

Abstract
Accumulating evidence indicates that interaction of stromal cell-derived factor 1 (SDF-1/CXCL12 [CXC motif, ligand 12]) with its cognate receptor, CXCR4 (CXC motif, receptor 4), generates signals that regulate hematopoietic progenitor cell (HPC) trafficking in the bone marrow. During granulocyte colony-stimulating factor (G-CSF)–induced HPC mobilization, CXCL12 protein expression in the bone marrow decreases. Herein, we show that in a series of transgenic mice carrying targeted mutations of their G-CSF receptor and displaying markedly different G-CSF–induced HPC mobilization responses, the decrease in bone marrow CXCL12 protein expression closely correlates with the degree of HPC mobilization. G-CSF treatment induced a decrease in bone marrow CXCL12 mRNA that closely mirrored the fall in CXCL12 protein. Cell sorting experiments showed that osteoblasts and to a lesser degree endothelial cells are the major sources of CXCL12 production in the bone marrow. Interestingly, osteoblast activity, as measured by histomorphometry and osteocalcin expression, is strongly down-regulated during G-CSF treatment. However, the G-CSF receptor is not expressed on osteoblasts; accordingly, G-CSF had no direct effect on osteoblast function. Collectively, these data suggest a model in which G-CSF, through an indirect mechanism, potently inhibits osteoblast activity resulting in decreased CXCL12 expression in the bone marrow. The consequent attenuation of CXCR4 signaling ultimately leads to HPC mobilization.
Introduction
The majority of hematopoietic progenitor cells (HPCs) reside in the bone marrow surrounded by a complex, highly organized microenvironment. Under normal conditions, a small number of HPCs are released into the peripheral blood. Agents with distinct cellular targets and biologic activities can induce the mobilization of HPCs into blood, including hematopoietic growth factors, chemotherapeutic agents, and chemokines.1,2 Recently, mobilized peripheral blood HPCs have become the principal cellular source for reconstitution of the hematopoietic system following myeloablative therapy. Currently, granulocyte colony-stimulating factor (G-CSF) is the most widely used agent to induce HPC mobilization due to its potency, predictability, and safety.3 However, the mechanisms responsible for G-CSF–induced HPC mobilization have not been defined.
We previously showed that G-CSF receptor (G-CSFR) expression on HPCs is not required for their mobilization by G-CSF, suggesting that G-CSF induces HPC mobilization indirectly through the generation of trans-acting signals.4 The nature of the transacting signals that mediate G-CSF–induced HPC mobilization is unknown; however, accumulating evidence suggests that interaction of CXCL12 (stromal-derived factor 1 [SDF-1]) with its cognate receptor, CXCR4 (CXC motif, receptor 4), may play an important role in regulating G-CSF–induced HPC mobilization. CXCL12 is a CXC chemokine constitutively produced in the bone marrow by stromal cells.5 Studies of CXCL12- or CXCR4-deficient mice have established that these genes are necessary for the normal migration of HPCs from the fetal liver to the bone marrow and in the efficient retention of myeloid precursors in the adult bone marrow.6,7 Moreover, treatment with AMD-3100, a specific antagonist of CXCR4, induces rapid and robust HPC mobilization in both humans and mice.8,9 Finally, we and others showed that CXCL12 protein expression in the bone marrow is significantly decreased following G-CSF treatment.10-12 Collectively, these data suggest a model in which disruption of CXCL12/CXCR4 signaling is a key step in G-CSF–induced HPC mobilization.
The mechanisms mediating the G-CSF–induced decrease in CXCL12 protein expression in the bone marrow have not been defined. Previous reports suggested that neutrophil elastase (NE) and cathepsin G (CG) might regulate CXCL12 protein expression in the bone marrow through proteolytic cleavage of CXCL12.10,11 However, mice genetically lacking NE and CG display normal G-CSF–induced HPC mobilization, and the expected decrease in bone marrow CXCL12 protein was observed.13 Thus, the G-CSF–induced decrease in CXCL12 protein expression in the bone marrow does not require these proteases. It is possible that other proteases can compensate for the loss of NE and CG. Alternatively, nonproteolytic mechanisms may regulate CXCL12 expression in the bone marrow during G-CSF–induced HPC mobilization.
In this study, we characterize G-CSF–induced HPC mobilization and CXCL12 expression in the bone marrow in a series of transgenic mice carrying targeted mutations of their G-CSFR. We provide further evidence that disruption of CXCL12/CXCR4 signaling in the bone marrow is a key step in HPC mobilization. G-CSF regulates CXCL12 expression in the bone marrow primarily at the mRNA level. Evidence is provided that G-CSF inhibits osteoblast number and activity through an indirect mechanism leading to decreased CXCL12 expression in the bone marrow.
Materials and methods
Mice
GEpoR-, d715-, and d715F-deficient mice were generated, as described previously.14-16 GEpoR, d715, and d715F mice were backcrossed 10 generations onto a C57BL/6 background. Six- to 10-week-old mice were used in all studies. Mice were housed in a specific pathogen-free environment. All experiments were approved by the Washington University Animal Studies Committee.
Mobilization protocols
G-CSF. Recombinant human G-CSF, a generous gift from Amgen (Thousand Oaks, CA), was diluted in phosphate-buffered saline (PBS) with 0.1% low endotoxin bovine serum albumin (Sigma, St Louis MO) and administered by daily subcutaneous injection at a dose of 250 μg/kg or 100 μg/kg per day for 5 days. Mice were analyzed 3 to 4 hours after the final G-CSF dose.
AMD3100. AMD3100, a generous gift from AnorMED (Vancouver, BC, Canada), was reconstituted in sterile PBS and administered as a single subcutaneous injection at a dose of 5 mg/kg. Mice were analyzed 3 hours after injection or at the indicated times.
Peripheral blood and bone marrow analysis
Blood was obtained by retro-orbital venous plexus sampling in polypropylene tubes containing EDTA (ethylenediaminetetraacetic acid). Complete blood counts were determined using a Hemavet automated cell counter (CDC Technologies, Oxford, CT). Bone marrow was harvested by flushing with α-modified Eagle medium (α-MEM) containing 10% fetal calf serum (FCS). Bone marrow extracellular fluid was obtained by flushing each femur with 1 mL ice-cold PBS without serum, and the supernatant was harvested after centrifugation at 400g for 3 minutes.
