

Abstract
Integrins mediate the adhesion of cells to each other and to the extracellular matrix during development, immunity, metastasis, thrombosis, and wound healing. Molecular defects in either the α- or β-subunit can disrupt integrin synthesis, assembly, and/or binding to adhesive ligands. This is exemplified by the bleeding disorder, Glanzmann thrombasthenia (GT), where abnormalities of the platelet-specific integrin, αIIbβ3, prevent platelet aggregation following vascular injury. We previously used a retrovirus vector containing a cDNA cassette encoding human integrin β3 to restore integrin αIIbβ3 on the surface of megakaryocytes derived from peripheral blood stem cells of GT patients. In the present study, bone marrow from β3-deficient (β3–/–) mice was transduced with the ITGβ3-cassette to investigate whether the platelet progeny could establish hemostasis in vivo. A lentivirus transfer vector equipped with the human ITGA2B gene promoter confined transgene expression to the platelet lineage. Human β3 formed a stable complex with murine αIIb, effectively restoring platelet function. Mice expressing significant levels of αIIbβ3 on circulating platelets exhibited improved bleeding times. Intravenous immunoglobulin effectively diminished platelet clearance in animals that developed an antibody response to αIIbβ3. These results indicate the feasibility of targeting platelets with genetic therapies for better management of patients with inherited bleeding disorders.
Introduction
Several hundred different proteins orchestrate the adhesion of platelets to the exposed extracellular matrices, signal transduction, aggregation, and clot retraction, leading to the formation of a platelet-plug that helps stop the flow of blood from a wound site. At least 5 members of the evolutionarily conserved integrin family of adhesion receptors are present on platelets to aid in these processes including α2β1, α5β1, α6β1, αvβ3, and αIIbβ3.1,2 The molecular structure was recently solved for one integrin, αvβ3,3 which directs binding of platelets and a variety of other cell types to vitronectin. Unlike αvβ3, integrin αIIbβ3 is expressed exclusively on megakaryocytes and platelets (≈ 80 000 copies per platelet)4 due to the presence of promoter regulatory elements that direct high-level, selective transcription of the ITGA2B gene early in megakaryocytopoiesis.5 αIIbβ3 mediates the interaction of activated platelets with multiple adhesive ligands, including fibrinogen, von Willebrand factor (VWF), fibronectin, thrombospondin, and collagen.2 Upon activation, αIIbβ3 changes its shape to bind its ligand with high affinity for effective platelet aggregation and retraction of a fibrin clot to seal a wound.6,7
Glanzmann thrombasthenia (GT) is a rare autosomal-recessive bleeding disorder resulting from genetic defects of either ITGA2B or ITGB3 that disrupt subunit synthesis, receptor assembly, and/or function, thus preventing αIIbβ3 from binding ligands essential for proper platelet aggregation.8 More than 100 distinct genetic defects have been characterized for GT, occurring with even distribution in both genes.9 The diagnosis of thrombasthenia, meaning “weak platelets,” is based on the demonstration of normal platelet levels, but abnormal platelet aggregation and clot retraction in response to physiologic agonists adenosine diphosphate (ADP), epinephrine, and thrombin.10,11 β3-deficient (β3–/–) mice exhibit a condition that is essentially identical to the phenotype for GT in humans where defective platelet function leads to prolonged bleeding.12 Of interest, β3–/– mice also display abnormalities in placental development, osteosclerosis,13 and increased tumor hypervascularization14 and growth,15 thus underscoring a vital role for αvβ3 in those processes.2
The current study was designed to improve our understanding relevant to the use of hematopoietic stem cells for gene therapy of hemorrhagic disorders. Information acquired from this work should be particularly useful for developing strategies to alleviate uncontrolled bleeding due to inherited platelet defects. Three issues were addressed: (1) Can mutant bone marrow stem cells be given adequate genetic information to allow megakaryocyte progeny to synthesize a transgene product that will help newly formed platelets to participate in normal hemostasis? (2) Will the product be maintained as a platelet-specific protein at therapeutic levels for a reasonable period of time? (3) Can the product be tolerated by the immune system or become a target for B- and T-cell–mediated immunity resulting in the premature destruction and clearing of the genetically altered megakaryocytes and platelets? The outcome from this study demonstrates the feasibility of platelet-specific gene therapy and paves the way for future studies in patients suffering from inherited bleeding disorders.
Materials and methods
Antibodies
A biotinylated antibody to murine αv (CD51), the phycoerythrin (PE)–conjugated antibody specific for human β3 (CD61), PE–anti–murine TER-119, and fluorescein isothiocyanate (FITC)–conjugated antibodies to the following murine proteins were used: αIIb (CD41), CD45 receptor (CD45R)/B220, Thy1.2 (CD90.2), Ly-6G, C, membrane attack complex type 1 (MAC-1), and isotype standards (PE–immunoglobulin G [IgG], FITC-IgG) (all from BD Biosciences, San Jose, CA). FITC-anti–human β3, a polyclonal antibody to murine VWF, and the isotype control were from Dako (Carpinteria, CA). An antibody (7E3)16 specific for the complex of human αIIbβ3 and αvβ3 was a gift from B. Coller (Rockefeller University, New York, NY). An antibody to an epitope exposed on the high affinity conformation of human β3 (D3)17 was a gift from L. Jennings (University of Tennessee, Memphis). An antibody to human αIIbβ3 complex (AP2)18 was from our Hybridoma Core Laboratory (Milwaukee, WI). PE-anti–murine αIIbβ3 activated complex specific (Jon/A) and FITC-rabbit anti–murine fibrinogen antibodies were from Emfret Analytics (Würzburg, Germany). An antibody to murine glycoprotein Ibα (GPIbα, PE-p0p4)19 was a gift from B. Nieswandt (University of Würzburg, Germany). FITC-F(ab′)2 goat anti–murine IgG Fc was from Jackson ImmunoResearch (West Grove, PA). A PE-antibiotin antibody was from Miltenyi Biotec (Gladbach, Germany).
Lentivirus
A fragment of the human ITGA2B gene promoter beginning at nucleotide –889 was previously used to drive transcription of cDNA encoding human β3 (gift from P. Newman, Blood Research Institute, Milwaukee, WI)20 within the Maloney murine leukemia oncoretrovirus (MuLV) vector, p-889PlA2β3, as described.21,22 To construct lentivirus plasmid pβ3-WPTS, the human ITGA2B gene promoter–driven ITGB3 cDNA cassette was excised from p-889PlA2β3 and subcloned into an HIV type-1 construct, pWPTS-GFP (gift from D. Trono, University of Geneva, Switzerland),23 replacing the elongation factor-1α promoter/GFP cDNA cassette. A lentivirus, p-889GFPII-WPTS, was also constructed for platelet-specific expression of a reporter gene, green fluorescent protein (GFP). The human ITGA2B gene promoter for GFP was amplified by polymerase chain reaction (PCR) from genomic DNA isolated from the human promegakaryocytic cell line, Dami, using nucleotides –889 to –872 (bold type) sense primer of ITGA2B (5′-TTACGCGTCGACAGATCTCCTTGCCACCTAGACC-3′) and antisense primer (5′-CGTCTTCCATGGTCCTTCTTCCACAACC-3′) encoding an NcoI restriction site and nucleotides +30 to +15 of ITGA2B and cloned into pGL3 (Promega, Madison, WI). The ITGA2B gene promoter sequence was confirmed by nucleotide analysis and the PCR product was excised from pGL3, and cDNA encoding a human ranilla GFP gene variant was excised from phrGFPII-1 (Stratagene, La Jolla, CA) and subcloned into (p)WPTS-GFP, replacing the elongation factor-1α promoter/GFP cDNA cassette.
