viability in the majority of primary patient AML cells. Primary patient AML blasts were isolated from bone marrow aspirate or peripheral blood samples by ficoll separation and treated in a dose-response series with gefitinib. Cell viability was evaluated at 6 days with an ATP-based assay and plotted as a percentage of control cells. Samples were evaluated in replicates of 4. Blasts from peripheral blood mononuclear cells (PBMC) from healthy human donors show markedly decreased response to gefitinib compared with AML samples. One illustrative example is included. FAB class and EC50 evaluation are reported in Table 1. Error bars indicate the standard deviation across 4 replicates.
Table 1.
Patient sample characteristics and in vitro gefitinib EC50
| Patient | Diagnosis | EC50, μM | Cytogenetic findings |
|---|---|---|---|
| 1 | M3-AML | 1.84 | 47, XX, +8, t(15;17) (q22;q21) |
| 2 | M5-AML | 1.00 | 45, XY, −7 |
| 3 | M4-AML | 3.94 | 46, XX, ? t(3;7) (q25-26; q35-36) [7]/46, XX [13]/46, XY [1] |
| 4 | M1-AML | 8.11 | 46, XY, der(6) ins(6;?) (q23;?) t(6;7) (q25;p13-14), der(7) add(7) (p13-14) del(7) 9 (q22q34), del(11) (q2?3), del(12) (q13q24) .ish der(6) (MLL+), del(11) (MLL−) |
| 5 | M1-AML | 1.21 | 45, XY, −7 [6]/46, XY [14] |
| 6 | M5-AML | 2.63 | 46, XX |
| 7 | M4-AML | 1.39 | 46, XY |
| 8 | M1-AML | 5.89 | 47, XY, +8 |
Primary patient leukemia blasts were collected by ficoll separation from bone marrow aspirate or peripheral blood. FAB subclass and cytogenetic findings are reported. The gefitinib concentration at which cell viability is 50% of DMSO-treated control cells (EC50) was determined at 6 days by an ATP-based assay performed in 4 replicates.
For patients from whom sufficient numbers of leukemic cells were available, we explored the effects of gefitinib on differentiation by evaluating morphologic and genome-wide gene expression changes. For example, in a patient with M3-AML with the t(15;17) and trisomy 8 (patient 1) and in a patient with M4-AML with normal cytogenetics (patient 7), gefitinib induced morphologic changes consistent with neutrophil differentiation (Figure 6A-D). Additionally, in a patient with M1-AML with monocytic features and trisomy 8 (patient 8), gefitinib induced morphologic evidence of macrophage differentiation (Figure 6E-F). Furthermore, microarray-based expression profiling indicated that 3-day exposure to 5 μM gefitinib induced a gene expression program consistent with neutrophil differentiation as assessed by the Mantel test (P = .001) for patient 1 and with a 3-day exposure to 2.5 μM gefitinib for patient 7 (P < .001). Inadequate RNA was available from patient 8. Similarly, a patient with M5-AML with a FMS-like tyrosine kinase-3 (FLT-3) mutation and monosomy 7 (patient 2) induced the gene expression program of myeloid differentiation in response to gefitinib (P < .001). These experiments indicate that clinically achievable doses of gefitinib induce bona fide myeloid differentiation of primary AML blasts in vitro. Detailed results are available in Table S1.
Figure 6.

Morphologic changes induced by gefitinib in primary patient AML blasts. May-Grünwald Giemsa staining of primary patient M3-AML blasts (patient 1) containing the t(15;17) and trisomy 8 treated in vitro for 3 days with (A) 0.01% DMSO and (B) 5 μM gefitinib, primary patient M4-AML blasts (patient 7) treated in vitro for 4 days with (C) 0.01% DMSO and (D) 2.5 μM gefitinib, and primary patient M1-AML blasts (patient 8) containing trisomy 8 treated in vitro for 7 days with (E) 0.01% DMSO and (F) 10 μM gefitinib. Images were acquired with an Olympus BH-2 microscope, × 100/1.25 magnification under oil, an Olympus Q-Color 5 digital camera, and Adobe Photoshop CS version 8.0 software.
Discussion
The road to developing new therapeutic agents for AML is a long and expensive one. The fact that AML is a relatively rare disease further adds to a market disincentive to develop new drugs for the disease. Accordingly, very few new agents have been FDA approved for patients with AML over the past decade. The problem is further compounded by the fact that for the majority of cases of AML, validated therapeutic targets are unknown and therefore conventional biochemical screens cannot be initiated.