CXCL12α ELISA
For enzyme-linked immunosorbent assays (ELISAs), 96-well plates were coated with 100 μL CXCL12 capture antibody (2 μg/mL) diluted in PBS and incubated overnight at room temperature. After incubation for 1 hour at room temperature with 300 μL blocking solution (1% bovine serum albumin [BSA], 5% sucrose, and 0.05% NaN3), 100 μL sample was added to each well and incubated for 2 hours at room temperature. After washing, 100 μL polyclonal biotinylated anti–human CXCL12 (250 ng/mL) in ELISA diluent (0.1% BSA, 0.05% Tween 20 in Tris [tris(hydroxymethyl-)aminomethane)]–buffered saline at pH7.3) was added to each well and incubated at room temperature for 2 hours. The reaction was developed by successive incubations with 1 μg/mL horseradish peroxidase streptavidin, substrate solution, and 50 μL2N H2SO4 to stop the reaction. A microplate reader set at 450 nm was used to determine optical density with readings at 550 nm subtracted from the results. Recombinant human CXCL12α was used to generate a standard curve. All ELISA reagents were purchased from R&D Systems (Minneapolis, MN).
Colony-forming cell assay
Blood, bone marrow, and spleen cells were harvested from mice using standard techniques and the number of nucleated cells in these tissues quantified using a Hemavet automated cell counter. We plated 10 to 20 μL blood, 1 × 105 nucleated spleen cells, or 2.0 × 104 nucleated bone marrow cells in 2.5 mL methylcellulose media supplemented with a cocktail of recombinant cytokines (MethoCult 3434; StemCell Technologies, Vancouver, BC, Canada). Cultures were plated in duplicate and placed in a humidified chamber with 6% CO2 at 37°C. Colonies containing at least 50 cells were counted on day 7 of culture.
Real-time quantitative RT-PCR
Femurs were flushed with a total of 2 ml TRIzol reagent (Invitrogen, Carlsbad, CA) followed by crushing of the remaining bone in TRIzol. RNA was isolated according to the manufacturer's instructions and resuspended in 150 μL RNase/DNase-free water. Real-time reverse transcriptasepolymerase chain reaction (RT-PCR) was performed using the TaqMan One-step RT-PCR Master Mix Reagents Kit (Applied Biosystems, Foster City, CA) on a GeneAmp 5700 Sequence Detection System (Applied Biosystems). The reaction mix consisted of 5 μL RNA, 12.5 μL RT-PCR mix, 200 nM forward primer, 200 nM reverse primer, 280 nM internal probe, and 0.625 μL Multiscribe reverse transcriptase and RNase inhibitor in a total reaction volume of 25 μL. Reactions were repeated in the absence of reverse transcriptase to confirm that DNA contamination was not present. RNA content was normalized to murine β-actin. PCR conditions were 48°C for 30 minutes and 95°C for 10 minutes, followed by 40 cycles of 95°C for 15 seconds and 60°C for 1 minute. Primers were: CXCL12 forward primer, 5′-GAGCCAACGTCAAGCATCTG-3′; CXCL12 reverse primer, 5′-CGGGTCAATGCACACTTGTC-3′; CXCL12 dT-FAM/TAMRA probe, 5′-TCCAAACTGTGCCCTTCAGATTGTTGC-3′; β-actin forward primer, 5′-ACCAACTGGGACGATATGGAGAAGA-3′; β-actin reverse primer, 5′-TACGACCAGAGGCATACAGGGACAA-3′; β-actin dT-FAM/TAMRA probe, 5′-AGCCATGTACGTAGCCATCCAGGCTG-3′; osteocalcin forward primer, 5′-TCTCTCTGCTCACTCTGCTGGCC-3′; osteocalcin reverse primer, 5′-TTTGTCAGACTCAGGGCCGC-3′; and osteocalcin dT-FAM/TAMRA probe, 5′-TGCGCTCTGTCTCTCTGACCTCACAGATGCCA-3′.
Cell sorting
Bone marrow cells were recovered from the femurs and tibia of mice by extensive flushing with 40 mL PBS. The femurs were then infused with PBS containing 50 mg/mL type IV collagenase (C5138, Sigma) and incubated at 37°C for 15 minutes. The collagenase-treated femurs were flushed again with PBS and cells pooled with the first flush fraction. Finally, the “empty” femurs were directly flushed with 1 mL TRIzol to recover RNA from cells firmly adherent to the bone matrix. The flushed cells were incubated with fluorescein isothiocyanate (FITC)–conjugated CD45 antibody and with the following panel of phycoerythrin (PE)–conjugated lineage-restricted antibodies: Gr-1 (granulocytes), B220 (B lymphocytes), CD3e (T lymphocytes), and Ter-119 (erythroid cells). All antibodies were from PharMingen (San Diego, CA). Cells were sorted on a MoFlo high-speed flow cytometer (Dako Cytomation, Fort Collins, CO). CXCL12 and β-actin mRNA were measured by quantitative real-time RT-PCR. To estimate the total CXCL12 mRNA contribution of each fraction, the number of cells in each fraction was multiplied by the amount of CXCL12 mRNA relative to β-actin mRNA found in that fraction. The number of cells in the TRIzol-flushed fraction was estimated using β-actin mRNA expression and was based on a standard curve showing that the level of β-actin mRNA correlated in a linear fashion with cell number (data not shown).
For stromal cell fractionation, femora, tibiae, and iliac crests were cleaned thoroughly to remove associated muscle tissue and then crushed in a mortar and pestle to release the marrow. Bone fragments were collected by filtration through a 40-μm cell strainer (BD Biosciences, San Jose, CA) and washed extensively in PBS with 2% FCS to remove nonadherent bone marrow cells. The bone fragments were further minced with a scalpel and then incubated at 37°C with a 3-mg/mL solution of type I collagenase (Worthington, Lakewood, NJ) in PBS for 40 minutes in a shaking waterbath. The resulting population of bone-derived cells was then depleted of residual hematopoietic cells by incubation with a cocktail of rat antimouse antibodies (B220, Mac-1, Gr-1, CD4, CD8, CD3, CD5, and Ter119) followed by incubation with anti–rat immunoglobulin-coupled Dynabeads (Dynal Biotech, Oslo Norway). Following lineage depletion, the cells were stained with a PE-conjugated anti-CD45, FITC-conjugated anti-CD31, biotinylated anti-CD51, and streptavidin-coupled allophycocyanin (all from PharMingen). The cells were separated using a FACSDiva high-speed cell sorter (BD Biosciences) into 3 fractions: endothelial cells (Lin– CD45–CD31+), osteoblasts (Lin– CD45–CD31–CD51+), and progenitor cells (Lin– CD45+). The purity of the endothelial and osteoblast fractions was confirmed by staining for von Willebrand factor or alkaline phosphatase, respectively (data not shown). Sorted cells were counted and then lysed in RNAZol (Iso-Tex Diagnostics, Friendswood, TX) or TRIzol for RNA isolation and subsequent real time RT-PCR analysis.
Osteoblast culture
Murine calvarial osteoblasts were obtained using minor modifications of published procedures.17 In brief, calvariae were removed aseptically from 3- to 4-day-old mice and incubated twice at 37°C for 10 minutes in PBS containing 4 mM EDTA and then subjected to repeated digestion for 10 minutes at 37°C with 200 U/mL type II collagenase (Worthington) in PBS. Products of early digestions were discarded, whereas later fractions (typically fractions 5-7) were collected by centrifugation and cultured in α-MEM containing 10% FCS and 1% penicillin/streptomycin. Cells were cultured until 80% confluent (undifferentiated osteoblasts). In some experiments, cells were then cultured in differentiation medium (α-MEM containing 10% FCS, 100 μg/mL ascorbic acid, and 5 mM β-glycerophosphate) for 1 week (differentiated osteoblasts).
