

Abstract
Craniosynostosis syndromes are autosomal dominant human skeletal diseases that result from various mutations in fibroblast growth factor receptor genes (Fgfrs). Apert syndrome (AS) is one of the most severe craniosynostosis syndromes and is associated with severe syndactyly of the hands and feet and with central nervous system malformations. AS is caused by specific missense mutations in one of two adjacent amino acid residues (S252W or P253R) in the highly conserved region linking Ig-like domains II and III of FGFR2. Here we demonstrate that these mutations break one of the cardinal rules governing ligand specificity of FGFR2. We show that the S252W mutation allows the mesenchymal splice form of FGFR2 (FGFR2c) to bind and to be activated by the mesenchymally expressed ligands FGF7 or FGF10 and the epithelial splice form of FGFR2 (FGFR2b) to be activated by FGF2, FGF6, and FGF9. These data demonstrate loss of ligand specificity of FGFR2 with retained ligand dependence for receptor activation. These data suggest that the severe phenotypes of AS likely result from ectopic ligand-dependent activation of FGFR2.
Keywords: FGF, FGF receptor, craniosynostosis, mutation, syndactyly
The importance of fibroblast growth factor receptor (FGFR) signaling in skeletal development has been illustrated by genetic studies of human skeletal disorders. A number of skeletal disorders, including various craniosynostosis and dwarfism syndromes, have been mapped to mutations in the genes encoding FGFRs (1–4). Dwarfing chondrodysplasia syndromes, including hypochondroplasia, achondroplasia, and thanatophoric dysplasia, result from missense mutations in FGFR3 and primarily affect bones undergoing endochondral ossification. Craniosynostosis syndromes, which include Apert syndrome (AS) (5), Crouzon syndrome (CS) (6, 7), CS with Acanthosis Nigricans (CSAN) (8), coronal craniosynostosis (CC) (9), Pfeiffer syndrome (PS) (10), Jackson–Weiss syndrome (JWS) (11), Antley–Bixler syndrome (12), and Beare–Stevenson cutis gyrata (13), share phenotypes that include premature closure of some cranial sutures but have distinct facial features, limb abnormalities, and, in some cases, central nervous system malformations (1, 3, 4, 14). With the exception of PS, CC, and CSAN, most craniosynostosis syndromes result from missense mutations in FGFR2. Unlike other craniosynostosis syndromes, AS is accompanied by severe syndactyly of the hands and feet (5, 15). These limb abnormalities are not observed in CS, in which the limbs are unaffected, in PS, which is characterized by broad thumbs and great toes, or in JWS, which has abnormalities localized to the feet (16). In addition to skeletal abnormalities, AS is also associated with severe central nervous system malformations and is often accompanied by mental retardation (17–20).
FGFRs are transmembrane receptor tyrosine kinase proteins that contain an extracellular ligand-binding domain, a single transmembrane domain, and an intracellular tyrosine kinase domain. The extracellular region, which contains two or three Ig-like domains, is important for FGF binding and the subsequent dimerization and activation of FGFRs. Mutations that cause craniosynostosis syndromes are found in three of the four members of FGFR family (FGFR1, -2, and -3). FGFR2 is the most frequently affected receptor with numerous mutations localized in or near the third Ig-like domain or in the sequence linking Ig-like domains II and III. One class of mutations causing CS, PS, or Jackson–Weiss syndrome involves the loss or addition of a cysteine residue (21–23). The consequence of this class of mutation on the function of FGFR activity is to create an unpaired-cysteine residue that facilitates the formation of intermolecular disulfide bonds causing ligand-independent dimerization, phosphorylation, and signaling (3, 21, 24). A similar point mutation in FGFR3, R248C, causes thanatophoric dysplasia and also results in constitutive ligand-independent signaling (24). Other mutations in close proximity to cysteine residues also lead to ligand-independent receptor activation (23).
AS mutations involve a missense substitution in one of two adjacent amino acid residues (S252W or P253R) localized in the highly conserved linker region between Ig-like domains II and III of FGFR2 (Fig. 1). Ig-like domains II and III and the interdomain linker comprise an FGF-binding site (25–27). Because these mutations do not lie near cysteine residues, it has been suggested that AS mutations may not result in constitutive activation of the receptor (23). One study demonstrated a change in FGFR2 affinity for FGF2 in AS by using an in vitro binding assay (28). However, the consequence of AS mutations on receptor activity in living cells has not been examined.
Figure 1.

Primary structure and sequence comparisons of FGFRs. (Upper) Structure of the FGFR showing the signal peptide, SP; acidic box, A; Ig-like domains, I, II, III; transmembrane domain, TM; and tyrosine kinase domain, TK. The stippled region in Ig-loop III is subject to tissue-specific alternative splicing. (Lower) Amino acid sequence comparison of the Ig-like domain II–III linker region of FGFRs 1–4. Identical amino acid residues are shaded. The serine–proline dipeptide involved in AS mutations is indicated by arrows. The position of conserved cysteine residues in Ig domains II and III is indicated.
