

Abstract
Staphylococcus aureus is a major human pathogen interfering with host-cell functions. Impaired wound healing is often observed in S aureus–infected wounds, yet, the underlying mechanisms are poorly defined. Here, we identify the extracellular adherence protein (Eap) of S aureus to be responsible for impaired wound healing. In a mouse wound-healing model wound closure was inhibited in the presence of wild-type S aureus and this effect was reversible when the wounds were incubated with an isogenic Eap-deficient strain. Isolated Eap also delayed wound closure. In the presence of Eap, recruitment of inflammatory cells to the wound site as well as neovascularization of the wound were prevented. In vitro, Eap significantly reduced intercellular adhesion molecule 1 (ICAM-1)–dependent leukocyte-endothelial interactions and diminished the consequent activation of the proinflammatory transcription factor nuclear factor κB (NFκB) in leukocytes associated with a decrease in expression of tissue factor. Moreover, Eap blocked αv-integrin–mediated endothelial-cell migration and capillary tube formation, and neovascularization in matrigels in vivo. Collectively, the potent anti-inflammatory and antiangiogenic properties of Eap provide an underlying mechanism that may explain the impaired wound healing in S aureus–infected wounds. Eap may also serve as a lead compound for new anti-inflammatory and antiangiogenic therapies in several pathologies.
Introduction
Staphylococcus aureus, and especially strains with resistance to antimicrobial agents, is an unabated challenge in community-acquired and nosocomial infections ranging from wound infections or osteomyelitis to life-threatening endocarditis, or septic shock.1,2 Wound infection with S aureus is frequently associated with impaired healing; yet, interference of S aureus with wound-healing mechanisms is poorly understood.3,4 S aureus expresses a number of bacterial cell wall–anchored adhesins mediating its adherence to host extracellular matrix (ECM) components.5 Moreover, S aureus produces and secretes proteins with ECM binding properties, such as coagulase,6 the extracellular fibrinogen binding protein,5 as well as Eap, also designated Map (major histocompatibility [MHC] class II analogous protein), a 60-kDa protein with a broad repertoire of interactions to host ECM components.7-9 Recently, we demonstrated direct interactions of Eap with the host adhesive proteins intercellular adhesion molecule 1 (ICAM-1), fibrinogen (FBG), and vitronectin (VN), resulting in the disruption of integrin-dependent leukocyte recruitment in vitro and in vivo, thereby serving as a potent anti-inflammatory factor.10 While Eap was also shown to exert immunomodulatory actions by interfering with T-cell functions,11 a possible interference of Eap with host wound healing is not defined.
Wound healing is a well-organized sequence of events involving inflammatory, proliferative, and maturation phases.12,13 (1) The inflammatory phase requires initial recruitment of neutrophils and later of macrophages that besides defending against invasive microbes, also release proinflammatory cytokines important for the subsequent activation of fibroblasts, keratinocytes, and endothelial cells in the proliferative phase of wound healing.12,13 Leukocyte recruitment requires a coordinated sequence of multistep adhesive events including selectin-mediated rolling, leukocyte activation leading to integrin-mediated firm adhesion, and diapedesis.14 During firm leukocyte adhesion to endothelial cells, leukocyte β2-integrins LFA-1 and Mac-1, as well as β1-integrins, interact with endothelial counterligands such as ICAM-1, or vascular-cell adhesion molecule 1 (VCAM-1).14-15 The activation of infiltrating leukocytes is linked to the modulation of gene expression of proinflammatory and procoagulant factors, such as interleukin-6 or tissue factor,16,17 mediated at least in part by the activation of the transcription factor nuclear factor κB (NFκB),18-20 which is a critical molecular bridge between cell exposure to inflammatory/infectious stimuli and gene expression.21 (2) During the proliferative phase, the migration and proliferation of keratinocytes, fibroblasts, and capillary endothelial cells results in re-epithelialization and tissue granulation in close context with neovascularization. Repair angiogenesis depends on the interplay between growth factors, such as the vascular endothelial growth factor (VEGF), as well as on adhesive contacts of the endothelial cells with the ECM.22,23 Emerging evidence also points to a crosstalk between inflammatory cells and angiogenesis, and particularly both proangiogenic and antiangiogenic activities of neutrophils and macrophages have been described.24 (3) In the maturation/remodeling phase, the completion of tissue repair is related to the proteolytic balance that regulates the degradation of excess collagen in the wound site.12,13
Wound healing is frequently delayed in S aureus–infected wounds.3,4 Our previous observations that identified Eap as an inhibitor of leukocyte recruitment10 prompted us to investigate whether Eap can influence the wound-healing process. We found Eap to exert anti-inflammatory actions by inhibiting leukocyte recruitment and activation, and strikingly, Eap was identified as a potent antiangiogenic factor in vitro and in vivo. Thus, by interfering with major steps of the wound-healing process, Eap appears to constitute a major bacterial component responsible for impaired wound healing.
Materials and methods
Materials
The following reagents were provided from these sources: blocking monoclonal antibody (mAb) IB4 against human CD18 (Alexis, Grünberg, Germany); mAb against tissue factor (W. Ruf, La Jolla, CA); mAb LM609 against αvβ3 and mAb 6S6 against CD29 (Chemicon, Hofheim, Germany); mAb against ICAM-1 (DAKO, Hamburg, Germany); rabbit polyclonal antibody against tissue factor (American Diagnostica, Heidelberg, Germany); rabbit anti-mouse myeloperoxidase (MPO) for Western blot analysis (Dunn Labortechnik, Asbach, Germany); rabbit anti–mouse MPO for immunohistochemistry (Abcam, Cambridge, MA); rat anti–mouse F4/80 (Acris, Hiddenhausen, Germany); rat anti–mouse CD31 (Pharmingen, Hamburg, Germany); respective isotype-matched control antibodies and secondary-conjugated antibodies (DAKO, Carpinteria, CA); recombinant ICAM-1, tumor necrosis factor–α (TNF-α), and VEGF (R&D Systems, Wiesbaden, Germany); and purified αvβ3-integrin and α5β1-integrin (Chemicon). FBG, fibronectin (FN), S aureus protein A and monocyte chemoattractant protein-1 (MCP-1) were from Sigma (Munich, Germany). VN was purified from human plasma and converted into the multimeric form as described.10 Polyclonal antibody against Eap was previously described.10
Bacteria and purification of Eap
S aureus strain Newman and the isogenic Eap-deficient strain AH12 were previously described.10 Eap from strain Newman was purified as described with modifications.10 Briefly, S aureus strain Newman was harvested following a 20-hour incubation in brain-heart infusion (BHI) medium (4 × 500 mL) at 37°C. The resultant 1 M lithium chloride–treated extract was dialysed against phosphate-buffered saline (PBS), concentrated and adsorbed onto SP Sepharose (Amersham-Pharmacia, Freiburg, Germany) in loading buffer (30 mM phosphate buffer [pH 7.0] 20 mM NaCl) at 4°C overnight. After stepwise elution with increasing NaCl concentrations, the pooled eluted fractions (between 0.6 and 0.8 M NaCl) were dialysed against 1:4 diluted PBS at 4°C and concentrated using Millipore centricon centrifugal filter devices (MWCO 30 kDa; Millipore, Bedford, MA). The rententate (containing 2 mg/mL Eap) was further purified by cation exchange chromatography on a Mono S 10/100 GL tricorn column using an ÄKTA fast-performance liquid chromatography system (Amersham-Pharmacia) operated with 10 mM Tris/HCl (pH 8.0) and an increasing linear NaCl gradient (0-1 M NaCl). Eap-containing fractions were purified on a Superdex 75 HR 10/30 gel filtration column (GE Healthcare, Uppsala, Sweden), equilibrated with Tris-buffered saline (TBS) at pH 7.4, and Eap-positive fractions were pooled, concentrated (1-1.5 mg/mL), sterile-filtered, and snap-frozen at –80°C until further use. Eap was devoid of detectable endotoxin.