Histomorphometry
Osteoblasts in the bone marrow were quantified by histomorphometry, as previously described.18 Briefly, femurs and tibiae were harvested, fixed overnight in 10% neutral formalin, decalcified by incubating in 14% EDTA at 4°C for 2 weeks, and then embedded in paraffin. To ensure that osteoclasts were excluded from the osteoblast count, deparaffinized sections were stained histochemically for tartrate-resistant acid phosphatase (TRAP) and counterstained with hematoxylin. Osteoblasts were counted in a blinded fashion in 4 to 6 200 × fields per section. In some cases, 2 sections 75 μm apart were taken from the same sample and osteoblast number averaged. The number of osteoblasts per millimeter bone perimeter (N.Ob/mm) was calculated using the OsteoMeasure Histomorphometry System (OsteoMetrics, Atlanta, GA).
Statistical analysis
Data are presented as mean plus or minus SEM or SD, as indicated in the text. Statistical significance was assessed using a 2-sided Student t test.
Results
The membrane-proximal region of the G-CSFR is sufficient to mediate HPC mobilization
To define the regions of the G-CSFR required for HPC mobilization, G-CSF–induced HPC mobilization was characterized in a series of transgenic mice expressing different targeted mutations of their G-CSFR (Figure 1A). The d715 G-CSFR mutation introduces a premature stop codon at nucleotide 2403, leading to truncation of the carboxy-terminal 96 amino acids of the G-CSFR. It is representative of G-CSFR mutations found in approximately 35% of patients with severe congenital neutropenia.19 Mice homozygous for the d715 G-CSFR mutation have normal basal hematopoiesis.15 In the d715F G-CSFR mutant, the sole remaining tyrosine (Y704) of d715 has been mutated to phenylalanine. Signal transducer and activator of transcription 3 (STAT-3) and STAT-5 activation by the d715F G-CSFR are markedly impaired.14 Homozygous d715F G-CSFR mutant mice display an isolated defect in granulopoiesis.14 In the GEpoR mutation, the entire cytoplasmic (signaling) domain of the G-CSFR is replaced with that of the erythropoietin receptor (EpoR).16 This chimeric receptor is predicted to bind G-CSF but transmit EpoR-specific signals. Homozygous GEpoR mice display peripheral neutropenia but have normal numbers of neutrophils in their bone marrow.16
Figure 1.

G-CSF–induced HPC mobilization in G-CSFR mutant mice. (A) Schematic of targeted G-CSFR mutations. Cytoplasmic tyrosines (Y) and the conserved box 1 and box 2 motifs are indicated. In the d715F mutant, the sole remaining tyrosine (Y704) of the G-CSFR has been mutated to phenylalanine (F). (B) Tissue distribution of HPCs following G-CSF treatment. Wild-type (WT) and G-CSFR mutant mice (n = 4, each) were treated with G-CSF (250 μg/kg/d) for 5 days and the number of CFU-Cs in blood, spleen, and bone marrow quantified 4 hours after the final dose of G-CSF. Data represent the mean ± SD. *P < .05 compared with G-CSF–treated WT mice.
The G-CSFR mutant mice, all inbred on a C57BL/6 background, were treated with G-CSF (250 μg/kg/d × 5 days) and the number of colony-forming cells (CFU-Cs) in the blood, spleen, and bone marrow measured (Figure 1B). A similar number of CFU-Cs was present at baseline in the bone marrow of all mice except for d715 mice, where a modest, but not significant, increase was observed. Compared with wild-type mice, HPC mobilization was significantly enhanced in d715 mice. Whereas a 15-fold increase from baseline in blood CFU-Cs was observed in wild-type mice, a 32-fold increase was observed in d715 mice. In contrast, HPC mobilization was severely impaired in GEpoR mice (1.7-fold increase in blood CFU-Cs from baseline), despite a normal number of CFU-Cs in the bone marrow. d715F mice displayed an intermediate phenotype. Although G-CSF induced a similar rise in blood and spleen CFU-Cs, the number of CFU-Cs in the bone marrow of d715F mice was significantly increased compared with wild-type mice. Similar results were observed after treating mice with 100 μg/kg/d G-CSF for 5 days (data not shown). These data show that the membrane-proximal 87 amino acids of the G-CSFR are sufficient to mediate G-CSF–induced HPC mobilization. Moreover, these data show that the signals generated by the GEpoR are not able to substitute for those of the G-CSFR to induce HPC mobilization.
Down-regulation of CXCL12α protein expression is a key event in G-CSF–induced HPC mobilization
Accumulating evidence suggests that CXCL12/CXCR4 signaling may be a key regulator of HPC trafficking in the bone marrow. We and others previously showed that CXCL12α protein expression in the bone marrow decreases during G-CSF–induced HPC mobilization.10-12 To extend these findings, we measured CXCL12α protein levels in the bone marrow of the G-CSFR mutant mice following G-CSF treatment (Figure 2A). As expected, G-CSF induced a significant decrease in CXCL12α protein expression in the bone marrow of wild-type mice. Likewise, a significant decrease in CXCL12α protein expression in the bone marrow of d715 and d715F mice was observed. In contrast, consistent with their impaired HPC mobilization phenotype, no significant change in CXCL12α protein expression was detected in GEpoR mice. In fact, a highly significant correlation was observed between the degree of HPC mobilization and the level of CXCL12α protein in the bone marrow (P < .001, Figure 2B).
Figure 2.

CXCL12α protein expression in the bone marrow following G-CSF treatment. (A) G-CSFR mutant mice (n = 7, each) were treated with G-CSF (100μg/kg/d) for 5 days and the amount of CXCL12α protein in the bone marrow extracellular fluid measured by ELISA. Data represent the mean ± SD. *P < .05 compared with untreated mice of the same genotype. (B) Plot of CXCL12α protein in the bone marrow versus the log of number of CFU-Cs in the blood on day 5 of G-CSF treatment (P < .001).
Recently, AMD3100, a selective CXCR4 antagonist capable of rapidly inducing HPC mobilization, was described.9 To determine whether disruption of CXCR4 signaling could rescue the HPC mobilization defect in GEpoR mice, we treated mice with AMD3100 and HPC mobilization was characterized. As reported previously, in wild-type mice, treatment with a single subcutaneous injection of AMD3100 induced a rapid increase in blood CFU-Cs that peaked 3 hours after injection (Figure 3).8 Interestingly, a similar increase in blood CFU-Cs was observed in GEpoR mice. Moreover, HPC mobilization by AMD3100 was found to be normal in G-CSFR–deficient mice (data not shown). These data show that AMD3100-induced HPC mobilization does not require G-CSFR signals.
