

Abstract
Canine leukocyte adhesion deficiency (CLAD) represents the canine counter-part of the human disease leukocyte adhesion deficiency (LAD). Defects in the leukocyte integrin CD18 adhesion molecule in both CLAD and LAD lead to recurrent, life-threatening bacterial infections. We evaluated ex vivo retroviral-mediated gene therapy in CLAD using 2 nonmyeloablative conditioning regimens—200 cGy total body irradiation (TBI) or 10 mg/kg busulfan—with or without posttransplantation immunosuppression. In 6 of 11 treated CLAD dogs, therapeutic levels of CD18+ leukocytes were achieved. Conditioning with either TBI or busulfan allowed long-term engraftment, and immunosuppression was not required for efficacy. The percentage of CD18+ leukocytes in the peripheral blood progressively increased over 6 to 8 months after infusion to levels ranging from 1.26% to 8.37% at 1-year follow-up in the 6 dogs. These levels resulted in reversal or moderation of the severe CLAD phenotype. Linear amplification–mediated polymerase chain reaction assays indicated polyclonality of insertion sites. These results describe ex vivo hematopoietic stem cell gene transfer in a disease-specific, large animal model using 2 clinically applicable conditioning regimens, and they provide support for the use of nonmyeloablative conditioning regimens in preclinical protocols of retroviral-mediated gene transfer for nonmalignant hematopoietic diseases such as LAD.
Introduction
Leukocyte adhesion deficiency (LAD) is a genetic immunodeficiency disease in which affected individuals suffer recurrent, life-threatening bacterial infections due to the inability of their neutrophils to adhere to blood vessel walls and migrate to sites of infection.1 The most common form of LAD, LAD-I, results from a deficiency in the surface expression of the CD11/CD18 leukocyte integrin complex as a result of mutations in the CD18 gene.2 Although myeloablative hematopoietic stem cell transplantation can cure LAD-I,3 the morbidity and mortality associated with myeloablative conditioning, coupled with the potential complication of graft-versus-host disease, has restricted its use.
Recently, we have explored the use of the homologous dog model, canine leukocyte adhesion deficiency (CLAD), to investigate alternative therapies for LAD-I.4,5 The dog model for LAD-I has several advantages over the mouse model. Dogs are larger, allowing for clinical interventions similar to those used in humans, and the genetic diversity of dogs is also similar.6 Our previous studies in CLAD dogs assessed the efficacy of matched littermate transplant using a nonmyeloablative regimen of 200 cGy total body irradiation (TBI),4,7 a dose currently in use in human clinical trials to reduce the morbidity and mortality in allogeneic hematopoietic stem cell transplantation. This regimen resulted in mixed chimerism that reversed the CLAD phenotype. These studies also established that a circulating donor peripheral-blood level of 250 to 500 CD18+ neutrophils/μL was sufficient to reverse the severe CLAD phenotype.7 In subsequent studies, we have demonstrated that levels of 50 to 100 CD18+ neutrophils/μL can convert the severe phenotype to a moderate phenotype in CLAD.8 Together, these findings suggest that gene therapy using autologous, CD18+ gene-corrected cells with a nonmyeloablative conditioning regimen might represent a viable therapeutic option in CLAD dogs, since only low levels of gene-corrected leukocytes have been achieved with gene therapy in genetic diseases in which gene replacement does not confer a selective advantage.
The failure of previous clinical trials involving phagocyte disorders (chronic granulomatous disease [CGD] and LAD-I) to achieve therapeutic levels of gene-corrected cells, despite high levels of gene-corrected cells observed in vitro, suggested conditioning was required for gene-corrected CD34+ cells to engraft.9,10 Although gene transfer experiments in normal mature dogs have achieved significant levels of marking, these levels have typically been in the context of myeloablative doses of TBI, such as 920 cGy.11,12 However, these doses are unlikely to be approved for gene therapy for nonmalignant diseases given the associated toxicities with high-dose TBI. Current clinical trials for adenosine deaminase (ADA)–deficient severe combined immunodeficiency (SCID) and patients with CGD have taken advantage of low doses of chemotherapy agents such as busulfan to provide a less toxic, nonmyeloablative conditioning regimen.13,14 The CLAD model provides a disease-specific, large animal model for testing the efficacy and safety of clinically applicable conditioning regimens prior to the infusion of autologous, CD18+ gene-corrected hematopoietic stem cells in LAD-I.
Materials and methods
Animals
NIH facilities for animal housing were accredited by the American Association for Accreditation of Laboratory Animal Care. Animal study protocols were approved by the NCI Institutional Animal Care and Use Committee. The studies were performed in accordance with the principles outlined in the Guide for the Care and Use of Laboratory Animals of the National Academy of Sciences, National Research Council. The breeding colony from which these animals were derived has been previously described.15
Monitoring of clinical course of CLAD animals
All animals were monitored by physical examination and temperature. CLAD dogs were treated prophylactically with oral amoxicillin/potassium clavulanate as previously described.4 Antibiotics were discontinued following engraftment of more than 1% circulating CD18+ leukocytes and the absence of symptoms of clinical disease. WBC counts and differentials were performed on peripheral-blood samples at a commercial laboratory (Antech Diagnostics, Lake Success, NY) or at the NIH Clinical Center laboratory. In some cases, pus was obtained by needle aspiration of the sites of palpable fluid collection in the soft tissues.
CD34+ collection
Bone marrow was collected from animals under general anesthesia by aspiration into heparinized syringes from the marrow cavities of the long bones and pelvis, and filtered as previously described.5 CD34+ cells were selected using an anti–canine CD34 antibody 1H6 (courtesy of Dr Richard Nash, Fred Hutchinson Cancer Research Center, Seattle, WA) as previously described.7 The CD34+ cells were cryopreserved (Plasmalyte A; 6.5% wt/vol HES Pentastarch; 5.8% wt/vol HSA; 4.8% vol/vol DMSO) using a controlled rate freezing device (Cryo 1° freezing container; Nalgene Nunc International, Rochester, NY) at –80°C for approximately 24 hours before transfer to liquid nitrogen storage.
