

Abstract
Class IA phosphatidylinositol-3 kinase (PI-3K) is a lipid kinase, which is activated in blood cells by hematopoietic growth factors. In vitro experiments using chemical inhibitors of PI-3K suggest that this kinase is potentially important for hematopoietic stem and progenitor cell (HSC/P) function, and recent studies identify PI-3K as a therapeutic target in treating different leukemias and lymphomas. However, the role of PI-3K in regulating fetal liver or adult hematopoiesis in vivo is unknown. Therefore, we examined PI-3K-deficient embryos generated by a targeted deletion of the p85α and p85β regulatory subunits of PI-3K (p85α-/-p85β+/-). The absolute frequency and number of hematopoietic progenitor cells were reduced in p85α-/- p85β+/- fetal livers compared with wild-type (WT) controls. Further, p85α-/-p85β+/- fetal liver hematopoietic stem cells (HSCs) had decreased multilineage repopulating ability in vivo compared with WT controls in competitive repopulation assays. Finally, purified p85α-/-p85β+/- c-kit+ cells had a decrease in proliferation in response to kit ligand (kitL), a growth factor important for controlling HSC function in vivo. Collectively, these data identify PI-3K as an important regulator of HSC function and potential therapeutic target in treating leukemic stem cells.
Introduction
Identification of signaling proteins, which control hematopoietic stem and progenitor cell (HSC/P) function, is critical for understanding steady-state hematopoiesis and hematologic diseases. Class IA phosphatidylinositol-3 kinase (PI-3K) is a lipid kinase, which is activated in blood cells by a variety of hematopoietic cell growth factors, which control HSC/P proliferation and survival.1-12 Studies using chemical inhibitors of class IA PI-3K activation in immortalized hematopoietic cell lines provide preliminary evidence that this kinase is important for HSC/P function.13-21 Further, recent genetic studies show that increased class IA PI-3K activation is essential for disease progression in murine models of myeloid leukemia and other hematologic malignancies.15,17,22-28 While these studies suggest that class IA PI-3K may function as a critical regulator of HSC/P function, no studies have formally determined the role of class IA PI-3K in controlling HSC/P function in vivo using established assays for testing HSC reconstituting ability. Thus, a rigorous, genetic evaluation of class IA PI-3K function in HSC/Ps in vivo is an important priority given its potential role as both a regulator of normal HSC function and a potential therapeutic target in treating specific hematologic diseases.
Class IA PI-3Ks (PI-3K) are heterodimers, which are composed of a p110-kDa catalytic subunit and a p85-kDa adaptor/regulatory subunit.29-32 The p110 subunit in PI-3K exists in complex with an adaptor unit that has 2 Src-homology-2 (SH2) domains.32 The adaptor subunits bind to phosphorylated tyrosine residues that are generated by activated receptor tyrosine kinases and various adaptor proteins, many of which have been shown to control the proliferation and survival of primitive hematopoietic progenitor cells.13-21 Tyrosine phosphorylation of these proteins allows translocation of the subunits to cytosolic membranes where lipid substrates reside.32 Mammals have 3 p110 isoforms (p110α, p110β, and p110δ), which are encoded by 3 separate genes, and at least 7 adaptor proteins, which are generated by expression and alternative splicing of 3 different genes: p85α, p85β, and P85.29-32
Many studies have used the PI-3K inhibitors, LY294002 and wortmannin, to investigate the role of class IA PI-3K in regulating hematopoietic cell proliferation, survival, and migration to specific growth factors.13-21,26,28,33 LY294002 and wortmannin are structurally unrelated, cell permeable, low-molecular-weight compounds that are, at low doses, rather specific PI-3K inhibitors.29-32 LY294002 inhibits by binding to the ATP-binding site of class IA PI-3K, and wortmannin binds irreversibly to the catalytic subunit of class IA PI-3K. However, a major limitation of these compounds is that they both show little selectivity within the PI-3K family, which contains multiple classes (class IA, IB, II, and III).29-32 Furthermore, most classes are inhibited at comparable concentrations with those required to inhibit class IA PI-3K.29-32 Protein kinases related to PI-3K for which information is available are also inhibited by LY294002 and wortmannin, and both compounds lose specificity at high concentrations.29-32 Further, administering either LY294002 or wortmannin in vivo to test the role of class IA PI-3K in controlling hematopoietic cell function is problematic because of the short half-lives, toxic effects, and the lack of specificity of these inhibitors in targeting different classes of PI-3K.29-32 Thus, while both wortmannin and LY294002 are major tools used to elucidate the biologic activation of PI-3K at the cellular level in short-term assays, it is possible that some of the effects are mediated through inhibition of related kinases.
To circumvent the experimental problems using PI-3K inhibitors and to investigate the function of class IA PI-3K in vivo, p85α knock-out mice were generated by Fruman et al.34 p85α-/- mice die within days after birth secondary to chylous ascites, hepatic necrosis, and respiratory failure.35 Despite the lethality of the genetic deletion, Fruman et al34 used the Rag2 blastocyst complementation system and found that B-cell development and function were impaired in the absence of p85α. In addition, other studies demonstrated that p85α-/- primary cells generated from embryonic day-13.5 fetal liver had decreased proliferation and migration in response to kit ligand (kitL) compared with wild-type mast cells.36 Finally, we recently showed that fetal liver-derived p85α-/- c-kit+ progenitor cells have decreased proliferation in response to kitL and erythropoietin and that p85α-/- macrophages have decreased migration and proliferation in response to macrophage-colony-stimulating factor (M-CSF).33,37 Collectively, these studies demonstrate both the importance of class IA PI-3K in regulating the function of multiple hematopoietic cell lineages, including lymphoid and myeloid cells, and the efficacy of using a genetic approach to investigate the function of class IA PI-3K in vivo.
In previous studies, class IA PI-3K activity was reduced by approximately 60% in different lineages of p85α-deficient cells in response to specific growth factors.34,36,37 The retention of residual class IA PI-3K was secondary to the compensatory up-regulation of the p85β subunit in p85α-deficient cells and the increased lipid kinase activity associated with the remaining p85β subunit.34,36 p85β-/- knock-out mice are viable and fertile with no obvious hematopoietic cell defects.38 Thus, we generated class IA PI-3K-deficient embryos with targeted deletions in both the p85α and p85β regulatory subunits (p85α-/-p85β+/-) to determine the role of class IA PI-3K in controlling steady-state hematopoiesis and HSC/P cell function. In these studies, our data show that genetic reduction of class IA PI-3K alters fetal hematopoiesis and diminishes multilineage HSC repopulating ability in vivo.
Materials and methods
Animals
p85α+/- and p85β-/- mice were obtained in a mixed C57BL/6J.129 background from Dr Lewis Cantley at Harvard University (Boston, MA) and were backcrossed for 12 generations into a C57BL/6J genetic strain. The p85α and p85β alleles were genotyped by polymerase chain reaction (PCR) as previously described.34,38 Congenic C57BL/6J (CD45.2+) and B6.SJL-PtrcaPep3b/BoyJ (B6.BoyJ, CD45.1+) mice were purchased from Jackson Laboratories (Bar Harbor, ME) for transplantation experiments. All studies were conducted with a protocol approved by the Indiana University Animal Care and Use Committee.