Cell culture
Myelo-monocytic THP-1 cells and human umbilical vein endothelial cells (HUVECs) were cultivated as described.25 Human neutrophils were isolated from peripheral blood as described.25
Leukocyte adhesion to endothelial cells and transendothelial migration
Adhesion of THP-1 cells or neutrophils to HUVEC monolayers and neutrophil transendothelial migration was tested exactly as described.25,26,27
Endothelial-cell migration
Endothelial-cell migration was tested using Transwell membranes (8-μm pore size and 6.5-mm diameter; Corning Costar) that were coated with VN, FBG, FN, or bovine serum albumin (BSA) (each at 10 μg/mL) exactly as described.28
In vitro ligand-receptor interactions
Binding of FBG, VN, or FN to immobilized integrins (10 μg/mL) was performed as described.25,29 Briefly, binding of biotin-FBG or VN (each at 2 μg/mL) to immobilized αvβ3-integrin or binding of FN to immobilized α5β1-integrin was performed in TBS containing 0.3% BSA, 0.05% Tween-20, and 1 mM Ca2+ in the absence or presence of Eap. After incubation for 2 hours at 22°C in each case, the respective antiligand antibodies (rabbit anti-VN and rabbit anti-FN) followed by addition of appropriate secondary peroxidase-conjugated antibodies were used. For detection of biotin-FBG, peroxidase-coupled streptavidin was used. After extensive washing the substrate ABTS (2,2′-azino-bis[3-ethylbenzthiazoline-6-sulfonic acid]) was added, and binding was quantitated at 405 nm. Nonspecific binding to BSA-coated wells was used as blank and was subtracted to calculate specific binding.
In vitro angiogenesis assay
In vitro angiogenesis in collagen gels was quantitated using endothelial-cell spheroids as described previously.30 In brief, spheroids containing 400 cells were generated overnight, after which they were embedded into collagen gels. A collagen stock solution was prepared prior to use by mixing 8 vol acidic collagen extract of rat tails (equilibrated to 2 mg/mL, 4°C) with 1 vol 10 × Earle balanced salt solution (EBSS; Gibco BRL, Eggenstein, Germany), and approximately 1 vol 0.2 N NaOH to adjust the pH to 7.4. This stock solution (0.5 mL) was mixed with 0.5 mL room-temperature medium (endothelial-cell basal medium [ECBM; PromoCell, Heidelberg, Germany] with 40% fetal calf serum [FCS; Biochrom, Berlin, Germany]) containing 0.5% (wt/vol) methylcellulose (to prevent sedimentation of spheroids prior to polymerization of the collagen gel) and 50 spheroids. The spheroid-containing gel was rapidly transferred into prewarmed 24-well plates and allowed to polymerize for 30 minutes, after which 0.1 mL ECBM with the corresponding test substance was laid on top of the gel. The gels were incubated at 37°C, 5% CO2, and 100% humidity. After 24 hours, in vitro angiogenesis was digitally quantitated by measuring the length of the sprouts that had grown out of each spheroid using the digital imaging software analysis (Soft Imaging Systems, Münster, Germany) analyzing at least 10 spheroids per experimental group and experiment.
Endothelial-cell proliferation
Endothelial-cell proliferation was determined by measuring total cell number exactly as described.31
Matrigel plug assay
After thawing on ice, matrigel (BD Bioscience, Bedford, MA) containing 30 IU/mL heparin (Roche, Mannheim, Germany), in the absence or presence of 100 ng/mL VEGF without or with Eap in a total volume of 400 μL was injected subcutaneously into the laterodorsal abdominal region of 8-week-old male C57Bl/6 mice. The matrigel samples were recovered 5 days after implantation for photo documentation. To quantify neovascularization the hemoglobin concentration in matrigel homogenizates was measured as described.32
Generation of cutaneous wounds
Wound-healing experiments were performed as described.27,33,34 Ten-week-old female mice were anesthetized and 2 round cutaneous wounds (diameter, 14 mm) were generated at both sides of the lower dorsal trunk after shaving the hair and disinfecting the skin. A full-thickness excisional wound was created by removal of the skin and panniculus carnosus. The wounds were separated by more than 1 cm skin. A semipermeable transparent dressing (Tegaderm; 3M Health Care, St Paul, MN) was placed over the wound. The different treatments or the inoculated bacteria(2.5 × 109/wound) were applied directly under the dressing at a volume of0.1 mL. This protocol was approved by the Govermental Office of Karlsruhe, Germany. To evaluate wound size, the vertical and horizontal diameters of each wound were recorded immediately after wounding at day 0 and at subsequent days thereafter in anesthetized mice. The mean of the wound diameters was used in the calculation of each wound area and wound size was calculated relative to the size at day 0.
In other experiments, wounds were extracted at days 0, 1, 3, and 5 after generation and processed for immunohistochemistry or Western blot analysis. For the preparation of total wound extract, wounds were homogenized using a pestle in liquid nitrogen and were then lysed in a buffer containing 50 mM Tris-HCl (pH 8.0), 150 mM NaCl, 0.02% sodium azide, 1% Triton X-100, 2 mM benzamidin, 20 μg/mL soybean inhibitor, 1 mM leupeptin, 1 μg/mL aprotinin, and 0.5 mM PMSF (phenylmethylsulphonylfluoride). After centrifugation (16 000g for 2 minutes at 4°C), lysates were cleared from insoluble material, followed by 4 freeze (–80°C)/thaw (37°C) cycles. Lysates were centrifuged (1 500g for 5 minutes at 4°C), the supernatant was collected and centrifuged again (16 000g for 1 minute at 4°C). The supernatant was used for Western blot analysis after determining and adjusting protein concentration. Western blot analysis for the detection of MPO or tissue factor was performed. The same protocol was used for the detection of Eap in S aureus–infected or noninfected wounds from human subjects.