As an alternate approach to the problem, we undertook a chemical screen based not on an isolated target but rather on a gene expression signature of the differentiated state, reasoning that any compound capable of inducing the gene expression program of neutrophilic differentiation would be of potential biologic and clinical interest. As with any chemical screening approach, the output of the primary screen rarely yields a compound that can be tested directly in humans. We therefore deliberately screened a library of compounds about which much was known with respect to biochemical properties and mechanism of action. The chemical identified in that screen, DAPH1, was one such compound. It is known to have EGFR kinase inhibitory activity, but its development as a drug ceased and it is no longer commercially available. Nevertheless, this functional knowledge of DAPH1 biochemical activity allowed us to move directly to the characterization of other EGFR family kinase inhibitors including gefitinib, which, given its FDA approval status, might be rapidly translated into the clinic.
Indeed we observed that gefitinib induces striking morphologic, biochemical, and gene expression evidence of myeloid differentiation both in AML cell lines and primary patient-derived AML blasts. As expected with terminal differentiating cells, gefitinib also inhibited the proliferation of AML blasts in vitro. The potential for gefitinib or other EGFR inhibitors as anti-AML agents has not been previously explored, likely in large measure because EGFR is not thought to be expressed in AML blasts. Our experiments confirm lack of expression of EGFR at the RNA or protein level, indicating that EGFR is not a target of gefitinib in these cells. Our observation that low micromolar concentrations of gefitinib are required to induce the differentiation effect is consistent with a non-EGFR target, given the well-documented 50% inhibitory concentration (IC50) of EGFR inhibition in the less than 50-nM range based on in vitro kinase profiling.18 The ability of gefitinib to inhibit the ERBB2 kinase has been well recognized. ERBB2 protein was undetectable in gefitinib-responsive AML cells, and the anti-ERBB2 antibody trastuzumab failed to recapitulate the differentiation effect.
These experiments strongly indicate that the differentiation-promoting effects of gefitinib occur through a non-EGFR, non-ERBB2 target, thereby implicating an as yet uncharacterized target of gefitinib. In that regard, a recent report described the systematic identification of protein targets of a panel of kinase inhibitors in clinical development.27 That report indicates, quite surprisingly, that most kinase inhibitors, including gefitinib, hit a large number of kinase targets, the majority of which were previously unknown. Of the 119 protein kinases evaluated, gefitinib, in particular, was shown to bind to at least 18 kinases at less than 10 μM. Each of these, either alone or in combination, should now be evaluated as the critical target through which the differentiative effects of gefitinib are exerted.
In parallel to such laboratory investigations of gefitinib mechanism of action in AML cells, we believe that the striking biologic effects of gefitinib on AML cells warrant immediate clinical translation. Gefitinib has been used to treat more than 100 000 patients with non-small cell lung cancer and has been FDA approved as third-line therapy for patients with locally advanced or metastatic disease. It is orally administered and has an excellent safety profile with no reported deleterious effects on normal hematopoiesis. Dose-limiting toxicity was most commonly diarrhea and rash.28 At the maximum tolerated dose (MTD; 700-1000 mg/d), the reported geometric mean Cmax was approximately 2.5 to 5 μM,24,25,29 in the range required to induce myeloid differentiation in vitro in our studies (mean responder EC50 2 μM). We therefore expect that treatment of patients at the MTD will be well tolerated and has the potential to induce AML differentiation in vivo. Given its outstanding safety profile, ease of administration, and compelling preclinical data, clinical trials testing gefitinib in relapsed or refractory patients with AML are therefore warranted.
Supplementary Material
Acknowledgments
We thank members of the laboratory of T.R.G., the Broad Institute, and, in particular, Ilene Galinsky, Peter Blanding, Richard Stone, Kelly Nazemi, Benjamin Ebert, Justin Lamb, Betty Li, David Dorfman, and Andrew Kung for their contributions.
Prepublished online as Blood First Edition Paper, July 5, 2005; DOI 10.1182/blood-2005-02-0488.
Supported by the National Institutes of Health (NIH), National Cancer Institute (NCI; 5K08 CA098444-03; K.S.), the Leukemia and Lymphoma Society, and Howard Hughes Medical Institute (T.R.G.).