The most significant determinant of ligand-binding specificity is tissue-specific alternative mRNA splicing in the exons encoding the carboxyl-terminal half of Ig-like domain III. For FGFR2, this splicing event creates two receptor isoforms, FGFR2b and FGFR2c, with exquisitely specific ligand-binding properties (29, 30). Expression of the two isoforms is regulated in a tissue-specific manner, with FGFR2b expression restricted to epithelial lineages and FGFR2c expression restricted to mesenchymal lineages (31, 32). Importantly, the mesenchymally expressed ligands, FGF7 and FGF10, can activate only FGFR2b, whereas FGF2, FGF4, FGF6, FGF8, and FGF9 are specific for FGFR2c (29, 33). In this report, we demonstrate that the S252W mutation in FGFR2 allows FGFR2c to be activated by FGF7 and FGF10, and FGFR2b to be activated by FGF2, FGF6, and FGF9, allowing autocrine signaling in tissues that express these ligands.
Materials and Methods
Reagents.
Human recombinant FGFs were from Peprotech (Rocky Hill, NJ). Anti-human placental alkaline phosphatase (hAP) agarose beads were from Sigma. d-luciferin was from Biosynth (Basel).
Receptor Expression Constructs.
AS mutations were created in the mouse Fgfr2c and human Fgfr2b cDNAs by using mismatched (lower-case) PCR primers: 5′-GTTGAACGGTggCCACACCGGC-3′ (S252W) containing a MscI site (underlined) and 5′-GTTGAACGGTCtagACACCGGC-3′ (P253R) containing a XbaI site (underlined). Flanking PCR primers spanning the ApaI site at the amino terminus and the ClaI site in the tyrosine kinase domain were used to amplify a fragment containing the AS mutations. The fragments were excised with ApaI and ClaI and ligated into the corresponding sites in the Fgfr2 cDNA.
To generate myc-tagged wild-type and mutant Fgfr2 cDNAs, the SphI-SpeI fragment containing the 3′ end of Fgfr2 was replaced with a SphI-XbaI fragment from human Fgfr2 containing sequences encoding a hexameric repeat of the myc epitope fused at the 3′ end (24). For transient or stable expression of myc-tagged wild-type and mutant FGFR2, a SpeI-XbaI fragment containing the full-length FGFR2 cDNA and myc epitope was cloned into the MIRB expression vector (24). The soluble receptor-binding protein fused to hAP was constructed as described (34).
Tissue Culture and Transfections.
MC3T3 cells were grown in MEM-α supplemented with 10% FBS/2 mM l-glutamine/100 units/ml of penicillin/100 units/ml of streptomycin. NIH 3T3 cells and COS-7 cells were grown in DMEM supplemented with 10% newborn bovine serum/2 mM l-glutamine/100 units/ml penicillin/100 units/ml streptomycin. A modified calcium phosphate precipitate method was used for transient transfection (35). For transient transfection assays using a luciferase reporter, 8 × 104 cells/well were plated into 12-well plates, and each well was transfected with a total of 1.1 μg plasmid DNA: 0.5 μg pMIRB/FGFR2c, 0.5 μg of osteocalcin FGF response element (pOCFRE)-luc (36), and 0.1 μg pSV40β-gal (CLONTECH catalog #6046-1). To generate MC3T3 cell lines that stably express FGFR2, 106 cells were transfected with 20 μg pMIRB/FGFR2c and selected in media containing 300 μg/ml G418 (GIBCO/BRL) for 10 days. Surviving cells were assayed for receptor protein expression by Western blot by using anti-Myc mAb antibody 9E10 (Santa Cruz Biotechnology) and maintained in media containing 300 μg/ml G418. BaF3 cells were grown, transfected, and selected as reported previously (34). Wild-type and mutant FGFR2c-AP fusion proteins were made by transiently transfecting COS-7 cells with 20 μg plasmid/106 cells by using the modified calcium phosphate precipitant method. The conditioned media were collected and quantified as described (34).
Luciferase Assays.
Twenty-four hours after transfection, cells were washed once with PBS and cultured in fresh media for 24 h. Before treatment with FGF2, transfected cells were serum starved for 12 h in DMEM containing 0.5% FBS. Heparin was added to a final concentration of 1 μg/ml and FGF2 or FGF7, to specified concentrations. After 12 h at 37°C, cells were lysed and subsequently assayed for luciferase activity as described (35). β-Galactosidase activity was determined with the Galacto-Light Plus system (Tropix, Bedford, MA). To normalize for transfection efficiencies, luciferase activity values from each transfection were normalized to the corresponding β-galactosidase activity.
Immunoprecipitations and Western blotting were carried out as described previously (24).
Ligand Binding and Proliferation Assays.
Ligand binding to soluble FGFRs was carried out as described previously (25, 34). The BaF3 cell mitogenic assay was carried out as described previously (34).
Data Analysis.
Competition curves for binding experiments were globally fit to Eq. 1, which describes a single-site binding isotherm scaled by a dilution factor that accounts for the fixed 125I-FGF concentration and increasing amounts of unlabeled FGF (unpublished work).
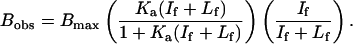 |
1 |
Bobs is the observed binding of 125I-FGF, Bmax is the total number of binding sites available, If is the free 125I-FGF concentration, Lf is the free cold FGF concentration, and Ka is the binding association constant. The values for (If + Lf) were determined by dividing the experimentally determined free radioligand concentration by the specific activity dilution factor. The final term in the equation is a dilution term that accounts for the decreasing signal because of a decrease in the specific activity of the 125I-FGF on addition of unlabeled FGF. Data sets were fitted by using the program scientist (Micromath Scientific Software, Salt Lake City, UT).