Fetal liver cell isolation
Multiple syngeneic FO founders were used to generate embryonic day-13.5 fetal livers from the 4 F2 p85α and p85β experimental genotypes as follows: FO = p85α+/-p85β+/+ × p85α+/+p85β-/-; F1 = p85α+/-p85β+/- × p85α+/-p85β+/-; and F2 = p85α+/+ p85β+/+ (WT), p85α-/-p85β+/+ (p85α-/-), p85α+/+p85β+/- (p85β+/-), p85α-/-p85β+/-.
p85α+/-p85β+/- pregnant females were killed by cervical dislocation on day 13.5 of gestation, and the embryos were removed through an anterior abdominal incision. After individual embryos were isolated, fetal livers were harvested and transferred to a 10 × 35-mm dish in Iscoves Modified Dulbecco Medium (IMDM; GIBCO, Gaithersburg, MD) supplemented with 20% fetal calf serum (FCS; Hyclone Laboratories, Logan, UT). Single-cell suspensions were prepared by pushing the hepatic tissues through progressively smaller needles (16-27 gauge). The remaining embryonic tissues were used for genotype analysis as previously described.39
C-kit+ cell isolation
Fetal liver cells isolated from WT, p85α-/-, p85β+/-, and p85α-/-p85β+/- embryos were incubated on ice with 5 μg fluorescein isothiocyanate (FITC)-conjugated c-kit monoclonal antibody (Pharmingen, San Diego, CA) per 106 cells for 30 minutes. Cells were washed and then purified by immunomagnetic bead enrichment as previously described.37 The purity of c-kit+ cells approximated 80% as determined by fluorescence cytometry (data not shown).
Hematopoietic progenitor colony assays
Clonogenic methylcellulose assays were plated in triplicate 35-mm plates (Becton Dickinson, Franklin Lakes, NJ) with either 40 000 low-density fetal liver cells/mL or 10 000 c-kit+ fetal liver cells/mL. Low-density fetal liver cells were obtained by Ficoll-density centrifugation as previously described.40 Cultures were established in 1% IMDM methylcellulose (Stem Cell Technologies, Vancouver, BC) with 30% FCS, 50 ng/mL recombinant murine kit ligand (kitL; Peprotech, Rocky Hill, NJ), 4 U/mL recombinant human erythropoietin (Amgen, Thousand Oaks, CA), 10 ng/mL recombinant murine granulocyte macrophage-colony-stimulating factor (GM-CSF; Peprotech), and 10 ng/mL recombinant murine thrombopoietin (TPO; Peprotech). Cultures were incubated in 5% CO2 at 37°C, and granulocyte and macrophage-colony-forming units (CFU-GMs), erythroid-burst-forming units (BFU-Es), granulocyte, erythroid, monocyte, and macrophage-colony-forming units (CFU-MIXs), and megakaryocyte-colony-forming units (CFU-Megs) were scored on day 7 of culture exactly as previously described.37,40 Erythroid-colony-forming units (CFU-Es) were scored on day 2 of culture as previously described.37 An unpaired Student t test was used to determine significant differences between experimental genotypes.
HPP-CFC and LPP-CFC assay
Purified c-kit+ fetal liver cells from the 4 experimental genotypes were cultured in triplicate 35-mm plates at a final concentration of 5 × 102 cells per milliliter. Cells were added to agar for growth of low-proliferating potential-colony-forming cells (LPP-CFCs) and high-proliferating potential-colony-forming cells (HPP-CFCs) in the presence of 10 ng/mL GM-CSF, 10 units/mL interleukin-3 (IL-3; Peprotech), 10 ng/mL kitL, and 50 ng/mL recombinant murine macrophage-colony-stimulating factor (M-CSF; R and D Research Laboratories, Minneapolis, MN) as previously described.40 Cultures were incubated at 8% CO2, 5% O2 and scored by indirect microscopy on day 7 for LPP-CFCs and day 14 for HPP-CFCs. A Zeiss Axiovert 25 microscope equipped with a Zeiss CP Apochromat 5 ×/0.40 objective lens (Zeiss, Oberkochen, Germany) was used to score colonies. For photographs, an RT color SPOT camera and SPOT Basic 4.1 imaging software (Diagnostic Instruments, Sterling Heights, MI) were used. An unpaired Student t test was used to determine significant differences between genotypes.
Thymidine incorporation assays
Fetal liver c-kit+ cells isolated from WT, p85α-/-, p85β+/-, and p85α-/- p85β+/- embryos were cultured in IMDM supplemented with 10% FCS and either 10 ng/mL GM-CSF, 10 ng/mL granulocyte-colony-stimulating factor (G-CSF; Peprotech), 10 units/mL IL-3, and 10 ng/mL kitL or no growth factors in a 37°C, 5% CO2 humidified incubator. Cells were cultured for 16 hours, and 1 μCi (0.037 MBq) tritiated thymidine (Perkin Elmer Life Sciences Products, Boston, MA) was added 5 hours prior to the harvest. Cells were lysed in 0.1 M sodium hydroxide for one hour. Lysates were collected into 5 mL liquid scintilant (Fisher Scientific, St Louis, MO), and β emission was measured as previously described.40 All conditions were performed in triplicate. An unpaired Student t test was used to determine significant differences between genotypes.
Western blot
Akt activation was determined by depriving c-kit+ cells of serum and growth factors for 16 to 20 hours, followed by stimulation with 10 ng/mL kitL. Cells were lysed in nonionic lysis buffer (20 mM Tris HCl [Sigma, St Louis, MO], 137 mM NaCl, 1 mM EGTA, 1% Triton X-100, 10% glycerol, 1.5 mM MgCl2, and complete protease inhibitors [Amersham Pharmacia Biotech, Piscataway, NJ]). Lysates were normalized for protein content using the bicinchoninic acid assay (Pierce Chemical, Rockford, IL). Lysates were boiled for 5 minutes, subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), and transferred to nitrocellulose. The membranes were blocked with PBS containing 5% blotting grade nonfat dry milk (Bio-Rad, Hercules, CA) for 1 hour. Membranes were incubated overnight at 4°C with anti-phospho-Akt (Cell Signaling, Beverly, MA). The secondary antibody used was anti-rabbit IgG conjugated to HRP (Amersham Pharmacia Biotech). Proteins were visualized by enhanced chemiluminescence (ECL; Amersham Pharmacia Biotech).
