

Abstract
Previous studies showed that chronic lymphocytic leukemia (CLL) cells exhibit certain mitochondrial abnormalities including mtDNA mutations, increased superoxide generation, and aberrant mitochondrial biogenesis, which are associated with impaired apoptosis and reduced sensitivity to fludarabine. Here we report that CLL cells and multiple myeloma cells are highly sensitive to brefeldin A, an inhibitor of endoplasmic reticulum (ER) to Golgi protein transport currently being developed as a novel anticancer agent in a prodrug formulation. Of importance, brefeldin A effectively induced apoptosis in fludarabine-refractory CLL cells. Disruption of protein trafficking by brefeldin A caused the sequestration of the prosurvival factors APRIL and VEGF in the ER, leading to abnormal ER swelling and a decrease in VEGF secretion. Such ER stress and blockage of secretory protein traffic eventually resulted in Golgi collapse, activation of caspases, and cell death. Notably, the cellular sensitivity to this compound appeared to be independent of p53 status. Taken together, these findings suggest that malignant B cells may be highly dependent on ER-Golgi protein transport and that targeting this process may be a promising therapeutic strategy for B-cell malignancies, especially for those that respond poorly to conventional treatments.
Introduction
Chronic lymphocytic leukemia (CLL) is a common adult leukemia with unique characteristics in that the disease progression involves the persistent accumulation of apparently quiescent B cells, primarily due to defects in apoptosis rather than increased cell proliferation.1 The apoptotic resistance of CLL cells has been attributed, in part, to high expression of antiapoptotic proteins such as Bcl-2, Bcl-XL, and Mcl-1.2-8 Our previous work demonstrated that CLL cells contain multiple mitochondrial defects including mtDNA mutations, aberrant mitochondrial biogenesis, and increased generation of the oxygen radical superoxide, which is predominantly produced in the mitochondria. These abnormalities are associated with alterations in sensitivity to anticancer agents, suggesting that they may compromise the execution of the mitochondrial apoptotic machinery.9-11
Fludarabine and alkylating agents have significantly improved clinical outcomes of CLL treatment. However, there are currently limited effective therapeutic options for patients that are refractory to these agents.1,12,13 Thus, identification and evaluation of novel agents for the treatment of refractory CLL are important and challenging tasks. While investigating the mechanistic basis of aberrant mitochondrial biogenesis activity in primary CLL cells, we observed that in addition to increased mitochondrial content, CLL cells also appear to have a more extensive endoplasmic reticulum (ER) network than normal B lymphocytes.10 The abundance of ER membranes in CLL cells has also been noted in earlier investigations and suggests that ER function may be very important for their survival. Consequently, this organelle may represent an attractive therapeutic target.14,15 Recent studies have identified a distinct apoptotic pathway that is triggered upon stress to the ER-Golgi network.16-19 Novel chemotherapeutic agents that stimulate this apoptotic pathway may be particularly effective against malignancies arising from cells with active secretory protein synthesis, such as B cells. In the current study, we analyzed the sensitivity of primary B-CLL cells and multiple myeloma cells to brefeldin A (BFA), an inhibitor of ER to Golgi protein transport that is currently under investigation as a novel anticancer agent in a prodrug form (breflate, NSC656202).20,21 BFA has direct inhibitory effects on ADP ribosylation factor (ARF), which is required for coatamer assembly onto Golgi membranes.22-26 Treatment of mammalian cells with brefeldin A induces both ER and Golgi stress due to its ability to cause a cytotoxic accumulation of proteins in the ER that would normally be trafficked through the Golgi to be processed for secretion.27 The stress to both organelles resulting from BFA treatment can ultimately culminate in the induction of apoptosis. Here we report that BFA effectively induces apoptosis in B-CLL cells and myeloma cells at submicromolar concentrations. Overall, the sensitivity of fludarabine-refractory B-CLL cells to brefeldin A is similar to that of cells from nonrefractory patients, indicating that the mechanisms of resistance to these agents do not overlap. Disruption of protein trafficking by BFA leads to activation of caspases-2, -8, -9, and -3, and sequestration of the prosurvival/chemoresistance factors APRIL and VEGF in the ER of B-CLL cells. Furthermore, BFA treatment results in a dose-dependent reduction in VEGF secretion in both primary B-CLL and multiple myeloma cells. Notably, the sensitivity of CLL cells to BFA appears to be independent of p53 status or the expression of the antiapoptotic proteins Bcl-2, Bcl-xL, Mcl-1, and XIAP, all of which have been linked to chemoresistance and poor clinical prognosis in CLL patients. Collectively, these findings suggest that inducing secretory stress by perturbing intracellular protein trafficking may be an effective therapeutic strategy for patients with B-cell malignancies that respond poorly to conventional anticancer agents.
Patients, materials, and methods
Cells and cell culture
Peripheral blood samples were obtained from CLL patients and healthy donors after informed consent according to the Declaration of Helsinki. Primary CLL cells and peripheral blood mononuclear cells (PBMCs) were isolated from blood specimens using a Ficoll density centrifugation method.28 Approval to use blood specimens was obtained from the University of Texas M. D. Anderson Cancer Center institutional review board for these studies. Cells were resuspended in RPMI 1640 medium supplemented with 10% fetal bovine serum and incubated at 37°C in a humidified incubator with 5% CO2. U266 and NCI-H929 multiple myeloma cells were obtained from American Type Culture Collection (ATCC; Manassas, VA) and cultured under the same conditions. Normal B lymphocytes were purified from whole blood using a negative selection method (RosetteSep B Cell Enrichment Cocktail; Stem Cell Technologies, Vancouver, BC).10,29 Enriched B cells were fixed for transmission electron microscopy as described in “Transmission electron microscopy.” Primary human fibroblasts were cultured in a 1:1 mixture of Medium 199 and MCDB medium supplemented with epidermal growth factor.
Determination of fludarabine IC50
Freshly isolated CLL cells were treated with various concentrations of 9-β-D-arabinofuranosyl-2-fluoro-adenine (F-ara-A, the nucleoside form of fludarabine for in vitro study; Sigma Chemical, St Louis, MO) ranging from 0.3 to 30 μM for 72 hours. IC50 was determined by calculating the concentration of F-ara-A that caused a 50% loss of viability measured by the MTT assay.
Measurement of apoptosis
The Active Caspase-3 Mab Apoptosis FITC kit and annexin V-FITC/propidium iodide (PI) double-staining were used in conjunction with flow cytometry to measure drug-induced cell death (Becton Dickinson, San Jose, CA). Cells were treated with various concentrations of F-ara-A or brefeldin A (Sigma Chemical) for the indicated times. Following drug treatment, cells were fixed and stained for active caspase-3 and analyzed using the FL-1H channel of a FACSCalibur flow cytometer (Becton Dickinson). The percentage of cells containing the active caspase-3 fragment was quantitated. Annexin V/PI fluorescence was also quantified by flow cytometry. The percentage of apoptotic cells was determined by adding the annexin V-positive/PI-negative cells (early apoptosis) and annexin V/PI double-positive cells (late apoptosis).30
Transmission electron microscopy
Normal B lymphocytes and primary CLL cells were fixed as previously described to examine ER content and drug-induced changes in ER morphology.31 Briefly, ultrathin sections were examined in a JEM 1010 transmission electron microscope (JEOL, Peabody, MA) at an accelerating voltage of 80 kV. Digital images were obtained using the AMT Imaging System (Advanced Microscopy Techniques, Danvers, MA).
Confocal microscopy
Cells were centrifuged onto glass slides (Cytospin 2; Thermo Shandon, Cheshire, United Kingdom) and fixed in methanol. The following antibodies and reagents were used for staining: caspase-2 (BD Biosciences, San Jose, CA), APRIL (R&D Systems, Minneapolis, MN), calreticulin (Stressgen Bioreagents, Victoria, BC), and VEGF (Santa Cruz Biotechnology, Santa Cruz, CA). The following fluorescently tagged secondary antibodies were used in the appropriate context: goat anti-mouse AlexaFluor 488 and goat anti-rabbit Texas Red. The Golgi apparatus was visualized with BODIPY-TR Ceramide and nuclei were counterstained with To-Pro-3 (all from Molecular Probes, Eugene, OR). Images were captured with a LSM 510 confocal microscope (Carl Zeiss, Oberkochen, Germany). Immunofluorescence was quantitated using Optimas software (Media Cybernetics, Silver Spring, MD).