Molecular Modeling.
Modeling of the structure of FGFR2c (S252W) and (P253R) was carried out by using the program o and selecting one of the preferred rotamers for the mutated side chains. The Protein Data Bank coordinates used were 1DJS [FGFR2c (26)] and 1QQK (FGF7; Ye, S., Luo, Y., Lu, W., Jones, R. B., Linhardt, R. J., Kan, M., Mckeehan, W. L. & Pelletier, H.). Fig. 5B was made by using the program ribbons (37).
Figure 5.

Molecular model showing potential interactions between the linker region amino acid residues W252 and R253 with FGF7. (A) View showing the position of FGF7 (gray with residue E91 shown in red) in the cleft between Ig-like domains II and III. (B) Expanded stereoscopic view of the boxed region in A. Backbone structure of FGFR2c is shown in gold. Both amino acid residues mutated in AS are shown together in purple. Other linker residues that hydrogen bond with βB-βC and βF-βG loops are shown in green. Amino acid residues in the βB-βC and βF-βG loops that hydrogen bond with the linker are shown in pink. Black lines indicate hydrogen bonds.
Results
Histological studies suggested that the AS phenotypes are caused by abnormal differentiation of mesenchyme-derived tissues that express the “c” splice form of FGFR2 (FGFR2c) (38, 39). To investigate the biochemical mechanism underlying AS, the S252W (AS) mutation was introduced into a full-length epitope-tagged Fgfr2c cDNA (Fig. 1). MC3T3 cells, a cell line derived from newborn murine calvaria, were transiently transfected with wild-type and mutant FGFR2c and treated with increasing concentrations of FGF2. FGFR signaling was quantified with a luciferase reporter gene driven by an FGF responsive enhancer derived from the osteocalcin promoter (OCFRE-luc), and transfection efficiency was normalized with a cotransfected β-galactosidase gene. After normalizing for β-galactosidase enzyme activity, the luciferase activity of cells expressing either wild-type or mutant FGFR2c was ligand responsive, and the activity of the mutant receptor was 1.5- to 4.1-fold higher than wild-type FGFR2c (Fig. 2A). Notably, FGFR2c (S252W) showed increased activity (4.1-fold) without addition of FGF2, indicating that the mutant receptor is either partially ligand independent or that the mutant receptor has increased sensitivity to a ligand expressed by MC3T3 cells.
Figure 2.

Analysis of FGFR signaling in MC3T3 cells. (A and B) Comparison of the osteocalcin FGF response element (OCFRE-luc) activity of cells transiently transfected with FGFR2c6Myc (wild type, open bars) or FGFR2c6Myc (S252W) (solid bars). (A) MC3T3 cells treated with indicated concentrations of FGF2; (B) MC3T3 cells treated with indicated concentrations of FGF7. OCFRE-luc, and pSVβ-gal were cotransfected as described in Materials and Methods. Both luciferase and β-galactosidase activities were quantified. All data were normalized to β-galactosidase activity and then plotted as fold induction over the wild type without FGF treatment. Fold induction was calculated by dividing the mean (±standard deviation) derived for each construct by the mean of the FGFR2c6Myc (wild type) without added FGF. (C and D) Phosphotyrosine analysis of MC3T3 cells stably expressing wild-type FGFR2c6myc or FGFR2c6Myc (S252W). (C) Cells treated with FGF2. (D) Cells treated with FGF7. Receptor proteins were immunoprecipitated with the anti-myc antibody and detected with antiphosphotyrosine antibody (C Upper; D Left). The expression level of receptor proteins was determined by reprobing the same blots with anti-myc antibody 9E10 (C Lower; D Right).
Because MC3T3 cells may express low levels of FGF2 (40), the assay was repeated in NIH 3T3 cells, which are thought to express little or no endogenous FGF2 (41). Surprisingly, the luciferase activity of cells transfected with FGFR2c (S252W) was much higher than that of cells transfected with wild-type FGFR2c and was not further increased by addition of exogenous FGF2 (data not shown). Because both wild-type and mutant receptors were ligand responsive in MC3T3 cells, these data suggested that the mutant FGFR2c could be selectively activated by another endogenous ligand expressed by NIH 3T3 cells. FGF7, known to be highly expressed in NIH 3T3 cells (41), became a candidate ligand to activate FGFR2c (S252W). To test this hypothesis, MC3T3 cells were again transfected with wild-type and mutant receptors and treated with increasing concentrations of FGF7. Wild-type FGFR2c showed no response to FGF7, whereas FGFR2c (S252W) showed a dose-dependent response to FGF7 (Fig. 2B). This observation is noteworthy because FGF7 is the most specific of the known FGF ligands and is only known to activate the epithelial splice form of FGFR2 (FGFR2b) (29, 30, 41).