Transplantation, white blood cell counts, and cell lineage analysis
Unfractionated fetal liver cells (CD45.2+, 1-2 × 106) from the 4 F2 experimental groups were resuspended in 450 μL IMDM supplemented with 20% FCS. Cells were transplanted into congenic lethally irradiated recipient mice (0.7 Gray + 0.4 Gray 4 hours later). The majority of recipients were C57BL/6J; however, to assess whether host hematopoiesis recovered during the study, some of the recipients were B6.BoyJ (CD45.1+). A total of 10 fetal livers per genotype were used for these studies. Mice were bled 1, 4, and 6 months following transplantation for peripheral blood counts and chimerism analyses. B6.BoyJ recipients had 97% to 100% donor-derived hematopoiesis, similar to previous studies from our laboratories.40-42 Peripheral blood counts including hematocrits, red blood cell counts (RBCs), white blood cell counts (WBCs), and absolute neutrophil, lymphocyte, and monocyte counts were measured using a Hemavet CDC Mascot cytometer (CDC Technologies, Oxford, CT). Peripheral blood smears were prepared from each sample with a modified Wright-Giemsa stain. Manual 100-cell leukocyte, lymphocyte, and monocyte differentials were performed on each sample. Absolute neutrophil, lymphocyte, and monocyte cell counts were calculated by multiplying the percentage of each cell type by the total WBC count. An unpaired Student t test was used to determine significant differences between genotypes.
Competitive repopulation assays
Competitive repopulation assays were conducted as previously described.43 Briefly, 1 × 106 unfractionated fetal liver cells from the 4 F2 experimental genotypes (CD45.2+) were mixed with a common pool of B6.BoyJ low-density bone marrow competitors (7.5 × 105 cells, CD45.1+). The cell mixture was resuspended in 0.5 mL IMDM containing 20% FCS and injected into the tail vein of 6 to 8 lethally irradiated recipient mice. Six mice received transplants of 7.5 × 105 competitor cells only (CD45.1+) to ensure that donor chimerism measurements (CD45.2+) were not contaminated by recovery of endogenous hematopoiesis from lethally irradiated C57BL/6J recipients (CD45.2+). Donor chimerism data for these mice ranged from 97% to 100%, demonstrating no to minimal recovery of host hematopoiesis. A total of 3 independent competitive repopulation transplant experiments were conducted.
Peripheral blood chimerism (CD45.2+ cells) was analyzed 1, 4, and 6 months following transplantation by fluorescence cytometry as previously described.43 In addition, donor contribution to multiple lineages was evaluated 6 months following transplantation using 2-color fluorescence cytometry as previously described.43 A total of 10 000 events were collected from each sample. All data were analyzed using CELLQuest software (Becton Dickinson, San Jose, CA). Instrument settings and gates used to analyze data were identical from month to month and between genotypes. An unpaired Student t test was used to determine significant differences between genotypes.
Relative repopulating ability of the donor cells compared with the competitor cell population was determined using a repopulating unit (RU) calculation described previously.43 This calculation allows quantitative comparison of repopulating ability between different test cell populations (WT, p85α-/-, p85β+/-, and p85α-/-p85β+/-). RUs transplanted into individual recipients at the 6-month time point were calculated for all 3 competitive repopulation experiments. Since a constant number of competitor cells (7.5 × 105) was used for all experiments, RU = 7.5 × measured donor chimerism/(100 - measured donor chimerism). Differences in RUs between experimental groups were compared using an unpaired Student t test.
Results
Genetic inactivation of the regulatory subunits of class IA PI-3K reduces absolute numbers and frequency of fetal liver hematopoietic progenitors
We previously demonstrated that genetic inactivation of p85α disrupts fetal erythropoiesis in vivo and reduces the absolute number and frequency of fetal liver erythroid progenitors.37 However, the effect of genetic inactivation of class IA PI-3K activity on both fetal and adult hematopoietic stem and progenitor cell function in vivo is unknown. Residual class IA PI-3K activity has been demonstrated in multiple p85α-/- cell lineages (30%-50%) due to a compensatory up-regulation of the p85β regulatory subunit.34,36 Therefore, to investigate the effect of genetic inactivation of class IA PI-3K activity on both fetal and adult hematopoiesis, we attempted to derive embryos deficient in both regulatory subunits of class IA PI-3K by intercrossing p85α+/- and p85β-/- mice.
Multiple F1 p85α+/-p85β+/- breeding pairs were mated, and no viable p85α-/-p85β-/- embryos were observed after day 10 of gestation. However, we were able to generate viable WT, p85α-/-, p85β+/-, and p85α-/-p85β+/- fetuses at day 13.5 for experiments. Similar to our previously reported data,37 p85α-/- embryos were pale with decreased total liver cellularity compared with WT and p85β+/- controls (data not shown). Moreover, heterozygous deletion of the p85β subunit (p85α-/-p85β+/-) in p85α-/- embryos resulted in small, pale embryos (data not shown), which had a further reduction in total liver cellularity compared with p85α-/- littermates (Figure 1). Thus, these observations suggested that genetic inactivation of the regulatory subunits of class IA PI-3K may alter fetal hematopoiesis in vivo.
Figure 1.

Genetic inactivation of the p85 regulatory subunits of class IA PI-3K decreases fetal liver cellularity in day-13.5 embryos. Individual embryos were isolated, fetal livers were harvested, and single-cell suspensions were prepared. Total liver cellularity was enumerated in 15 embryos/genotype. *P < .001; ** P < .005.
To test this hypothesis directly, we next measured the total number of multipotential and committed hematopoietic progenitors in day-13.5 fetal livers. Equal numbers of low-density fetal liver cells from the 4 experimental genotypes were plated in standard methylcellulose progenitor assays as previously described.40 As shown in Table 1, the absolute numbers of multipotential (CFU-MIX), erythroid (BFU-E and CFU-E), and myeloid (CFU-GM and CFU-Meg) progenitors were significantly decreased in both p85α-/- and p85α-/-p85β+/- fetal livers compared with WT and p85β+/- controls. Furthermore, p85α-/-p85β+/- fetal livers contained fewer multipotential, erythroid, and myeloid progenitors compared with p85α-/- fetal livers, suggesting that heterozygous inactivation of the p85β allele in p85α-/- fetal liver cells further reduces their clonogenic progenitor-forming ability (Table 1).
Table 1.
Total fetal liver hematopoietic progenitors
| Genotype | CFU-MIX, × 105 | BFU-E, × 103 | CFU-E, × 105 | CFU-GM, × 105 | CFU-MEG, × 103 |
|---|---|---|---|---|---|
| WT | 1.23 ± 0.003 | 47 ± 0.27 | 4.0 ± 0.005 | 2.5 ± 0.002 | 6.1 ± 0.03 |
| p85β+/- | 1.37 ± 0.002 | 41 ± 0.18 | 4.2 ± 0.003 | 3.0 ± 0.005 | 6.8 ± 0.05 |
| p85α-/- | 0.6 ± 0.005 | 20 ± 0.1* | 3.0 ± 0.001* | 1.5 ± 0.001* | 3.66 ± 0.06* |
| p85α-/-; p85β+/- | 0.1 ± 0.001*† | 2.8 ± 0.15*† | 1.1 ± 0.001*† | 0.21 ± 0.001*† | 0.7 ± 0.04*† |
Data are presented as the mean plus or minus the standard error of the mean.