Western blotting
Lysates were prepared as previously described.32 Equal amounts of protein were electrophoresed on sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) gels and transferred to nitrocellulose membranes. Membranes were probed with the following antibodies: Bcl-2 (Dako, Carpinteria, CA); Bcl-XL and XIAP (BD Biosciences); Mcl-1 (Santa Cruz Biotechnology); caspases-2, -8, and -9 and active caspase-3 (Cell Signaling, Waltham, MA); APRIL (R&D Systems); and actin (Sigma Chemical). Bands were visualized by chemiluminescent detection.
Enzyme-linked immunosorbent assay (ELISA)
Cells were treated for 16 hours with 30 or 100 ng/mL brefeldin A. ELISA assays were performed to quantitate the amount of VEGF secreted into the medium of control and drug-treated cells (R&D Systems) according to the manufacturer's recommended procedures.
Determination of p53 deletion in CLL cells
TP53 gene deletion in CLL cells was determined by fluorescent in situ hybridization (FISH) analysis of the chromosome 17p deletion. This assay was performed by the cytogenetics laboratory at the University of Texas M. D. Anderson Cancer Center. The procedures for the assay were described in detail previously.33
Statistical analyses
The statistical significance of the difference between control and drug-treated cells in their VEGF and APRIL immunofluorescence intensity and the difference in drug sensitivity of CLL cells with differential p53 status was determined by the Mann-Whitney test as previously described.10 A P value of less than .05 was considered statistically significant.
Results
Primary CLL cells from fludarabine-refractory patients are sensitive to brefeldin A-induced apoptosis
Our previous studies using transmission electron microscopy (TEM) showed that CLL cells contain abnormally high numbers of mitochondria compared with normal B lymphocytes. From the high-resolution TEM images, we also noticed that CLL cells appeared to contain a greater volume of endoplasmic reticulum network than normal B cells (Figure 1A-B). The profusion of ER membranes in CLL cells suggests the ER function may be important for survival and, thus, might serve as a potential therapeutic target. In order to explore this possibility, peripheral blood specimens were obtained from 30 CLL patients (15 nonrefractory, 15 fludarabine refractory; Table 1). We evaluated the sensitivity of primary CLL cells to fludarabine nucleoside (F-ara-A), a frontline agent in CLL therapy, in comparison with brefeldin A (BFA), which is the parent compound of the new anticancer agent breflate (NSC656202). BFA inhibits the transport of proteins from the ER to the Golgi apparatus due to its antagonistic effects on ADP ribosylation factor (ARF), which is required for coatamer assembly onto Golgi membranes.22-26 Primary CLL cells were treated for 48 hours with various concentrations of F-ara-A (range, 0.3-30 μM) and BFA (range, 10-1000 ng/mL or 35.7 nM-3.57 μM). Drug-induced apoptosis was assayed by measuring caspase-3 activation (Figure 1C-E) and double-staining with annexin V/PI (Figure 2). Primary cells from nonrefractory patients responded well to in vitro treatment with both F-ara-A and BFA, whereas cells from fludarabine-refractory patients were largely unaffected by in vitro treatment with F-ara-A, even at concentrations (> 10 μM) that exceed what is clinically achievable in the plasma at the end of an infusion.34-37 In contrast, BFA-treated cells from the same patients showed strong evidence of caspase-3 activation in response to a relatively low concentration of the compound (100 ng/mL, 357 nM).
Figure 1.

Brefeldin A induces apoptosis in fludarabine-refractory B-CLL cells. (A-B) Primary CLL cells exhibited a more extensive ER network than normal B lymphocytes. TEM was used to reveal the relative abundance of ER membranes (arrows) in primary CLL cells (A) and normal B lymphocytes (B). Original magnification, × 7500. Images were visualized using a JEM 1010 transmission electron microscope with a built-in camera and accelerating voltage of 80 KV (JEOL USA, Peabody, MA). A numeric aperture of 2.0 was used. (C-E) Fludarabine-refractory CLL cells were sensitive to BFA-induced apoptosis. CLL cells from 30 patients were treated in vitro with the indicated concentrations of fludarabine nucleoside (F-ara-A) or BFA for 48 hours. Drug-induced apoptosis was measured by flow cytometric analysis of activated caspase-3. Representative results are shown in panel C. The bar graphs in panels D and E show the percentage of cells with activated caspase-3 for nonrefractory patients (n = 15) and fludarabine-refractory CLL patients (n = 15), respectively. ▪ represent cells treated with F-ara-A;  depict cells treated with BFA. Error bars indicate standard error of the mean (SEM).
depict cells treated with BFA. Error bars indicate standard error of the mean (SEM).
Table 1.
Clinical parameters of CLL patients and in vitro cellular sensitivity to F-ara-A
| Patient no. | Age, y | RAI | Prior treatment | Refractory | IC50 F-ara-A | % 17p deletion |
|---|---|---|---|---|---|---|
| 1 | 47 | 4 | No | N/A | 6.6 | ND |
| 2 | 72 | 2 | No | N/A | 6.6 | NEG |
| 3 | 55 | 1 | No | N/A | 4.9 | NEG |
| 4 | 59 | 2 | No | N/A | 29.8 | 83.5 |
| 5 | 56 | 1 | No | N/A | 0.76 | ND |
| 6 | 56 | 1 | No | N/A | 0.91 | NEG |
| 7 | 65 | 1 | No | N/A | 0.29 | NEG |
| 8 | 44 | 4 | No | N/A | 2.06 | ND |
| 9 | 69 | 2 | No | N/A | 0.25 | NEG |
| 10 | 77 | 0 | FCR | No | 0.93 | ND |
| 11 | 68 | 2 | FCR | No | 8.90 | 18 |
| 12 | 68 | 4 | FAMP | No | 8.20 | ND |
| 13 | 62 | 4 | FCR | No | 0.84 | NEG |
| 14 | 60 | 3 | FCR | No | 3.54 | NEG |
| 15 | 32 | 0 | FCR | No | 0.85 | NEG |
| 16 | 61 | 4 | FAMP | Yes | > 30 | ND |
| 17 | 71 | 4 | FAMP | Yes | > 30 | 52.5 |
| 18 | 69 | 1 | FCR | Yes | > 30 | 78.5 |
| 19 | 70 | 4 | FAMP | Yes | > 30 | 85.5 |
| 20 | 56 | 1 | BMT, FAMP | Yes | 21.8 | 15.5 |
| 21 | 51 | 3 | CFAR | Yes | > 30 | 62.5 |
| 22 | 44 | 4 | FCR | Yes | > 30 | NEG |
| 23 | 54 | 1 | FCR | Yes | > 30 | ND |
| 24 | 74 | 1 | FC | Yes | 10.0 | ND |
| 25 | 80 | 2 | FAMP | Yes | > 30 | NEG |
| 26 | 72 | 4 | FND, FC, ESHAP, A, RIT | Yes | 23.2 | ND |
| 27 | 57 | 1 | FAMP, RIT | Yes | 30.0 | 9 |
| 28 | 69 | 3 | FR, BMT, CL | Yes | > 30 | ND |
| 29 | 61 | 4 | F, C, M, RIT, A, G | Yes | > 30 | NEG |
| 30 | 74 | 4 | FCR | Yes | > 30 | 77.5 |
IC50 values for F-ara-A (fludarabine) were determined by MTT assay as described in “Patients, materials, and methods.”
RAI indicates disease stage; N/A, not applicable; ND, not determined; NEG, negative for TP53 deletion; FCR, fludarabine, cyclophosphamide, and rituximab; FAMP, fludarabine; BMT, bone marrow transplantation; CFAR, cyclophosphamide, fludarabine, alemtuzumab, and rituximab; FC, fludarabine and cyclophosphamide; FND, fludarabine, mitoxantrone, dexamethasone; ESHAP, etoposide, methylpredniylosolone, cisplatin, and cytarabine; A, alemtuzumab; RIT, rituximab; CL, clofarabine; F, fludarabine; C, cyclophosphamide; M, mitoxantrone; and G, Geminex.
Figure 2.