Figure 3.

AMD3100 mobilization in GEpoR mice. Mice were treated with a single subcutaneous injection of AMD3100 (5 mg/kg). The number of CFU-Cs in the blood was measured over a 6-hour period (n = 3-4, each time point). Data represent the mean ± SD.
G-CSF regulates expression of CXCL12 mRNA in the bone marrow during HPC mobilization
Whereas previous studies have focused on the proteolytic cleavage of CXCL12, we considered an alternative mechanism to account for the decrease in CXCL12 protein in the bone marrow. We measured CXCL12 mRNA expression in the bone marrow during G-CSF treatment by directly flushing isolated femurs with TRIzol reagent to ensure that RNA was recovered from all cell types in the bone marrow. Real-time RT-PCR was performed for CXCL12 and mouse β-actin, as a control for RNA quality and content. CXCL12 mRNA progressively decreased during G-CSF treatment reaching a nadir on day 5 when HPC mobilization is maximal and returned to normal 2 days after discontinuing G-CSF (Figure 4A). The decrease in CXCL12 mRNA closely mirrored the decrease in CXCL12α protein expression in the bone marrow (Figure 4A). In fact, a strong correlation between CXCL12 mRNA and protein was observed (Figure 4B). It is possible that the marked myeloid expansion in the bone marrow induced by G-CSF may “dilute out” the CXCL12-expressing cells, thereby decreasing total CXCL12 mRNA levels in the bone marrow. To test this possibility, we examined CXCL12 mRNA expression in GEpoR mice. Importantly, G-CSF induces a similar increase in myeloid cells in the bone marrow in these mice compared with wild-type mice (the absolute number of neutrophils per femur in wild-type and GEpoR mice after G-CSF treatment was 15.4 ± 3.0 and 13.1 ± 1.8, respectively). However, despite comparable myeloid expansion, no significant decrease in CXCL12 mRNA was detected in GEpoR mice during G-CSF treatment (Figure 4C). Collectively, these data suggest that CXCL12 expression is regulated primarily at an mRNA level by G-CSF.
Figure 4.

CXCL12 mRNA expression during G-CSF–induced HPC mobilization. (A) WT mice were treated with G-CSF (100 μg/kg/d) for 5 days followed by a 2-day recovery period. The number of CFU-Cs in the blood (top panel) and CXCL12 protein expression in bone marrow extracellular fluid (middle panel) were measured at the indicated time points (n = 2, each). CXCL12 mRNA expression in the bone marrow was measured by directly flushing femurs with TRIzol and performing real-time RT-PCR on the recovered RNA. Shown is the relative amount of CXCL12 mRNA compared with β-actin mRNA (bottom panel). (B) Plot of CXCL12α protein versus CXCL12 mRNA (r2 = 0.56, P < .02). (C) WT and GEpoR mice (n = 6, each) were treated with G-CSF for 5 days and CXCL12 mRNA quantified. Data represent the mean ± SD. *P < .05 compared with day 0 or untreated mice.
Osteoblasts are the major source of CXCL12 in the bone marrow
Although controversial, current evidence suggests that CXCL12 is expressed in the bone marrow by osteoblasts, endothelial cells, and scattered stromal cells.5,20 Moreover, a recent report suggested that stem and progenitor cells may express CXCL12 at a low level.21 To determine which cell types in the bone marrow express CXCL12 mRNA and are down-regulated in response to G-CSF, mice were treated with G-CSF and bone marrow cells sorted into stromal cell (CD45– Lin–), progenitor-enriched (CD45+ Lin–), and mature hematopoietic cell (Lin+) fractions (Figure 5). In addition, TRIzol was injected directly into the flushed femurs to assess the contribution of cells remaining tightly associated with the bone matrix (“bone fraction” in Figure 5B). In untreated mice, the great majority of CXCL12 mRNA was found in the stromal cell and bone fractions (Figure 5B). Furthermore, CXCL12 expression in these fractions was decreased by G-CSF treatment. These results suggest that stromal cells are the major source of CXCL12 in the bone marrow and are down-regulated by G-CSF treatment.
Figure 5.

Regulation of bone marrow stromal cell activity during G-CSF–induced HPC mobilization. (A) Bone marrow cells were recovered from the femurs and tibiae of mice by flushing and treating with collagenase and then sorted into the indicated cell populations based on CD45 and lineage expression. Shown is a representative histogram. (B) To examine cells firmly adherent to the bone matrix, the flushed femurs were injected with TRIzol to obtain the “bone fraction.” Total CXCL12 mRNA in each cell population was estimated by multiplying the measured CXCL12 mRNA by the cell number in each cell fraction; the number of cells in the bone fraction was estimated based on β-actin mRNA levels. *P < .05. (C) Cells harvested from the bone fraction were sorted into the indicated cell populations (see “Materials and methods”) and CXCL12 mRNA expression relative to β-actin expression was measured. Data represent the mean ± SEM.
To define which stromal cell types express CXCL12, the boneadherent cell population was further fractionated into hematopoietic progenitor, mature osteoblast, and endothelial cell fractions (see “Materials and methods”). Low-level CXCL12 mRNA was again detected in the hematopoietic cell fraction (Figure 5C). Consistent with previous studies showing constitutive CXCL12 expression in bone marrow endothelial cells,22 a relatively high level of CXCL12 mRNA was detected in the endothelial cell fraction. However, the highest level of CXCL12 mRNA expression was detected in the mature osteoblast fraction. Relative to β-actin mRNA, mature osteoblast express 9.4-fold more CXCL12 mRNA than endothelial cells. These data suggest that the majority of CXCL12 in the bone marrow microenvironment is produced by osteoblasts.
G-CSF treatment potently inhibits osteoblast activity in the bone marrow
Surprisingly, despite the decrease in total bone marrow CXCL12 mRNA expression (Figure 5B), on a per cell basis no significant decrease in CXCL12 mRNA was detected in osteoblasts isolated from mice following G-CSF treatment (Figure 5C). These data raised the possibility that, rather than affecting SDF-1 expression per osteoblast, G-CSF regulated the number of osteoblasts in the bone marrow. To explore this possibility, osteoblast number in the bone marrow was measured by histomorphometry. Indeed, after 5 days of G-CSF treatment, a striking reduction in the number of endosteal osteoblasts was observed (Figure 6A-C). To confirm this observation, the expression of osteocalcin, a specific marker of mature osteoblasts, in the bone marrow during G-CSF treatment was assessed (Figure 6D). Notably, osteocalcin mRNA expression was sharply reduced during G-CSF treatment; a 47± 12-fold reduction in osteocalcin mRNA (relative to β-actin mRNA) was observed in the bone marrow of mice treated with G-CSF compared with untreated mice. Likewise, a significant decrease in serum osteocalcin protein was detected in G-CSF–treated mice (data not shown). This latter finding is consistent with a previous report showing that serum levels of osteocalcin decreased in patients during G-CSF treatment.23 Collectively, these data provide strong evidence that G-CSF treatment potently suppresses osteoblast activity in the bone marrow.