Western blot analysis
Western Blot analysis for the detection of tissue factor on THP-1 cells, or for the detection of tissue factor or MPO in wound extracts, was performed as described.27
Immunohistochemistry
At the time of death, immediately after final tracing of the wound edges, the entire wound was excised down to the fascia. Tissue was snap-frozen and stored at –80°C. For detection of neutrophils, macrophages, and vessels, antibodies to mouse MPO, F4/80, and platelet endothelial-cell adhesion molecule 1 (PECAM-1) were used, respectively. Thin cryostat sections (10 μm) were prepared from day-1 or day-5 wounds and were collected on slides. After fixing with ice-cold acetone for 10 minutes, blocking, and washings, sections were incubated with a rabbit anti-MPO (diluted 1:100), anti–mouse F4/80 (diluted 1:200), or biotinylated rat anti–mouse CD31 (diluted 1:200) for 2 hours at room temperature. Sections were then reacted with appropriate peroxidase-coupled secondary antibodies. Peroxidase activity was detected with AEC substrate. Consecutive sections that stained without first antibodies were used as negative controls. Slides were coverslipped and examined under a Zeiss microscope equipped with a Plan-Neofluar 10×/0.3 (Figure 4) or 20×/0.5 (Figures 1, 2) objective lens (Zeiss, Oberkochen, Germany). Photographs were collected with an ORCA ER camera (Hamamatsu, Hamamatsu City, Japan), and were acquired and analyzed with Metamorph software (Molecular Devices, Downingtown, PA). For the evaluation of blood vessels present in the wounds at day 5, 7 microscopic fields per wound and 8 wounds per treatment group were analyzed, and neovascularization was expressed as number of blood vessels per microscopic field.
Figure 4.

Eap inhibits neovascularization in wound healing. (A) Typical photomicrographs (× 100 magnification) of immunohistochemical staining for the detection of PECAM-1 (CD31) in association with blood vessels at day 5 following wound generation in the absence (buffer) or presence of 20 μg Eap is shown. Negative control (first antibody omitted) is also shown. (B) Statistical evaluation of the density of CD31-positive blood vessels (expressed as number of vessels/field) in wound sections at day 5 following wound generation in the absence (buffer) or presence of 20 μg Eap is shown. Evaluation of 7 fields per wound from 8 wounds per treatment group is shown. (C) Mice were anesthetized and fluorescent microbeads were injected. The recovered fluorescence per weight of wound tissue is shown, and data expressed relative to control (buffer-treated wounds) are means ± SD of 10 wounds. *P < .01 compared with control.
Figure 1.

Detection of Eap in wounds and effect of Eap on wound healing. (A) Eap detection in S aureus–infected wounds. (B) Eap was detected by Western blot analysis in extracts of noninfected (lane 1) or S aureus–infected (lane 2) wounds. Molecular mass markers are indicated along the right margin. (C) The course of wound closure is shown in the absence (buffer; •), or in the presence of wild-type S aureus (▪) or the Eap-deficient strain (□). *P < .05 compared with buffer-treated wounds; #P < .05 compared with wounds that received the Eap-deficient strain; **P < .05 compared with buffer-treated wounds. (D) The course of wound closure is shown in the absence (•) or presence of 20 μg Eap (▪) or protein A (▵). *P < .05 compared with buffer-treated wounds. Wound closure is expressed relative to the wound size of each wound at day 0 that represents 100%. Data are expressed as mean ± SD (n = 8).
Figure 2.

Inhibition of leukocyte infiltration into wounds by Eap. (A-B) Typical photomicrographs of immunohistochemical staining for the detection of neutrophils (anti-MPO) at day 1 following wound generation in the absence (A; buffer) or presence of 20 μg Eap (B) is shown. (C) The expression of MPO at day 1 following wound generation as assessed by Western blot is shown in the absence (□) or in the presence (▪) of Eap. The insert demonstrates a typical blot with staining for MPO. Densitometric data are expressed relative to control (buffer-treated wounds), and are means ± SD of 3 separate experiments. (D-E) Typical photomicrographs of immunohistochemical staining for the detection of macrophages (anti-F4/80) at day 5 following wound generation in the absence (D; buffer) or presence (E) of Eap is shown. (F) The expression of tissue factor at day 5 after wound generation as assessed by Western blot analysis is shown in the absence (□) or presence (▪) of Eap. The insert demonstrates a typical blot. Densitometric data are expressed relative to control (buffer-treated wounds), and are means ± SD of 3 separate experiments.
For the detection of Eap in human wounds, biopsies were taken from S aureus–infected or noninfected wounds; tissues were processed as described and Eap detection was performed with sheep polyclonal antibody against Eap. Written consent was obtained from all patients.
Quantification of blood flow using microspheres
For the measurement of blood flow, Fluospheres (Molecular Probes, Leiden, the Netherlands) were used as described.35 Briefly, at day 5 following wound generation mice were anesthetized and the chest was opened, followed by injection of 1.5 × 106 microspheres (diameter, 10 μm), dissolved in 0.5 mL of 0.9% NaCl into the left ventricle. Mice were killed 3 minutes after injection, and the wounds were extracted, weighed, and digested using 4 M KOH for 24 hours. Fluorescence per weight of tissue sample in each wound specimen was determined according to the manufacturer′s protocol.
Electrophoretic mobility shift assay (EMSA)
Nuclear proteins from cells were harvested and assayed for transcription factor binding activity using the NFκB consensus sequence 5′-GTTGAGGGGACTTTCCCAGGC-3′ as described.27 Specificity of binding was ascertained by competition with a 160-fold molar excess of unlabeled consensus oligonucleotides and by supershift experiments.
Statistical analysis
Data were compared using analysis of variance (ANOVA) with post-hoc analysis as appropriate; P values of less than .05 were regarded as significant.