Time- and concentration-dependent induction of apoptosis by brefeldin A in primary CLL cells in comparison with normal cells. (A-B) Time course of apoptosis induction by various concentrations of brefeldin A in primary CLL cells. Drug-induced apoptosis was quantitated by flow cytometric analysis of caspase-3 activation (A) and annexin V-FITC/PI double-staining (B). n = 3; error bars indicate SEM. (C-D) Normal human peripheral blood mononuclear cells (PBMCs) and fibroblasts were treated with the indicated concentrations of brefeldin A for 48 hours. Drug-induced apoptosis was quantitated by flow cytometric analysis of caspase-3 activation (C) and annexin V-FITC/PI double-staining (D). Error bars indicate the SEM.
Overall, the cells from fludarabine-refractory and nonrefractory patients responded similarly to BFA in spite of their intrinsic difference in sensitivities to F-ara-A (Table 1; Figure 1D-E). In fact, all specimens showed significant caspase-3 activation within the range of BFA concentrations evaluated even in cases where the highest concentration of fludarabine (30 μM) had no significant effect on cell viability or apoptosis induction. This indicates that the mechanisms of resistance to fludarabine and BFA are nonoverlapping.
Since B cells are actively engaged in the production and secretion of proteins that are important autocrine and paracrine regulators of homeostasis, we reasoned that perturbation of protein trafficking may be a generally effective therapeutic strategy for B-cell malignancies. To explore this possibility further, we examined the proapoptotic effects of BFA on 2 multiple myeloma cell lines. U266 cells underwent apoptosis in a manner similar to that observed in CLL cells, and NCI-H929 cells were even more sensitive to BFA (Figure S1; see the Supplemental Figure link at the top of the online article, at the Blood website). We next carried out time-course studies to investigate the kinetics of BFA-induced cell death in primary CLL cells and multiple myeloma cells. Cells were treated with BFA for various times ranging from 12 hours to 48 hours. Apoptosis induction was measured by active caspase-3 staining and annexin V/PI double-staining. As shown in Figure 2A-B, BFA-induced apoptosis was time dependent, with a substantial amount of cell death at 36 to 48 hours in CLL cells. A time- and concentration-dependent induction of apoptosis was also seen in myeloma cells (Figure S1). In contrast, peripheral blood mononuclear cells (PBMCs) from healthy donors and normal human fibroblasts exhibited significantly less sensitivity to BFA, as demonstrated by both caspase-3 activation (Figure 2C) and annexin V reactivity (Figure 2D). These results together suggest that BFA appears to have reasonable selectivity against malignant B cells.
Brefeldin A activates the Golgi resident caspase-2 and caspases-8, -9, and -3
Recent studies have defined a distinct apoptotic pathway in mammalian cells that is initiated by ER/Golgi stress and appears to function upstream of the mitochondrial cell death cascade.16-19 BFA is known to cause a collapse of the Golgi apparatus and a consequential accumulation of secretory proteins in the ER.27,38 The ability of BFA to cause direct Golgi stress prompted us to investigate the effects of this agent on caspase-2 in primary CLL cells. Caspase-2 can be distinguished from other caspases based on its subcellular localization pattern. In unstressed HeLa cells, thymocytes, and neuronal cells, caspase-2 localizes almost exclusively to the nuclear membrane and the Golgi apparatus.39-45 We first used confocal microscopy to evaluate the subcellular distribution of caspase-2 in primary CLL cells before and after incubation with BFA. In agreement with earlier reports in other cell types, caspase-2 appeared to be exclusively localized to the nuclear membrane and Golgi apparatus in CLL cells (Figure 3A). Following 24 hours of treatment with 100 ng/mL BFA, caspase-2 lost its normal subcellular localization pattern and appeared throughout the nucleus (Figure 3A). The Golgi apparatus in BFA-treated cells could no longer be visualized by the Golgi staining, indicating the collapse of this organelle. Furthermore, since caspase-2 could no longer be detected outside of the nucleus, it is likely that the Golgi pool of this protein translocated to the nucleus in response to the stress induced by this agent. We next examined the activation status of caspases-2, -8, -9, and -3 in CLL and U266 myeloma cells following treatment with BFA. Immunoblotting with an anti-capase-2 antibody demonstrated a significant decrease in the protein levels of procaspase-2 following BFA treatment, indicating proteolytic processing to its lower molecular weight active form. BFA treatment also led to the activation of caspases-8, -9, and -3 (Figure 3B). These results demonstrate that BFA can trigger activation of multiple caspase cascades and may explain, in part, why cells from patients with high levels of antiapoptotic proteins such as Bcl-2 and Bcl-XL can still be triggered to undergo apoptosis upon treatment with this agent.
Figure 3.

Activation of multiple caspases by brefeldin A. (A) Caspase-2 is localized to the Golgi apparatus and nuclear membrane in primary CLL cells. Control and brefeldin A-treated (100 ng/mL, 24h) B-CLL cells were cytocentrifuged onto glass slides, fixed, and stained with a monoclonal human anti-caspase-2 antibody and markers for the nucleus (To-Pro-3) and Golgi apparatus (BODIPY-TR-Ceramide). Subcellular localization of caspase-2 was visualized by confocal microscopy using an LSM 510 confocal microscope with a Plan-Neofluar dry 40 ×/0.75 objective lens and a built-in camera (Carl Zeiss, Thornwood, NY). (B) BFA treatment leads to activation of caspases-2, -8, -9, and -3. Lysates were prepared from cells with or without 100 ng/mL BFA treatment for 24 hours. Western blotting was used to evaluate the indicated caspases in primary CLL cells and U266 cells. The antibody used for caspase-3 recognizes the cleaved (active) form only. Actin was used as a loading control. NR = nonrefractory; R = fludarabine refractory.
Brefeldin A treatment causes severe ER dilation in primary CLL cells
The inhibitory effects of BFA on the Golgi apparatus ultimately lead to perturbation of normal ER function. In order to examine this aspect of BFA's mechanism of action, primary CLL cells from a representative fludarabine-sensitive patient were treated with 10 μM F-ara-A or 30-100 ng/mL BFA for 24 hours and fixed for analysis of changes in ER morphology by TEM. As shown in Figure 4A, BFA treatment caused a severe, concentration-dependent dilation of ER membranes. This dramatic change in ER morphology occurred prior to the onset of appreciable apoptosis (Figure 4B) and did not occur in fludarabine-treated cells, indicating that these effects were specific to BFA treatment and not a general feature of CLL cells undergoing apoptosis. Taken together, these data suggest that CLL cells may be intrinsically sensitive to this particular type of cellular stress.
Figure 4.

Brefeldin A causes severe dilation of ER membranes in primary CLL cells prior to the onset of apoptosis. (A) Primary CLL cells were treated with 10 μM F-ara-A or 30 ng/mL (107 nM) and 100 ng/mL (357 nM) BFA for 24 hours. Cells were fixed and ER morphology was analyzed by transmission electron microscopy as described in “Patients, materials, and methods.” Original magnification of all images is indicated. Images were obtained as for Figure 1A-B. (B) Brefeldin A induces changes in ER morphology prior to the onset of appreciable apoptosis. A portion of BFA-treated primary CLL cells used for transmission electron microscopy analysis was fixed for active caspase-3 staining to quantitate the percentage of apoptotic cells. Only a minor portion of brefeldin A-treated cells stained positively for caspase-3 at this time point (24h). Error bars indicate the standard deviation (n = 4).
Brefeldin A inhibits the secretion of Golgi-trafficked prosurvival secretory proteins
B cells secrete a number of proteins that regulate various biologic processes including maturation, differentiation, activation, and cellular viability.46 Considering this, perturbing the normal secretory function of these cells with an agent such as BFA may be an effective way to disrupt protein homeostasis to an extent that initiates the cell death process. A number of investigations have demonstrated that the secretory protein, vascular endothelial growth factor (VEGF), promotes the survival of CLL cells and may reduce their sensitivity to anticancer agents.47-55 These reports prompted us to investigate whether BFA could inhibit VEGF secretion as an additional mechanism to abrogate survival signaling. Confocal microscopy was used to examine the subcellular distribution and relative intracellular levels of VEGF in primary CLL cells with and without BFA treatment. As shown in Figure 5A-B, BFA treatment led to a significant increase in VEGF content in the cells (P < .01). The majority of the VEGF immunofluorescent signals colocalized with calreticulin, an ER resident protein. This suggested that the drug was blocking its secretion, resulting in an abnormal accumulation of VEGF in the ER. In order to confirm that BFA treatment inhibits VEGF secretion, the levels of VEGF in culture medium were quantitated by ELISA. As shown in Figure 5C, in primary CLL cells isolated from 2 patients, BFA treatment led to a dose-dependent reduction in VEGF secretion.