Figure 6.
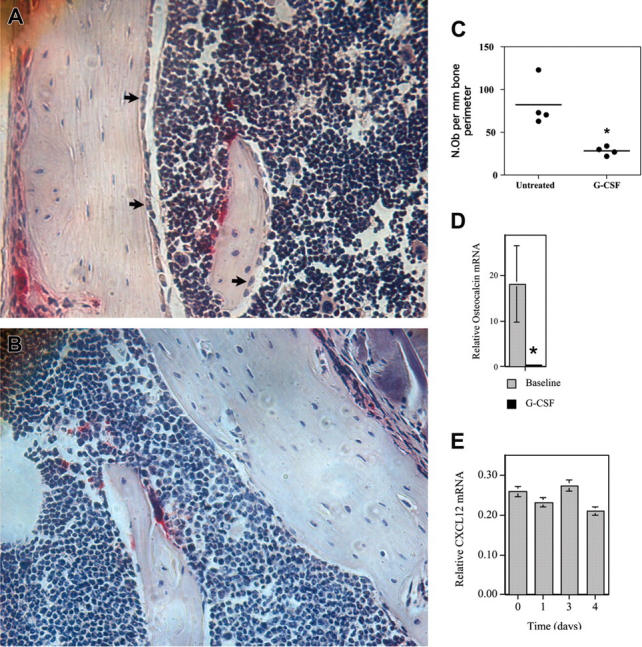
G-CSF inhibits osteoblast activity in the bone marrow. (A-D) WT mice were treated with G-CSF (125 μg/kg twice daily for 5 days) and osteoblast activity assessed. (A-B) Representative photomicrographs show endosteal osteoblasts (arrows) in untreated (A) or G-CSF–treated mice (B); original magnification × 400. (C) Quantification of osteoblast number by histomorphometry. The number of osteoblasts (N.Ob) per millimeter of bone perimeter is shown. (D) Bone marrow osteocalcin mRNA expression. Total bone marrow RNA was obtained by directly flushing femurs with TRIzol. The expression of osteocalcin mRNA relative to β-actin mRNA is shown. (E) Primary osteoblasts were cultured in the presence of 100 ng/mL G-CSF for the indicated time and CXCL12 mRNA quantified. Data represent the mean ± SEM. *P < .05. All images were obtained with a Nikon Eclipse E600 microscope using a Nikon PlanApo 20 ×/0.45 NA objective (Nikon, Melville, NY). The microscope was equipped with a Sony DXC S500 digital camera (Sony Electronics, Park Ridge, NJ), and images were captured using Kodak Imaging for Windows software (Eastman Software, Billerica, MA).
We next investigated whether G-CSF could directly regulate CXCL12 expression in cultures of primary murine osteoblasts. Osteoblasts were harvested from the calvariae of newborn mice and cultured in the presence or absence of G-CSF for 5 days. In some experiments, the osteoblasts were first cultured for 1 week in the presence of ascorbic acid and β-glycerophosphate to induce osteoblast differentiation. As expected, a high level of CXCL12 protein and mRNA expression was detected in cultures of undifferentiated and differentiated osteoblasts (Figure 6E and data not shown). However, G-CSF had no significant effect on CXCL12 expression. Moreover, no G-CSFR mRNA was detected using a sensitive RT-PCR assay (data not shown). These data suggest that G-CSF does not directly regulate CXCL12 expression in osteoblasts.
Discussion
Clinically, G-CSF is the most widely used agent to mobilize HPCs, yet the mechanisms mediating HPC mobilization by G-CSF are poorly understood. To begin to define the regions of the G-CSFR that mediate this response, we characterized HPC mobilization by G-CSF in a series of transgenic mice carrying different targeted G-CSFR mutations. HPC mobilization in d715 G-CSFR mice is significantly enhanced compared with wild-type mice, suggesting the presence of an inhibitory domain in the carboxy-terminal tail of the G-CSFR. Previous studies have shown that both receptor internalization24,25 and activation of negative regulators of signaling (including suppressor of cytokine signaling 3 [SOC3],26 SHP-1 [SH2-containing phosphatase-1],25 and SH2-contianing inositol phosphatase [SHIP]25) are defective with the d715 G-CSFR. Whether any of these signaling alterations is responsible for the increased mobilization response remains to be answered. Interestingly, the number of CFU-Cs in the bone marrow of G-CSF–treated d715F mice is increased compared with G-CSF–treated wild-type mice, despite comparable numbers of the CFU-Cs in the blood and bone marrow. These observations are consistent with a subtle defect in HPC mobilization in d715F mice. Nonetheless, these data suggest that STAT-3 and STAT-5 activation by the G-CSFR is not absolutely required for HPC mobilization because their activation by the d715F G-CSFR is markedly impaired.14 Interestingly, G-CSF–induced HPC mobilization is markedly impaired in GEpoR mice, despite a comparable (to wild-type mice) expansion in myeloid cells and HPCs in the bone marrow. Thus, signals generated by the chimeric GEpoR are able to efficiently transduce proliferative but not mobilization signals, suggesting an element of specificity in the mobilization signaling pathways. Of note, these data clearly demonstrate that increases in bone marrow cellularity and HPC content alone are not sufficient to induce HPC mobilization.
Accumulating evidence suggests that CXCL12/CXCR4 signaling plays a key role in regulating HPC trafficking in the bone marrow. Mice with targeted disruptions of CXCL12 or CXCR4 exhibit defective hematopoiesis in the bone marrow, possibly due to the failure of HPCs to migrate from the fetal liver to the bone marrow.7,27 Moreover, mice given transplants with CXCR4-deficient bone marrow cells show reduced engraftment and premature release of immature myeloid cells into the blood.7,27 Elevation of CXCL12 levels in the blood by administration of CXCL12 or by injection of an adenoviral vector expressing CXCL12 is associated with a significant mobilization of HPCs into the blood.28,29 Conversely, treatment with AMD3100, a selective antagonist of CXCR4, induces rapid and robust HPC mobilization in mice and humans.8,9 Finally, we and others previously showed that G-CSF treatment results in a significant decrease in CXCL12 protein levels in the bone marrow of wild-type mice.10-12 In the present study, we show that CXCL12 protein levels in the bone marrow after G-CSF treatment strongly correlate with HPC mobilization in the G-CSFR mutant mice. For example, the greatest decrease in CXCL12 protein expression in the bone marrow was observed in those mice displaying the most robust HPC mobilization, namely, the d715 G-CSFR mice. Perhaps most telling is the lack of a significant decrease in CXCL12 protein expression in the mobilizationdefective GEpoR mice (Figure 2). The availability of AMD3100, a selective CXCR4 antagonist, provided the opportunity to determine whether disruption of CXCR4 signaling could rescue the mobilization defect in GEpoR mice. Indeed, AMD3100-induced HPC mobilization in GEpoR mice was comparable to wild-type mice. Collectively, these data suggest that CXCL12 is an important retention signal for HPC in the bone marrow, and the data support a model in which disruption of CXCL12/CXCR4 signaling is a key step in G-CSF–induced HPC mobilization.