The effect of the S252W mutation on receptor activation was also assayed by examining receptor autophosphorylation in response to FGF ligands. MC3T3 cells, stably expressing either wild-type FGFR2c or FGFR2c (S252W), were serum starved and stimulated with FGF2 or FGF7. Myc-tagged FGFR2c was immunoprecipitated and assayed for phosphotyrosine content and receptor protein levels. Western blot analysis showed that without addition of exogenous ligand or at low FGF2 concentrations, tyrosine phosphorylation of wild-type FGFR2c was barely detectable, whereas the signal from FGFR2c (S252W) was more intense (Fig. 2C). At high concentrations of FGF2, phosphorylation of both wild-type and mutant FGFR2c was similar. In contrast to wild-type FGFR2c, which was not phosphorylated by addition of FGF7, FGFR2c (S252W) showed robust tyrosine phosphorylation in response to FGF7 (Fig. 2D).
AS mutations, located in the common linker region of FGFR2, will affect both b and c splice forms of the receptor. To examine the effect of the S252W mutation on either receptor splice form, we assayed ligand-dependent and ligand-independent FGFR activity in BaF3 cells. BaF3 cells are a lymphoid cell line, which are dependent on cytokine IL3 for growth and have no intrinsic response to FGF. However, when stably transfected to express an FGFR, BaF3 cells show a dose-dependent mitogenic response to FGF in the absence of IL3 (42). Nevertheless, because BaF3 cells express no endogenous FGF, the background activity in the absence of added FGF is very low regardless of whether the cells express an FGFR (29). Mitogenic assays in BaF3 cells are therefore a sensitive assay to detect ligand-independent receptor activity and can be used to examine the specificity of the mutant receptor toward different FGFs.
BaF3 cells, stably transfected with myc-tagged FGFR2b, FGFR2c, FGFR2b (S252W), or FGFR2c (S252W), were assayed with several members of the FGF family, (FGF1, FGF2, FGF6, FGF7, FGF9, and FGF10) for their ability to induce a mitogenic response (Fig. 3). Without added FGF, there was no difference in the mitogenic activity of BaF3 cells expressing wild-type or mutant receptors, demonstrating that the S252W mutation did not cause any ligand-independent receptor activation.
Figure 3.

Mitogenic activity of BaF3 cells expressing wild-type FGFR2 (circles) and S252W mutant FGFR2 (squares) after treatment with different FGFs. (A, C, E, and G) Comparison of wild-type and mutant FGFR2c. (B, D, F, and H) Comparison of wild-type and mutant FGFR2b. Each stably selected polyclonal cell pool was treated with FGF1, FGF2, FGF6 (not shown), FGF7, FGF9, and FGF10 (not shown), as indicated.
Comparison of wild-type and mutant FGFR2c showed a similar mitogenic response to FGF1 (Fig. 3A) or FGF6 (not shown) at all ligand concentrations examined, consistent with a previous observation that the binding affinity of FGFR2c to FGF1 or FGF6 was not affected by the S252W mutation (28). With FGF2 and FGF9, the mitogenic activity of cells expressing mutant FGFR2c was higher than that of cells expressing wild-type FGFR2c in the linear range of the assay (Fig. 3 C and G). Remarkably, FGF7 (Fig. 3E) and FGF10 (which is similar to FGF7 and a high-affinity ligand for FGFR2b; not shown) elicited a robust dose-dependent response on cells expressing FGFR2c (S252W) but showed no activity toward cells expressing wild-type FGFR2c. When cells expressing wild-type and mutant FGFR2b were compared, FGF1 (Fig. 3B), FGF7 (Fig. 3F), and FGF10 (not shown) showed similar activity. Interestingly, FGF2 (Fig. 3D), FGF6 (not shown), and FGF9 (Fig. 3H) elicited a dose-dependent response with FGFR2b (S252W) expressing cells but were inactive on cells expressing wild-type FGFR2b. Thus both mutant splice forms of FGFR2 appear to have lost ligand specificity but retain ligand dependence.
About 35% of patients presenting with AS have the P253R missense mutation. Because the (S252W) mutation rendered FGFR2 sensitive to a completely new subset of FGFs, it was important to determine whether the P253R mutation had a similar effect. To quantitatively compare these two mutations, we analyzed the ability of FGF2 and FGF7 to bind soluble forms of either wild-type FGFR2c, wild-type FGFR2b, FGFR2c (S252W), or FGFR2c (P253R) extracellular domains, fused to hAP. Binding affinities were determined by competition of iodinated ligands with increasing amounts of unlabeled ligand to generate a competition curve (Fig. 4). Competition curves from two to three experiments were then globally fitted to a single-site ligand-binding equation to accurately estimate the binding constant (Kd).
Figure 4.

Binding properties of wild-type and mutant FGFR2 extracellular domains fused to alkaline phosphatase. Competition binding curves are shown for bound 125I-FGF vs. total FGF. (A–C) FGF7 binding to FGFR2c (S252W), FGFR2c (P253R), and FGFR2b, respectively. (D–F) FGF2 binding to FGFR2c (S252W), FGFR2c (P253R), and FGFR2c, respectively.