Figure 5.

Brefeldin A inhibits VEGF secretion. (A) Brefeldin A treatment caused VEGF to accumulate in the ER of primary CLL cells. CLL cells were treated with 100 ng/mL BFA for 24 hours. Cells were cytocentrifuged onto glass slides and fixed as described in “Materials and methods.” Fixed cells were stained with an antibody against VEGF and markers for the ER (calreticulin) and nucleus (To-Pro-3). The fluorescence of stained cells was visualized by confocal microscopy. Images were obtained as for Figure 3A. (B) Quantitation of the brefeldin A-induced increase in VEGF immunofluorescence. The intensity of immunofluorescence of multiple cells was quantitated for the control and BFA-treated cells using Optimas software as described in “Patients, materials, and methods” (n = 8). Error bars indicate the SD. RFU indicates relative fluorescence units. (C) Brefeldin A causes a dose-dependent reduction in VEGF secretion. Primary CLL cells were treated with 30 and 100 ng/mL BFA for 16 hours. Secreted levels of VEGF were quantitated in the culture medium by ELISA as described in “Patients, materials, and methods.” Bars represent the average of 2 assays, which varied by less than 10%.
Another secretory protein that has received increasing attention in the field of CLL biology is APRIL (a proliferation-inducing ligand), a member of the tumor necrosis factor (TNF) superfamily. A unique feature of APRIL that distinguishes it from other members of the TNF superfamily is that it must be cleaved at specific arginine residues by a furin convertase in the Golgi in order to be secreted and biologically active.56 As such, we hypothesized that BFA treatment would cause an accumulation of a biologically inactive form of APRIL in the ER of CLL cells. To test this hypothesis, we analyzed control and BFA-treated primary CLL cells by confocal microscopy to examine the intracellular content and subcellular distribution of the APRIL protein. Control cells displayed low basal APRIL immunofluorescence. In contrast, BFA-treated cells showed a dramatic accumulation of APRIL, the bulk of which appeared to be localized to the ER (Figure 6A, P < .01). The increase in intracellular APRIL protein levels was confirmed by immunoblotting (Figure 6C). Since APRIL is known to play an important role in regulating CLL viability, it is likely that the inhibition of APRIL processing/trafficking by BFA also contributes to its potency in CLL cells. These data suggest that BFA's ability to abrogate survival signaling by inhibiting the secretion of prosurvival secretory factors may underlie its potent cytotoxic effects against malignant B cells.
Figure 6.

Induction of APRIL accumulation in the ER of B-CLL cells following brefeldin A treatment. (A) APRIL accumulates in the ER following treatment with brefeldin A. Primary CLL cells were treated with 100 ng/mL BFA for 24 hours. Following drug treatment, cells were cytocentrifuged onto glass slides and fixed as described in “Patients, materials, and methods.” Cells were stained with a monoclonal anti-APRIL antibody and markers for the ER (calreticulin) and nucleus (To-Pro-3). APRIL immunofluorescence was visualized by confocal microscopy. Images were obtained as for Figure 3A. (B) Quantitation of the BFA-induced increase in APRIL immunofluorescence. The intensity of fluorescence was quantitated using Optimas software as described in “Materials and methods.” n = 8 for each condition; error bars indicate the SD. (C) Brefeldin A causes an increase in the intracellular levels of the APRIL protein. Primary CLL cells were treated with 100 ng/mL BFA for 24 hours. Lysates from control and drug-treated cells were electrophoresed, and APRIL levels were assayed by immunoblotting with an APRIL-specific antibody. Actin was used as a loading control. NR = nonrefractory; R = refractory.
The sensitivity of CLL cells to brefeldin A is independent of p53 status and the expression of several antiapoptotic proteins
Because the loss of p53 (chromosome 17p deletion) is associated with a poor prognosis for disease progression and resistance to therapy,57-60 we evaluated whether p53 status affects the sensitivity of CLL cells to BFA. Of the 30 CLL patient samples used in this study, 20 patient samples had cytogenetic analyses for deletion of p53 at chromosome 17p by fluorescent in situ hybridization (FISH) analysis.33 Of these 20 patient samples, 11 were negative for 17p deletions and the remaining 9 were positive (Table 1). We then compared the sensitivity of CLL cells with and without 17p deletions to BFA and observed no significant difference between these 2 groups of samples (Figure 7A, P = .704). This suggests that loss of functional p53 in CLL cells does not affect the sensitivity to BFA. CLL cells also express high levels of many antiapoptotic proteins including Bcl-2, Bcl-XL, Mcl-1, and XIAP. The elevated expression of such proteins impairs the execution of the mitochondrial apoptotic machinery and, like loss of functional p53, has been linked to in vitro and in vivo chemoresistance and a generally poor clinical prognosis.2-8 We thus examined the relative protein expression of the antiapoptotic factors Bcl-2, Bcl-XL, Mcl-1, and XIAP in primary cells from 3 nonrefractory and 4 refractory CLL patients and U266 and NCI-H929 myeloma cells (Figure 7B). The expression of Bcl-2 and Bcl-XL was generally higher in the CLL patient specimens examined than in U266 and NCI-H929 cells. The percentage of active caspase-3-positive cells following 48 hours of treatment with 100 ng/mL (357 nM) BFA is noted for each sample in Figure 7B. Overall, high expression of these antiapoptotic proteins appears not to be associated with a reduction in in vitro sensitivity to BFA.
Figure 7.
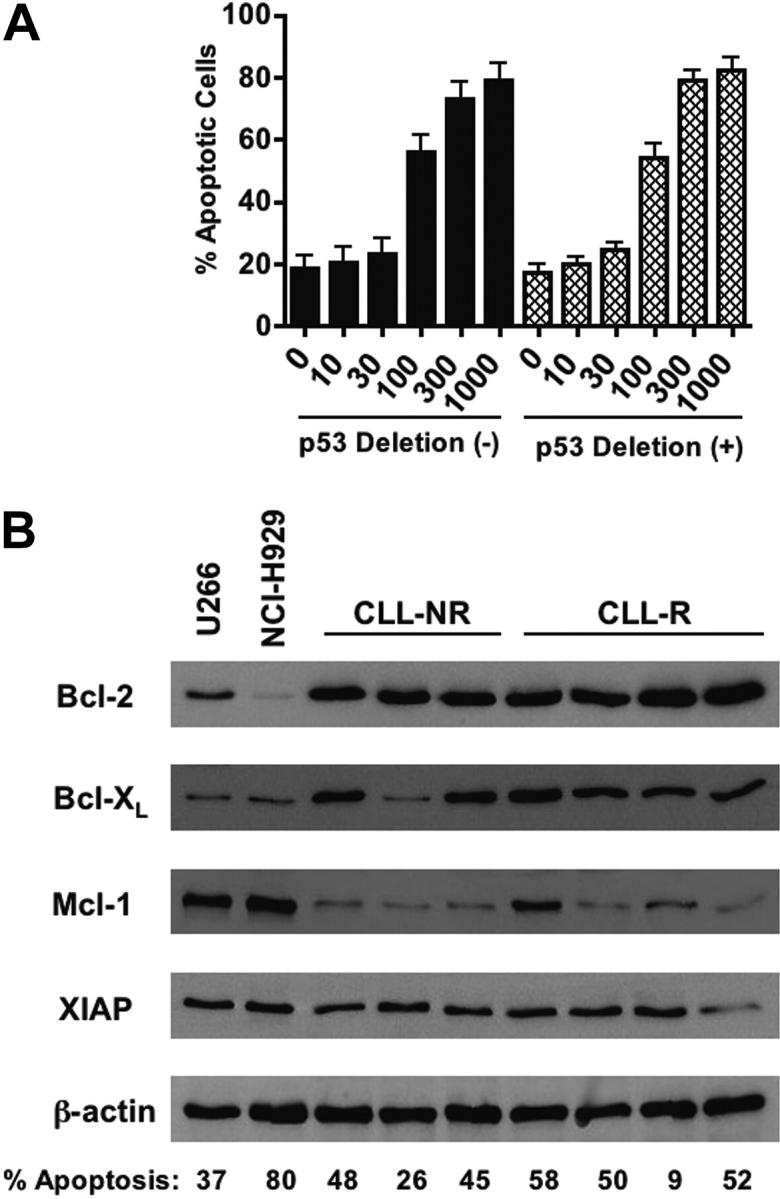
The sensitivity of primary B-CLL cells to brefeldin A is independent of p53 status or the relative expression of key antiapoptotic proteins. (A) Comparison of BFA-induced apoptosis (active caspase-3 staining) in CLL patient samples with (n = 9) or without (n = 11) p53 (17p) deletion. No statistically significant difference in sensitivity to brefeldin A was observed between the 2 groups (P = .704). Error bars indicate SEM. (B) Lysates were prepared from primary CLL cells and multiple myeloma cell lines U266 and NCI-H929. Equivalent amounts of protein from each sample were subjected to SDS-PAGE electrophoresis. Separated proteins were transferred to nitrocellulose membranes and probed for the antiapoptotic proteins Bcl-XL, Bcl-2, Mcl-1, and XIAP. Actin was used as a loading control. NR = nonrefractory; R = fludarabine refractory. The percentage of cells positive for active caspase-3 as assessed by flow cytometry after 48 hours of treatment with 100 ng/mL brefeldin A is noted for each sample. Values have been adjusted to exclude spontaneous apoptosis.