Recombinant lentivirus was generated from HEK 293T cells (6 × 106 cells/100-mm plate) cotransfected with calcium phosphate precipitation of 3 plasmids: (1) pβ3-WPTS or p-889GFPII-WPTS, and (2) a construct expressing the HIV-1 gag/pol, tat, and rev genes required for efficient lentivirus production, pCMVΔR8.91 (gift from D. Trono),24 and (3) a plasmid expressing the vesicular stomatitis virus envelope glycoprotein (G), pCI-VSV-G (gift from J. Olsen, University of North Carolina, Chapel Hill), at 15:15:7.5 μg/plate. Virus was collected and concentrated 500-fold by low-speed centrifugation and stocks were stored frozen at –80°C until used as previously described.21 Virus titer was determined by real-time quantitative reverse-transcriptase–PCR measuring copies of proviral DNA integrated into the genome of circulating murine mononuclear cells as recently described.25 Replication-competent virions were confirmed absent from viral stocks by using extended marker rescue assays.22
Murine bone marrow isolation, transduction, and transplantation
Bone marrow was isolated from β3–/–12 or β3+/+ mice and Ficoll-purified mononuclear cells were transduced at a multiplicity of infection (MOI) equal to 5 β3 virions per β3–/– cell or 3–889GFPII virions per β3+/+ cell over a 24-hour period as previously described.26 A dose of 9.0 × 106 cells was transplanted by tail-vein injection (500 μL/mouse) into each recipient (6-8 weeks old) conditioned with a lethal dose of 1100 cGy total body irradiation with a cesium irradiator (Shepherd Mark I, J. L. Shepherd, San Fernando, CA). Results shown with 6 mice in this study reflect a range of observations recorded with 21 mice that received a primary transplant of bone marrow transduced with a lentivirus construct containing the αIIb gene promoter–driven ITGB3 cDNA cassette. Fourteen mice received an initial transplant of the ITGA2B gene promoter–driven ITGB3 cDNA cassette within the MuLV oncoretrovirus backbone.22 ITGB3–transgene expression persisted for only 10 to 15 weeks after transplantation (likely resulting from inactivation of MuLV)27; therefore, the lentivirus backbone was substituted for MuLV to transfer the ITGB3 cassette into bone marrow transplanted into 21 additional mice to obtain more stable transgene expression.24 Gene expression and function were also examined with platelets collected from mice that received β3-lentivector–transduced bone marrow from one donor (mouse A) as a second- (2 mice) or third (4 mice)–generation transplant recipient. Animal studies complied with institutional guidelines approved by the Animal Care and Use Committee of the Medical College of Wisconsin's American Association for the Accreditation of Laboratory Animal Care–approved Biomedical Resource Center.
Blood collection
Mice were anesthetized with an inhalation anesthetic and blood (100 μL) was collected by tail-vein bleed into a microtube containing 1.0 mL Tyrode buffer with 0.13 M sodium citrate anticoagulant and 1 μg prostaglandin E1 (Sigma, St Louis, MO) similar to a previously described protocol.28 Blood was layered onto Fico/Lite for Platelets (Atlanta Biologicals, Norcross, GA) and centrifuged for 15 minutes at 700g. The plasma layer was collected and stored frozen at –80°C until used for plasma antibody analysis. Platelets were collected at the interface, mononuclear cells from the Ficoll layer and erythrocytes from the pellet. Cells were washed once with phosphate-buffered saline (PBS) buffer containing 0.5% bovine serum albumin (BSA) and 2 mM EDTA (ethylenediaminetetraacetic acid), and used directly for flow cytometry or platelet aggregation assays.
Flow cytometry
Washed platelets, leukocytes, and erythrocytes were blocked for 60 minutes in buffer with 10 μg murine IgG. Samples were then incubated for 30 minutes with PE- and/or FITC-conjugated antibodies that specifically recognize human β3 or murine cell-surface markers, diluted with 750 μL buffer and analyzed on a FACScan flow cytometer (Becton Dickinson, San Jose, CA). The data were analyzed with Win MDI software (Joseph Trotter, Scripps Research Institute, La Jolla, CA). Nonspecific staining was determined with isotype-matched control antibodies. αIIbβ3 receptor levels were calculated from the geometric mean fluorescent intensity (MFI) of FITC-murine αIIb on sample platelets divided by the MFI of αIIb on normal murine platelets, which was then multiplied by 100. A minimum of 2 × 104 events was collected for the expression analysis. In some experiments, platelets were pretreated with the fibrinogen mimetic peptide, GRGDW (2 mM, synthesized by our Protein Core Laboratory). Cytofix and PERM/WASH reagents (BD Biosciences) were used for experiments requiring intracellular detection of protein by flow cytometry.
Flow cytometric analysis was performed on washed platelets, leukocytes, and erythrocytes collected from mice that received –889GFPII-transduced marrow to determine if the human αIIb gene promoter directed lineage-specific expression of GFP (a minimum of 10 000 events were collected). To determine if human β3 expression was confined to the platelet lineage, approximately 500 to 2500 positive events were collected for each lineage-specific marker and a density plot was displayed to reveal the percentages of cells simultaneously staining positive with the marker and human β3.
Platelet aggregation
Platelets were isolated from blood with Fico/Lite and resuspended to 5 × 105/μL in modified Tyrode buffer (pH 7.4) containing 1 mM CaCl2 and 1 mM MgCl2. Samples (250 μL) were placed in an aggregometry cuvette and warmed to 37°C with stirring (1000 rpm). Aggregation was initiated by adding 0.15 mg/mL peak 1 soluble human fibrinogen (gift from M. Mosesson, Blood Research Institute, Milwaukee, WI)29 or soluble murine fibrinogen (Enzyme Research Laboratories, South Bend, IN), and a platelet activation agonist of 100 μM ADP was added individually or in cocktail with 20 μM epinephrine (BioData, Horsham, PA) and 250 μM thrombin receptor activating-peptide (proteinase-activated receptor 4 [PAR4], GYPGKF-NH2) synthesized by our Protein Core Laboratory. Aggregation was monitored on a platelet aggregation profiler-4 (PAP-4) and analyzed with data interface software (BioData). Studies to measure inhibition of platelet aggregation were performed by pretreating platelets with 7E3 for 30 minutes at 37°C followed by the addition of fibrinogen and agonist.
Pulmonary thromboembolism assay
To determine if platelet function was restored in vivo, mice were intravenously injected with 0.35 mg/g platelet agonist, ADP (25 mg/mL), in Tyrode buffer as previously described.30 Circulating platelet levels were measured with an Animal Blood Counter (Oxford Science, Oxford, CT) 1 hour before and 5 minutes after ADP treatment. Each animal was killed at 10 minutes after injection. The lungs were removed and immersed in 10% neutral-buffered formalin and embedded in paraffin, and sections were prepared in our histology core facility with Gomori trichrome stain to demonstrate differential staining of protein fibers (fibrotic tissue: muscle, collage fibers, fibrin clots) and erythrocytes. Lung sections were mounted on glass slides, examined with a light microscope (× 400 magnification), and photographed with a Kodak DC290 digital camera using Microscopy Documentation System 290 software (both from Eastman Kodak, Rochester, NY).
Bleeding time assay
The tail bleeding time was defined as the time required for a stream of bleeding to end from a severed tip of a tail that was immersed in 0.9% isotonic saline at 37°C as previously described.12 The average bleeding time was reported for β3–/– and β3+/+ controls, consisting of 20 mice per group. Tails were cauterized if bleeding proceeded beyond the experimental end point of 600 seconds.