Results
Inhibition of wound healing by Eap
S aureus–infected wounds often display impaired wound healing. As shown in Figure 1A and B, Eap was detected in S aureus–infected wounds from patients but not in noninfected wounds by immunohistochemistry and by Western blot. We have previously shown that Eap binds endothelial ICAM-1 and inhibits leukocyte recruitment.10 As inflammatory-cell infiltration is crucial in wound healing, we continued to define the functional role of Eap in wound healing. Excisional wounds were placed on the dorsal skin of mice. In the presence of wild-type S aureus a dramatic delay in wound healing was observed (Figure 1C). A significant delay in wound closure was observed already after the second day. In the presence of S aureus, wound closure was observed after 14 to 15 days, whereas control wounds healed after 10 to 11 days (Figure 1C). To define the role of Eap in the inhibition of wound healing by S aureus, we engaged an isogenic Eap-deficient strain. The inhibitory effect of S aureus in wound healing was reverted when the Eap-deficient strain was used (Figure 1C), indicating that Eap contributes to the S aureus–mediated delay in wound closure. Moreover, we applied isolated Eap locally to the wound on days 0, 1, 3, and 5. In Eap-treated wounds a significant delay in closure was observed after the second day compared with buffer-treated wounds. Thereafter, the wound areas of Eap-treated mice remained significantly larger until day 13 and healed with a delay of 3 days compared with buffer-treated wounds (Figure 1D). Inhibition of wound healing by Eap was also dose-dependent (10, 20, and 30 μg/wound were used; data with 20 μg/wound are shown). In contrast, S aureus protein A did not affect wound healing (Figure 1D). Moreover, coincubation of Eap with an antibody against Eap neutralized the inhibitory effect of Eap (not shown). These data indicate that Eap inhibits macroscopic wound healing.
Reduced leukocyte infiltration in wounds and leukocyte activation by Eap
In the inflammatory phase of wound healing, neutrophils and macrophages migrate into the wound site at different times following injury.12,13 One day after injury, neutrophil infiltration at the wound sites of Eap-treated mice was markedly lower, compared with control mice (Figure 2A-B). The detection of MPO by Western blot analysis was also decreased in Eap-treated mice (Figure 2C), indicating less infiltrated neutrophils in these wounds. Using the anti-F4/80 antibody that recognizes macrophages, the number of macrophages at day 5 was found to be significantly lower in Eap-treated wounds compared with buffer-treated wounds (Figure 2D-E). In contrast, in protein A–treated wounds no decrease in neutrophil or macrophage recruitment was observed (not shown). These data indicate that Eap inhibits leukocyte infiltration to wound sites. The infiltrating leukocytes in wounds release proinflammatory and procoagulant factors, such as tissue factor, that contribute to the repair mechanism. We therefore compared the expression of tissue factor in Eap- and buffer-treated wounds. At days 3 and 5 after injury reduced levels of tissue factor were found by Western blot analysis in Eap-treated wounds compared with control wounds (Figure 2F; data for day 3 not shown).
We previously demonstrated that Eap inhibits the interaction between ICAM-1 and its leukocyte counterreceptors Mac-1 and LFA-1.10 As Eap-treated wounds displayed reduced inflammatory-cell infiltration, we investigated whether Eap interferes with the adhesion of peripheral blood neutrophils or THP-1 cells to endothelial cells. The presence of Eap significantly reduced the β2-integrin–dependent adhesion of neutrophils to endothelial cells to the level of inhibition obtained with blocking mAb against ICAM-1 (Figure 3A). MCP-1–stimulated neutrophil transendothelial migration was also blocked by Eap (Figure 3B). Inhibition of neutrophil transendothelial migration by Eap could be due to its antiadhesive effect. In order to study whether Eap directly interferes with transendothelial migration, experiments were performed in 2 ways: (1) neutrophils and inhibitors were coincubated during the whole course of the transmigration experiment; and (2) neutrophils were incubated on endothelial cells for 20 minutes in the absence of competitors to facilitate their initial attachment on the endothelial surface, and thereafter inhibitors were added into the wells. CD18 and ICAM-1 participate in both neutrophil-endothelial adhesion and neutrophil transendothelial migration.36 Therefore, anti-CD18 and anti–ICAM-1 inhibited neutrophil diapedesis in both settings (ie, when coincubated with the neutrophils during the whole course of the transmigration assay), and when added to the wells after initial neutrophil attachment. Unsurprisingly, the degree of inhibition of transmigration by anti-CD18 and anti–ICAM-1 was slightly lower in the latter situation. In contrast, anti-CD29 blocked transmigration only when coincubated with the neutrophils, but not when added after the initial neutrophil attachment, suggesting a role of β1-integrin primarily in neutrophil adhesion. Consistent with our previous studies that identified Eap to block ICAM-1,10 Eap behaved similarly to anti-CD18 or anti–ICAM-1, which suggests that Eap interferes with both neutrophil adhesion and transmigration (Figure 3B). The effect of Eap on HUVEC-related neutrophil adhesion and their transmigration was also dose dependent (not shown). No effect of protein A on neutrophil-endothelial adhesion and neutrophil transmigration was found (Figure 3A-B). Similar results were obtained with THP-1 cells (not shown).
Figure 3.

Influence of Eap on leukocyte–endothelial-cell interactions and on the activity of NFκB. (A) Adhesion of human neutrophils to endothelial cells is shown in the absence (–) or presence of blocking mAb to CD18, ICAM-1, and CD31 as indicated (each 20 μg/mL), Eap, or protein A (each 20 μg/mL). Cell adhesion is expressed relative to control (in the absence of competitor). Data are means ± SD (n = 3) of a typical experiment; similar results were obtained in 3 separate experiments. (B) The transendothelial migration of human neutrophils in response toward 50 ng/mL MCP-1 is shown in the absence (–) or presence of mAb to CD18, mAb to ICAM-1, mAb to CD29 (each 20 μg/mL), Eap, or protein A (each 20 μg/mL). Neutrophils and inhibitors were coincubated during the whole course of the transmigration experiment (□), or neutrophils were incubated on the endothelial cells for 20 minutes in the absence of competitors in order to facilitate their initial attachment on the endothelial surface, and inhibitors were added thereafter into the wells (▪). Transmigration is presented as percent of control (in the absence of competitor). Data are means ± SD (n = 3) of a typical experiment; similar results were obtained in 3 separate experiments. (C) The DNA-binding activity of NFκB without or with TNFα or TNFα plus Eap in THP-1 cells is shown in the absence (□) or presence ( ) of ICAM-1 (10 μg/mL), as indicated. The insert demonstrates a typical EMSA for NFκB DNA-binding activity (1 indicates control; 2, TNF-α; 3, TNF-α+Eap; 4, ICAM-1; 5, ICAM-1+TNFα; and 6, ICAM-1+TNFα+Eap). (D) The expression of tissue factor without or with TNFα or TNFα plus Eap in THP-1 cells is shown in the absence (□) or presence () of ICAM-1 (10 μg/mL). Densitometric data are expressed relative to control (100% control is represented in the absence of stimuli or competitors), and are means ± SD of 2 separate experiments. *P < .05.