Discussion
B-CLL is presently an incurable malignancy that is characterized by the persistent accumulation of apparently mature, monoclonal B cells, which eventually develop resistance to conventional anticancer agents, especially in later stages of the disease. Chemoresistance has been attributed to a variety of factors such as high expression of antiapoptotic proteins, loss of functional p53, mitochondrial defects, and, possibly, in vivo exposure to exogenous prosurvival cytokines in the microenvironment.2-10,57-65 These observations have prompted rigorous characterization of the molecular and biochemical features of the disease to gain a better understanding of what additional factors impact chemosensitivity and, hopefully, to uncover novel therapeutic targets. Regardless of the specific mechanistic basis, these studies collectively suggest that a high proportion of CLL patients have defects in their mitochondrial apoptotic machinery that are linked to a poor in vivo response to conventional therapeutic anticancer agents. As such, novel strategies are clearly required to improve the clinical outcomes of patients with this malignancy.
For many years, it was widely believed that there were 2 major pathways of programmed cell death: the intrinsic pathway, which is mainly controlled at the level of the mitochondria, and the extrinsic pathway, which is regulated by the binding of specific death ligands to their receptors on the cell surface. A number of recent studies have provided direct evidence that programmed cell death cascades can be initiated at other sites within the cell, in particular, organelles such as the ER and Golgi apparatus.16 Considering that CLL cells, like many other malignant cells, have evolved mechanisms that render them less sensitive to chemostimulation of their mitochondrial death machinery, agents capable of initiating apoptosis at other organelle sites may have potential therapeutic promise. To explore this possibility, we evaluated the sensitivity of primary CLL cells to fludarabine, a frontline conventional anticancer agent, and brefeldin A, an inhibitor of ER to Golgi protein transport currently being developed as an anticancer agent in prodrug form (breflate, NSC656202) that induces ER and Golgi stress. We hypothesized that malignant B cells may be intrinsically sensitive to this type of stress due to their high level of secretory protein synthesis and dependency on the ER and Golgi to carry out such functions. Our data indicate that cells from fludarabine-refractory patients can be stimulated to undergo apoptosis with relatively low concentrations of BFA in spite of in vivo and in vitro resistance to fludarabine. This demonstrates that the mechanisms of resistance of these 2 agents do not overlap and moreover, that the cellular response to secretory stress remains intact in these cells. Notably, both multiple myeloma cell lines evaluated were also highly sensitive to BFA. This illustrates the potential efficacy of disrupting secretory homeostasis in malignant B cells as a therapeutic strategy. In fact, recent studies have demonstrated that XBP-1 and IRE1, both involved in the unfolded protein response (UPR) to ER stress, play critical roles in B-cell maturation. Mice deficient in XBP-1 display a nearly complete loss of plasma cells, and loss of its upstream regulator, IRE1, results in disruption of both the early and late stages of B lymphopoiesis.66-69 This indicates that ER homeostasis is essential for the development and survival of B cells and provides further rationale for the investigation of agents that target the ER-Golgi network for the treatment of B-cell malignancies.
Of interest, although refractory cells from certain patients were less sensitive to BFA than those from nonrefractory patients, the sensitivity of several fludarabine-refractory specimens to BFA was notably higher than that of the nonrefractory cells analyzed. The mechanistic basis for the heightened sensitivity of certain refractory cells to BFA is not known at the present time, but may be related to varying degrees of dependence on specific secretory factors whose secretion is blocked by this agent. Future investigations are needed to provide further insight into this phenomenon.
BFA appears to induce apoptosis in B-CLL cells through activation of caspase-2, the only caspase currently known to localize to the Golgi apparatus, and caspases -8, -9, and -3. Similarly to caspases-8 and -9, caspase-2 appears to function as an initiator caspase and has been suggested to commence apoptosis at the Golgi apparatus by cleaving the Golgi-resident protein, golgin-160. Upon activation, caspase-2 can translocate to mitochondria where it appears to play a role in direct membrane permeabilization and facilitates the release of cytochrome c and the proapoptotic protein Smac.39-45 A recent study demonstrated that mutation of the caspase cleavage sites of the caspase-2 substrate golgin-160 significantly reduced sensitivity to BFA and death receptor-mediated apoptosis.70 This suggests that caspase-2 plays an important role in the initiation of BFA-induced apoptosis and highlights a potential link between the ER-Golgi and death receptor apoptotic cascades. It is unclear at the present time if the observed activation of caspase-8 and caspase-9 is independent of caspase-2 or downstream of its activation. Since the Golgi apparatus serves as the major intracellular storage site for death receptors, it is possible that Golgi stress may lead to the release of these death receptors and the engagement of that apoptotic pathway.71-73
Notably, the sensitivity of CLL cells to this agent does not seem to be impacted by p53 (chromosome 17p) status or the expression levels of antiapoptotic proteins Bcl-2, Bcl-XI, Mcl-1, and XIAP, all of which have been associated with resistance to conventional chemotherapeutic agents and a poor clinical prognosis. The ability of BFA to induce apoptosis independent of p53 status has also been noted in other types of malignant cells.74,75 In contrast, overexpression of antiapoptotic proteins by gene transfer has been shown to reduce sensitivity to BFA in cell culture systems.76,77 The fact that we did not observe a correlation between high antiapoptotic protein expression and reduced sensitivity to BFA in this study could be due to a difference between endogenous expression levels and those achieved by gene transfer, or due to the functional effect of these proteins in different cellular contexts.
TEM analyses of ER morphology following brefeldin A treatment revealed that it caused massive, dose-dependent dilation of ER membranes, likely due to an accumulation of secretory proteins that would normally be trafficked through the Golgi to be processed for secretion. Since B cells are heavily engaged in secretory protein synthesis, agents such as BFA may be particularly effective at inducing a type of severe stress that results in the activation of apoptotic cascades. This is especially true considering that secretory proteins such as APRIL and VEGF play an important role in maintaining the viability of CLL cells and other malignant B cells and confer cellular resistance to cancer therapy.78-80 An earlier report indicated that APRIL mRNA levels are elevated in a subset of CLL patients.78 More recent studies have demonstrated that exogenous APRIL protects against both spontaneous and drug-induced apoptosis, that APRIL levels are significantly higher in the sera of CLL patients compared with healthy individuals, and that, moreover, APRIL transgenic mice develop a B-cell malignancy that resembles CLL.79,80 As a whole, these data indicate that APRIL likely plays a role in the pathologic process of CLL.