It is likely that multiple mechanisms contribute to the disruption of this signaling pathway. CD26 (dipeptidylpeptidase IV), a membranebound extracellular serine-protease expressed on a subset of HPCs, inactivates CXCL12 through proteolytic cleavage.30,31 Importantly, G-CSF–induced HPC mobilization is defective in CD26-deficient mice or in wild-type mice treated with a specific CD26 inhibitor.30,31 However, there is no evidence showing that CD26 activity is modulated during G-CSF treatment. In contrast, G-CSF treatment induces the release of a number of proteases into the bone marrow microenvironment, including NE, CG, and matrix metalloproteinase-9 (MMP-9).32 These proteases are able to cleave several adhesion molecules thought to play an important role in regulating HPC trafficking in the bone marrow, including c-Kit, vascular cell adhesion molecule 1 (VCAM-1), CXCR4, and CXCL12.10,11,33,34 In particular, NE and CG are able to cleave and inactivate CXCL12 in vitro.10,11 However, G-CSF–induced HPC mobilization and decrease in bone marrow CXCL12 protein are normal in NE × CG-deficient mice.13 Thus, there must be efficient NE- and CG-independent mechanisms to disrupt CXCL12/CXCR4 signaling during G-CSF–induced HPC mobilization.
As an alternative mechanism to proteolytic cleavage to regulate CXCL12 expression, we examined the effect of G-CSF treatment on the expression of CXCL12 mRNA in the bone marrow. We show that G-CSF treatment induces a decrease in bone marrow CXCL12 mRNA that mirrors the fall in CXCL12 protein. In fact, a strong correlation between CXCL12 protein and mRNA levels in the bone marrow was observed. This decrease in CXCL12 mRNA is not simply due to the dilution of CXCL12-expressing cells in the bone marrow during G-CSF treatment because no significant decrease in CXCL12 mRNA was observed in GEpoR mice, despite a similar expansion of myeloid cells in the bone marrow. These data suggest that during G-CSF–induced HPC mobilization, CXCL12 expression in the bone marrow is primarily regulated at the mRNA level.
The mechanism by which G-CSF regulates CXCL12 mRNA expression in the bone marrow is an important unanswered question. In particular, the cell types in the bone marrow that express CXCL12 and are regulated during G-CSF treatment are unknown. One report suggested that CXCL12 is primarily expressed by osteoblasts, endothelial cells, and scattered stromal cells in the mesenchyme.5 In contrast, Ara and colleagues, using a transgenic mouse in which the green fluorescent protein gene was inserted into the CXCL12 gene locus, reported that endothelial cells and osteoblasts in the bone marrow did not constitutively express CXCL12.35 Finally, a recent report suggested that a subset of hematopoietic progenitors produce a small amount of CXCL12.21
To address this question, we quantified CXCL12 mRNA expression in sorted bone marrow populations of mature hematopoietic cells, progenitor cells, and stromal cells. These data confirm that progenitor cells, defined as CD45+ Lin– cells, express a low level of CXCL12. Given the low level of expression and the relative scarcity of these cells, it is unlikely that hematopoietic progenitor cells contribute significantly to the bulk production of CXCL12 in the bone marrow. Nonetheless, it is possible that CXCL12 expression by progenitor cells may significantly regulate the trafficking of progenitors cells through an autocrine or paracrine mechanism; further study is needed to address this possibility. On the other hand, bone marrow stromal cells appear to be the major source of CXCL12 in the bone marrow. Within the stromal cell fraction, endothelial cells and mature osteoblasts express significant CXCL12 mRNA. Based on the high level of CXCL12 expression per cell and the relative abundance of osteoblasts within the bone marrow stromal cell fraction, we conclude that osteoblasts are the major source of CXCL12 in the bone marrow. Interestingly, CXCL12 mRNA expression per osteoblast did not change during G-CSF treatment. Rather, G-CSF appears to regulate CXCL12 mRNA expression in the bone marrow by decreasing osteoblast number.
Accumulating evidence indicates that osteoblasts play a key role in establishing and maintaining the stem cell niche in the bone marrow.20,36,37 In addition to CXCL12, osteoblasts express several genes thought to be important for stem cell function, including the notch ligand Jagged-1,37 a number of hematopoietic growth factors (eg, G-CSF),38 angiopoietin,39 and N-cadherin.36 Herein, we show that G-CSF potently inhibits mature osteoblast activity in the bone marrow. After 5 days of G-CSF treatment, mature osteoblast number in the bone marrow was reduced at least 3-fold. Moreover, osteocalcin mRNA expression in the bone marrow was reduced nearly 50-fold. The magnitude of the change in osteocalcin expression compared with the change in osteoblast number in the bone marrow suggests that G-CSF may regulate both osteoblast number and activity. Intriguingly, patients treated long-term with G-CSF develop marked osteopenia.40 In addition, transgenic mice overexpressing G-CSF develop osteopenia.41,42 Collectively, these data raise the possibility that G-CSF, by regulating osteoblast function, may have profound effects on the stem cell niche that ultimately contribute to HSC mobilization.
We previously showed by analysis of bone marrow chimeras between G-CSFR–deficient and wild-type mice that G-CSFR expression on bone marrow stromal cells was neither necessary nor sufficient to mediate G-CSF–induced hematopoietic progenitor cell mobilization.4 Consistent with this finding, in the present study we show that cultured primary osteoblasts do not express detectable G-CSFR using a sensitive RT-PCR assay. Moreover, G-CSF does not modulate CXCL12 expression in primary osteoblast cultures. Together, these data provide compelling evidence that G-CSF regulates osteoblast CXCL12 through an indirect mechanism.
In summary, this study provides additional evidence that strongly supports a model in which disruption of CXCL12/CXCR4 signaling is a key event in G-CSF–induced HPC mobilization. Osteoblasts appear to be the major source of CXCL12 production in the bone marrow. G-CSF treatment potently inhibits osteoblast activity in the bone marrow, thereby reducing CXCL12 expression. These data suggest a model (Figure 7) in which G-CSF initiates the mobilization cascade by stimulating an as yet unidentified G-CSFR–expressing cell population in the bone marrow. These cells then generate a trans-acting signal that suppresses osteoblast activity and, in particular, CXCL12 expression. The consequent decrease in CXCR4 signaling in hematopoietic progenitor cells then enhances their migration from the bone marrow, through unclear mechanisms. A better understanding of the mechanism by which G-CSF regulates CXCL12 mRNA expression may lead to the development of improved clinical protocols for stem cell mobilization in patients.