Packaging cell line production
Vector pMSCV-cCD18 was constructed by inserting the canine CD18 cDNA16 into pMSCV-neo (Clontech Laboratories, Mountain View, CA) with deletion of the internal pgk promoter and neomycin resistance gene. Vector pMSCV-cCD18 was cotransfected with plasmid pSV2neo using calcium phosphate precipitation (CalPhos Mammalian Transfection Kit; Clontech Laboratories) into the PG13 packaging cell line.17 Clones were picked by resistance to G418. Titers were assessed using transduction on the LAD Epstein-Barr virus (EBV)–immortalized B-cell line ZJ,18 with clone no. 15 demonstrating the highest transduction and therefore the highest titer.
Production of vector-containing media
PG13/MSCV-cCD18 producer cells were plated at 3.6 × 106 per 150 mm diameter dish in 30 mL Dulbecco modified Eagle medium (Invitrogen, Carlsbad, CA) supplemented with 10% fetal bovine serum (Gemini Bio-Products, Woodland, CA) and 100 U/mL each of penicillin and streptomycin (Hyclone, Logan, UT) and incubated at 37°C in 10% CO2. Fresh medium was added approximately 24 hours later and the cells were transferred to 33°C. The vector-containing media (VCM) was harvested 24 and 48 hours later, passed through a 0.22-μm polyethersulfone membrane filter (Corning Incorporated Life Sciences, Acton, MA) and frozen at –80°C until thawed for transduction. The titer of each stock was determined using the LAD EBV B-cell line ZJ.
Transduction of CLAD CD34+ cells
CD34+ cells were resuspended in StemSpan SFEM media (StemCell Technologies, Vancouver, BC, Canada) and 50 ng/mL canine interleukin-6 (cIL-6; R&D Systems, Minneapolis, MN), canine stem-cell factor (cSCF; Amgen, Thousand Oaks, CA), human thrombopoietin (hTPO; R&D Systems), and human Flt3-Ligand (hFlt3-L; Immunex/Amgen). Cells were prestimulated for approximately 12 to 18 hours. Non–tissue culture–treated T-75 flasks were coated with Retronectin (Cambrex BioScience, Walkersville, MD) at a concentration of 5 μg/cm2 per flask. Retroviral vector was precoated onto Retronectin-treated flasks by 2 exposures of VCM. After the second VCM precoating, prestimulated cells were resuspended in VCM and placed in the flasks at 37°C, 10% CO2. After approximately 16 to 24 hours, cells were removed from the flasks, centrifuged (400g, 25°C, 15 minutes) and resuspended in fresh VCM. After a second exposure to VCM, cells were harvested from the flasks, washed, and resuspended in a solution of Plasmalyte A+1% heat-inactivated autologous serum and 2 U/mL heparin.
Conditioning regimen and immunosuppression
Nine CLAD dogs received TBI at a nonmyeloablative dose of 200 cGy delivered from a 60Co source prior to the infusion of transduced cells either 24 hours prior to infusion (A1, A4, B1-B5; day –1), or 2 hours prior to infusion on the day of the transplantation (A1-A3; day 0). Dog A1 received 2 infusions of cells, with the second infusion occurring approximately 24 hours after the first infusion. Two CLAD dogs (C1, C2) received a nonmyeloablative dose of busulfan (Busulfex; ESP Pharma, Edison, NJ) at 10 mg/kg actual body weight infused intravenously over 1 hour on day –2. This busulfan dose has been shown to be nonmyeloablative in CLAD transplant studies.19 Transduced cells were infused intravenously over approximately 5 minutes. Posttransplantation immunosuppression (A1-A4) consisted of cyclosporine (CSP, Sandimmune; Novartis, East Hanover, NJ) and mycophenolate mofetil (MMF, CellCept; Roche, Nutley, NJ) as previously described.20
Flow cytometry of CD18+ levels following transplantation
To assess the percentage of CD18+ gene-corrected leukocytes after infusion, peripheral-blood leukocytes were collected from each recipient and analyzed for expression of surface CD18 by incubation with a mouse anti–human CD18 monoclonal antibody (MHM23; DakoCytomation, Carpinteria, CA) or with a mouse anti–canine CD18 monoclonal antibody (CA1.4E9; Serotec, Raleigh, NC). Leukocyte subsets were determined using scatter profiles, or subset specific antibodies as previously described.7 T-lymphocyte subsets were analyzed using antibodies to CD4 (YKIX302.9; Serotec), CD8 (YCATE 55.9; Serotec), and CD45RA (CA4.1D3; Serotec). To facilitate analysis, unconjugated antibodies were labeled in some cases with a Zenon labeling kit (Invitrogen/Molecular Probes, Eugene, OR) according to the manufacturer's directions. Cells were analyzed by flow cytometry on a Becton Dickinson FACSCalibur (San Jose, CA) using Cellquest software. Additional software analysis of the data were performed using FlowJo software (Tree Star, San Carlos, CA).
Cell adhesion assays
Fibrinogen (50 μL; Calbiochem/EMD Biosciences, La Jolla, CA) at 2 mg/mL in phosphate buffered saline (PBS) was coated onto thin-bottom 96-well microtiter plates (Costar 3603; Corning, Corning, NY) for at least 2 hours at room temperature (or 16 hours at 4°C). For blocking experiments, peripheral-blood leukocytes were preincubated with 4 μg anti-CD18 antibody 60.321 per 1 million cells. Leukocytes were labeled with the fluorescent dye Hoechst 33 342 (Invitrogen) to determine nucleated cells. Peripheral-blood leukocytes (500 000 cells in 50 μL) were added to fibrinogen-coated wells in either the presence or absence of 10 ng/mL PMA (phorbol 12-myristate 13-acetate) and incubated at 37°C for 20 to 30 minutes. Nonadherent leukocytes were removed by gentle washing 3 times with addition of PBS/1% human serum albumin (HSA) buffer followed by vacuum aspiration. The remaining adherent leukocytes were stained for the presence of surface CD18 by incubation with anti–canine CD18 antibody CA1.4E9 (labeled with AlexaFluor 488) for 30 minutes at 4°C, followed by washing with PBS/1% HSA buffer and centrifugation at 300g for 5 minutes. Wells were analyzed on an iCys Research Imaging Cytometer (CompuCyte, Cambridge, MA).