P < .02 compared with WT
P < .05 compared with p85α-/-
The reduction in absolute numbers of hematopoietic progenitors in p85α-/- and p85α-/-p85β+/- fetal livers could be secondary to an intrinsic hematopoietic progenitor defect, or alternatively, a defect in the fetal liver microenvironment. To directly test the effect of p85α and p85β deficiency on hematopoietic progenitor colony formation, we sorted equal numbers of fetal liver c-kit+ cells, which is a phenotypically defined cell population enriched for hematopoietic progenitors, from the 4 experimental genotypes and compared the ability of these cells to form hematopoietic cell colonies in clonogenic assays, similar to previous studies37,44 For these studies, the number of both committed and immature clonogenic progenitors derived from purified c-kit+ cells was quantitated using LPP-CFC and HPP-CFC assays. LPP-CFCs constitute committed myeloid progenitors, similar to CFU-GM colonies assayed in methylcellulose cultures (Table 1). HPP-CFCs are immature progenitors with self-renewal potential demonstrated by replating capacity. Despite exhibiting a decrease in total fetal liver cellularity, p85α-/- and p85α-/-p85β+/- fetal livers had an equivalent proportion of c-kit+ cells compared with WT and p85β+/- fetal livers (data not shown). Similar to the committed progenitor data shown in Table 1, there was a marked reduction in the number of LPP-CFCs and HPP-CFCs generated from both p85α-/- and p85α-/-p85β+/- c-kit+ cells compared with WT and p85β+/- controls (Figure 2A-B). Furthermore, p85α-/-p85β+/- c-kit+ cells formed fewer LPP-CFCs and HPP-CFCs compared with p85α-/- c-kit+ cells, suggesting that heterozygous inactivation of the p85β allele in p85α-/- c-kit+ cells further reduces their clonogenic progenitor-forming ability. Collectively, these data argue that an intrinsic p85α-/- and p85α-/-p85β+/- hematopoietic progenitor cell defect contributes, at least in part, to the reduction of immature and committed hematopoietic progenitors in day-13.5 fetal livers in vivo.
Figure 2.

Reduced LPP-CFC/HPP-CFC frequency and proliferation of p85α-/- and p85α-/-; p85β+/- c-kit+ fetal liver cells. c-kit+ cells were purified from WT, p85β+/-, p85α-/-, and p85α-/-p85β+/- fetal livers and plated in agar cultures containing growth factors that promote the growth of (A) LPP-CFCs and (B) HPP-CFCs. Data shown are the mean number of colonies per 4000 c-kit+ cells plated. *P < .05; n = 4. (C) Representative LPP-CFC photomicrographs generated from WT and p85α-/-; p85β+/- c-kit+ cells. Original magnification, × 40. (D) Proliferation of WT, p85β+/-, p85α-/-, and p85α-/-p85β+/- c-kit+ fetal liver cells in response to kitL. Freshly isolated c-kit+ fetal liver cells were plated in triplicate with no growth factors or 10 ng/mL KitL for 16 hours. Cells were pulsed with tritiated thymidine and harvested for measurement of β emission. Results represent the mean thymidine incorporation ± SEM of 9 independent experiments. *P < .05. (E) Akt activation of WT, p85β+/-, p85α-/-, and p85α-/-p85β+/- c-kit+ fetal liver cells in response to kitL. Western blots for Akt phosphorylation and total Akt are shown. Data are representative of 4 independent experiments. **P < .05 for p85α-/- vs p85α-/-p85β-/-.
p85α-/- and p85α-/-p85β+/- c-kit+ cells have decreased proliferation in response to kitL
Because these data establish a previously unrecognized role of the p85 regulatory subunits in regulating fetal hematopoiesis in vivo, we next tested whether the p85α-/- and p85α-/-p85β+/- hematopoietic progenitor cell phenotype could be linked to either a decrease in proliferation and/or survival of hematopoietic progenitors. We initially compared the percentage of freshly isolated c-kit+ cells undergoing apoptosis from day-13.5 fetal livers of the 4 experimental genotypes. Cells were stained with annexin to identify apoptotic cells. No differences in apoptosis of the c-kit+ cells isolated from the 4 experimental genotypes were observed (data not shown).
We next tested whether p85α-/- or p85α-/-p85β+/- deficiency would alter the proliferation of c-kit+ progenitor cells in response to specific growth factors. Of interest, while counting LPP-CFC colonies, it was noted that the size of individual colonies derived from p85α-/-p85β+/- c-kit+ cells was significantly smaller than WT controls (Figure 2C). These observations suggested that p85α-/-p85β+/- hematopoietic progenitor cells may have reduced proliferation in response to growth factors, which control progenitor cell proliferation. To formally test this hypothesis, c-kit+ cells derived from WT, p85α-/-, p85β+/-, and p85α-/-p85β+/- fetal livers were stimulated with either GM-CSF, G-CSF, IL-3, or kitL, which are growth factors important for HPP-CFC and LPP-CFC colony formation. Proliferation was measured using thymidine incorporation assay as previously described.37 No difference was observed in c-kit+ cell proliferation between the 4 experimental genotypes in response to either GM-CSF, G-CSF, or IL-3 (data not shown). However, p85α-/- and p85α-/-p85β+/- c-kit+ cells had a marked reduction in KitL-induced proliferation compared with WT and p85β+/- c-kit+ cells (Figure 2D). Activation of Akt is PI-3K dependent and provides a sensitive measure of PI-3K activity.45 Therefore, we measured Akt activation in all 4 experimental genotypes to determine whether decreased KitL-induced proliferation in p85α-/- and p85α-/-p85β+/- c-kit+ cells corresponded to decreased Akt activation. Both p85α-/- and p85α-/-p85β+/- c-kit+ cells had a decrease in Akt activation in response to KitL compared with WT and p85β+/- c-kit+ cells (Figure 2E). These data are consistent with previously published studies, which demonstrated that p85α-/- bone marrow-derived mast cells have decreased proliferation in response to KitL.36 Further, p85α-/- p85β+/- c-kit+ cells displayed a significant decrease in proliferation and Akt activation compared with p85α-/- c-kit+ cells in response to KitL (Figure 2D-E). Together these data support diminished cytokine-induced proliferation of p85α-/- and p85α-/- p85β+/- c-kit+ cells as a potential mechanism for the observed reduction in hematopoietic progenitor colony size and number.