The importance of the proangiogenic molecule VEGF as a modulator of the biology of CLL cells has also received increasing attention in recent years. Several investigations have demonstrated that B-CLL cells produce functional VEGF in vitro and in vivo, and, like APRIL, the serum levels of this protein are significantly elevated in the blood of CLL patients compared with healthy individuals. CLL cells also express all 3 VEGF receptors. The increased expression of VEGF receptors and high levels of VEGF in the serum have been linked to a poor clinical prognosis in CLL patients.47-55 More recent evidence suggests that VEGF may also be an important mediator of the antiapoptotic effect induced by the interaction of CD40 with its ligand, CD40L.81,82 The inhibitory effect of BFAon the processing of secretory proteins such as APRIL and VEGF likely abrogates their survival signals and contributes to the sensitivity of these malignant B cells to this agent.
Of interest, a recent study also demonstrated that CLL cells can be effectively killed by tetrocarcin A, an antibiotic compound that induces cell death through an ER stress-mediated mechanism, albeit one that is functionally distinct from that of brefeldin A.83 Tetrocarcin A also appears to kill CLL cells in a manner independent of Bcl-2 levels. Taken together these data suggest that agents that activate apoptotic cascades by inducing ER/secretory stress may represent a promising therapeutic strategy to exploit a vulnerable aspect of B-cell biology. Breflate, a prodrug form of brefeldin A, is currently in development as a novel anticancer agent.20,21 Our results indicate that breflate and other agents of this type may be particularly effective for the treatment of refractory B-cell malignancies such as B-CLL and multiple myeloma. Future preclinical studies are warranted to explore this exciting possibility.
Supplementary Material
Acknowledgments
The authors wish to thank Dr Jinsong Liu for providing human fibroblasts and Min Du for her assistance with obtaining blood from healthy donors.
Prepublished online as Blood First Edition Paper, September 6, 2005; DOI 10.1182/blood-2005-05-1923.
Supported in part by grants CA85563, CA100428, CA109041, CA081534, CA100632, and CA16672 from the National Institutes of Health. J.S.C. and S.T.N. are recipients of fellowships from the American Legion Auxiliary and the Sowell-Huggins Families.
J.S.C. designed and carried out the majority of the experiments and wrote the paper; S.T.N. participated in immunofluorescent staining and confocal microscopy studies; Y.V.K. was involved in patient selection and obtaining blood samples from patients with CLL for this study and assisted with clinical data analysis; K.D. performed transmission electron microscopy studies; D.J.M. provided intellectual input regarding experimental design and data interpretation; M.J.K. identified CLL patients for this study and participated in experimental design and data interpretation; and P.H. directed this study and participated in experimental design, data interpretation, and preparation of the paper.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 U.S.C. section 1734.
References
- 1.Caligaris-Cappio F, Hamblin TJ. B-cell chronic lymphocytic leukemia: a bird of a different feather. J Clin Oncol. 1999;17: 399-408. [DOI] [PubMed] [Google Scholar]
- 2.Kitada S, Zapata JM, Andreeff M, Reed JC. Bryostatin and CD40-ligand enhance apoptosis resistance and induce expression of cell survival genes in B-cell chronic lymphocytic leukaemia. Br J Haematol. 1999;106: 995-1004. [DOI] [PubMed] [Google Scholar]
- 3.Gottardi D, Alfarano A, De Leo AM, et al. In leukemic CD5+ B cells the expression of BCL-2 gene family is shifted toward protection from apoptosis. Br J Haematol. 1996;94: 612-618. [DOI] [PubMed] [Google Scholar]
- 4.Kitada S, Andersen J, Akar S, et al. Expression of apoptosis-regulating proteins in chronic lymphocytic leukemia: correlations with in vitro and in vivo chemoresponses. Blood. 1998;91: 3379-3389. [PubMed] [Google Scholar]
- 5.Munzert G, Kirchner D, Stobbe H, et al. Tumor necrosis factor receptor-associated factor 1 gene overexpression in B-cell chronic lymphocytic leukemia: analysis of NF-kappa B/Rel-regulated inhibitors of apoptosis. Blood. 2002;100: 3749-3756. [DOI] [PubMed] [Google Scholar]
- 6.Bannerji R, Kitada S, Flinn IW, et al. Apoptotic-regulatory and complement-protecting protein expression in chronic lymphocytic leukemia: relationship to in vivo rituximab resistance. J Clin Oncol. 2003;21: 1466-1471. [DOI] [PubMed] [Google Scholar]
- 7.Moyshynska O, Sankaran K, Pahwa P, Saxena A. Prognostic significance of a short sequence insertion in the MCL-1 promoter in chronic lymphocytic leukemia. J Natl Cancer Inst. 2003;96: 673-682. [DOI] [PubMed] [Google Scholar]
- 8.Saxena A, Viswanathan S, Moyshynska O, Tandon P, Sankaran K, Sheridan DP. Mcl-1 and Bcl-2/Bax ratio are associated with treatment response but not with Rai stage in B-cell chronic lymphocytic leukemia. Am J Hematol. 2004;75: 22-33. [DOI] [PubMed] [Google Scholar]
- 9.Carew JS, Zhou Y, Albitar M, Carew JD, Keating MJ, Huang P. Mitochondrial DNA mutations in primary leukemia cells after chemotherapy: clinical significance and therapeutic implications. Leukemia. 2003;17: 1437-1447. [DOI] [PubMed] [Google Scholar]
- 10.Carew JS, Nawrocki ST, Xu RH, et al. Increased mitochondrial biogenesis in primary leukemia cells: the role of endogenous nitric oxide and impact on sensitivity to fludarabine. Leukemia. 2004;18: 1934-1940. [DOI] [PubMed] [Google Scholar]
- 11.Zhou Y, Hileman EO, Plunkett W, Keating MJ, Huang P. Free radical stress in chronic lymphocytic leukemia cells and its role in cellular sensitivity to ROS-generating anticancer agents. Blood. 2003;101: 4098-4104. [DOI] [PubMed] [Google Scholar]
- 12.Kay NE, Hamblin TJ, Jelinek DF, et al. Chronic lymphocytic leukemia. Hematology (Am Soc Hematol Educ Program). 2002: 193-213. [DOI] [PubMed]
- 13.Bannerji R, Byrd JC. Update on the biology of chronic lymphocytic leukemia. Curr Opin Oncol. 2000;12: 22-29. [DOI] [PubMed] [Google Scholar]
- 14.Rubartelli A, Sitia R, Zicca A, Grossi CE, Ferrarini M. Differentiation of chronic lymphocytic leukemia cells: correlation between the synthesis and secretion of immunoglobulins and the ultrastructure of the malignant cells. Blood. 1983;62: 495-504. [PubMed] [Google Scholar]
- 15.Rubartelli A, Sitia R, Grossi CE, Ferrarini M. Maturation of chronic lymphocytic leukemia B cells: correlation between the capacity of responding T-cell factors in vitro and the stage of maturation reached in vivo. Clin Immunol Immunopathol. 1985;34: 296-303. [DOI] [PubMed] [Google Scholar]
- 16.Ferri KF, Kroemer G. Organelle-specific initiation of cell death pathways. Nat Cell Biol. 2001;3: E255-E263. [DOI] [PubMed] [Google Scholar]
- 17.Rao RV, Castro-Obregon S, Frankowski H, et al. Coupling endoplasmic reticulum stress to the cell death program: an Apaf-1-independent intrinsic pathway. J Biol Chem. 2002;277: 21836-21842. [DOI] [PubMed] [Google Scholar]
- 18.Rutkowski DT, Kaufman RJ. A trip to the ER: coping with stress. Trends Cell Biol. 2004;14: 20-28. [DOI] [PubMed] [Google Scholar]
- 19.Kadowaki H, Nishitoh H, Ichijo H. Survival and apoptosis signals in ER stress: the role of protein kinases. J Chem Neuroanat. 2004;28: 93-100. [DOI] [PubMed] [Google Scholar]
- 20.Phillips LR, Wolfe TL, Malspeis L, Supko JG. Analysis of brefeldin A and the prodrug breflate in plasma by gas chromatography with mass selective detection. J Pharm Biomed Anal. 1998;16: 1301-1309. [DOI] [PubMed] [Google Scholar]
- 21.Developmental Therapeutics Program, National Cancer Institute. http://dtp.nci.nih.gov/docs/dtp_search.html. Accessed May 1, 2005.