5′-LTR integration site analysis using linear amplification–mediated polymerase chain reaction (LAM-PCR)
Genomic DNA was extracted from peripheral-blood leukocytes obtained from the dogs at selected time points after gene therapy. LAM-PCR was performed essentially as described,22 with the following changes. The junction sequence between the viral long-terminal repeat (LTR) and the host genome was linear amplified using 0.25 pmol 5′-biotinylated primer LTRa with either Platinum Taq polymerase (Invitrogen) or Qiagen Taq polymerase (Qiagen, Valencia, CA). Products were digested with 10 U TasI endonuclease (Fermentas, Hanover, MD), then ligated to a double-stranded asymmetric linker cassette containing 4 U Fast-Link DNA Ligase (Epicentre Biotechnologies, Madison, WI) at room temperature for 15 minutes. After separation from magnetic beads, the DNA was used for PCR amplification (95°C for 5 minutes; 35 cycles of 95°C for 15 seconds, 58°C for 30 seconds, 72°C for 45 seconds; 72°C for 10 minutes) containing 2.5 U Platinum Taq or Qiagen Taq and 12.5 pmol each of vector-specific primer (5′-GTCTCTCTGTTCCTAACC-3′) and linker cassette-specific primer (5′-GACCCGGGAGATCTGAATTC-3′). Eight percent of the previous PCR reaction was then used as template for a second, nested PCR amplification using 12.5 pmol each of nested primers (5′-GCTAGCTTGCCAAACCTA-3′ and 5′-AGTGGCACAGCAGTTAGG-3′), using conditions identical to the first PCR. PCR reactions were purified using the Qiaquick PCR Purification Kit (Qiagen, Valencia, CA) and a portion was analyzed on a Spreadex EL1200 high-resolution gel (Elchrom Scientific, Cham, Switzerland).
Results
Construction of a retroviral vector expressing canine CD18
To generate a high-titer retroviral vector capable of transducing canine hematopoietic stem cells, the canine CD18 cDNA was cloned into a murine stem-cell virus (MSCV) retroviral vector backbone23 to produce the vector MSCV-cCD18 (Figure 1). The retroviral vector was pseudotyped with a gibbon ape leukemia virus (GALV) envelope using the PG13 packing cell line as described.17 Transduction of CLAD CD34+ cells by the PG13/MSCV-cCD18 vector corrected the CLAD CD18 defect in vitro, demonstrating that the C36S CD18 mutation in CLAD does not represent a dominant-negative mutation (data not shown).
Figure 1.

Diagram of experimental design. Bone marrow (BM) was harvested from CLAD dogs and enriched for CD34+ cells. CD34+ cells were used immediately, or cryopreserved and thawed 2 to 3 weeks later. After transduction, cells were harvested, washed, and reinfused into CLAD pups that had received conditioning with either 200 cGy TBI on day 0 or –1 (n = 9), or 10 mg/kg busulfan intravenously on day –2(n = 2). Four dogs (A1-A4) received posttransplantation immunosuppression with CSP starting on day –1 and continuing until day +65 (35 days at 30 mg/kg, 30 days at 15 mg/kg), along with MMF from day 0 to day +28 (at 20 mg/kg). Seven dogs received no posttransplantation immunosuppression. LTR, retroviral long terminal repeat; Ψ+, extended packaging signal; gln tRNA PBS, glutamine tRNA primer binding site; PCMV, PCC4-cell passaged myeloproliferative sarcoma virus.
Transduction of CLAD CD34+ hematopoietic stem cells for infusion
To demonstrate the ability of the PG13/MSCV-cCD18 vector to correct the CLAD CD18 defect in vivo, autologous CLAD bone marrow CD34+ cells were transduced and reinfused into nonmyeloablated animals. A schematic of the procedure is shown in Figure 1.
Since the CD18 mutation prevents surface expression, it was unclear whether the infusion of CD18-bearing cells would lead to an immune response against CD18. Therefore, the first 4 dogs (A1-A4) received postinfusion immunosuppression with CSP and MMF (Figure 1). Once it was established that graft rejection had not developed in the first group of dogs, the second group of dogs (B1-B5) received 200 cGy TBI, but no postinfusion immunosuppression. The third group of dogs (C1, C2) received busulfan intravenously as an alternate source of nonmyeloablative conditioning, and no postinfusion immunosuppression. All CLAD dogs were treated before 5.5 months of age because of the natural history of the disease; CLAD-affected animals universally succumb to infection before 6 months of age.24
The transduction efficiency ranged from 11.6% to 33.4%, resulting in a range in the number of CD18+CD34+ cells infused (Table 1). Dog A1 received 2 infusions with an averaged transduction efficiency of 17.8%. The dog with the highest number of infused CD18+CD34+ cells, C1, received 6.1 × 105 CD18+CD34+ cells/kg. In contrast, dog B1 received only 0.8 × 105 CD18+CD34+ cells/kg. Despite this range there was no direct correlation between the number of infused CD18+CD34+ cells and the level of CD18+ leukocytes at 1 year after infusion.
Table 1.
Characteristics of CLAD dogs receiving CD34+/CD18+ gene-corrected cells
| Name | Age at infusion, mo | Transduction efficiency, % | CD18+ CD34+/kg, ×106 | Conditioning regimen | Immunosuppression | % CD18+ leukocytes |
|---|---|---|---|---|---|---|
| A1-Bailey* | 4.9 | 17.8 | 0.20 | TBI | CSP and MMF | 3.44, at 21 mo |
| A2-Rudolph† | 4.8 | 14.3 | 0.41 | TBI | CSP and MMF | 1.3, at 21 mo |
| A3-Blitzen† | 4.6 | 24.5 | 0.55 | TBI | CSP and MMF | 0.72, at 13 mo |
| A4-Sambuca‡ | 5.2 | 33.4 | 0.26 | TBI | CSP and MMF | 0.35, at 6 mo |
| B1-Capt Hook | 2.7 | 15.8 | 0.08 | TBI | None | 3.59, at 21 mo |
| B2-Black Pearl | 3.7 | 21.1 | 0.17 | TBI | None | 2.42, at 21 mo |
| B3-Sonic§ | 2.3 | 14.2 | 0.38 | TBI | None | 0.75, at 4 mo |
| B4-Storm§ | 2.1 | 11.6 | 0.16 | TBI | None | 0.43, at 4 mo |
| B5-Blackbeard‡ | 3.7 | 19.3 | 0.09 | TBI | None | 0.044, at 3.5 mo |
| C1-Maverick | 2.6 | 11.6 | 0.61 | Busulfan | None | 7.62, at 15 mo |
| C2-Mystic§ | 2.8 | 11.6 | 0.55 | Busulfan | None | 0.35, at 3 mo |
% CD18+ leukocytes indicates the percentage of CD18+ leukocytes in the peripheral blood at the latest follow-up timepoint available; TBI, total body irradiation; CSP, cyclosporine; MMF, mycophenolate mofetil.