FGFR2b bound FGF7 with high affinity (Kd = 310 pM), whereas, as expected, wild-type FGFR2c did not bind FGF7. Compared with FGFR2b, the affinity of FGFR2c (S252W) and FGFR2c (P253R) was 24- and 1.5-fold lower, respectively (Table 1). These data demonstrate that both the S252W and P253R mutations confer high-affinity binding of FGF7 to FGFR2c. Unlike with FGF7, FGFR2c, FGFR2c (S252W), and FGFR2c (P253R), all bound FGF2 with similar affinity (within 2-fold; Table 1). This is in contrast to the data of Anderson et al. (28), where they observed a 2- to 6-fold increase in affinity of FGF2 for mutant FGFR2c.
Table 1.
Calculated binding constants from competition binding data
| Ligand | Receptor | Mutation | Kd, nM | n* |
|---|---|---|---|---|
| FGF7 | FGFR2b | Wild type | 0.31 ± 0.03 | 3 |
| FGF7 | FGFR2c | Wild type | NB† | |
| FGF7 | FGFR2c | S252W | 7.31 ± 0.60 | 3 |
| FGF7 | FGFR2c | P253R | 0.47 ± 0.06 | 2 |
| FGF2 | FGFR2c | Wild type | 1.12 ± 0.09 | 2 |
| FGF2 | FGFR2c | S252W | 0.74 ± 0.05 | 2 |
| FGF2 | FGFR2c | P253R | 1.20 ± 0.20 | 2 |
Number of curves that were globally fitted.
NB, no binding was observed.
Discussion
FGF7 and FGF10 are normally expressed in mesenchymal tissue and bind only the epithelial splice form of FGFR2 (FGFR2b) (29, 33). Mutations allowing FGFR2c to be activated by these ligands could permit autocrine signaling within mesenchymal tissue where Fgfr2c expression and signaling is normally very restricted. Similarly, mutations allowing FGFR2b to be activated by FGF2, FGF6, or FGF9 could lead to aberrant signaling in epithelial tissues. The establishment of ligand-dependent autocrine signaling pathways could explain the severity and unique features of AS compared with other craniosynostosis syndromes.
Most cases of AS are caused by a missense mutation at either serine 252 or proline 253, in the highly conserved region linking the second and third Ig-like domains of FGFR2. Recently, very rare cases of AS have been identified in which de novo Alu-insertions have occurred upstream or within the alternatively spliced exon encoding “c” specific sequence (exon 9). Molecular studies showed that these Alu-insertions affect the alternative splicing of Fgfr2, resulting in ectopic expression of FGFR2b in mesenchyme-derived cell lineages that normally express FGFR2c (43). Ectopic expression of FGFR2b could render mesenchymal cells sensitive to locally expressed ligand (FGF7 or FGF10). The findings reported here, that the S252W or P253R mutations in FGFR2 allow FGFR2c to be activated by FGF7 or FGF10, strongly support the hypothesis that AS results from aberrant activation of mesenchymally expressed FGFR2 by mesenchymally expressed FGF(s) and explain why the rare Alu insertion mutations (43) and the AS missense mutations result in similar phenotypes.
Both biochemical and structural studies (25–27) have identified the contribution of Ig-like domains II and III and the interdomain linker region to an FGF-binding site. Although the effect of the AS mutations on receptor structure is not known, it is likely that the AS mutations alter the conformation of the linker region and change the relative orientation of Ig-like domains II and III. This could uncouple the mechanisms that link receptor structure to ligand-binding specificity. Additionally, these mutations could affect ligand binding through direct side chain interactions. Molecular modeling (Fig. 5) based on the crystal structures of FGFR2c (26) and FGF7 [S. Ye, Y. Luo, W. Lu, R. B. Jones, R. J. Linhardt, M. Kan, W. L. Mckeehan, and H. Pelletier (1999) Protein Data Base ID code 1QQK] indicates that AS mutations could disrupt the hydrogen bonding network between the linker and two of the loops (βB-βC, βE-βF) in Ig-like domain III that participate in ligand binding (26). Furthermore, the arginine side chain in the P253R mutation would be positioned to interact with E91 (E145 in full-length FGF7) in the FGF7 structure [S. Ye, Y. Luo, W. Lu, R. B. Jones, R. J. Linhardt, M. Kan, W. L. Mckeehan, and H. Pelletier (1999) Protein Data Base ID code 1QQK] (Fig. 5) or E158 in FGF10. This potential interaction may explain the higher affinity of the P253R mutant receptor for FGF7 and subtle differences in phenotype associated with these two mutations (44). However, the significance of the phenotypic consequences of these two mutations remains controversial (44, 45).
Mesenchymal Pathology in AS.
FGF signaling plays an important role during cranial development. In situ hybridization studies localize Fgfr2 expression to proliferating stem cells in developing cranial sutures. Fgfr2 expression is down-regulated when differentiation begins (46, 47). When FGF-soaked beads were placed over a developing suture in vitro, cell differentiation increased, and suture closure was accelerated, suggesting that FGFR activity is critical for maintaining the balance between cell proliferation and differentiation and that increased FGFR2 activity could result in premature fusion of the sutures (47). The phenotype of CS nicely fits a model in which mutations that cause ligand-independent receptor dimerization and constitutive receptor signaling lead to premature fusion of the sutures.