p85α-/- and p85α-/-p85β+/- fetal liver cells are capable of supporting steady-state hematopoiesis in adult recipients
Our data demonstrate that genetic reduction of class IA PI-3K activity perturbs fetal hematopoiesis in vivo. To determine whether genetic inactivation of the p85 regulatory subunits of class IA PI-3K would alter steady-state hematopoiesis in adult mice, we transplanted both p85α-/- and p85α-/-p85β+/- fetal liver cells and appropriate experimental control cells into lethally irradiated WT mice. Peripheral blood counts of mice that underwent transplantation were measured 6 months after transplantation. Total white blood cell counts (×103/μL) were decreased in mice reconstituted with p85α-/- and p85α-/-p85β+/- fetal liver cells compared with mice reconstituted with WT and p85β+/- cells (WT = 28 ± 1.5; p85β+/- = 23.7 ± 1.9; p85α-/- = 14.2 ± 1.5*;p85α-/-p85β+/- = 15.3* ± 1.0; n = 10 mice/genotype; *P < .001 vs WT). As previously observed, total white blood cell counts are higher in mice reconstituted with WT fetal liver cells compared with WT mice that did not undergo transplantation.40,42 Detailed analyses revealed that the lower total white blood cell count in recipients of transplanted p85α-/- and p85α-/- p85β+/- fetal liver cells was secondary to a decrease in the absolute number of lymphocytes (× 103/μL) (WT = 21.2 ± 1.4; p85β+/- = 17.4 ± 1.3; p85α-/- = 6.3 ± 1.0*; p85α-/-p85β+/- = 8.44* ± 0.5; n = 10 mice/genotype; *P < .001 vs WT), which is consistent with previous studies demonstrating that genetic disruption of p85α perturbs B-cell development and function in vivo using the Rag2 blastocyst complementation method.34 However, no differences were noted in the absolute peripheral counts of the other hematopoietic lineages tested including neutrophils, platelets, red blood cells, monocytes, basophils, and eosinophils (data not shown). To examine whether differences existed in the percentage of phenotypically defined stem cell populations, we evaluated the bone marrow of reconstituted mice for Sca-1+c-kit+lin- (KSL) cells using multicolor fluorescence cytometry. The proportion of KSL cells was not different between mice that received transplants of WT and p85α-/-p85β+/- fetal liver cells (WT = 0.016% ± 0.007% and p85α-/-p85β+/- = 0.026% ± 0.02%; n = 3 mice/genotype). Collectively, these data demonstrate that fetal liver cells from p85α-/- and p85α-/-p85β+/- embryos cannot sustain lymphopoiesis but are capable of supporting steady-state production of the other hematopoietic cell lineages tested in adult mice.
p85α-/- and p85α-/-; p85β+/- fetal liver cells have decreased competitive repopulating ability in vivo
The murine c-kit receptor and its ligand, KitL, are components of a signaling pathway that is essential for murine hematopoiesis and maintenance of the repopulating ability of HSCs.46-48 Given that p85α-/- and p85α-/-p85β+/- c-kit+ cells have decreased proliferation in response to kitL and form fewer HPP-CFCs in vitro, we next tested whether genetic inactivation of the p85 regulatory subunits of class IA PI-3K function would diminish multilineage HSC repopulating ability. Fetal liver cells from day-13.5 WT, p85α-/-, p85β+/-, and p85α-/-p85β+/- embryos (CD45.2+) were used as test cells in competitive repopulation assays. Test cells from each of the 4 experimental genotypes were mixed with a common pool of competitor cells (CD45.1+) before being transplanted into lethally irradiated recipient mice. Peripheral blood chimerism was evaluated by fluorescence cytometry as previously described.43 Three independent competitive repopulation transplant experiments were conducted with 6 to 8 recipients/test cell group. Mean donor chimerism of mice that underwent transplantation from a representative experiment at 1, 4, and 6 months following transplantation is shown in Figure 3A. Consistent with a defect in the progenitor compartment, short-term repopulating ability of p85α-/- and p85α-/-p85β+/- fetal liver cells was significantly lower than WT and p85β+/- controls 1 month following transplantation (Figure 3A). In addition, long-term repopulating ability of p85α-/- and p85α-/-p85β+/- fetal liver cells was reduced 2- to 3-fold compared with WT and p85β+/- controls 4 to 6 months following transplantation (Figure 3A). Further, donor chimerism of mice that received transplants of p85α-/-p85β+/- test cells was consistently lower than mice that received transplants of p85α-/- test cells at all time points evaluated (Figure 3A). Similar results were obtained in the other 2 competitive repopulation experiments conducted (data not shown).
Figure 3.
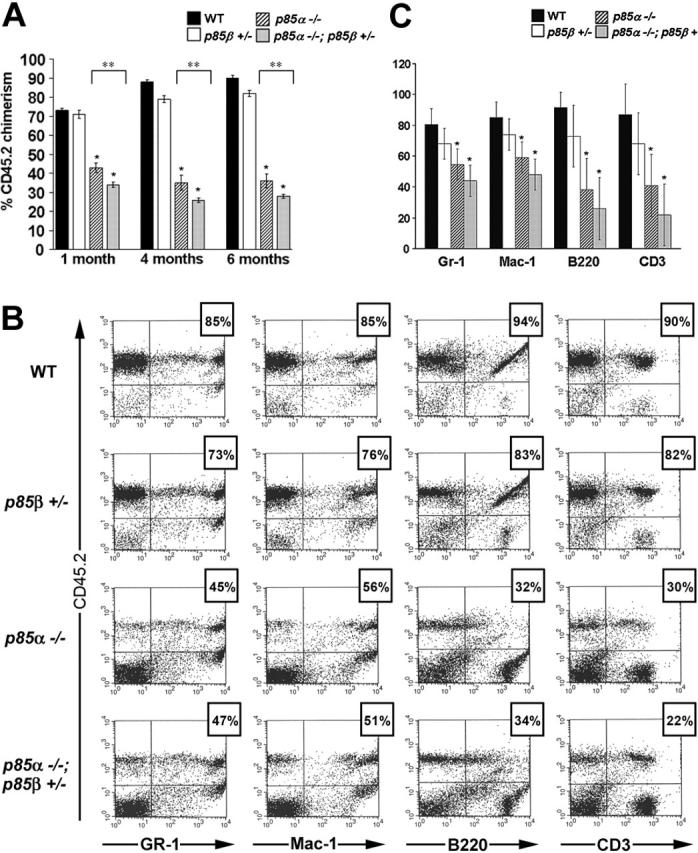
Impaired multilineage HSC repopulating ability in p85α-/-; p85β+/- fetal liver cells. (A) Mean donor chimerism 1, 4, and 6 months following transplantation. *P < .01 compared with WT; **P < .05. (B) Representative dot plots from multilineage analyses. The percentage above each dot plot represents the calculated donor contribution to an individual lineage. Donor contribution to individual lineages was calculated by dividing CD45.2+lineage+ by total lineage+ cells (UR/[UR+LR]). (C) Donor contribution to myeloid and lymphoid lineages in long-term reconstituted mice. *P < .01 compared with WT; **P < .05. Error bars represent the standard error of the mean.
To assess donor cell contribution to myeloid and lymphoid lineages in mice that underwent transplantation, multilineage analyses were conducted by detecting simultaneous expression of CD45.2 and specific lineage markers as previously described.43 The summary of multilineage analyses is illustrated in Figure 3B-C. Donor cells from p85α-/- and p85α-/-p85β+/- fetal livers were capable of multilineage repopulation, albeit at a consistently lower level than WT and p85β+/- controls (Figure 3B-C). Although steady-state hematopoiesis data revealed a significant reduction in absolute lymphocyte count in mice reconstituted with p85α-/- and p85α-/-p85β+/- fetal liver cells, competitive repopulation experiments detected only a modest reduction in the contribution of p85α-/- and p85α-/-p85β+/- donor cells to T-cell (CD3+) and B-cell (B220+) populations compared with myeloid cells (Gr1 and Mac1) (Figure 3B-C). Of importance, this observation suggests that the overall decrease in donor chimerism of mice that received transplants of p85α-/- or p85α-/-p85β+/- fetal liver cells cannot be explained in total by a loss of donor lymphocytes, but represents reduced HSC reconstituting ability.