- 22.Donaldson JG, Kahn RA, Lippincott-Schwartz J, Klausner RD. Binding of ARF and beta-COP to Golgi membranes: possible regulation by a trimeric G protein. Science. 1991;254: 1197-1199. [DOI] [PubMed] [Google Scholar]
- 23.Donaldson JG, Cassel D, Kahn RA, Klausner RD. ADP-ribosylation factor, a small GTP-binding protein, is required for binding of the coatamer protein beta-COP to Golgi membranes. Proc Natl Acad Sci U S A. 1992;89: 6408-6412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Donaldson JG, Finazzi D, Klausner RD. Brefeldin A inhibits Golgi membrane-catalyzed exchange of guanine nucleotide onto ARF protein. Nature. 1992;360: 350-352. [DOI] [PubMed] [Google Scholar]
- 25.Helms JB, Rothman JE. Inhibtion by brefeldin A of a Golgi membrane enzyme that catalyzes exchange of guanine nucleotide bound to ARF. Nature. 1992;360: 352-354. [DOI] [PubMed] [Google Scholar]
- 26.Orci L, Palmer DJ, Ravazzola M, Perrelet A, Amherdt M, Rothman JE. Budding from Golgi membranes requires the coatamer complex of nonclathrin coar proteins. Nature. 1993;362: 648-652. [DOI] [PubMed] [Google Scholar]
- 27.Fujiwara T, Oda K, Yokota S, Takatsuki A, Ikehara Y. Brefeldin A causes disassembly of the Golgi complex and accumulation of secretory proteins in the endoplasmic reticulum. J Biol Chem. 1988; 263: 18545-18552. [PubMed] [Google Scholar]
- 28.Huang P, Robertson LE, Wright S, Plunkett W. High molecular weight DNA fragmentation: a critical event in nucleoside analogue-induced apoptosis in leukemia cells. Clin Cancer Res. 1995;9: 1005-1013. [PubMed] [Google Scholar]
- 29.Tsukada N, Burger JA, Zvaifler NJ, Kipps TJ. Distinctive features of “nurselike” cells that differentiate in the context of chronic lymphocytic leukemia. Blood. 2002;99: 1030-1037. [DOI] [PubMed] [Google Scholar]
- 30.Pelicano H, Feng L, Zhou Y, et al. Inhibition of mitochondrial respiration: a novel strategy to enhance drug-induced apoptosis in human leukemia cells by a reactive oxygen species-mediated mechanism. J Biol Chem. 2003;278: 37832-37839. [DOI] [PubMed] [Google Scholar]
- 31.Muller HK, Bucana CD, Kripke ML, Cox PA, Saijo S, Strickland FM. Ultraviolet irradiation of murine skin alters cluster formation between lymph node dendritic cells and specific T lymphocytes. Cell Immunol. 1994;157: 263-276. [DOI] [PubMed] [Google Scholar]
- 32.Nawrocki ST, Sweeney-Gotsch B, Takamori R, McConkey DJ. The proteasome inhibitor bortezomib enhances the activity of docetaxel in orthotopic human pancreatic tumor xenografts. Mol Cancer Ther. 2004;3: 59-70. [PubMed] [Google Scholar]
- 33.Glassman AB, Hayes KJ. The value of fluorescence in situ hybridization in the diagnosis and prognosis of chronic lymphocytic leukemia. Cancer Genet Cytogenet. 2005;158: 88-91. [DOI] [PubMed] [Google Scholar]
- 34.Danhauser L, Plunkett W, Lilliemark J, Gandhi V, Iacoboni S, Keating MJ. Comparison between the plasma and intracellular pharmacology of 1-beta-D-arabinofuranocytosine and 9-beta-D-arabinofuranosyl-2-fluoroadenine 5'-monophosphate in patients with relapsed leukemia. Leukemia. 1987; 1: 638-643. [PubMed] [Google Scholar]
- 35.Danhauser L, Plunkett W, Keating M, Cabanillas F. 9-beta-D-arabinofuranosyl-2-fluoroadenine 5'-monophosphate pharmacokinetics in plasma and tumor cells of patients with relapsed leukemia and lymphoma. Cancer Chemother Pharmacol. 1986;18: 145-152. [DOI] [PubMed] [Google Scholar]
- 36.Gandhi V, Kemena A, Keating MJ, Plunkett W. Cellular pharmacology of fludarabine triphosphate in chronic lymphocytic leukemia cells during fludarabine therapy. Leuk Lymphoma. 1993; 10: 49-56. [DOI] [PubMed] [Google Scholar]
- 37.Gandhi V, Plunkett W. Cellular and clinical pharmacology of fludarabine. Clin Pharamacokinet. 2002;41: 93-103. [DOI] [PubMed] [Google Scholar]
- 38.Ulmer JB, Palade GE. Effects of Brefeldin A on the Golgi complex, endoplasmic reticulum and viral envelope glycoproteins in murine erythroleukemia cells. Eur J Cell Biol. 1991;54: 38-54. [PubMed] [Google Scholar]
- 39.Mancini M, Machamer CE, Roy S, et al. Caspase-2 is localized at the Golgi complex and cleaves golgin-160 during apoptosis. J Cell Biol. 2000;149: 603-612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lassus P, Opitz-Araya X, Lazebnik Y. Requirement for caspase-2 in stress-induced apoptosis before mitochondrial permeabilization. Science. 2002;297: 1352-1354. [DOI] [PubMed] [Google Scholar]
- 41.Read SH, Baliga BC, Ekert PG, Vaux DL, Kumar S. A novel Apaf-1-independent putative caspase-2 activation complex. J Cell Biol. 2002; 159: 739-745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tinel A, Tschopp J. The PIDDosome, a protein complex implicated in activation of caspase-2 in response to genotoxic stress. Science. 2004;304: 843-846. [DOI] [PubMed] [Google Scholar]
- 43.Robertson JD, Gogvadze V, Kropotov A, Vakifahmetoglu H, Zhivotovsky B, Orrenius S. Processed caspase-2 can induce mitochondria-mediated apoptosis independently of its enzymatic activity. EMBO Rep. 2004;5: 643-648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Baliga BC, Read SH, Kumar S. The biochemical mechanism of caspase-2 activation. Cell Death Differ. 2004;11: 1234-1241. [DOI] [PubMed] [Google Scholar]
- 45.Enoksson M, Robertson JD, Gogvadze V, et al. Caspase-2 permeabilizes the outer mitochondrial membrane and disrupts the binding of cytochrome c to anionic phospholipids. J Biol Chem. 2004;279: 49575-49578. [DOI] [PubMed] [Google Scholar]
- 46.Gass JN, Gunn KE, Sriburi R, Brewer JW. Stressed out B cells? Plasma cell differentiation and the unfolded protein response. Trends Immunol. 2004;25: 17-24. [DOI] [PubMed] [Google Scholar]
- 47.Chen H, Trewecke AT, West DC, et al. In vitro and in vivo production of vascular endothelial growth factor by chronic lymphocytic leukemia cells. Blood. 2000;96: 3181-3187. [PubMed] [Google Scholar]
- 48.Aguayo A, Kantarjian H, Manshouri T, et al. Angiogenesis in acute and chronic leukemias and myelodisplastic syndromes. Blood. 2000;96: 2240-2245. [PubMed] [Google Scholar]
- 49.Ferrajoli A, Manshouri T, Estrov Z, et al. High levels of vascular endothelial gropwth factor receptor-2 correlate with shortened survival in chronic lymphocytic leukemia. Clin Cancer Res. 2001;7: 795-799. [PubMed] [Google Scholar]
- 50.Kay NE, Bone ND, Tschumper RC, et al. B-CLL cells are capable of synthesis and secretion of both pro- and anti-angiogenic molecules. Leukemia. 2002;16: 911-919. [DOI] [PubMed] [Google Scholar]
- 51.Faderl S, Keating MJ, Do KA, et al. Expression profile of 11 proteins and their prognostic significance in patients with chronic lymphocytic leukemia (CLL). Leukemia. 2002;16: 1045-1052. [DOI] [PubMed] [Google Scholar]
- 52.Kay NE. The angiogenic status of B-CLL cells B cells: the role of VEGF receptors. Leuk Res. 2004;28: 221-222. [DOI] [PubMed] [Google Scholar]