Figure 7.

Model of G-CSF–induced HPC mobilization. Osteoblasts constitutively produce large amounts of CXCL12, providing an important retention signal for HPCs in the bone marrow. G-CSF initiates the mobilization cascade by stimulating a population of G-CSFR+ cells in the bone marrow. These cells, in turn, negatively regulate osteoblast number and activity, resulting in decreased CXCL12 expression in the bone marrow. The consequent decrease in CXCR4 signaling in HPCs leads to their migration from the bone marrow to blood.
Acknowledgments
The authors thank Jill Mayer and Katherine Stumpf for their expert technical assistance. We also thank Deborah Novack and Pat Kellar for their expert assistance in performing the osteoblast histomorphometry. Finally, the authors thank Amgen and AnorMed for generously providing the G-CSF and AMD-3100, respectively.
Prepublished online as Blood First Edition Paper, July 21, 2005; DOI 10.1182/blood-2004-01-0272.
Supported by grants from the National Institutes of Health Heart, Lung and Blood Institute (R01 HL60772-01A1, D.C.L.; T32 HL 07088-23, C.L.S.) and from the National Health and Medical Research Council of Australia (288701, J.-P.L.).
C.L.S. and M.J.S. performed and analyzed most of the data and wrote the majority of the manuscript; F.L. performed and analyzed most of the mobilization experiments and wrote the relevant experimental sections; B.S. and P.J.S. performed the stromal cell sorting experiments and wrote the relevant sections; I.W. and J.-P.L. performed the qRT-PCR analysis of SDF-1 expression in sorted stromal cell populations and wrote the relevant sections; J.C. and F.P.R. performed the experiments with primary osteoblasts and wrote the relevant sections; and D.C.L., the senior author, supervised all experiments and was responsible for the final manuscript.
C.L.S. and M.J.C. contributed equally to this study.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 U.S.C. section 1734.
References
- 1.Thomas J, Liu F, Link DC. Mechanisms of mobilization of hematopoietic progenitors with granulocyte colony-stimulating factor. Curr Opin Hematol. 2002;9: 183-189. [DOI] [PubMed] [Google Scholar]
- 2.To LB, Haylock DN, Simmons PJ, Juttner CA. The biology and clinical uses of blood stem cells. Blood. 1997;89: 2233-2258. [PubMed] [Google Scholar]
- 3.Pelus LM, Horowitz D, Cooper SC, King AG. Peripheral blood stem cell mobilization. A role for CXC chemokines. Crit Rev Oncol Hematol. 2002;43: 257-275. [DOI] [PubMed] [Google Scholar]
- 4.Liu F, Poursine-Laurent J, Link DC. Expression of the G-CSF receptor on hematopoietic progenitor cells is not required for their mobilization by GCSF. Blood. 2000;95: 3025-3031. [PubMed] [Google Scholar]
- 5.Ponomaryov T, Peled A, Petit I, et al. Induction of the chemokine stromal-derived factor-1 following DNA damage improves human stem cell function. J Clin Invest. 2000;106: 1331-1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nagasawa T, Hirota S, Tachibana K, et al. Defects of B-cell lymphopoiesis and bone-marrow myelopoiesis in mice lacking the CXC chemokine PBSF/SDF-1. Nature. 1996;382: 635-638. [DOI] [PubMed] [Google Scholar]
- 7.Ma Q, Jones D, Springer TA. The chemokine receptor CXCR4 is required for the retention of B lineage and granulocytic precursors within the bone marrow microenvironment. Immunity. 1999;10: 463-471. [DOI] [PubMed] [Google Scholar]
- 8.Broxmeyer HE, Hangoc G, Cooper S, Li X, Bridger G, Clapp D. Interference of the SDF-1/CXCR4 axis in mice with AMD3100 induces rapid high level mobilization of hematopoietic progenitor cells, and AMD3100 acts synergistically with G-CSF and MIP-1α to mobilize progenitors [abstract]. Blood. 2001;98: 811a. [Google Scholar]
- 9.Liles WC, Broxmeyer HE, Rodger E, et al. Mobilization of hematopoietic progenitor cells in healthy volunteers by AMD3100, a CXCR4 antagonist. Blood. 2003;102: 2728-2730. [DOI] [PubMed] [Google Scholar]
- 10.Levesque JP, Hendy J, Takamatsu Y, Simmons PJ, Bendall LJ. Disruption of the CXCR4/CXCL12 chemotactic interaction during hematopoietic stem cell mobilization induced by GCSF or cyclophosphamide. J Clin Invest. 2003;111: 187-196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Petit I, Szyper-Kravitz M, Nagler A, et al. G-CSF induces stem cell mobilization by decreasing bone marrow SDF-1 and up-regulating CXCR4. Nat Immunol. 2002;3: 687-694. [DOI] [PubMed] [Google Scholar]
- 12.Semerad CL, Liu F, Gregory AD, Stumpf K, Link DC. G-CSF is an essential regulator of neutrophil trafficking from the bone marrow to the blood. Immunity. 2002;17: 413-423. [DOI] [PubMed] [Google Scholar]
- 13.Levesque JP, Liu F, Simmons PJ, et al. Characterization of hematopoietic progenitor mobilization in protease-deficient mice. Blood. 2004;104: 65-72. [DOI] [PubMed] [Google Scholar]
- 14.McLemore ML, Grewal S, Liu F, et al. STAT-3 activation is required for normal G-CSF-dependent proliferation and granulocytic differentiation. Immunity. 2001;14: 193-204. [DOI] [PubMed] [Google Scholar]
- 15.McLemore ML, Poursine-Laurent J, Link DC. Increased granulocyte colony-stimulating factor responsiveness but normal resting granulopoiesis in mice carrying a targeted granulocyte colony-stimulating factor receptor mutation derived from a patient with severe congenital neutropenia. J Clin Invest. 1998;102: 483-492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Semerad CL, Poursine-Laurent J, Liu F, Link DC. A role for G-CSF receptor signaling in the regulation of hematopoietic cell function but not lineage commitment or differentiation. Immunity. 1999;11: 153-161. [DOI] [PubMed] [Google Scholar]
- 17.Aronow MA, Gerstenfeld LC, Owen TA, Tassinari MS, Stein GS, Lian JB. Factors that promote progressive development of the osteoblast phenotype in cultured fetal rat calvaria cells. J Cell Physiol. 1990;143: 213-221. [DOI] [PubMed] [Google Scholar]
- 18.Parfitt AM, Drezner MK, Glorieux FH, et al. Bone histomorphometry: standardization of nomenclature, symbols, and units. Report of the ASBMR Histomorphometry Nomenclature Committee. J Bone Miner Res. 1987;2: 595-610. [DOI] [PubMed] [Google Scholar]