Assay to detect an immune response to αIIbβ3
Flow cytometry was used to determine if murine plasma contained antibodies that recognized proteins on the surface of platelets from mice and humans (provided by B. Curtis, BloodCenter of Wisconsin, Milwaukee). Platelets were washed and incubated with 1:10 diluted murine plasma and an FITC-conjugated F(ab′)2 goat anti–murine IgG Fc (1:100 diluted) and examined by flow cytometry as previously described.31 An antibody to the human αIIbβ3 complex (AP2) was used as a positive control for human platelets, and an antibody to murine GPIbα was used as a positive control for recognition of mouse platelets. Buffer served as a negative control and buffer-diluted plasma from control mice served as an internal negative control. The ratio of antibody binding to platelets was determined by dividing the MFI of platelets with mouse (A) plasma by the MFI of platelets with control buffer as previously described.31 A ratio above 2.0 denotes a positive immune response to platelet proteins. Human platelet studies complied with institutional guidelines approved by the Human Research Review Committee of the Medical College of Wisconsin.
Intravenous immunoglobulin
Mice received an intravenous injection of 0.5 mg/g body weight of immunoglobulin G (Sigma) once per day for 3 consecutive days as previously described.32
Results
Platelet-specific expression of integrin β3
To investigate the feasibility of gene therapy for correcting molecular defects in platelets, we targeted expression of the human integrin β3-subunit in megakaryocytes of β3–/– mice affected with GT. In Figure 1A, cDNA encoding human β3 was subcloned into a vector derived from an HIV type-1 lentivirus.23 The enhancer/promoter of the viral 3′-long terminal repeat (LTR) was removed from this construct to allow the vector to self-inactivate (SIN), and the human ITGA2B gene promoter was used to direct megakaryocyte-specific synthesis of β3. This promoter binds factors (GATA and Ets) that mediate high-level gene transcription, which is confined to megakaryocytes due to the presence of a repressor region that inhibits gene expression in other lineages.5 Mononuclear cells were isolated from bone marrow of β3–/– mice, transduced with β3 lentivirus (MOI 5), and transplanted into lethally irradiated β3–/– littermates (9 × 106 cells/mouse) as a model for autologous hematopoietic stem cell gene therapy for inherited bleeding disorders.
Figure 1.
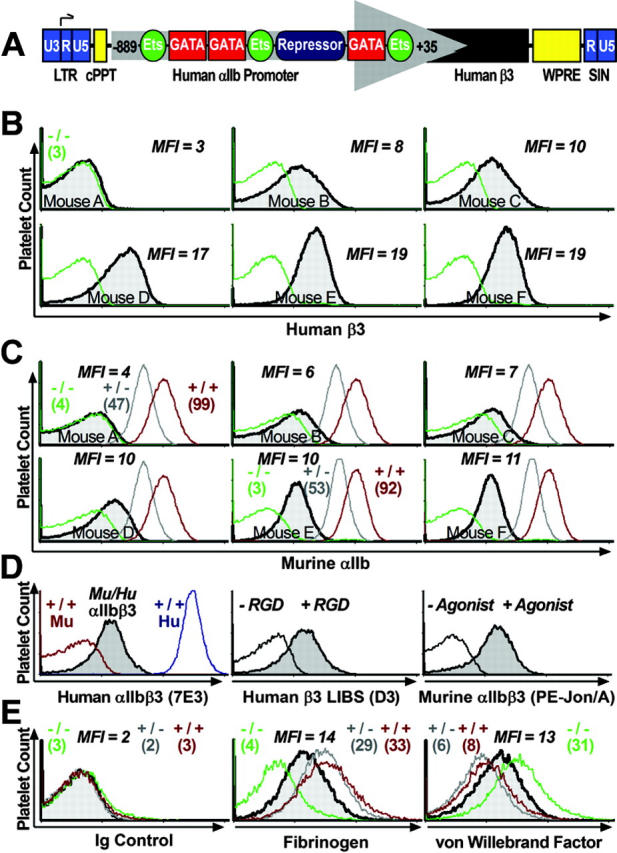
Expression of a functional, hybrid murine αIIb-human β3 integrin complex. (A) Schematic diagram of lentivirus vector β3-WPTS. The enhancer/promoter of the viral 3′-long terminal repeat (LTR) was removed to allow the vector to self-inactivate (SIN), and the human ITGA2B gene promoter (nucleotides –889 to +35) was used to direct megakaryocyte-specific synthesis of human β3 in mice. The promoter binds GATA and Ets for high-level gene transcription in megakaryocytes, and there is also a repressor region that inhibits gene expression in other lineages. The woodchuck hepatitis virus postregulatory element (WPRE) and the central polypurine tract (cPPT) were used to enhance the efficiency of transgene expression. (B) β3-WPTS–transduced bone marrow was transplanted into β3–/– mice (as described in “Materials and methods”). Flow cytometric histograms of murine platelets isolated from circulating whole blood of transplant recipients A to F showed that 5 mice exhibited significant levels of β3 on their platelet surface (shaded peak) compared with MFI levels for platelets from a β3–/– control (overlay histogram) using a PE-conjugated antibody to human β3. (C) Flow cytometric analysis detected integrin αIIb on the surface of platelets from recipients A to F with an FITC-conjugated antibody to murine αIIb. Histograms showed that mice B to F expressed αIIb on platelets (shaded peak) at intermediate MFI levels compared with the levels (in parentheses) on platelets from β3–/–, β3+/–, and β3+/+ controls. (D, left) Flow cytometric analysis revealed that Alexa 488–conjugated antibody (7E3) specific for human β3 in complex with αIIb or αv reacted positively with platelets from a β3–/– mouse expressing human β3 in complex with murine αIIb (shaded peak) in comparison with murine β3+/+ platelets serving as a negative control and human β3+/+ platelets used as a positive control. (Middle) Platelets from the mouse in the left panel were used to show that a fibrinogen mimetic peptide containing Arg-Gly-Asp (+RGD) could induce murine platelets expressing human β3 (shaded peak) to bind a monoclonal antibody (D3) (plus PE-F(ab′)2 goat anti–murine IgG Fc secondary antibody) that recognizes a ligand induced binding site (LIBS) exposed only on the high-affinity conformation of human β3. The platelets failed to bind D3 in the absence of RGD peptide (–RGD). (Right) Histogram demonstrating that an antibody (PE-Jon/A) specific for the high-affinity conformation of murine αIIbβ3 reacted positively with platelets expressing human β3 from the mouse described in the left and middle panels (shaded peak) following treatment with a cocktail of physiologic agonist of platelet activation (ADP, epinephrine, PAR4). Quiescent platelets were not recognized by PE-Jon/A in the absence of agonist (–Agonist). Results shown were observed in at least 2 separate experiments analyzing platelets from 3 separate mice that expressed human β3 at similar MFI levels. (E) Platelets from the mouse described in panel D were fixed and permeabilized to perform quantitative analysis with rabbit polyclonal antibodies to detect the intracellular storage of major ligands for αIIbβ3, fibrinogen, and VWF. (Left) Histogram shows that a nonreactive Alexa 647 rabbit polyclonal antibody did not react with murine platelets expressing human β3 (shaded peak) nor did it stain platelets from β3–/–, β3+/–, and β3+/+ controls. A nonreactive FITC-Ig showed identical results (not shown). (Middle, right) Histograms reveal that an FITC-antibody to fibrinogen (middle) and an Alexa 647–antibody to VWF (right) recognized platelets from the mouse expressing human β3 (shaded peak) at intermediate MFI levels compared with the level (in parentheses) in platelets from β3–/–, β3+/–, and β3+/+ controls. Results shown were observed in at least 2 separate experiments analyzing platelets from 3 separate mice that expressed human β3 at similar MFI levels.
Human β3 was detected on the surface of circulating blood platelets collected from 6 transplant recipients using immunocytometric analysis (Figure 1B; mice A-F, shaded peak) beginning at 3 weeks after transplantation. Platelets collectively stained positive with an antibody that specifically recognized human β3 with an MFI of approximately 3 to 6 times the level on platelets from β3–/– mice (Figure 1B, overlay histogram), indicating that most of the platelets from mice B to F were expressing human β3. The mice expressed different levels of integrin β3 on the platelet surface, as demonstrated with an incremental increase in the MFI from mice A to F.