) of ICAM-1 (10 μg/mL), as indicated. The insert demonstrates a typical EMSA for NFκB DNA-binding activity (1 indicates control; 2, TNF-α; 3, TNF-α+Eap; 4, ICAM-1; 5, ICAM-1+TNFα; and 6, ICAM-1+TNFα+Eap). (D) The expression of tissue factor without or with TNFα or TNFα plus Eap in THP-1 cells is shown in the absence (□) or presence () of ICAM-1 (10 μg/mL). Densitometric data are expressed relative to control (100% control is represented in the absence of stimuli or competitors), and are means ± SD of 2 separate experiments. *P < .05.
The transcription factor NFκB links inflammatory stimuli and gene expression in leukocytes and endothelial cells,21 and ligation and activation of leukocyte integrins as it takes place during adhesive interactions can result in the activation of NFκB.19,20 We therefore tested whether the anti-inflammatory effect of Eap extends toward differences in NFκB-binding activity, as assessed by EMSA in THP-1 cells. As shown in Figure 3C, ligation of β2-integrins with recombinant ICAM-1 increased TNFα–induced NFκB activity. This effect was prevented by Eap, but not by protein A (not shown). NFκB is central in the regulation of the expression of inflammatory mediators in leukocytes such as tissue factor, and TNFα-induced tissue factor expression in monocytes is upregulated upon β2-integrin ligation with ICAM-1.18 Coincubation of THP-1 cells with Eap prevented the ICAM-1–induced tissue factor expression in THP-1 cells (Figure 3D). Taken together, these data clearly demonstrate that Eap exerts anti-inflammatory properties by blocking ICAM-1–dependent leukocyte adhesion to and transmigration through the endothelium and reducing the ICAM-1–mediated up-regulation of the activity of NFκB with respective consequences for the expression of proinflammatory and procoagulant molecules in vitro and in vivo.
Impaired angiogenesis in Eap-treated wounds
During the proliferative phase of wound healing, neovascularization is indispensable for the generation of granulation tissue. Immunohistochemical inspection of Eap-treated wounds displayed a dramatic inhibition of neovascularization, as the number of CD31-positive vessels at day 5 after injury was reduced in Eap-treated wounds compared with buffer control (Figure 4A-B). No effect of protein A in wound neovascularization was observed (not shown). In order to assess and quantify angiogenesis in more detail, we examined the perfusion of wounds using fluorescent microspheres. In line with the previous findings, Eap treatment significantly reduced blood flow in wounds by 40% to 50% (Figure 4C). Thus, Eap exerts strong antiangiogenic activity, impairing neovascularization in the proliferative phase of wound repair.
Inhibition of αv-integrin–dependent endothelial-cell migration by Eap
As Eap binds to different ECM proteins, we hypothesized that it might interfere with interactions between integrins and matrix proteins that are crucial in angiogenesis. Endothelial cells engage different integrins to migrate onto matrix proteins such as VN, FBG, or FN. Whereas endothelial-cell migration toward VN and FBG is dependent predominantly on αv-integrins, α5β1-integrin mediates migration toward FN, as shown by blockade with specific antibodies (Figure 5A). Eap, but not protein A, dose-dependently blocked αv-integrin–dependent migration toward VN or FBG, both under basal conditions or under stimulation with VEGF (only data in the presence of VEGF are shown in Figure 5A). Only a very moderate but not significant inhibitory effect of Eap on the α5β1-integrin–dependent migration of endothelial cells toward FN was observed.
Figure 5.

Eap interferes with αvβ3-integrin–dependent cell function. (A) VEGF (20 ng/mL)–stimulated migration of HUVECs toward VN, FBG, or FN (each 5 μg/mL) is shown in the absence (▪) or presence of the blocking mAb against αvβ3-integrin, LM609 (for VN and FBG), the blocking mAb against β1-integrin, 6S6 (for FN) (20 μg/mL; □), or in the presence of Eap (;20 μg/mL), or protein A ( ;20 μg/mL). Cell migration is expressed relative to control, which is represented as cell migration in the absence of any stimulus or competitor. Data are means ± SD of 4 experiments performed in triplicate. *P < .05 compared with VEGF-stimulated migration in the absence of competitor. ns indicates nonsignificant. (B-D) Binding of VN to immobilized αvβ3-integrin (B), binding of FBG to immobilized αvβ3-integrin (C), and binding of FN to immobilized α5β1-integrin(D) was performed in the absence (–; ▪) or presence of blocking mAb against αvβ3-integrin, LM609 (for VN and FBG), the blocking mAb against β1-integrin, 6S6 (for FN) (20 μg/mL; □), or increasing concentrations of Eap () as indicated. Specific binding is expressed as absorbance at 405 nm. Data are means ± SD (n = 3) of a typical experiment; similar results were observed in 3 separate experiments.
;20 μg/mL). Cell migration is expressed relative to control, which is represented as cell migration in the absence of any stimulus or competitor. Data are means ± SD of 4 experiments performed in triplicate. *P < .05 compared with VEGF-stimulated migration in the absence of competitor. ns indicates nonsignificant. (B-D) Binding of VN to immobilized αvβ3-integrin (B), binding of FBG to immobilized αvβ3-integrin (C), and binding of FN to immobilized α5β1-integrin(D) was performed in the absence (–; ▪) or presence of blocking mAb against αvβ3-integrin, LM609 (for VN and FBG), the blocking mAb against β1-integrin, 6S6 (for FN) (20 μg/mL; □), or increasing concentrations of Eap () as indicated. Specific binding is expressed as absorbance at 405 nm. Data are means ± SD (n = 3) of a typical experiment; similar results were observed in 3 separate experiments.
In agreement with our previous observations that Eap directly interacts with VN or FBG,10 Eap was found to block the binding of both ECM proteins to immobilized αvβ3-integrin in a dose-dependent manner. In contrast, although Eap also binds to FN,10 the interaction between FN and α5β1-integrin was not affected by Eap (Figure 5B-D), indicating that Eap specifically affects αv-integrin–dependent interactions that are crucial for angiogenesis.37
Inhibition of endothelial proliferation, capillary sprout formation, and in vivo angiogenesis by Eap
VEGF-stimulated increase in endothelial-cell proliferation plays a crucial role during pathologic neovascularization in wound healing. VEGF-stimulated HUVEC proliferation was inhibited in the presence of Eap (Figure 6A) in a dose-dependent manner, whereas protein A was not effective (not shown). In a capillary-like sprout formation in vitro assay that examines the invasive and migratory potential of endothelial cells in a 3D cell-spheroid system, Eap dose-dependently reduced the VEGF-induced capillary-like sprouting (Figure 6B). The inhibitory concentration of 50% (IC50) of Eap in the 3D angiogenesis assay is 10.7 μg/mL (95% confidence interval from 5.9 to 19.6 μg/mL), very well within the range of Eap found in the supernatants of S aureus in culture.8 Moreover, Eap inhibited sprout formation to the same extent as established αv-integrin inhibitors, such as the mAb LM609 or cyclic RGD peptides do, whereas protein A had no inhibitory effect (Figure 6C).