Dog received 2 infusions. Transduction efficiency was calculated as weighted average of both infusions. Indicated CD18+CD34+/kg number is the combined total of both infusions.
Euthanized at end of study.
Dogs were reconditioned and reinfused with additional transduced cells. Both dogs subsequently died or were euthanized due to complications of repeat conditioning.
Dogs euthanized from complications of CLAD or conditioning.
Flow cytometric analysis of CD18+ cells in the peripheral blood after infusion
To assess the ability of the transduced cells to contribute to hematopoiesis, surface expression of CD18 was assessed on peripheral-blood leukocytes prior to infusion and at monthly time points following infusion. The kinetics of the CD18+ leukocyte levels for the 6 long-term surviving CLAD dogs following the infusion of the transduced cells are shown (Figure 2). In contrast to CLAD allo-transplant studies, where near maximum levels of CD18+ leukocytes were achieved within 1 to 3 months after transplantation,4 the number of retrovirally transduced CD18+ leukocytes in the peripheral-blood increased more gradually (Figure 2). This finding also contrasts with the retroviral marking studies in myeloablated dogs where maximum engraftment typically occurs within the first 2 months following infusion.12,25
Figure 2.

Flow cytometric analysis of CD18+ leukocytes in the peripheral blood following infusion of gene-corrected cells. Peripheral-blood leukocytes from the 6 CLAD dogs surviving long-term were isolated, stained with an anti-CD18 antibody, and examined by flow cytometry. The percentage of CD18+ leukocytes was determined in comparison to isotype stained controls. Prior to the infusion of the gene-corrected CD34+ cells, there were no CD18+ leukocytes (not different from isotype control staining) in the peripheral blood. The CD18+ leukocyte percentages over time in the peripheral blood are shown.
CLAD dogs from all 3 cohorts became long-term survivors following the infusion of autologous, CD18 gene-corrected cells (Table 1). Three of the 4 dogs in the first cohort survived for at least 1 year after infusion, with the highest circulating level of CD18+ leukocytes (3.44%) observed in dog A1 at 21 months after infusion (Table 1). In the second cohort, 2 of the 5 CLAD dogs were alive 21 months after infusion, with CD18+ leukocyte levels of 3.59% in dog B1 and 2.42% in dog B2. The achievement of these levels in the absence of rejection indicated that immunosuppression was not required for long-term engraftment. In the third cohort, which consisted of 10 mg/kg busulfan conditioning, 1 of the 2 CLAD dogs (C1) had the highest CD18+ level (8.37%) observed in any of the treated dogs to date (Figure 2), highlighting the potential of busulfan to replace TBI as a conditioning agent.
Increasing contribution of CD18+ lymphocytes in the peripheral blood
To examine the time-course of CD18+ leukocyte subset engraftment, we assessed the levels of CD18+ granulocyte, monocyte, and lymphocyte populations at monthly intervals after infusion in the 6 long-term survivors (Figure 3). Although several animals demonstrated detectable levels of gene-corrected myeloid cells, particularly neutrophils, at 1 month after infusion, the highest levels of gene-corrected cells occurred at later time points, typically 7 to 9 months after infusion. The kinetics of engraftment of both granulocyte and monocyte populations were similar (Figure 3A-B), likely reflecting the fact that both types of cells derive from the same progenitor population. In contrast to the myeloid-cell compartment, the lymphocyte population in the peripheral-blood continued to increase with time (Figure 3C), reaching a plateau of 8% to 20% in 3 animals (B1, B2, C1) at approximately 12 months after infusion. Dog B1 has remained near 20% at 21 months after infusion. The CD18+ lymphocyte population was comprised primarily of CD3+ T-lymphocyte cells, with a smaller contribution of CD21+ B lymphocytes (data not shown). The elevated lymphocyte level is similar to the situation in our dogs that underwent allogeneic transplantation, where the CD18+ lymphocyte levels gradually increase until they reach a plateau at approximately 1 year after transplantation, at which time the CD18+ donor lymphocyte percentages in the peripheral blood were significantly greater than the CD18+ donor myeloid-cell percentages.4,7 Analysis of T-lymphocyte markers indicated a strong bias toward loss of CD45RA expression, indicating an activated/memory phenotype for CD3+CD18+ cells. Examination of CD45RA expression on peripheral-blood CD3+ T-lymphocytes of 4 dogs (A1, B1, B2, C1 at 15-22 months after infusion) indicated an average of 21.4% of CD18+ CD3+CD45RA–/lo cells, in contrast to an average of 8.6% CD18–CD3+CD45RA–/lo cells. A similar examination of the CD4 and CD8 phenotypes of the lymphocyte compartment of the same 4 dogs did not reveal a preference for the CD18+ lymphocytes, although CD18– lymphocytes from all 4 dogs did show a bias toward a CD4 phenotype (data not shown). Curiously, nearly all CD18+ CD3+CD45RA–/lo cells were of the CD4+ phenotype, although this was also true of cells from a normal dog.
Figure 3.

Assessment of CD18+ peripheral-blood leukocyte subsets in the 6 treated CLAD dogs with 1-year follow-up. The contribution of each subset was assessed in each of the 6 long-term treated dogs by flow cytometric analysis of CD18 expression on (A) granulocytes, (B) CD14+ monocytes, and (C) lymphocytes. Each subset was analyzed using a specific subset antibody, or was determined by the absence of CD14 staining and side scatter (SSC) profile (high SSC, granulocytes; low SSC, lymphocytes), along with an anti-CD18 antibody.
Preferential adhesion of activated CD18+ leukocytes
Although flow cytometric analysis demonstrated restoration of CD18 expression on the gene-corrected leukocyte surfaces in all the dogs treated with gene therapy, demonstration of functional correction of the adhesion defect required in vitro adhesion assays. Peripheral-blood leukocytes were isolated from 5 treated CLAD dogs (A1, A2, B1, B2, C1), and unstimulated (without PMA) or stimulated (with PMA) leukocytes were added to wells coated with fibrinogen, a substrate for CD11/CD18 binding.26 In the absence of stimulation by PMA, a well-described activator of CD11/CD18, the cells remained rounded (Figure 4A, top panel) with few CD18+ cells present. The remaining CD18–leukocytes most likely adhered to fibrinogen through the VLA-5 integrin.27 Upon stimulation by PMA, there was increased adhesion and spreading of activated CD18+ leukocytes on the fibrinogen substrate (Figure 4A, middle panel). This adherence of activated CD18+ leukocytes to fibrinogen was blocked by preincubation with an anti-CD18 antibody 60.3, which is known to block CD11/CD18-mediated leukocyte adhesion (Figure 4A, bottom panel).28
Figure 4.