Consistent with the levels of αIIb expressed on human platelets,11 cytometric analysis in Figure 1C (mice A and E, controls) revealed that an antibody specific for murine αIIb bound to β3–/– platelets with an MFI less than 5% of the level for platelets from normal β3+/+ mice, while heterozygous β3+/– platelets expressed αIIb at approximately 50% of normal levels. Murine αIIb was newly detected on the surface of circulating blood platelets from transplant recipients (Figure 1C; mice B-F, shaded peak), indicating the successful formation of a hybrid murine αIIb–human β3 integrin complex. There was a moderate density of αIIbβ3 receptors on the surface of platelets from animals that underwent transplantation, as indicated by detection of murine αIIb with an MFI equal to approximately 6% to 12% of the level on platelets from normal β3+/+ mice. Immunocytometric analysis failed to detect significant levels of integrin αv on the surface of platelets from β3–/– and β3+/+ control mice. αv was also not detected on murine platelets expressing human β3, indicting that murine platelets express integrin β3 primarily in complex with murine αIIb (data not shown).
In Figure 1D, we observed that an antibody (7E3)16 specific for human integrin β3 in complex with αIIb or αv reacted only with normal human platelets and murine platelets expressing human β3 (left panel), thus indicating that murine αIIb formed a hybrid complex with human β3. Further studies suggest that the hybrid integrin complex can be induced to undergo a conformational change consistent with outside-in integrin signaling, since platelets from mice that underwent transplantation bound an antibody (D3)17 that reacts specifically with an epitope exposed on the high affinity conformation of human β3 following treatment of platelets with a fibrinogen mimetic peptide containing Arg-Gly-Asp (Figure 1D, middle). An antibody (Jon/A-PE)33 specific for the active conformation of murine αIIbβ3 recognized the hybrid complex only on platelets treated with a cocktail of physiologic agonists of platelet activation, suggesting that the receptor can undergo conformation change associated with agonist induced inside-out integrin signaling (Figure 1D, right).
β3–/– mice exhibit a condition commonly found in GT characterized by the inability of patient platelets to store a major ligand for αIIbβ3, fibrinogen.11 This is demonstrated in Figure 1E, where the immunohistogram of fixed/permeabilized platelets from a β3–/– mouse displayed a low level of fibrinogen compared with β3+/– and β3+/+ controls having appreciable levels of platelet fibrinogen (middle panel). Remarkably, β3–/– platelets induced to express moderate levels of murine αIIb-human β3 (shown in Figure 1D) showed a renewed ability to store platelet fibrinogen at 3-fold higher levels compared with β3–/– control platelets (Figure 1E, middle). Surprisingly, when fluorescence immunocytometric analysis was used to detect another major ligand for αIIbβ3, VWF, within platelets from β3–/–, β3+/–, and β3+/+ controls, we discovered that β3–/– mice had 4 times the normal level of platelet VWF (Figure 1E, right). As observed for fibrinogen, the level of platelet VWF became normalized when β3–/– platelets were induced to express human β3 (Figure 1E, right).
A lentivirus vector encoding GFP under the transcriptional control of the human ITGA2B gene promoter was transduced into murine bone marrow and transplanted into lethally irradiated β3+/+ mice to determine if the human ITGA2B gene promoter confined transgene expression to the murine platelet lineage (Figure 2A). Flow cytometric analysis performed on platelets isolated from the peripheral blood of one mouse showed significant levels of GFP with an MFI equal to 5 times the level detected in platelets from an animal that did not receive a transplant of GFP-transduced marrow (Figure 2A, top row). In contrast, cells exhibiting the forward- and side-scattering properties of white (Figure 2A, middle row) and red (Figure 2A, bottom row) blood cells failed to demonstrate measurable levels of GFP as evidenced by the overlapping fluorescent histograms of cells from the transplant recipient with the negative control, suggesting that the human ITGA2B promoter targeted transgene expression specifically to murine platelets.
Figure 2.
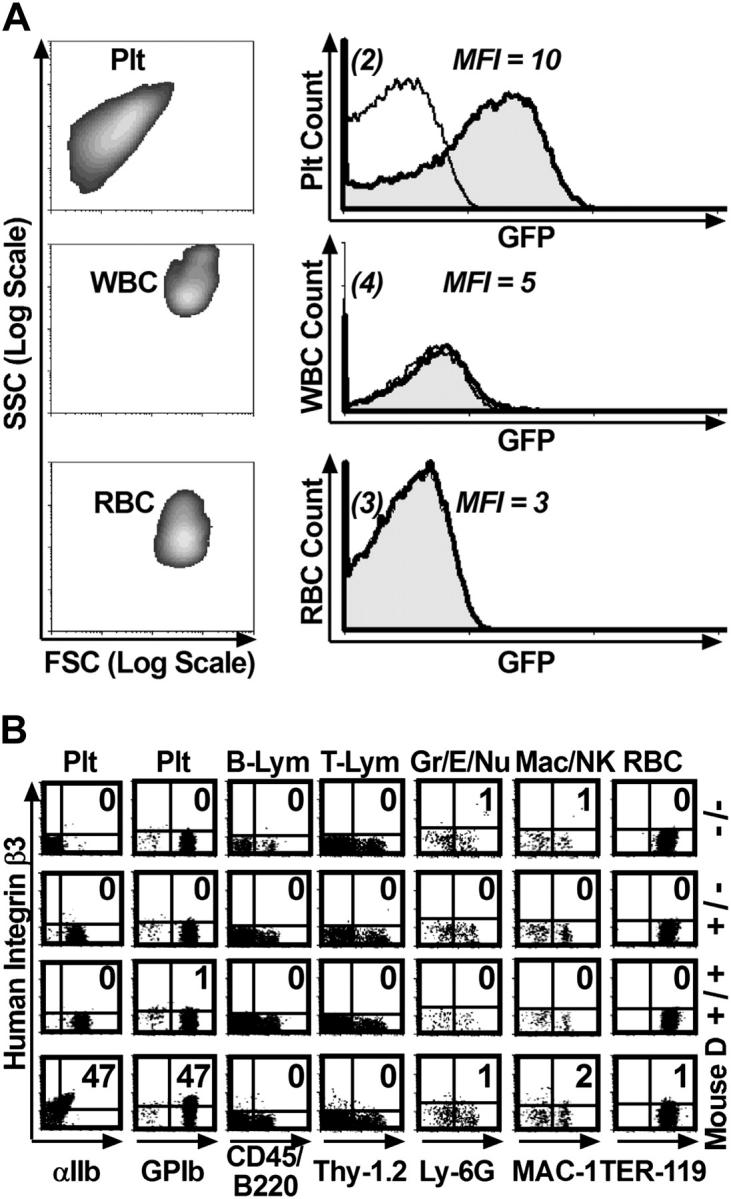
The human αIIb gene promoter confined transgene expression to platelets. (A) The human αIIb promoter confined expression of a GFP reporter gene within the platelet lineage. Entities exhibiting the forward (FSC) and side (SSC) scattering properties of platelets (Plt), white blood cells (WBC), and red blood cells (RBC) isolated from circulating whole blood of a β3+/+ mouse that received a transplant of –889GFPII-transduced bone marrow (column 1, density plot) were used to construct flow cytometric histograms comparing untransduced and GFP-transduced lineages (column 2). The transplant recipient showed significant levels of GFP in platelets (row 1, column 2, shaded peak) compared with the MFI for platelets from a β3+/+ control that did not receive a bone marrow transplant (open overlay histogram). In contrast, GFP was not detected above background levels within the WBCs or RBCs of the mouse that underwent transplantation. The result shown is representative of the outcome from analysis of peripheral blood collected from one mouse on 5 separate occasions. (B) Two-color flow cytometric analysis showed that the human αIIb promoter targeted expression of human β3 to platelets. A panel of antibodies that react with surface markers (x-axis, bottom) of specific murine cell lineages (x-axis, top) was used in conjunction with an antibody to human β3 (y-axis). The percentage of cells coexpressing both markers is indicated in each density plot (top right quadrant). Human β3 was not detected in cells from β3–/–, β3+/–, and β3+/+ controls (rows 1-3), while mouse D had significant levels of β3 detectable only in platelets (row 4). Plt indicates platelet; B-Lym, B lymphocyte; T-Lym, T lymphocyte; Gr/E/Nu, granulocyte/eosinophil/neutrophil; Mac/NK, macrophage/natural killer cell; and RBC, red blood cell. The result shown is representative of the outcome observed in 4 experiments that analyzed peripheral blood collected from 2 mice on 2 separate occasions.