- 53.Bairey O, Boycov O, Kaganovsky E, Zimra Y, Shaklai M, Rabizadeh E. All three receptors for vascular endothelial growth factor (VEGF) are expressed on B-chronic lymphocytic leukemia (CLL) cells. Leuk Res. 2004;28: 243-248. [DOI] [PubMed] [Google Scholar]
- 54.Lee YK, Bone ND, Strege AK, Shanafelt TD, Jelinek DF, Kay NE. VEGF receptor phosphorylation status and apoptosis is modulated by a green tea component, epigallocatechin-3-gallate (EGCG), in B-cell chronic lymphocytic leukemia. Blood. 2004;104: 788-794. [DOI] [PubMed] [Google Scholar]
- 55.Lee YK, Shanafelt TD, Bone ND, Strege AK, Jelinek DF, Kay NE. VEGF receptors on chronic lymphocytic leukemia (CLL) B cells interact with STAT 1 and 3: implication for apoptosis resistance. Leukemia. 2005;19: 513-523. [DOI] [PubMed] [Google Scholar]
- 56.Lopez-Fraga M, Fernandez R, Albar JP, Hahne M. Biologically active APRIL is secreted following intracellular processing in the Golgi apparatus by furin convertase. EMBO Rep. 2001;2: 945-951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wattel E, Preudhomme C, Hecquet B, et al. p53 mutations are associated with resistance to chemotherapy and short survival in hematologic malignancies. Blood. 1994;84: 3148-3157. [PubMed] [Google Scholar]
- 58.Cordone I, Masi S, Mauro FR, et al. p53 expression in B-cell chronic lymphocytic leukemia: a marker of disease progression and poor prognosis. Blood. 1998;91: 4342-4349. [PubMed] [Google Scholar]
- 59.Sturm I, Bosanquet AG, Hermann S, Guner D, Dorken B, Daniel PT. Mutation of p53 and consecutive selective drug resistance in B-CLL occurs as a consequence of prior DNA-damaging therapy. Cell Death Differ. 2003;10: 477-484. [DOI] [PubMed] [Google Scholar]
- 60.Giles F, Bekele BN, O'Brien S, et al. A prognostic model for survival in chronic lymphocytic leukemia based on p53 espression. Br J Haematol. 2003;121: 578-585. [DOI] [PubMed] [Google Scholar]
- 61.Ghia P, Caligaris-Cappio F. The indisplensable role of microenvironment in the natural history of low-grade B-cell neoplasms. Adv Cancer Res. 2000;79: 157-173. [DOI] [PubMed] [Google Scholar]
- 62.Ghia P, Granziero L, Chilosi M, Caligaris-Cappio F. Chronic B cell malignancies and bone marrow microenvironment. Semin Cancer Biol. 2002;12: 149-155. [DOI] [PubMed] [Google Scholar]
- 63.Pederson IM, Kitada S, Leoni LM, et al. Protection of CLL B cells by a follicular dendritic cell line is dependent on induction of Mcl-1. Blood. 2002; 100: 1795-1801. [PubMed] [Google Scholar]
- 64.Cuni S, Perez-Aciego P, Perez-Chacon G, et al. A sustained activation of PI3K/NF-kappaB pathway is critical for the survival of chronic lymphocytic leukemia B cells. Leukemia. 2004;18: 1391-1400. [DOI] [PubMed] [Google Scholar]
- 65.Munk Pederson I, Reed J. Microenvironmental interactions and survival of CLL B-cells. Leuk Lymphoma. 2004;45: 2365-2372. [DOI] [PubMed] [Google Scholar]
- 66.Reimold AM, Iwakoshi NN, Manis J, et al. Plasma cell differentiation requires the transcription factor XBP-1. Nature. 2001;412: 300-307. [DOI] [PubMed] [Google Scholar]
- 67.Calfon M, Zeng H, Urano F, et al. IRE1 couples endoplasmic reticulum overload to secretory capacity by processing XBP-1 mRNA. Nature. 2002;415: 92-96. [DOI] [PubMed] [Google Scholar]
- 68.Zhang K, Wong HN, Song B, Miller CN, Scheuner D, Kaufman RJ. The unfolded protein response sensor IRE1a is required at 2 distinct steps in B cell lymphopoiesis. J Clin Invest. 2005;115: 268-281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Iwakoshi NN, Lee AH, Vallabbajosyula P, Otipoby KL, Rajewsky K, Glimcher LH. Plasma cell differentation and the unfolded protein response intersect at the transcription factor XBP-1. Nat Immunol. 2003;4: 321-329. [DOI] [PubMed] [Google Scholar]
- 70.Maag RS, Mancini M, Rosen A, Machamer CE. Caspase-resistant golgin-160 disrupts apoptosis induced by secretory pathway stress and ligation of death receptors. Mol Biol Cell. 2005;16: 3019-3027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Jones SJ, Ledgerwood EC, Prins JB, et al. TNF recruits TRADD to the plasma membrane but not the trans-Golgi network, the prinicipal subcellular location of TNF-R1. J Immunol. 1999;162: 1042-1048. [PubMed] [Google Scholar]
- 72.Zhang XD, Franco AV, Nguyen T, Gray CP, Hersey P. Differential localization and regulation of death and decoy receptors for TNF-related apoptosis-inducing ligand (TRAIL) in human melanoma cells. J Immunol. 2000;164: 3961-3970. [DOI] [PubMed] [Google Scholar]
- 73.Storey H, Stewart A, Vandenabeele P, Luzio JP. The p55 tumour necrosis factor receptor TNFR1 contains a trans-Golgi network localization signal in the C-terminal region of its cytoplasmic tail. Biochem J. 2002;366: 15-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Shao RG, Shimizu T, Pommier Y. Brefeldin A is a potent inducer of apoptosis in human cancer cells independently of p53. Exp Cell Res. 1996;227: 190-196. [DOI] [PubMed] [Google Scholar]
- 75.Pommepuy I, Terro F, Petit B, et al. Brefeldin A induces apoptosis and cell cycle blockade in glioblastoma cell lines. Oncology. 2003;64: 459-467. [DOI] [PubMed] [Google Scholar]
- 76.Guo H, Tittle TV, Allen H, Maziarz RT. Brefeldin A-mediated apoptosis requires the activation of caspases and is inhibited by Bcl-2. Exp Cell Res. 1998;245: 57-68. [DOI] [PubMed] [Google Scholar]
- 77.Contreras JL, Smyth CA, Bilbao G, et al. Coupling endoplasmic reticulum stress to cell death program in isolated human pancreatic islets: effects of gene transfer of Bcl-2. Transpl Int. 2003; 16: 537-542. [DOI] [PubMed] [Google Scholar]
- 78.Novak AJ, Bram RJ, Kay NE, Jelinek DF. Aberrant expression of B-lymphocyte stimulator by B chronic lymphocytic leukemia cells: a mechanism for survival. Blood. 2002;100: 2973-2979. [DOI] [PubMed] [Google Scholar]
- 79.Kern C, Cornuel J-F, Billard C, et al. Involvement of BAFF and APRIL in the resistance to apoptosis of B-CLL through an autocrine pathway. Blood. 2004;103: 679-688. [DOI] [PubMed] [Google Scholar]
- 80.Planelles L, Carvatho-Pinto CE, Hardenberg G, et al. APRIL promotes B-1 cell-associated neoplasm. Cancer Cell. 2004;6: 399-408. [DOI] [PubMed] [Google Scholar]
- 81.Kay NE, Wasil T. Survival of chronic lymphocytic leukemia cells: CD40L and the vascular endothelial growth factor (VEGF) connection. Leukemia. 2005;19: 531-532. [DOI] [PubMed] [Google Scholar]
- 82.Farahani M, Trewecke AT, Toh CH, et al. Autocrine VEGF mediates the antiapoptotic effect of CD154 on CLL cells. Leukemia. 2005;19: 524-530. [DOI] [PubMed] [Google Scholar]
- 83.Anether G, Tinhofer I, Senfter M, Greil R. Tetrocarcin-A-induced ER stress mediates apoptosis in B-CLL cells via a bcl-2-independent pathway. Blood. 2003;101: 4561-4568. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}