Although AS has been classified as a craniosynostosis syndrome, clinical studies indicated that AS is very different from other craniosynostosis syndromes. Comparison of the skulls of AS and CS in infancy revealed that in AS, the coronal sutures were always prematurely fused and the midline sutures (metopic and sagittal) were absent and replaced by a widely patent midline calvarial defect that eventually filled in by coalescence of bony islands (48). The lambdoid and squamosal sutures formed relatively normally. In contrast, in CS there was no midline defect, and all sutures were prematurely fused. Clinical studies demonstrate that cartilaginous abnormalities in the cranial base are a primary cause of abnormal cranial development in AS (49). These abnormalities affect brain growth and subsequent calvarial bone growth. In AS, the frontal and parietal bone centers become displaced, resulting in their direct fusion at the position of the coronal suture (50). In CS, calvarial bone centers are not displaced, and the primary defect appears to be intrinsic to the sutures, resulting in early fusion of all sutures.
Another unique phenotype of AS is severe syndactyly of the hands and feet, which is not observed in CS and other craniosynostosis syndromes. AS limbs show skeletal defects, including osseous fusion of the digits and phalangeal joints and the presence of ectopic cartilage in periarticular tissues and flexor tendons (51). This suggests that the limb phenotypes in AS originate from abnormalities in the condensation stage of development caused by ectopic FGFR signaling.
Cartilage is formed through differentiation of condensed mesenchyme regulated by epithelial–mesenchymal interactions during early stages of embryonic development. In situ hybridization studies show that Fgfr2c is highly expressed in precartilage cell condensations and that Fgf7 is highly expressed in the surrounding mesenchyme (52). Changing the ligand-binding specificity of FGFR2c to render it capable of binding FGF7 could allow the FGFR2-expressing cells to respond to proximally produced ligand. Unlike in CS, where FGFR2 is uniformly activated in a ligand-independent manner, in AS, FGFR2 should be ectopically activated in a gradient extending from the source of FGF7 in surrounding mesenchyme toward the center of the mesenchymal condensation. These distinct modes of FGFR2 activation, as well as possible differences in the level of FGFR2 activation, could account for some of the phenotypic differences between AS and CS.
Altered ligand-binding specificity of FGFR2b may also contribute to the severe phenotypes of AS. Fgfr2b is expressed in the apical ectodermal ridge (AER) during limb development and in the osteogenic front of the cranial sutures (53, 54). Ectopic activation of FGFR2b signaling by FGF2 expression in sutural mesenchyme (46) or FGF4, 8, 9, or 17 expression in the AER (55) may contribute to craniosynostosis and syndactyly, respectively.
Acknowledgments
This work was funded by National Institutes of Health Grant HD35692 (D.M.O.), American Chemical Society Grant RPG-97–165-01-GMC (G.W.), Washington University Digestive Diseases Research Core Center Grant P30-DK52574, and a generous gift from the Alice and Julius Kantor Charitable Trust.
Abbreviations
- FGFR
fibroblast growth factor receptor
- AS
Apert syndrome
- CS
Crouzon syndrome
- hAP
human placental alkaline phosphatase
References
- 1.Naski M C, Ornitz D M. Front Biosci. 1998;3:D781–D794. doi: 10.2741/a321. [DOI] [PubMed] [Google Scholar]
- 2.Ornitz D M. In: Skeletal Growth Factors. Canalis E, editor. Philadelphia: Lippincott; 2000. pp. 197–209. [Google Scholar]
- 3.Muenke M, Schell U. Trends Genet. 1995;11:308–313. doi: 10.1016/s0168-9525(00)89088-5. [DOI] [PubMed] [Google Scholar]
- 4.Webster M K, Donoghue D J. Trends Genet. 1997;13:178–182. doi: 10.1016/s0168-9525(97)01131-1. [DOI] [PubMed] [Google Scholar]
- 5.Wilkie A O M, Slaney S F, Oldridge M, Poole M D, Ashworth G J, Hockley A D, Hayward R D, David D J, Pulleyn L J, Rutland P, et al. Nat Genet. 1995;9:165–172. doi: 10.1038/ng0295-165. [DOI] [PubMed] [Google Scholar]