To determine if human β3 was expressed exclusively on platelets, flow cytometric analysis was performed on whole blood isolated from β3–/– mice that received transplants of β3-transduced bone marrow (Figure 2B). Peripheral blood was collected from mouse D, which had one of the highest expression levels of β3 on its platelets (shown in Figure 1B). Blood cells were simultaneously stained with PE- and FITC-conjugated antibodies, one specific for human β3 (Figure 2B, y-axis) and the other a marker for a distinct murine cell type (Figure 2B, x-axis). Approximately 500 to 2500 positive events were collected for each lineage-specific marker. A cell type was considered to express human β3 if a significant percentage of cells simultaneously stained positive with both antibodies (Figure 2B, upper right quadrant of each density plot). As expected, platelets from controls expressed murine αIIb at different levels (identical to Figure 1C), while another platelet-specific marker, GPIb, as well as markers for all other cell lineages were expressed uniformly in control mice (Figure 2B, column 1-7, rows 1-3). Furthermore, the results showed that human β3 was not detected in association with the surface marker for any cell type from β3–/–, β3+/–, and β3+/+ control mice (Figure 2B, columns 1-7, rows 1-3). In contrast, mouse D had a high percentage of platelets coexpressing human β3 with murine αIIb and GPIb (47%; Figure 2B, columns 1-2, row 4), while human β3 was not detected above background levels on any other cell lineage (Figure 2B, columns 3-7, row 4).
Correction of integrin/platelet function
The hallmark of GT is a failure of platelets to aggregate upon stimulation with physiologic agonists of platelet activation.11 As a test for restored function, peripheral blood platelets were collected from each mouse to determine if the murine/human hybrid αIIbβ3 integrin complex could become activated, bind fibrinogen, and form platelet aggregates ex vivo. Platelet aggregation was induced by the addition of a cocktail of platelet activation agonists (ADP, epinephrine, and PAR4) in the presence of human fibrinogen. Results shown in Figure 3A demonstrate that platelets from mice A to D as well as β3–/–, β3+/–, and β3+/+ control samples aggregated in direct correlation with the level of β3 on their platelets (determined in Figure 1). Platelets from mouse A failed to aggregate (–14%), whereas mice B (42%), C (46%), and D (52%) each showed restored aggregation in accord with their increasing levels of αIIbβ3 on platelets. Platelets from experimental mice also aggregated in the presence of murine fibrinogen (not shown). In addition, samples could be induced to aggregate with a single agonist (ADP) as well as using the cocktail of physiologic agonists (not shown). When a mixture of platelets from mice E and F was pretreated with the complex-specific antibody, 7E3, we observed that agonist-induced platelet aggregation was increasingly inhibited with higher concentrations of 7E3 (0-50 μg/mL), indicating that the hybrid αIIbβ3 complex played a direct role in platelet aggregation (Figure 3B).
Figure 3.

Platelet function was restored in recipients of β3-transduced marrow. (A) Aggregation was measured ex vivo following incubation of washed platelets with fibrinogen and a cocktail of activation agonists (ADP, epinephrine, and PAR4). Platelets from β3-transduced marrow recipients A to D as well as β3–/–, β3+/–, and β3+/+ control samples aggregated in direct correlation with the level of β3 on their platelets. These results were observed for each mouse in at least 2 separate experiments. (B) Aggregation was measured ex vivo following a 30-minute pretreatment of platelets at 37°C with αIIbβ3 and αvβ3 complex–specific antibody, 7E3 (known to inhibit platelet aggregation),16 followed by incubation of washed platelets with human fibrinogen and a cocktail of platelet activation agonist (ADP, epinephrine, and PAR4). Shown is the aggregation profile of a mixture of platelets from β3-transduced marrow recipients E to F, which was increasingly inhibited with higher concentrations of 7E3 (0-50 μg/mL). This result represents the outcome of 3 separate experiments. Aggregation was not inhibited with nonspecific mouse Ig. (C) In vivo platelet function was examined by light microscopic analysis of fixed lung tissue stained with trichrome following intravenous injection of a platelet agonist (ADP) into mice (magnification, 400×). Thromboemboli (blue, arrows) formed in the pulmonary blood vessels (BV) of β3+/– and β3+/+ controls, while platelets in β3–/– animals were unable to form emboli. In contrast to results with β3–/– mice, platelets within transplant recipient C formed emboli that occluded the pulmonary blood vessels. A indicates alveolus; AD, alveolar duct; and TB, terminal bronchiole. This result represents the outcome observed after viewing several sections of each lung from 8 controls (3 β3–/–, 2 β3+/–, and 3 β3+/+ mice) and 3 mice expressing human β3. Images were captured with a Nikon Eclipse TS100 microscope (Nikon, Tokyo, Japan) using a 40×/0.55 numeric aperture objective.
Platelet function was next examined in vivo to determine whether platelets expressing the hybrid αIIbβ3 complex could form aggregates within the dynamic environment of the vasculature. Previous investigations used intravenously injected agonists of platelet activation to induce the formation of pulmonary thromboemboli in mice as a model to test the efficacy of antithrombotic agents.34 In the current study, infusion of ADP led to the formation of platelet emboli in the lungs of β3+/+ and β3+/– mice as shown in Figure 3C, where paraffin-embedded sections of fixed tissue were treated with trichrome. This stain differentially displays protein fibers (fibrotic tissue: muscle, collagen fibers, fibrin clots) and erythrocytes to allow identification of occluded blood vessels (BVs) (Figure 3C, arrows) among the terminal bronchiole (TB), alveoli (A), and alveolar ducts (AD). Platelets from β3–/– mice are unable to form aggregates; therefore, their pulmonary blood vessels were free of emboli following ADP treatment, and erythrocytes flowed through the BV without evidence of obstruction. In striking contrast, infusion of ADP into mouse C resulted in the formation of pulmonary thromboemboli with occluded vessels similar to lung tissue from ADP-challenged β3+/– and β3+/+ controls, demonstrating restored platelet function within the animal that underwent transplantation. Analysis of circulating peripheral blood in a subset of mice revealed normal platelet counts for all animals 1 hour prior to ADP treatment. Five minutes after ADP infusion, platelet levels decreased by approximately 50% within β3+/+ mice, while levels within β3+/– mice and β3–/– mice expressing human β3 decreased by approximately 30%. In contrast, platelet levels did not decrease within β3–/– mice treated with ADP. This outcome is consistent with the results of the histochemical analysis (Figure 3C).
A tail bleeding time assay was used to test whether platelets were able to mediate in vivo aggregation and clot retraction necessary to seal a wound. As previously reported,12 we also measured short bleeding times in normal β3+/+ mice (≈ 97 seconds, n = 20) when a distal 3-mm segment of tail was removed and immersed in saline at 37°C, and all of our β3–/– animals (n = 20) required cauterization of their wound to prevent bleeding far beyond the experimental end point of 600 seconds. Similar to the β3–/– controls, mice A and B that underwent transplantation were unable to heal their wound within 600 seconds. However, mouse B did exhibit reduced blood flow beginning at 420 seconds, indicating that intermediate levels of expression of αIIbβ3 on platelets slightly alleviated bleeding associated with GT. Remarkably, the 4 transplant recipients (mice C-F) with the highest level of human β3 on their platelets (Figure 1) stopped bleeding (at 500, 599, 581, and 455 seconds) prior to the experimental end point, thus demonstrating functional correction of the GT phenotype.