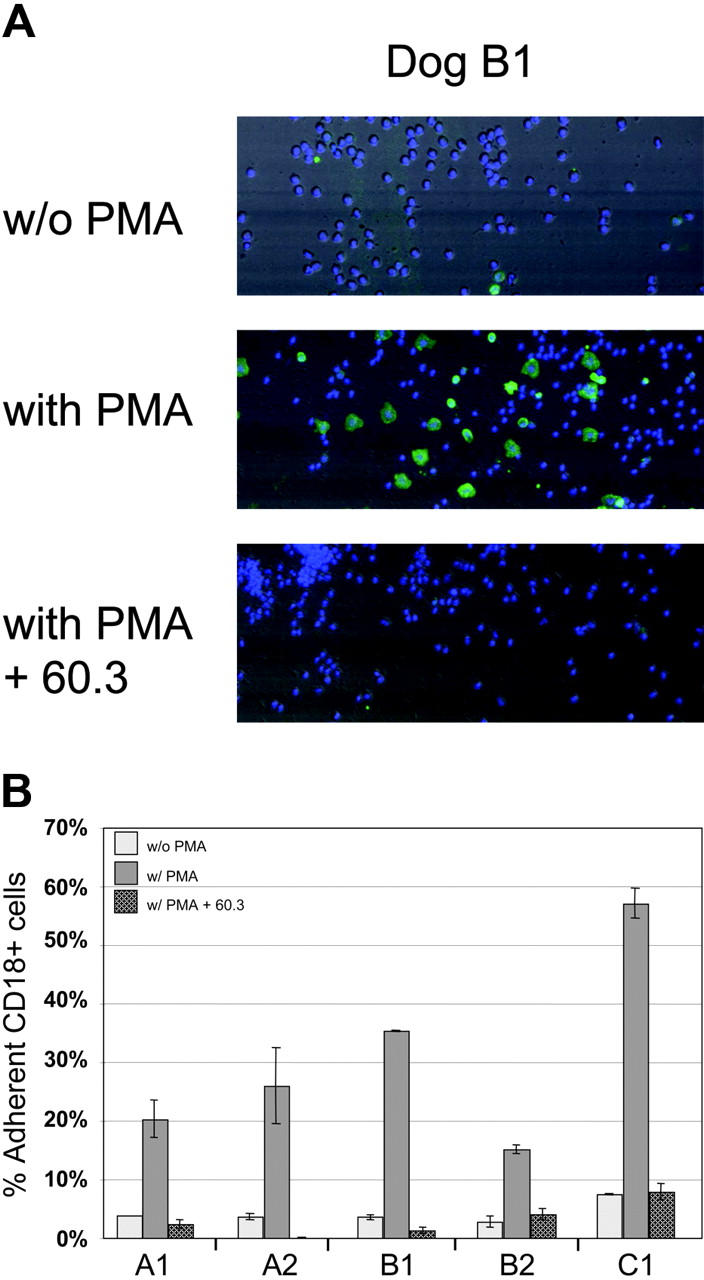
In vitro detection of adhesion by activated CD18+ leukocytes. Peripheral-blood leukocytes from dogs A1, A2, B1, B2, and C1 were added to fibrinogen-coated wells with or without stimulation by PMA or blocking by the anti-CD18 antibody 60.3 antibody. (A) Photo-multiplier tube (PMT) reconstructed images of field images (500 μm × 192 μm) of representative areas of the wells from dog B1 using scatter, CD18 green fluorescence, and blue fluorescence for Hoechst-stained nucleated cells. Without PMA stimulation, few of the adherent leukocytes are CD18+ (without PMA; top panel). Activated CD18+ leukocytes are present following PMA stimulation (with PMA; middle panel). Blocking of PMA-stimulated CD18+ leukocytes was accomplished using the anti-CD18 monoclonal antibody 60.3 (with PMA + 60.3; bottom panel). (B) Cell adherence was assessed using total peripheral-blood leukocytes from 5 treated CLAD dogs. The number of adherent leukocytes that are CD18+, divided by the total number of adherent CD18+ and CD18– leukocytes present, indicates the percentage of adherent leukocytes that are CD18+. The percentage of nonspecific adherence is influenced by washing stringency, hence the ratio of the stimulated-to-nonstimulated percentages is a more accurate indicator of preferential adhesion than the total number of cells binding. The average of 2 experiments is shown with the range indicated.
Peripheral-blood leukocytes from 5 treated CLAD dogs were compared in the adhesion assay described. In the absence of PMA, low percentages of CD18+ leukocytes adhered (Figure 4B), consistent with the requirement of stimulation for CD18-dependent adhesion. Following PMA stimulation, there was a 5- to 9-fold increase in the percentage of adherent leukocytes that were CD18+ (Figure 4B). This increased adhesion in response to PMA could be blocked by the anti-CD18 antibody 60.3, demonstrating that the adhesion of the activated, CD18+ gene-corrected leukocytes was CD18 dependent.
In vivo function of CD18+ leukocytes
The development of an inflammatory exudate in 2 CLAD dogs (A3, A4) shortly after infusion of gene-corrected cells provided the opportunity to examine the function of CD18+ gene-corrected leukocytes in vivo, namely the ability of CD18+ leukocytes to preferentially migrate into external tissues. Leukocytes from the peripheral blood and pus from 2 treated CLAD dogs were examined for CD18 expression. In dog A3 the percentage of CD18+ leukocytes in the peripheral blood was 0.21% at 3 weeks after infusion, whereas the percentage of CD18+ leukocytes in the inflammatory exudate was 11.8% (Figure 5, top panels). Similarly, in dog A4 the percentage of CD18+ cells in the peripheral blood was 0.16% at 9 weeks after infusion, whereas the percentage of CD18+ leukocytes in the inflammatory exudate was 2.3% (Figure 5, bottom panels). Thus, similar to results in the allo-transplantation studies with saliva,7 leukocytes expressing CD18 demonstrated a preferential migration into external sites, especially an inflammatory exudate (Figure 5).
Figure 5.