- 19.Germeshausen M, Schulze H, Ballmaier M, Zeidler C, Welte K. Mutations in the gene encoding neutrophil elastase (ELA2) are not sufficient to cause the phenotype of congenital neutropenia. Br J Haematol. 2001;115: 222-224. [DOI] [PubMed] [Google Scholar]
- 20.Taichman RS. Blood and bone: two tissues whose fates are intertwined to create the hematopoietic stem cell niche. Blood. 2005;105: 2631-2639. [DOI] [PubMed] [Google Scholar]
- 21.Lataillade JJ, Clay D, Bourin P, et al. Stromal cell-derived factor 1 regulates primitive hematopoiesis by suppressing apoptosis and by promoting G(0)/G(1) transition in CD34(+) cells: evidence for an autocrine/paracrine mechanism. Blood. 2002;99: 1117-1129. [DOI] [PubMed] [Google Scholar]
- 22.Ceradini DJ, Kulkarni AR, Callaghan MJ, et al. Progenitor cell trafficking is regulated by hypoxic gradients through HIF-1 induction of SDF-1. Nat Med. 2004;10: 858-864. [DOI] [PubMed] [Google Scholar]
- 23.Takamatsu Y, Simmons PJ, Moore RJ, Morris HA, To LB, Levesque JP. Osteoclast-mediated bone resorption is stimulated during short-term administration of granulocyte colony-stimulating factor but is not responsible for hematopoietic progenitor cell mobilization. Blood. 1998;92: 3465-3473. [PubMed] [Google Scholar]
- 24.Hermans MH, Antonissen C, Ward AC, Mayen AE, Ploemacher RE, Touw IP. Sustained receptor activation and hyperproliferation in response to granulocyte colony-stimulating factor (G-CSF) in mice with a severe congenital neutropenia/acute myeloid leukemia-derived mutation in the G-CSF receptor gene. J Exp Med. 1999;189: 683-692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hunter MG, Avalos BR. Phosphatidylinositol 3′-kinase and SH2-containing inositol phosphatase (SHIP) are recruited by distinct positive and negative growth-regulatory domains in the granulocyte colony-stimulating factor receptor. J Immunol. 1998;160: 4979-4987. [PubMed] [Google Scholar]
- 26.Hortner M, Nielsch U, Mayr LM, Johnston JA, Heinrich PC, Haan S. Suppressor of cytokine signaling-3 is recruited to the activated granulocytecolony stimulating factor receptor and modulates its signal transduction. J Immunol. 2002;169: 1219-1227. [DOI] [PubMed] [Google Scholar]
- 27.Kawabata K, Ujikawa M, Egawa T, et al. A cellautonomous requirement for CXCR4 in long-term lymphoid and myeloid reconstitution. Proc Natl Acad Sci U S A. 1999;96: 5663-5667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hattori K, Heissig B, Tashiro K, et al. Plasma elevation of stromal cell-derived factor-1 induces mobilization of mature and immature hematopoietic progenitor and stem cells. Blood. 2001;97: 3354-3360. [DOI] [PubMed] [Google Scholar]
- 29.Moore MA, Hattori K, Heissig B, et al. Mobilization of endothelial and hematopoietic stem and progenitor cells by adenovector-mediated elevation of serum levels of SDF-1, VEGF, and angiopoietin-1. Ann N Y Acad Sci. 2001;938: 36-45; discussion 45-37. [DOI] [PubMed] [Google Scholar]
- 30.Christopherson KW 2nd, Cooper S, Broxmeyer HE. Cell surface peptidase CD26/DPPIV mediates G-CSF mobilization of mouse progenitor cells. Blood. 2003;101: 4680-4686. [DOI] [PubMed] [Google Scholar]
- 31.Christopherson KW, Cooper S, Hangoc G, Broxmeyer HE. CD26 is essential for normal GCSF-induced progenitor cell mobilization as determined by CD26–/– mice. Exp Hematol. 2003;31: 1126-1134. [DOI] [PubMed] [Google Scholar]
- 32.Levesque JP, Hendy J, Takamatsu Y, Williams B, Winkler IG, Simmons PJ. Mobilization by either cyclophosphamide or granulocyte colony-stimulating factor transforms the bone marrow into a highly proteolytic environment. Exp Hematol. 2002;30: 440-449. [DOI] [PubMed] [Google Scholar]
- 33.Levesque JP, Takamatsu Y, Nilsson SK, Haylock DN, Simmons PJ. Vascular cell adhesion molecule-1 (CD106) is cleaved by neutrophil proteases in the bone marrow following hematopoietic progenitor cell mobilization by granulocyte colony-stimulating factor. Blood. 2001;98: 1289-1297. [DOI] [PubMed] [Google Scholar]
- 34.Heissig B, Hattori K, Dias S, et al. Recruitment of stem and progenitor cells from the bone marrow niche requires MMP-9 mediated release of kitligand. Cell. 2002;109: 625-637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ara T, Tokoyoda K, Sugiyama T, Egawa T, Kawabata K, Nagasawa T. Long-term hematopoietic stem cells require stromal cell-derived factor-1 for colonizing bone marrow during ontogeny. Immunity. 2003;19: 257-267. [DOI] [PubMed] [Google Scholar]
- 36.Zhang J, Niu C, Ye L, et al. Identification of the haematopoietic stem cell niche and control of the niche size. Nature. 2003;425: 836-841. [DOI] [PubMed] [Google Scholar]
- 37.Calvi LM, Adams GB, Weibrecht KW, et al. Osteoblastic cells regulate the haematopoietic stem cell niche. Nature. 2003;425: 841-846. [DOI] [PubMed] [Google Scholar]
- 38.Taichman RS, Emerson SG. Human osteoblasts support hematopoiesis through the production of granulocyte colony-stimulating factor. J Exp Med. 1994;179: 1677-1682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Arai F, Hirao A, Ohmura M, et al. Tie2/angiopoietin-1 signaling regulates hematopoietic stem cell quiescence in the bone marrow niche. Cell. 2004;118: 149-161. [DOI] [PubMed] [Google Scholar]
- 40.Sekhar RV, Culbert S, Hoots WK, Klein MJ, Zietz H, Vassilopoulou-Sellin R. Severe osteopenia in a young boy with Kostmann's congenital neutropenia treated with granulocyte colony-stimulating factor: suggested therapeutic approach. Pediatrics. 2001;108: E54. [DOI] [PubMed] [Google Scholar]
- 41.Lee MY, Fukunaga R, Lee TJ, Lottsfeldt JL, Nagata S. Bone modulation in sustained hematopoietic stimulation in mice. Blood. 1991;77: 2135-2141. [PubMed] [Google Scholar]
- 42.Kokai Y, Wada T, Oda T, et al. Overexpression of granulocyte colony-stimulating factor induces severe osteopenia in developing mice that is partially prevented by a diet containing vitamin K2 (menatetrenone). Bone. 2002;30: 880-885. [DOI] [PubMed] [Google Scholar]