Antibody response to αIIbβ3 was diminished with intravenous immunoglobulin
To determine if the transplant recipients developed a humoral antibody response to the newly expressed β3, normal human platelets were incubated with plasma from each mouse, and the presence of platelet-bound antibody was assessed by flow cytometry using fluorescent anti–mouse Ig. Buffer without plasma was used as a negative control, and a monoclonal antibody to the human integrin αIIbβ3 complex (AP2) served as a positive control. Histograms in Figure 4A showed that (1:10 diluted) plasma from mouse A had a relatively high affinity and mouse B a low affinity for normal human platelets, while plasmas from mice C and D did not react with human platelets. Further analysis in Figure 4B revealed that plasma from mouse A also bound to murine platelets expressing murine β3 at normal (+/+) and partial (+/–) levels as well as murine β3–/– platelets expressing human β3. However, plasma from mouse A did not react with platelets from a β3–/– mouse, indicating that the antibody response in mouse A was specific for the αIIbβ3 complex and/or the individual integrin subunits.
Figure 4.

IVIG treatment diminished an antibody response to αIIbβ3. (A) Flow cytometric histograms showed that plasma from mice A and B contained Ig antibodies (shaded peak) that reacted with normal human platelets. Displayed is the relative fluorescence intensity of human platelets incubated with 1:10 diluted murine plasma and an FITC-conjugated F(ab′)2 goat anti–murine IgG Fc secondary antibody. Platelets incubated with secondary antibody and dilution buffer served as a negative control, while a monoclonal antibody to the human αIIbβ3 complex (AP2) was used as a positive control. This result was observed 4 times using platelets from 2 separate human donors analyzed on 2 separate occasions. (B) As in panel A, fluorescence analysis showed that 1:10 diluted plasma from mouse A also reacted with platelets from normal β3+/+, heterozygous β3+/– mice as well as platelets from another human β3-transduced transplant recipient (shaded peak). In striking contrast, plasma did not react with platelets isolated from a β3–/– mouse. Dilution buffer served as the negative control and a PE-conjugated monoclonal antibody to murine GPIbα was used as a positive control. (C) IVIG (0.5 mg/g body weight) was injected each day for 3 days into mouse A. Flow cytometry was then performed with human platelets incubated in plasma from mouse A and secondary antibody as described in panel A. The level of plasma Ig binding to human platelets before and after IVIG treatment of mouse A (black line) was determined by dividing the MFI of platelets incubated with mouse A plasma by the MFI of platelets treated with negative control buffer. The ratio decreased below 2.0 (dotted line), indicating a negligible affinity of plasma Ig for platelet proteins. The overlay graph shows an increase in platelets expressing αIIbβ3 following IVIG treatment of mouse A (orange line). The MFI ratio was calculated from flow-cytometric histograms detecting the binding of an FITC-conjugated antibody against murine αIIb to platelets from mouse A versus antibody binding to β3–/– platelets. These results represent the outcome of IVIG treatment for 3 mice with detectable plasma Ig to human platelets. (D) Following IVIG treatment, mouse A had restored platelet function in an aggregation assay performed at 27 weeks after transplantation. This result was observed using platelets from mouse A and platelets isolated from mice that received a transplant of bone marrow derived from mouse A as second- and third-generation recipients. (E) As in panel C, flow cytometric analysis using the MFI ratio of platelets binding an FITC-conjugated antibody to murine αIIb demonstrated long-term (32 weeks), stable expression of αIIbβ3 on the surface of platelets from mouse A after IVIG and mice B and D. Note: mouse C was killed for the in vivo platelet function assay at week 5.
Intravenous immunoglobulin (IVIG) has been used in people to attenuate an acquired immune response to integrin αIIbβ3.35 Therefore, mouse A was treated with IVIG to investigate whether its immune response to the newly formed αIIbβ3 complex could be alleviated. Flow cytometric analysis detected an immediate reduction in the level of plasma Ig from mouse A that bound to human platelets following 3 injections of IVIG (0.5 mg/g) (Figure 4C, black line). As a result, there was also a marked rise in the level of αIIbβ3 expressed on platelets from mouse A (Figure 4C, orange line), which stabilized at 20 weeks after transplantation (9 weeks after IVIG). A platelet aggregation assay performed at 27 weeks after transplantation showed that function was also restored in platelets from mouse A (Figure 4D).
Transgenic expression of αIIbβ3 was monitored in mice A to D for 32 weeks after transplantation to determine the efficiency and longevity of lentivirus transduction. As shown in Figure 4E, expression of human β3 was stable for untreated mice B and D and after IVIG for mouse A. Further analysis revealed that αIIbβ3 expression levels and aggregation profiles have remained stable for platelets derived from bone marrow that has been serially transplanted from mouse A into 3 subsequent generations of β3–/– mice (not shown).
Discussion
There are several megakaryocyte-specific gene promoters that could potentially direct transgene transcription including the following: members of the GP1BA-5-9 complex,36 ITGA2B,37 GP6,38 MPL,39 and platelet factor 4.40 These promoters bind GATA-1, Ets (Fli-1), and FOG-1 factors that induce transcription in early and mid stages of megakaryocytopoiesis.41,42 When human CD34+ peripheral blood stem cells were transduced with a retrovirus construct controlled by an 889-nucleotide fragment of the human αIIb promoter, we previously observed lineage-specific transgene expression of a reporter gene in cultured megakaryocytes of healthy individuals21 and ex vivo correction of the GT phenotype following the expression of the human integrin β3-subunit in megakaryocytes derived from GT patients.22 Since nucleotide sequence analysis revealed a high degree of homology between the gene promoters for human ITGA2B and murine Itga2b,43 and recent studies demonstrated that the human promoter fragment could drive “species-independent” megakaryocyte-specific gene expression in transgenic mice,44 this study was performed to test whether a lentivirus transfer vector under the transcriptional control of the human ITGA2B gene promoter could direct expression of the human integrin β3 in megakaryocytes and platelets of β3–/– mice. The results of this investigation newly demonstrate that the ITGA2B promoter directed expression of human β3 resulting in the formation of a stable, functional hybrid murine αIIb-human β3 integrin complex, which was confined to the surface of murine peripheral blood platelets (Figures 1, 2). Storage of fibrinogen was re-established when β3–/– platelets expressed human β3 (Figure 1). Of interest, VWF was detected at 3- to 4-fold higher levels within platelets of β3–/– mice compared with β3+/– and β3+/+ controls. This parallels the finding of a recent report describing increased levels of another α-granule protein, tumor growth factor β1 (TGF-β1), within β3–/– platelets.45 As observed for fibrinogen, VWF levels returned closer to normal values when β3–/– platelets were induced to express human β3 (Figure 1). This indicates that modulation of αIIbβ3 receptor levels may directly or indirectly affect the storage of other platelet proteins, although the mechanism and its potential effect on hemostasis and whether this occurs in human GT patients require further study. αIIbβ3 was detected at subnormal levels on the surface of platelets from mice that received transplants of β3-transduced marrow (ranging from 6% to 12% of normal αIIbβ3 receptor density) (Figures 1, 2). This result is remarkable since most of these animals displayed restored platelet function (Figure 3). The hybrid αIIbβ3 complex appeared to play a direct role in the aggregation of activated platelets since monoclonal antibody 7E316 inhibited aggregation of β3-transduced murine platelets, as occurs when 7E3 binds specifically to αIIbβ3 and αvβ3 causing reduced ligand recognition of normal human platelets. Results from bleeding time assays suggest that it is critical to obtain at least 7% of normal levels of integrin αIIbβ3 receptors per platelet to achieve effective wound healing in the β3–/– mouse since platelets with lower receptor levels were unable to control bleeding. Future studies to improve gene therapy for GT will likely explore methods to increase platelet receptor levels by use of recently characterized distal regulatory elements shown to enhance the ITGA2B promoter fragment's ability to drive high-level, megakaryocyte-specific transgene expression.46
IVIG effectively diminished platelet clearance and led to restored platelet function in a mouse (A) that developed an antibody response to αIIbβ3 (Figure 4). This issue is critical because human gene therapy for GT could lead to the formation of (1) inhibitory antibodies to αIIbβ3 that prevent platelet aggregation, and/or (2) an immune response that results in clearance of transduced platelets and thrombocytopenia as seen for GT patients that become refractory to transfused platelets from healthy donors.11 Integrin αIIbβ3 is a major antigen for alloimmune-, autoimmune-, and isoimmune-mediated thrombocytopenias, where antibody-coated or immune complex–coated platelets are destroyed prematurely by the reticuloendothelial system resulting in peripheral blood thrombocytopenia.35 Patients with immune-mediated platelet clearance have been successfully treated with corticosteroids, IVIG, and splenectomy.47 Therefore, it seemed reasonable to try to alleviate this condition in the experimental mice with one of these therapies. Following IVIG, plasma from mouse A showed an immediate drop in the level of Ig binding to platelets, which correlated with an increase in platelets expressing αIIbβ3 (Figure 4). This is consistent with results of IVIG use in humans48 and further suggests that this may be a useful model to investigate IVIG's mechanism of action.49
Results from this study indicate the likelihood for platelets to deliver additional therapeutic agents to the site of a vascular injury for treatment of other platelet defects and more common bleeding disorders such as hemophilia26 or to protect the platelet lineage from the harmful effects of cancer chemotherapies.50
Acknowledgments
We appreciate the technical assistance of Andrew Lochowitz and Brian Curtis (BloodCenter of Wisconsin, Milwaukee). Takara Bio Inc (Otsu, Shinga, Japan) kindly supplied RetroNectin for this study.