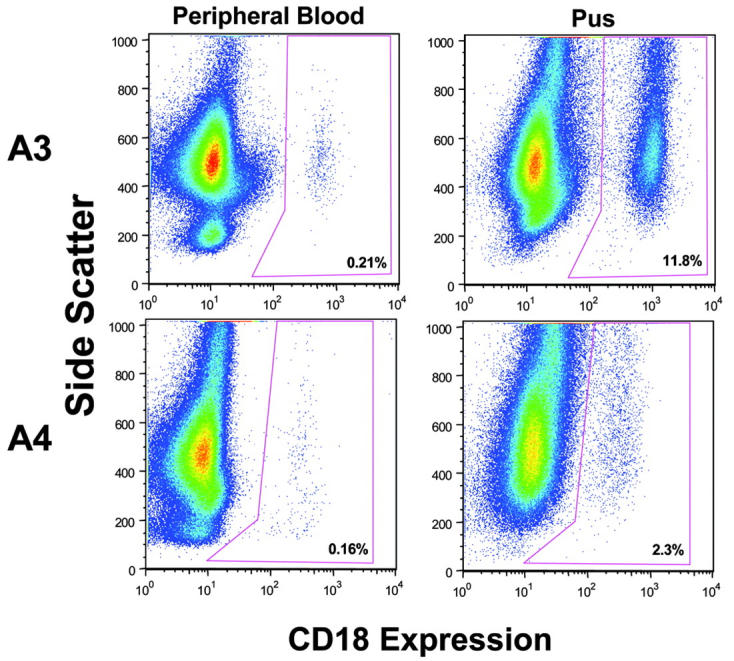
Selective migration of CD18+ leukocytes in vivo. Analysis of CD18+ leukocytes in the peripheral-blood and pus collected at the same time in 2 CLAD dogs after infusion of CD18+ gene-corrected cells demonstrated in vivo migration of CD18+ leukocytes. Leukocytes were isolated from the peripheral blood and pus from infected tissue (dog A3, week 3; dog A4, week 9) and stained for CD18. Dotplots represent CD18 expression (x-axis) versus side-scatter (y-axis). Percentages of CD18+ leukocytes are shown at the lower right corner.
Improvement of the CLAD phenotype with CD18+ gene-corrected cells
Since the in vitro and in vivo assays demonstrated normal phenotypic function of CD18+ gene-corrected cells, we examined whether the levels of circulating CD18+ leukocytes were sufficient to improve or reverse the clinical phenotype in the treated dogs. Previous studies in CLAD dogs that underwent allogeneic transplantation indicated that a low percentage of circulating CD18+ leukocytes (2%-5%) could reverse the disease phenotype.7
We examined the clinical course of the 6 dogs with 1-year follow-up by assessment of episodes of fever requiring clinical intervention and days spent in intensive care. The onset of craniomandibular osteopathy (CMO) and hypertropic osteodystrophy (HOD), which are noninfectious, nonneoplastic proliferative bone diseases occurring with a high incidence in CLAD dogs, were also noted.29 All 6 CLAD dogs manifested severe HOD or CMO prior to the infusion of gene-corrected cells with multiple episodes of fever with pain requiring intensive care (Figure 6). In the 4 dogs with the highest level of CD18+ leukocyte levels (A1, B1, B2, C1), few episodes of fever have occurred since attainment of their CD18+ gene-corrected cells at a CD18+ ANC of 100 cells/μL (correlating very closely to a 1% CD18+ leukocyte level), and all 4 have had reversal of their severe CLAD phenotype. None of these 4 dogs had recurrence of CMO or HOD since shortly after reaching a 100 CD18+ ANC. All 4 dogs also had fewer episodes of severe leukocytosis (> 30 K/μL), a hallmark of overt infection, and all 4 dogs had near normalization of their WBC counts (13 k/μL-23 K/μL at the latest timepoints) and are off prophylactic antibiotics. The 2 dogs with the lowest CD18 levels (A2, A3) also had improvement of their clinical phenotype, as demonstrated by their survival beyond 6 months of age, and by fewer episodes of fever and clinical signs of CLAD after attaining 100 ± 5 CD18+ ANC (Figure 6). Both dogs achieved a CD18+ ANC of 50 at 12 to 16 weeks after transplantation. This level has been show previously to convert the severe CLAD phenotype to a moderate phenotype7,8 and likely contributed to their increased survival. Analysis of all 6 dogs indicated a significant change in the average number of days of clinical signs after reaching an ANC of 100 ± 5 CD18+ neutrophils/μL(P = .007).
Figure 6.

Clinical course of 6 long-term surviving CLAD dogs following gene therapy. The clinical course of the 6 long-term CLAD survivors treated with gene therapy is shown prior to and following infusion of gene-corrected cells. The line and arrow indicate the period of follow-up for each dog. Episodes of fever (≥ 102.4°F) necessitating a more aggressive treatment (increased antibiotics/fluids) are shown by the vertical lines. The shaded boxes indicate periods during which the animal was in intensive care. The appearance of an episode of HOD (lameness/joint swelling/leg pain) or CMO (bony jaw/cranial swelling/soreness) is indicated by the closed diamonds. The day of infusion (day 0) is indicated. Attainment of a CD18+ ANC level of 100 ± 5 cells/μL is indicated by the inverted pyramid. Time in days in relationship to the infusion is shown on the x-axis in the panel.
Five CLAD dogs did not have reversal of the phenotype with gene therapy. Dog B3 was euthanized due to exocrine pancreatic insufficiency, a sequelae from conditioning with radiation. Dog B4 had a very low level of engraftment, suffered chronic enteritis, and had severe, obstructive mesenteric lymphadenopathy. Dog C2 suffered from severe CMO, which compromised her ability to open her mouth and eat. Two of the dogs (A4, B5) had a very low level of CD18+ neutrophils following gene therapy and had recurrent symptoms of CLAD. Both dogs received a second round of conditioning and infusion of CD18+ gene-corrected cells to boost their CD18+ circulating leukocyte levels. However, both dogs subsequently became pancytopenic and were euthanized or died of complications from radiation-induced enteritis within weeks of the reconditioning.