The online version of this article contains a data supplement.
An Inside Blood analysis of this article appears at the front of this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 U.S.C. section 1734.
References
- 1.Waxman S. Differentiation therapy in acute myelogenous leukemia (non-APL). Leukemia. 2000;14: 491-496. [DOI] [PubMed] [Google Scholar]
- 2.Miller WH Jr, Waxman S. Differentiation induction as a treatment for hematologic malignancies. Oncogene. 2002;21: 3496-3506. [DOI] [PubMed] [Google Scholar]
- 3.Chen ZX, Xue YQ, Zhang R, et al. A clinical and experimental study on all-trans retinoic acid-treated acute promyelocytic leukemia patients. Blood. 1991;78: 1413-1419. [PubMed] [Google Scholar]
- 4.Fenaux P, Chastang C, Chevret S, et al. A randomized comparison of all transretinoic acid (ATRA) followed by chemotherapy and ATRA plus chemotherapy and the role of maintenance therapy in newly diagnosed acute promyelocytic leukemia. The European APL Group. Blood. 1999;94: 1192-1200. [PubMed] [Google Scholar]
- 5.Fenaux P, Chevret S, Guerci A, et al. Long-term follow-up confirms the benefit of all-trans retinoic acid in acute promyelocytic leukemia. European APL group. Leukemia. 2000;14: 1371-1377. [DOI] [PubMed] [Google Scholar]
- 6.Smith ML, Cavenagh JD, Lister TA, Fitzgibbon J. Mutation of CEBPA in familial acute myeloid leukemia. N Engl J Med. 2004;351: 2403-2407. [DOI] [PubMed] [Google Scholar]
- 7.Frohling S, Schlenk RF, Stolze I, et al. CEBPA mutations in younger adults with acute myeloid leukemia and normal cytogenetics: prognostic relevance and analysis of cooperating mutations. J Clin Oncol. 2004;22: 624-633. [DOI] [PubMed] [Google Scholar]
- 8.Mueller BU, Pabst T, Osato M, et al. Heterozygous PU.1 mutations are associated with acute myeloid leukemia [letter]. Blood. 2003;101: 2074. [DOI] [PubMed] [Google Scholar]
- 9.Xu G, Nagano M, Kanezaki R, et al. Frequent mutations in the GATA-1 gene in the transient myeloproliferative disorder of Down syndrome. Blood. 2003;102: 2960-2968. [DOI] [PubMed] [Google Scholar]
- 10.Stegmaier K, Ross KN, Colavito SA, O'Malley S, Stockwell BR, Golub TR. Gene expression-based high-throughput screening (GE-HTS) and application to leukemia differentiation. Nat Genet. 2004;36: 257-263. [DOI] [PubMed] [Google Scholar]
- 11.Trinks U, Buchdunger E, Furet P, et al. Dianilinophthalimides: potent and selective, ATP-competitive inhibitors of the EGF-receptor protein tyrosine kinase. J Med Chem. 1994;37: 1015-1027. [DOI] [PubMed] [Google Scholar]
- 12.Buchdunger E, Trinks U, Mett H, et al. 4,5-dianilinophthalimide: a protein-tyrosine kinase inhibitor with selectivity for the epidermal growth factor receptor signal transduction pathway and potent in vivo antitumor activity. Proc Natl Acad Sci U S A. 1994;91: 2334-2338. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 13.Sokoloski JA, Blair OC, Sartorelli AC. Alterations in glycoprotein synthesis and guanosine triphosphate levels associated with the differentiation of HL-60 leukemia cells produced by inhibitors of inosine 5′-phosphate dehydrogenase. Cancer Res. 1986;46: 2314-2319. [PubMed] [Google Scholar]
- 14.Golub TR, Slonim DK, Tamayo P, et al. Molecular classification of cancer: class discovery and class prediction by gene expression monitoring. Science. 1999;286: 531-537. [DOI] [PubMed] [Google Scholar]
- 15.Stegmaier K, Corsello SM, Ross KN, Wong JS, DeAngelo DJ, Golub TR. Broad Institute cancer program publication: Gefitinib (Iressa) induces myeloid differentiation of acute myeloid leukemia. http://www.broad.mit.edu/cancer/pub/AML_gefitinib. Accessed on May 20, 2005. [DOI] [PMC free article] [PubMed]