The current address of G.C.W. is as follows: Blood Research Institute, BloodCenter of Wisconsin, 8727 Watertown Plank Rd, Milwaukee, WI 53226-3548.
Prepublished online as Blood First Edition Paper, June 21, 2005; DOI 10.1182/blood-2004-12-4619.
Supported by grants HL-68138 (D.A.W.), PO1-HL66105 (R.O.H.), and PO1-HL45100 (G.C.W.) from the National Institutes of Health, and by an American Heart Association Award (Northland Affiliate) BGIA 0060437Z (D.A.W.) and a gift from the Children's Hospital Foundation (D.A.W.), Midwest Athletes against Childhood Cancer (MACC) Fund (D.A.W.), and Glanzmann Research Foundation (D.A.W.).
An Inside Blood analysis of this article appears in the front of this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 U.S.C. section 1734.
References
- 1.Hynes RO, Zhao Q. The evolution of cell adhesion. J Cell Biol. 2000;150: F89-F96. [DOI] [PubMed] [Google Scholar]
- 2.Hynes RO. Integrins: bidirectional, allosteric signaling machines. Cell. 2002;110: 673-687. [DOI] [PubMed] [Google Scholar]
- 3.Xiong JP, Stehle T, Diefenbach B, et al. Crystal structure of the extracellular segment of integrin alpha Vbeta3. Science. 2001;294: 339-345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wagner CL, Mascelli MA, Neblock DS, Weisman HF, Coller BS, Jordan RE. Analysis of GPIIb/IIIa receptor number by quantification of 7E3 binding to human platelets. Blood. 1996;88: 907-914. [PubMed] [Google Scholar]
- 5.Prandini MH, Martin F, Thevenon D, Uzan G. The tissue-specific transcriptional regulation of the megakaryocytic glycoprotein IIb gene is controlled by interactions between a repressor and positive cis-acting elements. Blood. 1996;88: 2062-2070. [PubMed] [Google Scholar]
- 6.Takagi J, Petre BM, Walz T, Springer TA. Global conformational rearrangements in integrin extracellular domains in outside-in and inside-out signaling. Cell. 2002;110: 599-611. [DOI] [PubMed] [Google Scholar]
- 7.Li R, Mitra N, Gratkowski H, et al. Activation of integrin alphaIIbbeta3 by modulation of transmembrane helix associations. Science. 2003;300: 795-798. [DOI] [PubMed] [Google Scholar]
- 8.Phillips DR, Jenkins CS, Luscher EF, Larrieu M. Molecular differences of exposed surface proteins on thrombasthenic platelet plasma membranes. Nature. 1975;257: 599-600. [DOI] [PubMed] [Google Scholar]
- 9.French DL, Coller BS. Hematologically important mutations: Glanzmann thrombasthenia. Blood Cells Mol Dis. 1997;23: 39-51. [DOI] [PubMed] [Google Scholar]
- 10.Glanzmann E. Hereditäre hämorrhagische Thrombasthenie: ein Beitrag zur Pathologie der Blutplättchen. J Kinderkr. 1918;88: 113-141. [Google Scholar]
- 11.George JN, Caen JP, Nurden AT. Glanzmann's thrombasthenia: the spectrum of clinical disease. Blood. 1990;75: 1383-1395. [PubMed] [Google Scholar]
- 12.Hodivala-Dilke KM, McHugh KP, Tsakiris DA, et al. Beta3-integrin-deficient mice are a model for Glanzmann thrombasthenia showing placental defects and reduced survival. J Clin Invest. 1999;103: 229-238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McHugh KP, Hodivala-Dilke K, Zheng MH, et al. Mice lacking beta3 integrins are osteosclerotic because of dysfunctional osteoclasts. J Clin Invest. 2000;105: 433-440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Reynolds LE, Wyder L, Lively JC, et al. Enhanced pathological angiogenesis in mice lacking beta3 integrin or beta3 and beta5 integrins. Nat Med. 2002;8: 27-34. [DOI] [PubMed] [Google Scholar]
- 15.Taverna D, Moher H, Crowley D, Borsig L, Varki A, Hynes RO. Increased primary tumor growth in mice null for beta3- or beta3/beta5-integrins or selectins. Proc Natl Acad Sci U S A. 2004;101: 763-768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Artoni A, Li J, Mitchell B, et al. Integrin beta3 regions controlling binding of murine mAb 7E3: implications for the mechanism of integrin alphaIIbbeta3 activation. Proc Natl Acad Sci U S A. 2004;101: 13114-13120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jennings LK, Haga JH, Slack SM. Differential expression of a ligand induced binding site (LIBS) by GPIIb-IIIa ligand recognition peptides and parenteral antagonists. Thromb Haemost. 2000;84: 1095-1102. [PubMed] [Google Scholar]
- 18.Pidard D, Montgomery RR, Bennett JS, Kunicki TJ. Interaction of AP-2, a monoclonal antibody specific for the human platelet glycoprotein IIb-IIIa complex, with intact platelets. J Biol Chem. 1983;258: 12582-12586. [PubMed] [Google Scholar]
- 19.Bergmeier W, Rackebrandt K, Schroder W, Zirngibl H, Nieswandt B. Structural and functional characterization of the mouse von Willebrand factor receptor GPIb-IX with novel monoclonal antibodies. Blood. 2000;95: 886-893. [PubMed] [Google Scholar]
- 20.Goldberger A, Kolodziej M, Poncz M, Bennett JS, Newman PJ. Effect of single amino acid substitutions on the formation of the PlA and Bak alloantigenic epitopes. Blood. 1991;78: 681-687. [PubMed] [Google Scholar]
- 21.Wilcox DA, Olsen JC, Ishizawa L, Griffith M, White GC II. Integrin alphaIIb promoter-targeted expression of gene products in megakaryocytes derived from retrovirus-transduced human hematopoietic cells. Proc Natl Acad Sci U S A. 1999;96: 9654-9659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wilcox DA, Olsen JC, Ishizawa L, et al. Megakaryocyte-targeted synthesis of the integrin beta(3)-subunit results in the phenotypic correction of Glanzmann thrombasthenia. Blood. 2000;95: 3645-3651. [PubMed] [Google Scholar]
- 23.Wiznerowicz M, Trono D. Conditional suppression of cellular genes: lentivirus vector-mediated drug-inducible RNA interference. J Virol. 2003;77: 8957-8961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zufferey R, Nagy D, Mandel RJ, Naldini L, Trono D. Multiply attenuated lentiviral vector achieves efficient gene delivery in vivo. Nat Biotechnol. 1997;15: 871-875. [DOI] [PubMed] [Google Scholar]