To quantitatively compare the repopulating ability between experimental groups, repopulating unit (RU) activity was calculated as previously described.43 Table 2 reports RU data for the 3 independent experiments. In all experiments, p85α-/- and p85α-/- p85β+/- fetal liver cells had fewer RUs per 1 × 106 test cells transplanted compared with WT and p85β+/- cells (Table 2). By combining RU data from all 3 experiments, it was determined that p85α-/- and p85α-/-p85β+/- fetal liver cells exhibited 11% and 6% of WT RUs, respectively. This calculation reflects RU frequency within the total number of test cells transplanted. Of importance, these data, together with the observation that total fetal liver cellularity in p85α-/- and p85α-/-p85β+/- embryos is reduced (Figure 1), support a profound loss of HSC repopulating ability in both p85α-/- and p85α-/-p85β+/- fetal livers. Collectively, these data demonstrate that class IA PI-3K is a critical regulator of multilineage HSC repopulating ability.
Table 2.
RU data
| Experiment no. | WT | p85β+/- | p85α-/- | p85α-/-p85β+/- |
|---|---|---|---|---|
| 1 | 30.3 ± 6.4 | 25.8 ± 3.7 | 4.4 ± 0.5* | 2.9 ± 0.6*† |
| 2 | 61.9 ± 8.2 | 44.3 ± 8.3 | 4.6 ± 0.6* | 1.9 ± 0.2*† |
| 3 | 32.9 ± 4.3 | 24.9 ± 2.4 | 4.4 ± 0.3* | 3.2 ± 0.5*† |
P < .02 compared with WT
P < .05 compared with p85α-/-
Discussion
Multiple growth factors implicated in the regulation of HSC/P proliferation and survival activate the class IA PI-3K-Akt-mTOR pathway.13-20,22-27,33,49-54 Further, this pathway is hyperactivated by a variety of transforming oncogenes in leukemias and lymphomas.15,17,22-28 Therefore, determination of how class IA PI-3K regulates HSC/P function is important for understanding both normal and pathologic hematopoiesis.
In this report, we provide data showing that class IA PI-3K is required to maintain fetal hematopoiesis and multilineage HSC repopulating ability by examining the phenotype of mice lacking the regulatory subunits of class IA PI-3K. Genetic reduction of the p85 regulatory subunits of class I PI-3K (p85α-/- and p85α-/- A p85β+/-) diminished the absolute number of multipotential, erythroid, and myeloid clonogenic progenitors in day-13.5 fetal livers. Moreover, c-kit+ cells harvested from p85α-/- and p85α-/- p85β+/- fetal livers exhibited a significant reduction in the total number and frequency of HPP-CFCs and LPP-CFCs. Together these data argue that an intrinsic p85α-/- and p85α-/-p85β+/- hematopoietic progenitor cell defect contributes, at least in part, to defective hematopoiesis in day-13.5 fetal livers in vivo.
Given our observations in p85α-/- and p85α-/-p85β+/- fetal livers, it was critical to evaluate whether function of the HSC compartment was also impaired. Engraftment of fetal liver cells into lethally irradiated recipients demonstrated that p85α-/- and p85α-/-p85β+/- fetal liver cells are capable of supporting adult steady-state hematopoiesis. However, lymphopoiesis was not sustained to wild-type levels, which is consistent with previous data showing that B-cell development requires intact PI-3K function. Further, using well-established competitive repopulation assays we demonstrate a marked reduction in the ability of p85α-/- and p85α-/-p85β+/- fetal liver cells to sustain multilineage repopulation compared with WT controls. In contrast to engraftment experiments, these studies did not reveal a dramatic defect in lymphoid reconstitution compared with myeloid reconstitution in transplant recipients of either p85α-/- or p85α-/-p85β+/- donor cells. Given that the only difference between the competitive repopulation studies and the engraftment experiments is cotransplantation of WT competitor cells, the WT cells present in the graft must contribute to the support of p85α-/- and p85α-/-p85β+/- donor cell lymphoid differentiation. Collectively, these data provide the first evidence that class IA PI-3K is required to sustain normal HSC reconstituting ability in vivo.
HSCs harvested from mice with mutations at the dominant white spotting (W) and Steel (Sl) loci, which encode for the murine c-kit receptor and its ligand, kitL, respectively, have decreased repopulating ability in vivo.55 Consistent with the phenotype of W or Sl mice, we demonstrate that both p85α-/- and p85α-/-p85β+/- c-kit+ cells have decreased proliferation in response to kitL, providing a potential mechanism for the decreased repopulating ability of p85α-/- and p85α-/-p85β+/- HSCs.
Two different groups have previously generated knock-in mice, which harbor a single point mutation in the p85α binding site of the c-kit receptor (Y719 mice)53,56 and steady-state hematopoiesis was unaffected in these mutant mice. However, HSC repopulating ability was not formally tested in these studies. The difference in the hematopoietic phenotypes of knock-in mice harboring the Y719 mutation and mice that received transplants of either p85α-/- or p85α-/-p85β+/- fetal liver stem cells raises the question of whether class IA PI-3K functions as a molecular switch for multiple growth factors to activate discrete signaling pathways, which control the proliferation and survival of HSC/Ps. Specifically, recent in vitro studies have shown that vascular endothelial growth factor 2 (VEGF-2), angiopoietin 1 (Ang-1), and stromal-derived factor 1 (SDF-1) control the proliferation and survival of HSC/Ps via activation of PI-3K.17,19,57,58 Therefore, it is intriguing to speculate that VEGF-2, Ang-1, SDF-1, KitL, and potentially other growth factors activate class IA PI-3K at least in part to control the function of HSCs. In summary, these studies identify PI-3K as an important regulator of HSC function and a potential therapeutic target in treating leukemic stem cells, which are dependent on PI-3K signaling for their proliferation.
Acknowledgments
We thank Janice L. Walls for administrative assistance on this article preparation.
Prepublished online as Blood First Edition Paper, October 20, 2005; DOI 10.1182/blood-2005-05-1985.
Supported by grants 1 KO8 CA096579-01 (D.A.I.), KO8 HLDK04071-01 (L.S.H.), R01 HL077175 (L.S.H.), RO1 HL075816 (R.K.), and P30 CA82709 (D.A.I., L.S.H., and R.K.), and by the Riley Children's Foundation (D.A.I., L.S.H., and R.K.). D.A.I. is a recipient of a Basil O'Connor Award from the March of Dimes (5-FY02-254).
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 U.S.C. section 1734.