Figure 6.

Effect of Eap on endothelial-cell proliferation and capillary sprout formation. (A) HUVEC proliferation. HUVECs were incubated without (□) or with (▪) VEGF (10 ng/mL) in the absence (–) or presence of increasing concentrations of Eap as indicated. Proliferation of HUVECs is expressed as a percentage of control, defined as cell proliferation in the absence of any stimulus or competitor. Data are means ± SD (n = 3) of 1 experiment typical of 3 separate experiments performed. (B-C) Capillary sprout formation. (B) HUVECs were incubated for 24 hours in the absence (–; □) or presence (▪) of VEGF (25 ng/mL) with or without increasing concentrations of Eap, as indicated. Capillary-like tube formation is expressed as capillary sprout length in micrometers. (C) HUVECs were incubated for 24 hours in the presence of VEGF (25 ng/mL) with or without (–) Eap, protein A (each 20 μg/mL), cyclic RGD peptide (cRGD; 20 μg/mL), or the mAb against αvβ3-integrin, LM609 (20 μg/mL), as indicated. Capillary-like tube formation is expressed relative to control, which is represented as sprout formation in the presence of VEGF and in the absence of any competitor. Data are means ± SD (n = 10) of a representative experiment. Similar results were obtained in 3 independent experiments.
Finally, the antiangiogenic activity of Eap in vivo was investigated using the matrigel plug assay. Matrigel in the absence or presence of Eap was injected subcutaneously into the laterodorsal abdominal region of mice and recovered 5 days later for analysis. VEGF-induced new-vessel formation was inhibited in matrigels exposed to Eap (Figure 7A), indicating that Eap exerts a strong antiangiogenic activity in vivo. By determining the hemoglobin concentration in the matrigels we found that 10 μg and 30 μg of Eap inhibited angiogenesis in the matrigel assay by 40% and 70%, respectively (Figure 7B).
Figure 7.
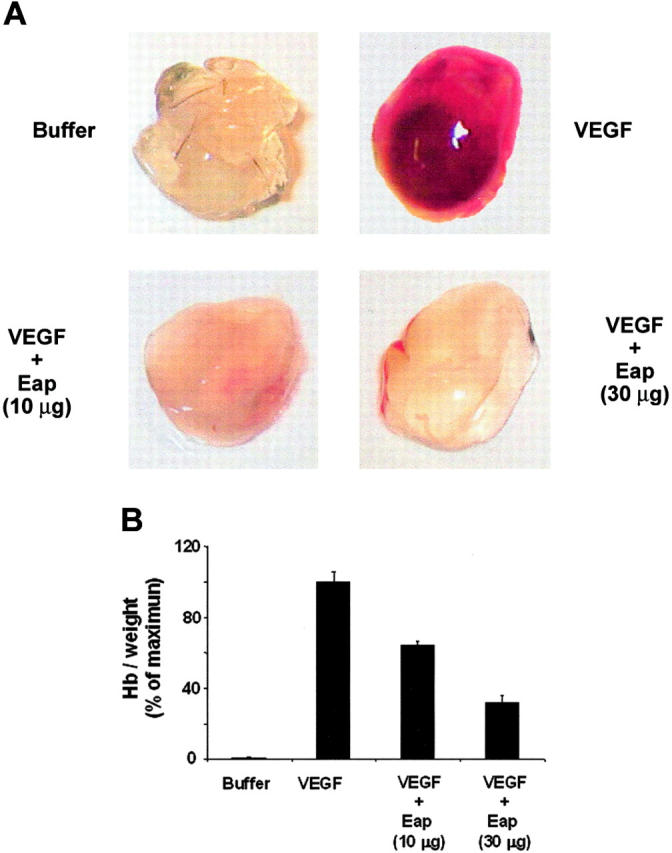
Inhibition of angiogenesis by Eap in the in vivo matrigel plug assay. (A) Neovascularization in the matrigel plug assay was performed without (buffer) or with VEGF (100 ng/mL) in the absence or presence of 10 μg or 30 μg Eap as indicated. Photographs of a typical experiment performed in triplicates are shown; similar results were obtained in 3 separate experiments. (B) The quantitation of neovascularization in the matrigels was performed by measuring the hemoglobin concentration. Hemoglobin concentration was expressed as milligrams of hemoglobin per grams of wet tissue. Data are expressed as a percentage of the maximum (VEGF treatment in the absence of competitors). Data are means ± SD (n = 3) of a typical experiment; similar results were observed in 3 separate experiments.
Discussion
Impaired wound healing is frequently seen in S aureus–infected chronic wounds. The present study clearly demonstrates for the first time that S aureus Eap delays wound closure due to its potent anti-inflammatory and antiangiogenic activities. The ability of Eap to interact with adhesion molecules and especially with endothelial ICAM-1 as well as with adhesive proteins in the ECM, thereby blocking integrin-mediated adhesive and migratory interactions of both inflammatory and endothelial cells, provides the common pathway for the anti-inflammatory and antiangiogenic actions of Eap. To our knowledge, the inhibition of neovascularization by Eap acting in concert with the anti-inflammatory properties of the molecule described here defines an entirely novel mechanism of inhibition of wound healing by S aureus.
Upon skin injury, initial clot formation and local inflammation characterized by infiltration of neutrophils and macrophages is followed by the proliferative phase, in which neovascularization of the granulation tissue is indispensable.12,13 The discovered features demonstrate that Eap specifically interferes with the inflammatory phase and blocks angiogenesis in the proliferative phase. First, in the presence of isolated Eap or wild-type S aureus wound closure was markedly delayed. The delay in wound healing due to S aureus presence was reversible when an isogenic Eap-deficient strain was engaged, clearly demonstrating that inhibition of wound healing in S aureus–infected wounds may be at least in part attributed to Eap. However, our findings also imply that other S aureus factors may be involved in delayed wound healing. Second, the subsequent analysis of Eap-treated wounds demonstrated reduced infiltration of neutrophils and macrophages, and consequently, reduced expression of the proinflammatory and procoagulant tissue factor. These data were supported by in vitro findings that Eap inhibits ICAM-1–dependent leukocyte endothelial adhesion and transendothelial migration and the ICAM-1–mediated up-regulation of NFκB activity and the consequent expression of tissue factor. Thus, Eap blocks ICAM-1–dependent leukocyte-endothelial interactions and ICAM-1–dependent leukocyte activation and the present data clearly extend previous findings by our group and others.10,38 Third, further analysis of wound tissue surprisingly revealed that Eap-treated wounds displayed dramatically impaired neovascularization. This profound reduction in neovascularization by Eap may explain the prolonged inhibitory effect of Eap on wound repair. Of note, the concentrations of Eap engaged in our studies lie very well within the range of the amounts of Eap found in the supernatants of S aureus in culture.8
During angiogenesis, ECM-associated proteins such as VN or FBG are incorporated into a provisional adhesive fibrillar network that regulates diverse endothelial-cell functions including growth and migration, and cellular adhesion–dependent signals are transmitted predominantly through αv-integrins.37 While Eap efficiently blocked endothelial-cell migration primarily by interfering with αv-integrins and their ligands VN and FBG, α5β1-dependent migration in response to FN was hardly affected by Eap. These findings demonstrate a specific role of Eap as a potent inhibitor of the αv-integrin–ligand system, very reminiscent of the blocking function of other αv-integrin inhibitors. This anti–αv-integrin activity of Eap thereby influences different angiogenesis-related functions of endothelial cells as was evidenced in Eap-mediated inhibition of capillary-like tube formation in 3-dimensional matrices or the blockade of neovascularization in matrigel plug assay in vivo. Furthermore, Eap dose-dependently inhibited VEGF-stimulated endothelial-cell proliferation; however the underlying mechanism is not revealed by our present results and should be addressed in a future study.