Clonality testing of leukocytes indicates maintenance of polyclonality
To determine the potential genotoxicity following the infusion of gene-corrected hematopoietic cells, especially in the context of increasing numbers of CD18+ lymphocytes, we examined the peripheral-blood leukocytes for evidence of clonality over time in the 6 dogs with the longest follow-up (A1-A3, B1, B2, C1). DNA isolated from leukocytes at 6 months and 12 months was amplified by LAM-PCR30 to examine clonality of retroviral integrations (Figure 7). The results demonstrate polyclonality at both time points, with no predominant clone or clones.
Figure 7.

Clonality analysis by linear amplification–mediated PCR (LAM-PCR). Genomic DNA was isolated from peripheral-blood leukocytes of 6 dogs at 2 time points (6 and 12 months after infusion) and subjected to LAM-PCR to examine clonality of retroviral insertions. The presence of a band likely indicates a unique insertion site. Samples were electrophoresed on a Spreadex gel and photographed by a digital camera. M indicates marker; bp, base pairs.
Discussion
We describe results with 11 CLAD pups who received autologous, CD18-transduced hematopoietic stem cells preceded by a clinically applicable, nonmyeloablative conditioning regimen of 200 cGy TBI or 10 mg/kg busulfan. The requirement for postinfusion immunosuppression was also assessed. Both reduced-intensity regimens resulted in the long-term engraftment of CD18+ gene-corrected stem cells with reversal or marked improvement of the CLAD clinical phenotype in 6 of the 11 treated pups with CLAD. Postinfusion immunosuppression was not required for reversal of the phenotype. Six dogs survived more than 12 months following infusion of autologous gene-corrected cells at 2 to 5 months of age. By comparison, untreated CLAD dogs universally die or are euthanized due to intractable infections by 6 months of age.4,24 This study represents the first successful application of a nonmyeloablative conditioning regimen preceding ex vivo gene transfer into hematopoietic stem cells in a disease-specific, large animal model.
Previous human gene therapy protocols involving ex vivo transduction of hematopoietic stem cells have not used homologous, large animal disease models prior to the initiation of human studies. Although marking studies in large animals such as dogs or nonhuman primates have been invaluable for optimization of transduction conditions and testing of new vectors, they unfortunately do not address the role of therapeutic genes for particular diseases.
The CLAD disease model has a number of important features to recommend it as a model genetic hematopoietic disease for preclinical studies. First, the distinct clinical phenotype in CLAD provides a clear end-point against which therapeutic intervention can be assessed, namely the reversal of the clinical syndrome of life-threatening infections. Second, the ability to detect CD18+ leukocytes in the peripheral blood by flow cytometry enables repeated and quantitative measurements of the level of corrected leukocytes using anti-CD18 antibodies. Third, the canine model is well established in the field of bone marrow transplantation,31,32 and studies in dogs have resulted in the direct translation of research data to the human clinical setting.
In our gene transfer studies in the CLAD model, animals were treated with a nonmyeloablative dose of 200 cGy TBI or 10 mg/kg busulfan, instead of a fully ablative 920 cGy dose used by other investigators.33,34 Part of the rationale for using a myeloablative dose as conditioning in previous marking studies in large animals, in addition to creating space, is to prevent the occurrence of an immune response against a transferred foreign marker gene. However, in one study despite the use of an ablative dose of 920 cGy TBI, an immune response against a foreign protein, green fluorescent protein, persisted.25
We initially pursued a nonmyeloablative regimen with immunosuppression consisting of CSP and MMF in the first 4 treated CLAD animals to prevent a potential lymphocyte-mediated immune response against CD18. This immunosuppressive regimen was based on a regimen used in our previous CLAD transplant studies.4,5,7 The results from our current studies indicate that in the gene-corrected autologous setting in CLAD, the presence of CD18 does not cause an immune response resulting in graft rejection.
The second issue that arises using a nonmyeloablative regimen is the stability of the transplant. Recent work by Dunbar and colleagues35 suggests clonal fluctuation of stem cells in nonhuman primates given a nonmyeloablative dose of 300 cGy TBI. Previous marking experiments in dogs and baboons using myeloablative conditioning showed a rapid increase in marked leukocytes in the first weeks after infusion, with the highest levels immediately after infusion.33,36,37 Since nonmyeloablative conditioning regimens typically do not lead to a significant drop in leukocyte counts, particularly of the myeloid compartment, this likely leads to competition between the infused cells and the remaining host cells. The myeloid compartment of our treated CLAD dogs appears to undergo some fluctuation with time.
The increasing contribution of CD18+ lymphocytes to the overall CD18+ leukocyte pool within the peripheral blood was also investigated. These increasing levels are similar to the kinetics seen in dogs with CLAD that have undergone transplantation, highlighting the important immune function of CD18 on lymphocytes. Since CD11/CD18 is an important costimulatory molecule for T-helper function, we assessed whether this could represent increased activation of CD18+ T cells. Analysis of the T-cell activation/memory marker CD45RA indicated a strong bias of the CD18+ T-lymphocyte populations toward an activated/memory CD45RA–/lo phenotype as opposed to the CD45RAhi naive phenotype. There was also no apparent bias of the CD18+ T lymphocytes in expression of CD4 or CD8 in contrast to CD18– T lymphocytes, which had greater CD4+ percentages. Since total T-cell numbers are increasing with time after transplantation, CD18+ T cells are contributing more to the overall CD18+ leukocyte levels.
The increasing levels of CD18+ leukocytes, particularly lymphocytes, also raised the possibility of cooperation with an oncogene activated by insertional mutagenesis, as shown by the leukemias in 3 X-linked–SCID patients infused with IL-2Rγ+ gene-corrected cells.38 Recent studies by Dave and colleagues39 suggest that the IL-2Rγ gene itself may be an oncogene. In comparison to the IL-2Rγ protein, the CD11/CD18 heterodimer may provide a lesser role in the development of lymphocytes, since the CD18+ T-lymphocyte levels in dogs with CLAD that underwent transplantation reach a plateau over time. In contrast, X-SCID transplant recipients have nearly 100% donor-derived IL-2Rγ+ T lymphocytes shortly after transplantation due to strong selection. To address the concern of increasing CD18+ lymphocyte levels in CLAD dogs, we tested DNA isolated from peripheral-blood leukocytes from all 6 long-term CLAD dogs for clonality by LAM-PCR. The results indicate maintenance of polyclonality, with no evidence to point toward a vector-mediated lymphoproliferative disease.
