

Abstract
Plasminogen (Plg)–deficient mice experience wasting and have decreased longevity due to disseminated fibrin deposition. We generated mice with combined deficiencies of Plg and coagulation factor IX (fIX) or XI (fXI) to determine the effects on the Plg null phenotype. Mice lacking Plg and fIX (Plg–/–/fIX–/–) have lower mortality at age 6 months than Plg–/–/fIX+/+ mice (15% and 67%, respectively) and less severe wasting, consistent with the importance of fIX in fibrin formation. In contrast, combined Plg and fXI deficiency (Plg–/–/fXI–/–) reduces life span (more than 90% mortality at 6 months) and is associated with leukocyte infiltration of the lungs and pulmonary fibrosis. These abnormalities are not seen in Plg–/– or Plg–/–/fIX–/– animals. Activated fXI is thought to function primarily as a fIX activator; however, our observation suggests that fXI may have functions in regulation of inflammation or tissue repair distinct from its role in coagulation.
Introduction
The serine protease plasmin plays a major role in regulating hemostasis through proteolytic degradation of fibrin.1 Studies using mice lacking the plasmin precursor plasminogen (Plg) have identified additional plasmin functions in inflammation, cell migration, and extracellular matrix degradation that have implications for a variety of pathologic processes.2 Plg null (Plg–/–) mice grow normally for the first 2 months of life and then develop a syndrome characterized by weight loss, poor wound healing, and premature death.3,4 Disseminated fibrin deposition underlies this phenotype, and mice lacking both Plg and fibrinogen do not display these abnormalities.5 It seems reasonable to hypothesize that factors that reduce thrombin generation and subsequent fibrin formation would have a beneficial effect on the Plg–/– phenotype.
Factor IX (fIX) and factor XI (fXI) are zymogen precursors of coagulation proteases (fIXa and fXIa, respectively) that have important effects on fibrin formation and maintenance.6 Both proteases have been implicated in vascular thrombosis in humans7,8 and animal models.9-12 FIX, the protein missing or defective in patients with hemophilia B, is converted to fIXa by the factor VIIa/tissue factor complex or by fXIa 6,13 and is probably required for a normal response to most hemostatic challenges. FXIa is needed in certain situations to supplement fIX activation14 and appears to be most important for hemostasis in tissues with high fibrinolytic activity.15,16 We created double homozygous null animals lacking Plg and either fIX or fXI and report on the effects of fIX or fXI deficiency on the wasting syndrome and longevity in Plg–/– mice.
Study design
All genotypes were backcrossed to the C57BL/6 strain through at least 7 generations. Plg+/– mice were kindly provided by Jay Degen (University of Cincinnati),3 and fIX null (fIX–/–) mice were a gift from Darrell Stafford (University of North Carolina, Chapel Hill).17 FXI null (fXI–/–) mice have been described.18 All mice were housed in microisolator cages on a 12-hour light-dark cycle, with controlled temperature/humidity and free access to food and water. The Institutional Animal Care and Use Committee of Vanderbilt University approved all procedures. Initially, the Plg+/– genotype was introduced onto the fIX or fXI null backgrounds. Plg+/–/fIX–/– females and Plg+/–/fIX– males were mated to generate Plg++, Plg+/–, and Plg–/– genotypes on the fIX null background. Similar crosses were made with Plg+/–/FXI–/– animals. Plg genotype was determined by polymerase chain reaction (PCR). Primer 5′-TGTGGGCTCAAAGATGGAAC (intron 2) was paired with 5′-GACAAGGGGACTCGCTGGATG (exon 2) or 5′-GTGCGAGGCCAGAGGCCACTT (targeting cassette) to detect wild-type or null alleles, respectively.5 Animals were weighed each week on a top-loading balance (model 400D; Denver Instrument, Arvada, CO). Mice were killed at age 6 weeks (4 mice per genotype), and tissues were fixed in 10% formalin. Lungs and tracheae were removed as a single unit and inflated by intratracheal infusion of formalin. Thin sections were stained with hematoxylin and eosin or Masson trichrome stain. Immunostaining for fibrin was performed with polyclonal goat-anti–mouse fibrinogen/fibrin immunoglobulin G (IgG) using a Vectastain immunohistochemistry kit (Vector Laboratories, Burlingame, CA).
Pulmonary fibrosis was induced by intratracheal bleomycin administration, and lungs were prepared for analysis as previously described.19 Fibrosis was assessed blindly by 2 investigators. Ten sequential, nonoverlapping fields (300 ×) were scored for each animal usinga0to4 point system (0, normal architecture; 1, increased thickness of 50% or more of interalveolar septa; 2, thickening of more than 50% of interalveolar septa without fibrotic foci; 3, thickening of interalveolar septa with isolated fibrotic foci; and 4, multiple fibrotic foci with total or subtotal distortion of parenchymal architecture). Mean score for all fields was calculated and the average for the 2 investigators taken as the score for each animal.