Eap may inhibit angiogenesis through a combination of several complementary mechanisms, culminating in blockade of αv-integrin function, that were identified in this study. As inflammatory cells are fine regulators of angiogenesis by providing growth factors and matrix metalloproteinases,24 the anti-inflammatory role of Eap may indirectly contribute to its antiangiogenic action as well. Moreover, tissue factor is not only a major initiator of hemostasis following vascular injury, but also a well-recognized proangiogenic factor.17 Eap-mediated inhibition of tissue factor expression can thereby down-regulate its angiogenic potential. Tissue factor expression in leukocytes depends in great parts on the activity of the transcription factor NFκB. In line with the Eap-mediated down-regulation of tissue factor expression, NFκB activation in monocytes was inhibited by Eap as well. Thus, the anti-inflammatory action of Eap extends to events downstream to integrin-dependent adhesion, such as blockade of NFκB activity. To our knowledge this is the first demonstration that an S aureus product may prevent NFκB activation, and that Eap might counteract the effects of peptidoglycan, which stimulates NFκB activation via toll-like receptor 239 and promotes wound repair.40
In addition to blocking leukocyte extravasation, Eap also interferes with T-cell functions, thereby affecting the course of chronic infections such as arthritis or osteomyelitis.11 Other immunomodulating properties of S aureus may add to the function of Eap described here. For example, Eap may cooperate with the chemotaxis-inhibitory protein of S aureus that blocks chemoattractant-induced neutrophil and monocyte migration into the infection site.41 However, these previous studies were limited to the immunomodulatory actions of Eap,10,11 but are extended here to inhibition of crucial endothelial-cell functions by Eap. The importance and implications of the observed antiangiogenic function of Eap are many-fold. Besides being responsible for the impaired wound healing in chronically S aureus–infected wounds or ulcera,3,4 as clearly demonstrated in the present study, the antiangiogenic properties of Eap may enable the development of a novel class of antiangiogenesis compounds to be engaged against uncontrolled neovascularization in cancer and other pathologies and studies are in progress to test these concepts.
Acknowledgments
The authors thank Yvonne Mueller and Hanno Welker for technical assistance; Dr Michael Kruhlak for help with microscopy studies; and Dr D. Linder for aminoterminal sequencing.
Prepublished online as Blood First Edition Paper, November 29, 2005; DOI 10.1182/blood-2005-08-3140.
Supported by the Intramural Research Program of the NIH, NCI (T.C.); and by grants from the Deutsche Forschungsgemeinschaft to T.C. (Ch279/2-1, Ch279/2-2 and SFB405), K.T.P. (Pr327/18-1), M. Herrmann (H3 1850/6-1), and M. Hussain (SFB492).
A.N.A., M.E., V.V.O., D.S., H.G.A., S.A.E., T.L., H.W., and H.P.H. performed research; A.S., U.S., and M. Hussain provided vital reagents; P.P.N. participated in designing research; M. Herrmann provided vital reagents, participated in designing research and wrote the manuscript; and K.T.P. and T.C. designed research and wrote manuscript.
K.T.P. and T.C. contributed equally to the work.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 U.S.C. section 1734.
References
- 1.Lowy FD. Staphylococcus aureus infections. N Engl J Med. 2003;339: 520-532. [DOI] [PubMed] [Google Scholar]
- 2.Waldvogel FA. New resistance in Staphylococcus aureus. N Engl J Med. 1999;340: 556-557. [DOI] [PubMed] [Google Scholar]
- 3.Madsen SM, Westh H, Danielsen L, Rosdahl VT. Bacterial colonization and healing of venous leg ulcers. APMIS. 1996;104: 895-899. [DOI] [PubMed] [Google Scholar]
- 4.Grimble SA, Magee TR, Galland RB. Methicillin resistant Staphylococcus aureus in patients undergoing major amputation. Eur J Vasc Endovasc Surg. 2001;22: 215-218. [DOI] [PubMed] [Google Scholar]
- 5.Flock JI. Extracellular-matrix-binding proteins as targets for the prevention of Staphylococcus aureus infections. Mol Med Today. 1999;12: 532-537. [DOI] [PubMed] [Google Scholar]
- 6.Sawai T, Tomono K, Yanagihara K, et al. Role of coagulase in a murine model of hematogenous pulmonary infection induced by intravenous injection of Staphylococcus aureus enmeshed in agar beads. Infect Immun. 1997;65: 466-471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McGavin MH, Krajewska-Pietrasik D, Rydén C, Höök M. Dentification of a Staphylococcus aureus extracellular matrix-binding protein with broad specificity. Infect Immun. 1993;61: 2479-2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Palma M, Haggar A, Flock JI. Adherence of Staphylococcus aureus is enhanced by an endogenous secreted protein with broad binding activity. J Bacteriol. 1999;181: 2840-2845.10217776 [Google Scholar]
- 9.Harraghy N, Hussain M, Haggar A, et al. The adhesive and immunomodulating properties of the multifunctional Staphylococcus aureus protein Eap. Microbiology. 2003;149: 2701-2707. [DOI] [PubMed] [Google Scholar]
- 10.Chavakis T, Hussain M, Kanse SM, et al. Staphylococcus aureus extracellular adherence protein (Eap) serves as anti-inflammatory factor by inhibiting the recruitment of host leukocytes. Nat Med. 2002;8: 687-693. [DOI] [PubMed] [Google Scholar]
- 11.Lee LY, Miyamoto YJ, McIntyre BW, et al. The Staphylococcus aureus Map protein is an immunomodulator that interferes with T cell-mediated responses. J Clin Invest. 2002;110: 1461-1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Martin P. Wound healing—aiming for perfect skin regeneration. Science. 1997;276: 75-81. [DOI] [PubMed] [Google Scholar]