References
- 1.Domen J, Weissman IL. Hematopoietic stem cells need two signals to prevent apoptosis: BCL-2 can provide one of these, Kitl/c-Kit signaling the other. J Exp Med. 2000;192: 1707-1718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Liu Q, Sasaki T, Kozieradzki I, et al. SHIP is a negative regulator of growth factor receptor-mediated PKB/Akt activation and myeloid cell survival. Genes Dev. 1999;13: 786-791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Skorski T, Bellacosa A, Nieborowska-Skorska M, et al. Transformation of hematopoietic cells by BCR/ABL requires activation of a PI-3k/Akt-dependent pathway. EMBO J. 1997;16: 6151-6161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kogan SC, Brown DE, Shultz DB, et al. BCL-2 cooperates with promyelocytic leukemia retinoic acid receptor alpha chimeric protein (PMLRARa) to block neutrophil differentiation and initiate acute leukemia. J Exp Med. 2001;193: 531-543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bouillet P, Huang DC, O'Reilly LA, et al. The role of the pro-apoptotic Bcl-2 family member bim in physiological cell death. Ann N Y Acad Sci. 2000; 926: 83-89. [DOI] [PubMed] [Google Scholar]
- 6.Shinjyo T, Kuribara R, Inukai T, et al. Downregulation of Bim, a proapoptotic relative of Bcl-2, is a pivotal step in cytokine-initiated survival signaling in murine hematopoietic progenitors. Mol Cell Biol. 2001;21: 854-864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bouillet P, Metcalf D, Huang DC, et al. Proapoptotic Bcl-2 relative Bim required for certain apoptotic responses, leukocyte homeostasis, and to preclude autoimmunity. Science. 1999;286: 1735-1738. [DOI] [PubMed] [Google Scholar]
- 8.Strasser A, Puthalakath H, Bouillet P, et al. The role of bim, a proapoptotic BH3-only member of the Bcl-2 family in cell-death control. Ann N Y Acad Sci. 2000;917: 541-548. [DOI] [PubMed] [Google Scholar]
- 9.Traver D, Akashi K, Weissman IL, Lagasse E. Mice defective in two apoptosis pathways in the myeloid lineage develop acute myeloblastic leukemia. Immunity. 1998;9: 47-57. [DOI] [PubMed] [Google Scholar]
- 10.Dijkers PF, Medemadagger RH, Lammers JW, Koenderman L, Coffer PJ. Expression of the pro-apoptotic Bcl-2 family member Bim is regulated by the forkhead transcription factor FKHR-L1. Curr Biol. 2000;10: 1201-1204. [DOI] [PubMed] [Google Scholar]
- 11.Domen J, Cheshier SH, Weissman IL. The role of apoptosis in the regulation of hematopoietic stem cells: overexpression of Bcl-2 increases both their number and repopulation potential. J Exp Med. 2000;191: 253-264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jiang X, Stuible M, Chalandon Y, et al. Evidence for a positive role of SHIP in the BCR-ABL-mediated transformation of primitive murine hematopoietic cells and in human chronic myeloid leukemia. Blood. 2003;102: 2976-2984. [DOI] [PubMed] [Google Scholar]
- 13.Lennartsson J, Shivakrupa R, Linnekin D. Synergistic growth of stem cell factor and granulocyte macrophage colony-stimulating factor involves kinase-dependent and -independent contributions from c-Kit. J Biol Chem. 2004;279: 44544-44553. [DOI] [PubMed] [Google Scholar]
- 14.Matsunaga T, Takemoto N, Sato T, et al. Interaction between leukemic-cell VLA-4 and stromal fibronectin is a decisive factor for minimal residual disease of acute myelogenous leukemia. Nat Med. 2003;9: 1158-1165. [DOI] [PubMed] [Google Scholar]
- 15.Chian R, Young S, Danilkovitch-Miagkova A, et al. Phosphatidylinositol 3 kinase contributes to the transformation of hematopoietic cells by the D816V c-Kit mutant. Blood. 2001;98: 1365-1373. [DOI] [PubMed] [Google Scholar]
- 16.Wandzioch E, Edling CE, Palmer RH, Carlsson L, Hallberg B. Activation of the MAP kinase pathway by c-Kit is PI-3 kinase dependent in hematopoietic progenitor/stem cell lines. Blood. 2004;104: 51-57. [DOI] [PubMed] [Google Scholar]
- 17.Larrivee B, Lane DR, Pollet I, Olive PL, Humphries RK, Karsan A. Vascular endothelial growth factor receptor-2 induces survival of hematopoietic progenitor cells. J Biol Chem. 2003; 278: 22006-22013. [DOI] [PubMed] [Google Scholar]
- 18.Heldin CH. Signal transduction: multiple pathways, multiple options for therapy. Stem Cells. 2001;19: 295-303. [DOI] [PubMed] [Google Scholar]
- 19.Lataillade JJ, Clay D, Bourin P, et al. Stromal cell-derived factor 1 regulates primitive hematopoiesis by suppressing apoptosis and by promoting G(0)/G(1) transition in CD34(+) cells: evidence for an autocrine/paracrine mechanism. Blood. 2002;99: 1117-1129. [DOI] [PubMed] [Google Scholar]
- 20.Zhao S, Zoller K, Masuko M, et al. JAK2, complemented by a second signal from c-kit or flt-3, triggers extensive self-renewal of primary multipotential hemopoietic cells. EMBO J. 2002;21: 2159-2167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hahn M, Li W, Yu C, Rahmani M, Dent P, Grant S. Rapamycin and UCN-01 synergistically induce apoptosis in human leukemia cells through a process that is regulated by the Raf-1/MEK/ERK, Akt, and JNK signal transduction pathways. Mol Cancer Ther. 2005;4: 457-470. [DOI] [PubMed] [Google Scholar]
- 22.Sattler M, Mohi MG, Pride YB, et al. Critical role for Gab2 in transformation by BCR/ABL. Cancer Cell. 2002;1: 479-492. [DOI] [PubMed] [Google Scholar]
- 23.Kirito K, Fox N, Kaushansky K. Thrombopoietin induces HOXA9 nuclear transport in immature hematopoietic cells: potential mechanism by which the hormone favorably affects hematopoietic stem cells. Mol Cell Biol. 2004;24: 6751-6762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Thompson JE, Thompson CB. Putting the rap on Akt. J Clin Oncol. 2004;22: 4217-4226. [DOI] [PubMed] [Google Scholar]
- 25.Recher C, Beyne-Rauzy O, Demur C, et al. Anti-leukemic activity of rapamycin in acute myeloid leukemia. Blood. 2005;105: 2527-2534. [DOI] [PubMed] [Google Scholar]
- 26.Kharas MG, Fruman DA. ABL oncogenes and phosphoinositide 3-kinase: mechanism of activation and downstream effectors. Cancer Res. 2005;65: 2047-2053. [DOI] [PubMed] [Google Scholar]
- 27.Mohi MG, Boulton C, Gu TL, et al. Combination of rapamycin and protein tyrosine kinase (PTK) inhibitors for the treatment of leukemias caused by oncogenic PTKs. Proc Natl Acad Sci U S A. 2004; 101: 3130-3135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wymann MP, Zvelebil M, Laffargue M. Phospho-inositide 3-kinase signalling: which way to target? Trends Pharmacol Sci. 2003;24: 366-376. [DOI] [PubMed] [Google Scholar]
- 29.Vanhaesebroeck B, Waterfield MD. Signaling by distinct classes of phosphoinositide 3-kinases. Exp Cell Res. 1999;253: 239-254. [DOI] [PubMed] [Google Scholar]
- 30.Wymann MP, Pirola L. Structure and function of phosphoinositide 3-kinases. Biochim Biophys Acta. 1998;1436: 127-150. [DOI] [PubMed] [Google Scholar]
- 31.Stein RC, Waterfield MD. PI3-kinase inhibition: a target for drug development? Mol Med Today. 2000;6: 347-357. [DOI] [PubMed] [Google Scholar]
- 32.Cockcroft S. Biology of Phosphoinositides. New York, NY: Oxford University Press; 2000.