Results and discussion
Plg+/– and wild-type mice grew similarly, while Plg–/– animals developed weight loss starting at about 8 weeks of age (Figure 1A), with more than 50% of animals dead by 6 months (Figure 1B) and few surviving 1 year. Mice with the Plg+/– genotype on fIX or fXI null backgrounds have normal growth and longevity and, with the exception of a bleeding tendency in fIX null mice, appear healthy. Crossing Plg+/– mice on fIX or fXI null backgrounds gave roughly the expected 1:2:1 ratio for the Plg+/+, Plg+/–, and Plg–/– genotypes (21:69:28 for fIX–/–, 24:54:26 for fXI–/–), indicating no increase in intrauterine mortality for double homozygous null animals.
Figure 1.

Effects of fIX or fXI deficiency on growth and longevity in Plg–/– mice. (A) Plg null allele and weight. Shown are average weights for 9 Plg+/+ (□), 19 Plg+/– (▵), and 14 Plg–/– (○) mice followed from birth to age 6 months. Error bars are not shown because of space limitations in the figure. (B) Plg null allele and survival. Shown is survival for Plg+/+ (solid line), Plg+/– (dotted line), and Plg–/– (dashed line) mice in panel A. (C) Effect of fIX or fXI deficiency on weight in Plg–/– mice. Shown are average weights for 19 wild-type (□) and 14 Plg–/– (○) mice (as in panel A) and 21 Plg+/+/fIX–/– (▴), 24 Plg+/+/fXI–/– (♦), 24 Plg–/–/fIX–/– (♦), and 25 Plg–/–/fXI–/– (▴) mice followed from birth to age 6 months. (D) Effect of fIX or fXI deficiency on survival in Plg–/– mice. Survival is shown for wild-type, Plg+/+/fIX–/–, and Plg+/+/fXI–/– mice (all represented by solid line) and Plg–/– (long-dashed line), Plg–/–/fIX–/– (short-dashed line), and Plg–/–/fXI–/– (dotted line) mice. Average weights in panels A and C are for surviving animals at each time point. Animals with the most significant weight loss die early and are not counted at subsequent time points. Lung fibrosis induced by 0.04 units (E) or 0.08 units (F) of intratracheal bleomycin. Lung fibrosis scores ± SEM were obtained 3 weeks after administration of bleomycin for wild-type (n = 6) and fXI–/– (n = 5) mice, as described in “Study design.” Results for Plg–/– mice (n = 6) are shown for comparison in the 0.04 unit experiment.
Plg–/–/fIX–/– mice have a milder phenotype than Plg–/–/fIX+/+ mice, with less pronounced weight loss (Figure 1C) and 86% 6-month survival (Figure 1D). This is consistent with an important role for fIX in thrombin generation and fibrin deposition in Plg–/– mice, although histologic analysis did not clearly demonstrate differences in extent of fibrin deposition between Plg–/–/fIX–/– and Plg–/–/fIX+/+ animals at age 6 weeks (data not shown). Notably, fIX–/– mice were larger than age-matched wild-type or fXI–/– animals (Figure 1C). Because fXI deficiency is associated with milder bleeding than fIX deficiency, fXI deficiency might be expected to have less, or no, impact on the Plg–/– phenotype. Surprisingly, Plg–/–/fXI–/– mice fell behind their Plg+/–/fXI–/– and Plg+/+/fXI–/– littermates, in terms of growth, earlier than was observed for Plg–/– mice (Figure 1C). Mortality for the Plg–/–/fXI–/– mice was 50% by age 12 weeks and 90% at 6 months (Figure 1D). There were striking difference in lung morphology between wild-type, Plg–/–/fXI+/+, and Plg–/–/fXI–/– mice (Figure 2). Plg–/– mice developed scattered patches of fibrin deposition in lung parenchyma (Figure 2B,2E), but most of the lung appeared normal, with relatively little collagen formation (Figure 2H).3,4 Similar histology was noted in Plg–/–/fIX–/– animals (data not shown). In contrast, a diffuse leukocyte infiltration developed in lungs of Plg–/–/fXI–/– mice (primarily mononuclear cells and some neutrophils; Figure 2C), with significant collagen formation (Figure 2I). No differences in fibrin deposition were obvious between Plg–/–/fXI–/– and Plg–/–/fXI+/+ mice in any tissue at 6 weeks of age (data not shown).