- 6.Reardon W, Winter R M, Rutland P, Pulleyn L J, Jones B M, Malcolm S. Nat Genet. 1994;8:98–103. doi: 10.1038/ng0994-98. [DOI] [PubMed] [Google Scholar]
- 7.Rutland P, Pulleyn L J, Reardon W, Baraitser M, Hayward R, Jones B, Malcolm S, Winter R M, Oldridge M, Slaney S F, et al. Nat Genet. 1995;9:173–176. doi: 10.1038/ng0295-173. [DOI] [PubMed] [Google Scholar]
- 8.Meyers G A, Orlow S J, Munro I R, Przylepa K A, Jabs E W. Nat Genet. 1995;11:462–464. doi: 10.1038/ng1295-462. [DOI] [PubMed] [Google Scholar]
- 9.Muenke M, Gripp K W, McDonald-McGinn D M, Gaudenz K, Whitaker L A, Bartlett S P, Markowitz R I, Robin N H, Nwokoro N, Mulvihill J J, et al. Am J Hum Genet. 1997;60:555–564. [PMC free article] [PubMed] [Google Scholar]
- 10.Lajeunie E, Ma H W, Bonaventure J, Munnich A, LeMerrer M. Nat Genet. 1995;9:108. doi: 10.1038/ng0295-108. [DOI] [PubMed] [Google Scholar]
- 11.Jabs E W, Li X, Scott A F, Meyers G, Chen W, Eccles M, Mao J, Charnas L R, Jackson C E, Jaye M. Nat Genet. 1994;8:275–279. doi: 10.1038/ng1194-275. [DOI] [PubMed] [Google Scholar]
- 12.Chun K, Siegel-Bartelt J, Chitayat D, Phillips J, Ray P N. Am J Med Genet. 1998;77:219–224. doi: 10.1002/(sici)1096-8628(19980518)77:3<219::aid-ajmg6>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- 13.Przylepa K A, Paznekas W, Zhang M, Golabi M, Bias W, Bamshad M J, Carey J C, Hall B D, Stevenson R, Orlow S, et al. Nat Genet. 1996;13:492–494. doi: 10.1038/ng0896-492. [DOI] [PubMed] [Google Scholar]
- 14.McKeehan W L, Wang F, Kan M. Prog Nucleic Acid Res Mol Biol. 1998;59:135–176. doi: 10.1016/s0079-6603(08)61031-4. [DOI] [PubMed] [Google Scholar]
- 15.Anderson P J, Hall C M, Evans R D, Hayward R D, Jones B M. J Pediatr Orthop. 1999;19:504–507. doi: 10.1097/00004694-199907000-00015. [DOI] [PubMed] [Google Scholar]
- 16.McKusick V A. Mendelian Inheritance in Man. 8th Ed. Baltimore: Johns Hopkins Univ. Press; 1988. [Google Scholar]
- 17.Cohen M M, Jr, Kreiborg S. Am J Dis Child. 1993;147:989–993. doi: 10.1001/archpedi.1993.02160330079025. [DOI] [PubMed] [Google Scholar]
- 18.Cohen M M, Jr, Kreiborg S. Am J Med Genet. 1990;35:36–45. doi: 10.1002/ajmg.1320350108. [DOI] [PubMed] [Google Scholar]
- 19.Maksem J A, Roessmann U. Acta Neuropathol. 1979;48:59–61. doi: 10.1007/BF00691792. [DOI] [PubMed] [Google Scholar]
- 20.Sarimski K. Genet Couns. 1997;8:317–322. [PubMed] [Google Scholar]
- 21.Wilkie A O M, Morriss-Kay G M, Jones E Y, Heath J K. Curr Biol. 1995;5:500–507. doi: 10.1016/s0960-9822(95)00102-3. [DOI] [PubMed] [Google Scholar]
- 22.Galvin B D, Hart K C, Meyer A N, Webster M K, Donoghue D J. Proc Natl Acad Sci USA. 1996;93:7894–7899. doi: 10.1073/pnas.93.15.7894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Robertson S C, Meyer A N, Hart K C, Galvin B D, Webster M K, Donoghue D J. Proc Natl Acad Sci USA. 1998;95:4567–4572. doi: 10.1073/pnas.95.8.4567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Naski M C, Wang Q, Xu J, Ornitz D M. Nat Genet. 1996;13:233–237. doi: 10.1038/ng0696-233. [DOI] [PubMed] [Google Scholar]
- 25.Chellaiah A, Yuan W L, Chellaiah M, Ornitz D M. J Biol Chem. 1999;274:34785–34794. doi: 10.1074/jbc.274.49.34785. [DOI] [PubMed] [Google Scholar]
- 26.Stauber D J, DiGabriele A D, Hendrickson W A. Proc Natl Acad Sci USA. 2000;97:49–54. doi: 10.1073/pnas.97.1.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Plotnikov A N, Schlessinger J, Hubbard S R, Mohammadi M. Cell. 1999;98:641–650. doi: 10.1016/s0092-8674(00)80051-3. [DOI] [PubMed] [Google Scholar]
- 28.Anderson J, Burns H D, Enriquez-Harris P, Wilkie A O M, Heath J K. Hum Mol Genet. 1998;7:1475–1483. doi: 10.1093/hmg/7.9.1475. [DOI] [PubMed] [Google Scholar]
- 29.Ornitz D M, Xu J, Colvin J S, McEwen D G, MacArthur C A, Coulier F, Gao G, Goldfarb M. J Biol Chem. 1996;271:15292–15297. doi: 10.1074/jbc.271.25.15292. [DOI] [PubMed] [Google Scholar]