- 16.Mantel N. The detection of disease clustering and a generalized regression approach. Cancer Res. 1967;27: 209-220. [PubMed] [Google Scholar]
- 17.Theilgaard-Monch K, Jacobsen LC, Borup R, et al. The transcriptional program of terminal granulocytic differentiation. Blood. 2005;105: 1785-1796. [DOI] [PubMed] [Google Scholar]
- 18.Wakeling AE, Guy SP, Woodburn JR, et al. ZD1839 (Iressa): an orally active inhibitor of epidermal growth factor signaling with potential for cancer therapy. Cancer Res. 2002;62: 5749-5754. [PubMed] [Google Scholar]
- 19.Barker AJ, Gibson KH, Grundy W, et al. Studies leading to the identification of ZD1839 (IRESSA): an orally active, selective epidermal growth factor receptor tyrosine kinase inhibitor targeted to the treatment of cancer. Bioorg Med Chem Lett. 2001;11: 1911-1914. [DOI] [PubMed] [Google Scholar]
- 20.Chen LL, Gansbacher B, Gilboa E, et al. Retroviral gene transfer of epidermal growth factor receptor into HL60 cells results in a partial block of retinoic acid-induced granulocytic differentiation. Cell Growth Differ. 1993;4: 769-776. [PubMed] [Google Scholar]
- 21.Walz TM, Malm C, Wasteson A. Expression of the transforming growth factor alpha protooncogene in differentiating human promyelocytic leukemia (HL-60) cells. Cancer Res. 1993;53: 191-196. [PubMed] [Google Scholar]
- 22.Moulder SL, Yakes FM, Muthuswamy SK, Bianco R, Simpson JF, Arteaga CL. Epidermal growth factor receptor (HER1) tyrosine kinase inhibitor ZD1839 (Iressa) inhibits HER2/neu (erbB2)-over-expressing breast cancer cells in vitro and in vivo. Cancer Res. 2001;61: 8887-8895. [PubMed] [Google Scholar]
- 23.Buhring HJ, Sures I, Jallal B, et al. The receptor tyrosine kinase p185HER2 is expressed on a subset of B-lymphoid blasts from patients with acute lymphoblastic leukemia and chronic myelogenous leukemia. Blood. 1995;86: 1916-1923. [PubMed] [Google Scholar]
- 24.Baselga J, Rischin D, Ranson M, et al. Phase I safety, pharmacokinetic, and pharmacodynamic trial of ZD1839, a selective oral epidermal growth factor receptor tyrosine kinase inhibitor, in patients with five selected solid tumor types. J Clin Oncol. 2002;20: 4292-4302. [DOI] [PubMed] [Google Scholar]
- 25.Ranson M, Hammond LA, Ferry D, et al. ZD1839, a selective oral epidermal growth factor receptor tyrosine kinase inhibitor, is well tolerated and active in patients with solid, malignant tumors: results of a phase I trial. J Clin Oncol. 2002;20: 2240-2250. [DOI] [PubMed] [Google Scholar]
- 26.Herbst RS, Maddox AM, Rothenberg ML, et al. Selective oral epidermal growth factor receptor tyrosine kinase inhibitor ZD1839 is generally well-tolerated and has activity in non-small-cell lung cancer and other solid tumors: results of a phase I trial. J Clin Oncol. 2002;20: 3815-3825. [DOI] [PubMed] [Google Scholar]
- 27.Fabian MA, Biggs WH III, Treiber DK, et al. A small molecule-kinase interaction map for clinical kinase inhibitors. Nat Biotechnol. 2005;23: 329-336. [DOI] [PubMed] [Google Scholar]
- 28.Cohen MH, Williams GA, Sridhara R, et al. United States Food and Drug Administration Drug Approval summary: Gefitinib (ZD1839; Iressa) tablets. Clin Cancer Res. 2004;10: 1212-1218. [DOI] [PubMed] [Google Scholar]
- 29.Nakagawa K, Tamura T, Negoro S, et al. Phase I pharmacokinetic trial of the selective oral epidermal growth factor receptor tyrosine kinase inhibitor gefitinib (`Iressa', ZD1839) in Japanese patients with solid malignant tumors. Ann Oncol. 2003;14: 922-930. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.