Figure 2.
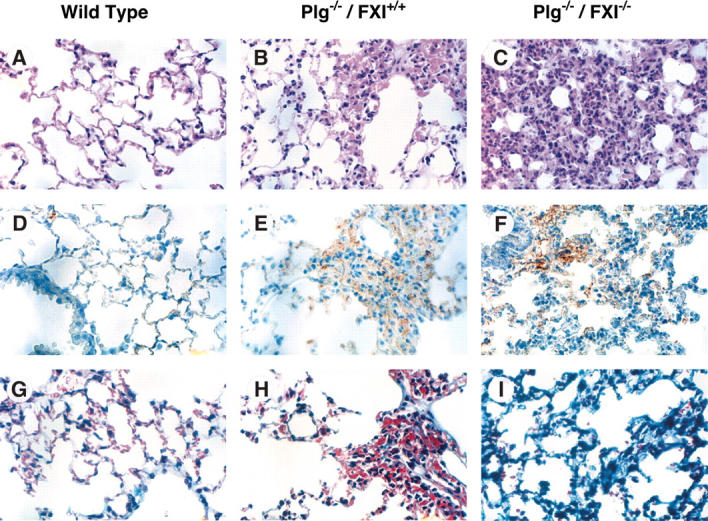
Photomicrographs of murine lung from wild-type, Plg–/–, and Plg–/–/fXI–/– mice. Shown are sections of lung from 6-week-old mice at × 40 magnification. Panels A, D, and G are from wild-type mice; B, E, and H from Plg–/–/fXI+/+ mice; and C, F, and I from Plg–/–/fXI–/– mice. (A-C) Hematoxylin and eosin. (D-F) Immunostaining with a polyclonal anti–murine fibrinogen/fibrin antibody. (G-I) Masson trichrome stain for collagen. The fields in panels B, E, and H were chosen to show a typical lung lesion in Plg–/– mice. These lesions are patchy, and most pulmonary architecture appears normal. In contrast, the leukocyte infiltration shown in panels C, F, and I for Plg–/–/fXI–/– is typically diffuse by age 6 weeks. Images were taken with an Olympus BX41 microscope (Olympus, Tokyo, Japan) fitted with a DP70 digital camera, using a 40 ×/1.7 NA UPlanF1 objective and Olympus DP controller software (version 1.2.1.108).
The pulmonary process in Plg–/–/fXI–/– mice is not seen in fXI-deficient mice or humans or Plg–/– mice. Plg–/– mice are more sensitive to bleomycin-induced pulmonary fibrosis than wild-type mice, suggesting a predisposition to fibrosis with injury.20 We noted greater pulmonary fibrosis in Plg–/– mice compared with wild-type mice (fibrosis scores 1.43 ± 0.24 and 0.98 ± 0.37, respectively) after intratracheal infusion of 0.04 units of bleomycin (Figure 1E), while fXI–/– mice (fibrosis score 0.76 ± 0.07) were similar to wild-type animals. Wild-type and fXI–/– mice were also similar at 0.08 units of bleomycin (Figure 1F; 1.69 ± 0.38 and 1.69 ± 0.24, respectively). The results suggest that fXI deficiency, by itself, does not predispose animals to pulmonary fibrosis.
The beneficial effect of fIX deficiency on the wasting syndrome caused by Plg deficiency in mice is consistent with the observed importance of fIX to thrombin generation and fibrin formation in humans. The pulmonary process in Plg–/–/fXI–/– mice, on the other hand, is difficult to reconcile with a role for fXIa that is restricted to fIX activation. The strikingly different effects of fIX and fXI deficiency on the Plg–/– phenotype suggest that a reduction in thrombin generation may not be the cause for the Plg–/–/fXI–/– phenotype. While the etiology of the pulmonary changes in Plg–/–/fXI–/– mice remains to be elucidated, the findings suggest that fXIa has functions unrelated to fIX activation, possibly in regulation of inflammation or wound repair.
Acknowledgments
The authors thank Jean McClure for preparing the manuscript figures.
Prepublished online as Blood First Edition Paper, June 28, 2005; DOI 10.1182/blood-2005-02-0577.
Supported by grants HL58837 (D.G.) and HL68121 (T.S.B.) from the National Heart, Lung, and Blood Institute, an Established Investigator Award from the American Heart Association (D.G.), and the Parker B. Francis Fellowship program (W.E.L.).
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 U.S.C. section 1734.
References
- 1.Collen D, Lijnen H. Fibrinolysis and the control of hemostasis. In: Stamatoyannopoulos G, Nienhuis AW, Majerus PW, Varmus H, eds. The Molecular Basis of Blood Disease. Philadelphia, PA: WB Saunders; 1994: 725-752.