- 30.Miki T, Bottaro D P, Fleming T P, Smith C L, Burgess W H, Chan A M-L, Aaronson S A. Proc Natl Acad Sci USA. 1992;89:246–250. doi: 10.1073/pnas.89.1.246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shi D L, Launay C, Fromentoux V, Feige J J, Boucaut J C. Dev Biol. 1994;164:173–182. doi: 10.1006/dbio.1994.1189. [DOI] [PubMed] [Google Scholar]
- 32.Orr-Urtreger A, Givol D, Yayon A, Yarden Y, Lonai P. Development (Cambridge, UK) 1991;113:1419–1434. doi: 10.1242/dev.113.4.1419. [DOI] [PubMed] [Google Scholar]
- 33.Igarashi M, Finch P W, Aaronson S A. J Biol Chem. 1998;273:13230–13235. doi: 10.1074/jbc.273.21.13230. [DOI] [PubMed] [Google Scholar]
- 34.Ornitz D M, Yayon A, Flanagan J G, Svahn C M, Levi E, Leder P. Mol Cell Biol. 1992;12:240–247. doi: 10.1128/mcb.12.1.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McEwen D G, Ornitz D M. J Biol Chem. 1998;273:5349–5357. doi: 10.1074/jbc.273.9.5349. [DOI] [PubMed] [Google Scholar]
- 36.Newberry E P, Boudreaux J M, Towler D A. Mol Endocrinol. 1996;10:1029–1040. doi: 10.1210/mend.10.8.8843419. [DOI] [PubMed] [Google Scholar]
- 37.Carson M. Methods Enzymol. 1997;277:493–505. [PubMed] [Google Scholar]
- 38.Lomri A, Lemonnier J, Hott M, de Parseval N, Lajeunie E, Munnich A, Renier D, Marie P J. J Clin Invest. 1998;101:1310–1317. [PMC free article] [PubMed] [Google Scholar]
- 39.Lomri A, Lemonnier J, de Pollack C, Hott M, de Parseval N, Lajeunie E, Munnich A, Renier D, Marie P J. J Bone Miner Res. 1997;12:S125. [Google Scholar]
- 40.Hurley M M, Abreu C, Gronowicz G, Kawaguchi H, Lorenzo J. J Biol Chem. 1994;269:9392–9396. [PubMed] [Google Scholar]
- 41.Miki T, Fleming T P, Bottaro D P, Rubin J S, Ron D, Aaronson S A. Science. 1991;251:72–75. doi: 10.1126/science.1846048. [DOI] [PubMed] [Google Scholar]
- 42.Ornitz D M, Leder P. J Biol Chem. 1992;267:16305–16311. [PubMed] [Google Scholar]
- 43.Oldridge M, Zackai E H, McDonald-McGinn D M, Iseki S, Morriss-Kay G M, Twigg S R, Johnson D, Wall S A, Jiang W, Theda C, et al. Am J Hum Genet. 1999;64:446–461. doi: 10.1086/302245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Slaney S F, Oldridge M, Hurst J A, Moriss-Kay G M, Hall C M, Poole M D, Wilkie A O. Am J Hum Genet. 1996;58:923–932. [PMC free article] [PubMed] [Google Scholar]
- 45.Park W J, Theda C, Maestri N E, Meyers G A, Fryburg J S, Dufresne C, Cohen M M, Jr, Jabs E W. Am J Hum Genet. 1995;57:321–328. [PMC free article] [PubMed] [Google Scholar]
- 46.Iseki S, Wilkie A O M, Heath J K, Ishimaru T, Eto K, Morrisskay G M. Development (Cambridge, UK) 1997;124:3375–3384. doi: 10.1242/dev.124.17.3375. [DOI] [PubMed] [Google Scholar]
- 47.Iseki S, Wilkie A O, Morriss-Kay G M. Development (Cambridge, UK) 1999;126:5611–5620. doi: 10.1242/dev.126.24.5611. [DOI] [PubMed] [Google Scholar]
- 48.Kreiborg S, Cohen M M., Jr Acta Odontol Scand. 1998;56:339–341. doi: 10.1080/000163598428275. [DOI] [PubMed] [Google Scholar]
- 49.Kreiborg S, Marsh J L, Cohen M M, Jr, Liversage M, Pedersen H, Skovby F, Borgesen S E, Vannier M W. J Craniomaxillofac Surg. 1993;21:181–188. doi: 10.1016/s1010-5182(05)80478-0. [DOI] [PubMed] [Google Scholar]
- 50.Mathijssen I M, Vaandrager J M, van der Meulen J C, Pieterman H, Zonneveld F W, Kreiborg S, Vermeij-Keers C. Plast Reconstr Surg. 1996;98:17–26. doi: 10.1097/00006534-199607000-00004. [DOI] [PubMed] [Google Scholar]
- 51.Holten I W, Smith A W, Bourne A J, David D J. Plast Reconstr Surg. 1997;99:1681–1687. doi: 10.1097/00006534-199705000-00031. [DOI] [PubMed] [Google Scholar]
- 52.Finch P W, Cunha G R, Rubin J S, Wong J, Ron D. Dev Dyn. 1995;203:223–240. doi: 10.1002/aja.1002030210. [DOI] [PubMed] [Google Scholar]
- 53.Rice D P C, Aberg T, Chan Y S, Tang Z Q, Kettunen P J, Pakarinen L, Maxson R E, Thesleff I. Development (Cambridge, UK) 2000;127:1845–1855. doi: 10.1242/dev.127.9.1845. [DOI] [PubMed] [Google Scholar]
- 54.Martin G R. Genes Dev. 1998;12:1571–1586. doi: 10.1101/gad.12.11.1571. [DOI] [PubMed] [Google Scholar]
- 55.Sun X, Lewandoski M, Meyers E N, Liu Y H, Maxson R E, Jr, Martin G R. Nat Genet. 2000;25:83–86. doi: 10.1038/75644. [DOI] [PubMed] [Google Scholar]