- 33.Munugalavadla V, Borneo J, Ingram DA, Kapur R. p85α subunit of class IA PI-3 kinase is crucial for macrophage growth and migration. Prepublished on March 15, 2005, as DOI 10.1182/blood-2004-10-4041. (Now available as Blood. 2005;106: 103-109.) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fruman DA, Snapper SB, Yballe CM, et al. Impaired B cell development and proliferation in absence of phosphoinositide 3-kinase p85a. Science. 1999;283: 393-397. [DOI] [PubMed] [Google Scholar]
- 35.Fruman DA, Mauvais-Jarvis F, Pollard DA, et al. Hypoglycaemia, liver necrosis and perinatal death in mice lacking all isoforms of phosphoinositide 3-kinase p85a. Nat Genet. 2000;26: 379-382. [DOI] [PubMed] [Google Scholar]
- 36.Lu-Kuo JM, Fruman DA, Joyal DM, Cantley LC, Katz HR. Impaired kit- but not FcepsilonRI-initiated mast cell activation in the absence of phosphoinositide 3-kinase p85a gene products. J Biol Chem. 2000;275: 6022-6029. [DOI] [PubMed] [Google Scholar]
- 37.Huddleston H, Tan B, Yang FC, et al. Functional p85alpha gene is required for normal murine fetal erythropoiesis. Blood. 2003;102: 142-145. [DOI] [PubMed] [Google Scholar]
- 38.Ueki K, Yballe CM, Brachmann SM, et al. Increased insulin sensitivity in mice lacking p85beta subunit of phosphoinositide 3-kinase. Proc Natl Acad Sci U S A. 2002;99: 419-424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yang FC, Ingram DA, Chen S, et al. Neurofibromin-deficient Schwann cells secrete a potent migratory stimulus for Nf1+/- mast cells. J Clin Invest. 2003;112: 1851-1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ingram DA, Wenning MJ, Shannon K, Clapp DW. Leukemic potential of doubly mutant Nf1 and Wv hematopoietic cells. Blood. 2003;101: 1984-1986. [DOI] [PubMed] [Google Scholar]
- 41.Haneline LS, Li X, Ciccone SL, et al. Retroviral-mediated expression of recombinant Fancc enhances the repopulating ability of Fancc-/- hematopoietic stem cells and decreases the risk of clonal evolution. Blood. 2003;101: 1299-1307. [DOI] [PubMed] [Google Scholar]
- 42.Zhang Y, Taylor B, Shannon K, Clapp DW. Quantitative effects of Nf1 inactivation on in vivo hematopoiesis. J Clin Invest. 2001;108: 709-715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Haneline LS, Gobbett TA, Ramani R, et al. Loss of FancC function results in decreased hematopoietic stem cell repopulating ability. Blood. 1999; 94: 1-8. [PubMed] [Google Scholar]
- 44.Khalaf WF, White H, Wenning MJ, Orazi A, Kapur R, Ingram DA. K-Ras is essential for normal fetal liver erythropoiesis. Blood. 2005;105: 3538-3541. [DOI] [PubMed] [Google Scholar]
- 45.Alessi DR, Cohen P. Mechanism of activation and function of protein kinase B. Curr Opin Genet Dev. 1998;8: 55-62. [DOI] [PubMed] [Google Scholar]
- 46.Nocka K, Tan JC, Chiu E, et al. Molecular bases of dominant negative and loss of function mutations at the murine c-kit/white spotting locus: W37, Wv, W41 and W. EMBO J. 1990;9: 1805-1813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Blouin. Clinical Disorders and Experimental Models of Erythropoietic Failure. Boca Raton, FL: CRC press; 1993.
- 48.Miller CL, Rebel VI, Helgason CD, Lansdorp PM, Eaves CJ. Impaired steel factor responsiveness differentially affects the detection and long-term maintenance of fetal liver hematopoietic stem cells in vivo. Blood. 1997;89: 1214-1223. [PubMed] [Google Scholar]
- 49.Linnekin D. Early signaling pathways activated by c-Kit in hematopoietic cells. Int J Biochem Cell Biol. 1999;31: 1053-1074. [DOI] [PubMed] [Google Scholar]
- 50.Kobayashi N, Saeki K, Yuo A. Granulocyte-macrophage colony-stimulating factor and interleukin-3 induce cell cycle progression through the synthesis of c-Myc protein by internal ribosome entry site-mediated translation via phosphatidylinositol 3-kinase pathway in human factor-dependent leukemic cells. Blood. 2003;102: 3186-3195. [DOI] [PubMed] [Google Scholar]
- 51.Helgason CD, Antonchuk J, Bodner C, Humphries RK. Homeostasis and regeneration of the hematopoietic stem cell pool are altered in SHIP-deficient mice. Blood. 2003;102: 3541-3547. [DOI] [PubMed] [Google Scholar]
- 52.Agosti V, Corbacioglu S, Ehlers I, et al. Critical role for Kit-mediated Src kinase but not PI 3-kinase signaling in pro T and pro B cell development. J Exp Med. 2004;199: 867-878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kissel H, Timokhina I, Hardy MP, et al. Point mutation in kit receptor tyrosine kinase reveals essential roles for kit signaling in spermatogenesis and oogenesis without affecting other kit responses. EMBO J. 2000;19: 1312-1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hammerman PS, Fox CJ, Birnbaum MJ, Thompson CB. The Pim and Akt oncogenes are independent regulators of hematopoietic cell growth and survival. Prepublished on February 10, 2005, as DOI 10.1182/blood-2004-09-3706. (Now available as Blood. 2005;105: 4477-4483.) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rebel VI, Miller CL, Eaves CJ, Lansdorp PM. The repopulation potential of fetal liver hematopoietic stem cells in mice exceeds that of their liver adult bone marrow counterparts. Blood. 1996;87: 3500-3507. [PubMed] [Google Scholar]
- 56.Blume-Jensen P, Jiang G, Hyman R, Lee KF, O'Gorman S, Hunter T. Kit/stem cell factor receptor-induced activation of phosphatidylinositol 3'-kinase is essential for male fertility. Nat Genet. 2000;24: 157-162. [DOI] [PubMed] [Google Scholar]
- 57.Arai F, Hirao A, Ohmura M, et al. Tie2/angiopoietin-1 signaling regulates hematopoietic stem cell quiescence in the bone marrow niche. Cell. 2004; 118: 149-161. [DOI] [PubMed] [Google Scholar]
- 58.Wakabayashi M, Miwa H, Shikami M, et al. Autocrine pathway of angiopoietins-Tie2 system in AML cells: association with phosphatidyl-inositol 3 kinase. Hematol J. 2004;5: 353-360. [DOI] [PubMed] [Google Scholar]