- 2.Plow E, Hoover-Plow J. The functions of plasminogen in cardiovascular disease. Trends Cardiovasc Med. 2004;14: 180-186. [DOI] [PubMed] [Google Scholar]
- 3.Bugge TH, Flick MJ, Daugherty CC, Degen JL. Plasminogen deficiency causes severe thrombosis but is compatible with development and reproduction. Genes Dev. 1995;9: 794-807. [DOI] [PubMed] [Google Scholar]
- 4.Ploplis V, Carmeliet P, Vazirzadeh S, et al. Effects of disruption of the plasminogen gene on thrombosis, growth, and health in mice. Circulation. 1995;92: 2585-2593. [DOI] [PubMed] [Google Scholar]
- 5.Bugge TH, Kombrinck KW, Flick MJ, Daugherty CC, Danton MS, Degen JL. Loss of fibrinogen rescues mice from the pleiotropic effects of plasminogen deficiency. Cell. 1996;87: 709-719. [DOI] [PubMed] [Google Scholar]
- 6.Davie EW, Fujikawa K, Kisiel W. The coagulation cascade: initiation, maintenance and regulation. Biochemistry. 1991;30: 10363-10370. [DOI] [PubMed] [Google Scholar]
- 7.Meijers JC, Tekelenburg WL, Bouma BN, Bertina RM, Rosendaal FR. High levels of coagulation factor XI as a risk factor for venous thrombosis. N Engl J Med. 2000;342: 696-701. [DOI] [PubMed] [Google Scholar]
- 8.van Hylckama-Vlieg A, van der Linden IK, Bertina RM, Rosendaal FR. High levels of factor IX increase the risk of venous thrombosis. Blood. 2000;95: 3678-3682. [PubMed] [Google Scholar]
- 9.Minnema MC, Friederick PW, Levi M, et al. Enhancement of rabbit jugular vein thrombolysis by neutralization of factor XI. In vivo evidence for a role of factor XI as an anti-fibrinolytic factor. J Clin Invest. 1998;101: 10-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rosen ED, Gailani D, Castellino FJ. Factor XI is essential for thrombus formation following FeCl3-induced injury of the carotid artery in the mouse. Thromb Haemost. 2002;87: 774-776. [PubMed] [Google Scholar]
- 11.Gruber A, Hanson SR. Factor XI-dependence of surface- and tissue factor-initiated thrombus formation in primates. Blood. 2003;102: 953-955. [DOI] [PubMed] [Google Scholar]
- 12.Wang X, Cheng Q, Xu L, et al. Effects of factor IX or factor XI deficiency on ferric chloride-induced carotid artery occlusion in mice. J Thromb Haemost. 2005;3: 695-702. [DOI] [PubMed] [Google Scholar]
- 13.Broze GJ, Girard TJ, Novotny WF. Regulation of coagulation by a multivalent Kunitz-type inhibitor. Biochemistry. 1990;29: 7539-7546. [DOI] [PubMed] [Google Scholar]
- 14.Broze GJ, Gailani D. The role of factor XI in coagulation. Thromb Haemost. 1993;70: 72-74. [PubMed] [Google Scholar]
- 15.Asakai R, Chung DW, Davie EW, Seligsohn U. Factor XI deficiency in Ashkenazi Jews in Israel. N Engl J Med. 1991;325: 153-158. [DOI] [PubMed] [Google Scholar]
- 16.Seligsohn U. Factor XI deficiency. Thromb Haemost. 1993;70: 68-70. [PubMed] [Google Scholar]
- 17.Lin HF, Maeda N, Smithies O, Straight DL, Stafford DW. A coagulation factor IX-deficient mouse model for human hemophilia B. Blood. 1997;90: 3962-3966. [PubMed] [Google Scholar]
- 18.Gailani D, Lasky NM, Broze GJ. A murine model of factor XI deficiency. Blood Coagul Fibrinolysis. 1997;8: 134-144. [DOI] [PubMed] [Google Scholar]
- 19.Lawson WE, Polosukhin VV, Zoia O, et al. Characterization of fibroblast specific protein 1 in pulmonary fibrosis. Am J Respir Crit Care Med. 2005;171: 899-907. [DOI] [PubMed] [Google Scholar]
- 20.Swaisgood CM, French EL, Noga C, Simon RH, Ploplis VA. The development of bleomycin-induced pulmonary fibrosis in mice deficient for components of the fibrinolytic system. Am J Pathol. 2000;157: 177-187. [DOI] [PMC free article] [PubMed] [Google Scholar]