- 25.Lizee G, Aerts JL, Gonzales MI, Chinnasamy N, Morgan RA, Topalian SL. Real-time quantitative reverse transcriptase-polymerase chain reaction as a method for determining lentiviral vector titers and measuring transgene expression. Hum Gene Ther. 2003;14: 497-507. [DOI] [PubMed] [Google Scholar]
- 26.Wilcox DA, Shi Q, Nurden P, et al. Induction of megakaryocytes to synthesize and store a releasable pool of human factor VIII. J Thromb Haemost. 2003;1: 2477-2489. [DOI] [PubMed] [Google Scholar]
- 27.Klug CA, Cheshier S, Weissman IL. Inactivation of a GFP retrovirus occurs at multiple levels in long-term repopulating stem cells and their differentiated progeny. Blood. 2000;96: 894-901. [PubMed] [Google Scholar]
- 28.Tibbles HE, Vassilev A, Wendorf H, et al. Role of a JAK3-dependent biochemical signaling pathway in platelet activation and aggregation. J Biol Chem. 2001;276: 17815-17822. [DOI] [PubMed] [Google Scholar]
- 29.Mosesson MW, Finlayson JS. Biochemical and chromatographic studies of certain activities associated with human fibrinogen preparations. J Clin Invest. 1963;42: 747-755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kigasawa K, Saitoh K, Iwadate K, Ohkubo K, Irino O. A method for monitoring ADP-induced thromboembolism in mice. Thromb Res. 1984;35: 311-318. [DOI] [PubMed] [Google Scholar]
- 31.Harrison CR, Curtis BR, McFarland JG, Huff RW, Aster RH. Severe neonatal alloimmune thrombocytopenia caused by antibodies to human platelet antigen 3a (Baka) detectable only in whole platelet assays. Transfusion. 2003;43: 1398-1402. [DOI] [PubMed] [Google Scholar]
- 32.Hansen RJ, Balthasar JP. Effects of intravenous immunoglobulin on platelet count and antiplatelet antibody disposition in a rat model of immune thrombocytopenia. Blood. 2002;100: 2087-2093. [PubMed] [Google Scholar]
- 33.Bergmeier W, Schulte V, Brockhoff G, Bier U, Zirngibl H, Nieswandt B. Flow cytometric detection of activated mouse integrin alphaIIbbeta3 with a novel monoclonal antibody. Cytometry. 2002;48: 80-86. [DOI] [PubMed] [Google Scholar]
- 34.Hsiao G, Yen MH, Lee YM, Sheu JR. Antithrombotic effect of PMC, a potent alpha-tocopherol analogue on platelet plug formation in vivo. Br J Haematol. 2002;117: 699-704. [DOI] [PubMed] [Google Scholar]
- 35.Newman PJ, Valentin N. Human platelet alloantigens: recent findings, new perspectives. Thromb Haemost. 1995;74: 234-239. [PubMed] [Google Scholar]
- 36.Roth GJ, Yagi M, Bastian LS. The platelet glycoprotein Ib-V-IX system: regulation of gene expression. Stem Cells. 1996;14: 188-193. [DOI] [PubMed] [Google Scholar]
- 37.Uzan G, Prenant M, Prandini MH, Martin F, Marguerie G. Tissue-specific expression of the platelet GPIIb gene. J Biol Chem. 1991;266: 8932-8939. [PubMed] [Google Scholar]
- 38.Holmes ML, Bartle N, Eisbacher M, Chong BH. Cloning and analysis of the thrombopoietin-induced megakaryocyte-specific glycoprotein VI promoter and its regulation by GATA-1, Fli-1, and Sp1. J Biol Chem. 2002;277: 48333-48341. [DOI] [PubMed] [Google Scholar]
- 39.Kaushansky K, Drachman JG. The molecular and cellular biology of thrombopoietin: the primary regulator of platelet production. Oncogene. 2002;21: 3359-3367. [DOI] [PubMed] [Google Scholar]
- 40.Doi T, Greenberg SM, Rosenberg RD. Structure of the rat platelet factor 4 gene: a marker for megakaryocyte differentiation. Mol Cell Biol. 1987;7: 898-904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Romeo PH, Prandini MH, Joulin V, et al. Megakaryocytic and erythrocytic lineages share specific transcription factors. Nature. 1990;344: 447-449. [DOI] [PubMed] [Google Scholar]
- 42.Wang X, Crispino JD, Letting DL, Nakazawa M, Poncz M, Blobel GA. Control of megakaryocyte-specific gene expression by GATA-1 and FOG-1: role of Ets transcription factors. EMBO J. 2002;21: 5225-5234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Block KL, Ravid K, Phung QH, Poncz M. Characterization of regulatory elements in the 5′-flanking region of the rat GPIIb gene by studies in a primary rat marrow culture system. Blood. 1994;84: 3385-3393. [PubMed] [Google Scholar]
- 44.Tronik-Le Roux D, Roullot V, Schweitzer A, Berthier R, Marguerie G. Suppression of erythromegakaryocytopoiesis and the induction of reversible thrombocytopenia in mice transgenic for the thymidine kinase gene targeted by the platelet glycoprotein alpha IIb promoter. J Exp Med. 1995;181: 2141-2151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Reynolds LE, Conti FJ, Lucas M, et al. Accelerated re-epithelialization in beta3-integrin-deficient-mice is associated with enhanced TGF-beta1 signaling. Nat Med. 2005;11: 167-174. [DOI] [PubMed] [Google Scholar]
- 46.Thornton MA, Zhang C, Kowalska MA, Poncz M. Identification of distal regulatory regions in the human alpha IIb gene locus necessary for consistent, high-level megakaryocyte expression. Blood. 2002;100: 3588-3596. [DOI] [PubMed] [Google Scholar]
- 47.Stasi R, Provan D. Management of immune thrombocytopenic purpura in adults. Mayo Clin Proc. 2004;79: 504-522. [DOI] [PubMed] [Google Scholar]
- 48.Berchtold P, Dale GL, Tani P, McMillan R. Inhibition of autoantibody binding to platelet glycoprotein IIb/IIIa by anti-idiotypic antibodies in intravenous gammaglobulin. Blood. 1989;74: 2414-2417. [PubMed] [Google Scholar]
- 49.Hansen RJ, Balthasar JP. Mechanisms of IVIG action in immune thrombocytopenic purpura. Clin Lab. 2004;50: 133-140. [PubMed] [Google Scholar]
- 50.Sawai N, Zhou S, Vanin EF, Houghton P, Brent TP, Sorrentino BP. Protection and in vivo selection of hematopoietic stem cells using temozolomide, O6-benzylguanine, and an alkyltransferase-expressing retroviral vector. Mol Ther. 2001;3: 78-87. [DOI] [PubMed] [Google Scholar]