- 13.Singer AJ, Clark RA. Cutaneous wound healing. N Engl J Med. 1999;341: 738-746. [DOI] [PubMed] [Google Scholar]
- 14.Springer TA. Traffic signals for lymphocyte recirculation and leukocyte emigration: the multistep paradigm. Cell. 1994;76: 301-314. [DOI] [PubMed] [Google Scholar]
- 15.Chavakis T, Preissner KT, Santoso S. Leukocyte trans-endothelial migration: JAMs add new pieces to the puzzle. Thromb Haemost. 2003;89: 13-17. [PubMed] [Google Scholar]
- 16.Lin ZQ, Kondo T, Ishida Y, Takayasu T, Mukaida N. Essential involvement of IL-6 in the skin wound-healing process as evidenced by delayed wound healing in IL-6-deficient mice. J Leukoc Biol. 2003;73: 713-721. [DOI] [PubMed] [Google Scholar]
- 17.Mackman N. Role of tissue factor in hemostasis, thrombosis and vascular development. Arterioscler Thromb Vasc Biol. 2004;24: 1015-1022. [DOI] [PubMed] [Google Scholar]
- 18.Fan ST, Edgington TS. Coupling of the adhesive receptor CD11b/CD18 to functional enhancement of effector macrophage tissue factor response. J Clin Invest. 1991;87: 50-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kim CH, Lee KH, Lee CT, et al. Aggregation of β2 integrins activates human neutrophils through the IκB/NF-κB pathway. J Leukoc Biol. 2004;75: 286-292. [DOI] [PubMed] [Google Scholar]
- 20.Schmal H, Czermak BJ, Lentsch AB, et al. Soluble ICAM-1 activates lung macrophages and enhances lung injury. J Immunol. 1998;161: 3685-3693. [PubMed] [Google Scholar]
- 21.Hatada EN, Krappmann D, Scheidereit C. NF-κB and the innate immune response. Curr Opin Immunol. 2000;12: 52-58. [DOI] [PubMed] [Google Scholar]
- 22.Carmeliet P. Angiogenesis in health and disease. Nat Med. 2003;9: 653-660. [DOI] [PubMed] [Google Scholar]
- 23.Strombald S, Cheresh DA. Integrins, angiogenesis and vascular cell survival. Chem Biol. 1996;3: 881-885. [DOI] [PubMed] [Google Scholar]
- 24.Frantz S, Vincent KA, Feron O, Kelly RA. Innate immunity and angiogenesis. Circ Res. 2005;96: 15-26. [DOI] [PubMed] [Google Scholar]
- 25.Chavakis T, Bierhaus A, Al-Fakhri N, et al. The pattern recognition receptor (RAGE) is a counter-receptor for leukocyte integrins: a novel pathway for inflammatory cell recruitment. J Exp Med. 2003;198: 1507-1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chavakis T, Keiper T, Matz-Westphal R, et al. The junctional adhesion molecule-C promotes neutrophil transendothelial migration in vitro and in vivo. J Biol Chem. 2004;279: 55602-55608. [DOI] [PubMed] [Google Scholar]
- 27.Chavakis T, Athanasopoulos A, Rhee JS, et al. Angiostatin is a novel anti-inflammatory factor by inhibiting leukocyte recruitment. Blood. 2005;105: 1036-1043. [DOI] [PubMed] [Google Scholar]
- 28.Chavakis T, Cines DB, Rhee JS, et al. Regulation of neovascularization by human neutrophil peptides (alpha-defensins): a link between inflammation and angiogenesis. FASEB J. 2004;18: 1306-1308. [DOI] [PubMed] [Google Scholar]
- 29.Chavakis T, Kanse SM, Lupu F, et al. Different mechanisms define the antiadhesive function of high molecular weight kininogen in integrin- and urokinase receptor-dependent interactions. Blood. 2000;96: 514-522. [PubMed] [Google Scholar]
- 30.Korff T, Augustin HG. Tensional forces in fibrillar extracellular matrices control directional capillary sprouting. J Cell Sci. 1999;112: 3249-3258. [DOI] [PubMed] [Google Scholar]
- 31.Economopoulou M, Bdeir K, Cines DB, et al. Inhibition of pathological retinal neovascularization by α-defensins. Blood. 2005;106: 3831-3838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Barcelos LS, Talvani A, Teixeira AS, et al. Impaired inflammatory angiogenesis, but not leukocyte influx, in mice lacking TNFR1. J Leukoc Biol. 2005;78: 352-358. [DOI] [PubMed] [Google Scholar]
- 33.Mori R, Kondo T, Nishie T, Ohshima T, Asano M. Impairment of skin wound healing in beta-1,4-galactosyltransferase-deficient mice with reduced leukocyte recruitment. Am J Pathol. 2004;164: 303-314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Goova MT, Li J, Kislinger T, et al. Blockade of receptor for advanced glycation end-products restores effective wound healing in diabetic mice. Am J Pathol. 2001;159: 513-525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Van Oosterhout MFM, Prinzen FW, Sakurada S, Glenny RW, Hales JRS. Fluorescent microspheres are superior to radioactive microspheres in chronic blood flow measurements. Am J Physiol Heart Circ Physiol. 1998;275: H110-H115. [DOI] [PubMed] [Google Scholar]
- 36.Shaw SK, Ma S, Kim MB, et al. Coordinated redistribution of leukocyte LFA-1 and endothelial cell ICAM-1 accompany neutrophil transmigration. J Exp Med. 2004;200: 1571-1580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Eliceiri BP, Cheresh DA. Adhesion events in angiogenesis. Curr Opin Cell Biol. 2001;13: 563-568. [DOI] [PubMed] [Google Scholar]
- 38.Haggar A, Ehrnfelt C, Holgersson J, Flock JI. The extracellular adherence protein from Staphylococcus aureus inhibits neutrophil binding to endothelial cells. Infect Immun. 2004;72: 6164-6167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lien E, Ingalls RR. Toll-like receptors. Crit Care Med. 2002;30: S1-S11. [PubMed] [Google Scholar]
- 40.Kilcullen JK, Ly QP, Chang TH, Levenson SM, Steinberg JJ. Nonviable Staphylococcus aureus and its peptidoglycan stimulate macrophage recruitment, angiogenesis, fibroplasia, and collagen accumulation in wounded rats. Wound Rep Reg. 1998;6: 149-156. [DOI] [PubMed] [Google Scholar]
- 41.de Haas CJC, Veldkamp KE, Peschel A, et al. Chemotaxis inhibitory protein of Staphylococcus aureus, a bacterial antiinflammatory agent. J Exp Med. 2004;199: 687-695. [DOI] [PMC free article] [PubMed] [Google Scholar]