In summary, the CLAD model facilitates the testing of efficacy and safety of novel therapeutic approaches in an appropriate, large animal model prior to their application in humans with the disease. Our results demonstrate that a nonmyeloablative conditioning regimen can allow sufficient engraftment of autologous, transduced CD34+ stem cells to reverse or improve the CLAD disease phenotype. Long-term follow-up will determine the ability of the engrafted cells to maintain a clinically relevant level of CD18+ leukocytes, and will allow for assessment of the safety of CD18-containing retroviral vectors. These studies are an important first step in the pathway toward future gene therapy studies in human LAD patients.
Acknowledgments
We wish to thank William Telford and Veena Kapoor, NCI, for flow cytometry assistance; Rima Adler, NHLBI, for assistance with the LAM-PCR assays; and Anastasia Sowers, NCI, for assistance with irradiation of the CLAD dogs.
Prepublished online as Blood First Edition Paper, July 25, 2006; DOI 10.1182/blood-2006-03-006908.
Supported by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research.
The authors declare no competing financial interests.
Presented in part in abstract form at the 46th and 47th annual meetings of the American Society of Hematology, San Diego, CA, December 6, 2004, and Atlanta, GA, December 12, 2005.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
References
- 1.Anderson DC, Schmalsteig FC, Finegold MJ, et al. The severe and moderate phenotypes of heritable Mac-1, LFA-1 deficiency: their quantitative definition and relation to leukocyte dysfunction and clinical features. J Infect Dis. 1985;152: 668-689. [DOI] [PubMed] [Google Scholar]
- 2.Kishimoto TK, Hollander N, Roberts TM, Anderson DC, Springer TA. Heterogenous mutations in the β subunit common to the LFA-1, Mac-1, and p150,95 glycoproteins cause leukocyte adhesion deficiency. Cell. 1987;50: 193-202. [DOI] [PubMed] [Google Scholar]
- 3.Thomas C, Le Deist F, Cavazzana-Calvo M, et al. Results of allogeneic bone marrow transplantation in patients with leukocyte adhesion deficiency. Blood. 1995;86: 1629-1635. [PubMed] [Google Scholar]
- 4.Bauer TR Jr, Gu YC, Tuschong LM, et al. Nonmyeloablative hematopoietic stem cell transplantation corrects the disease phenotype in the canine model of leukocyte adhesion deficiency. Exp Hematol. 2005;33: 706-712. [DOI] [PubMed] [Google Scholar]
- 5.Creevy KE, Bauer Jr TR, Tuschong LM, et al. Mixed chimeric hematopoietic stem cell transplant reverses the disease phenotype in canine leukocyte adhesion deficiency. Vet Immunol Immunopathol. 2003;95: 113-121. [DOI] [PubMed] [Google Scholar]
- 6.Lindblad-Toh K, Wade CM, Mikkelsen TS, et al. Genome sequence, comparative analysis and haplotype structure of the domestic dog. Nature. 2005;438: 803-819. [DOI] [PubMed] [Google Scholar]
- 7.Bauer TR Jr, Creevy KE, Gu YC, et al. Very low levels of donor CD18+ neutrophils following allogeneic hematopoietic stem cell transplantation reverse the disease phenotype in canine leukocyte adhesion deficiency. Blood. 2004;103: 3582-3589. [DOI] [PubMed] [Google Scholar]
- 8.Gu YC, Bauer TR, Sokolic RA, et al. Conversion of the severe to the moderate disease phenotype with donor leukocyte microchimerism in canine leukocyte adhesion deficiency. Bone Marrow Transplant. 2006;37: 607-614. [DOI] [PubMed] [Google Scholar]
- 9.Barese CN, Goebel WS, Dinauer MC. Gene therapy for chronic granulomatous disease. Expert Opin Biol Ther. 2004;4: 1423-1434. [DOI] [PubMed] [Google Scholar]
- 10.Bauer Jr TR, Hickstein DD. Gene therapy for leukocyte adhesion deficiency. Curr Opin Mol Ther. 2000;2: 383-388. [PubMed] [Google Scholar]
- 11.Horn PA, Keyser KA, Peterson LJ, et al. Efficient lentiviral gene transfer to canine repopulating cells using an overnight transduction protocol. Blood. 2004;103: 3710-3716. [DOI] [PubMed] [Google Scholar]
- 12.Thomasson B, Peterson L, Thompson J, Goerner M, Kiem HP. Direct comparison of steady-state marrow, primed marrow, and mobilized peripheral blood for transduction of hematopoietic stem cells in dogs. Hum Gene Ther. 2003;14: 1683-1686. [DOI] [PubMed] [Google Scholar]
- 13.Aiuti A, Ficara F, Cattaneo F, Bordignon C, Roncarolo MG. Gene therapy for adenosine deaminase deficiency. Curr Opin Allergy Clin Immunol. 2003;3: 461-466. [DOI] [PubMed] [Google Scholar]
- 14.Ott MG, Schmidt M, Schwarzwaelder K, et al. Correction of X-linked chronic granulomatous disease by gene therapy, augmented by insertional activation of MDS1-EVI1, PRDM16 or SETBP1. Nat Med. 2006;12: 401-409. [DOI] [PubMed] [Google Scholar]
- 15.Creevy KE, Bauer TR Jr, Tuschong LM, et al. Canine leukocyte adhesion deficiency colony for investigation of novel hematopoietic therapies. Vet Immunol Immunopathol. 2003;94: 11-22. [DOI] [PubMed] [Google Scholar]
- 16.Kijas JMH, Bauer TR Jr, Gäfvert S, et al. A missense mutation in the β-2 integrin gene (ITGB2) causes canine leukocyte adhesion deficiency. Genomics. 1999;61: 101-107. [DOI] [PubMed] [Google Scholar]
- 17.Miller AD, Garcia JV, von Suhr N, Lynch CM, Wilson C, Eiden MV. Construction and properties of retrovirus packaging cells based on gibbon ape leukemia virus. J Virol. 1991;65: 2220-2224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bauer Jr TR, Miller AD, Hickstein DD. Improved transfer of the leukocyte integrin CD18 subunit into hematopoietic cell lines by using retroviral vectors having a gibbon ape leukemia virus envelope. Blood. 1995;86: 2379-2387. [PubMed] [Google Scholar]
- 19.Sokolic RA, Bauer TR, Gu YC, et al. Nonmyeloablative conditioning with busulfan before matched littermate bone marrow transplantation results in reversal of the disease phenotype in canine leukocyte adhesion deficiency. Biol Blood Marrow Transplant. 2005;11: 755-763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Storb R, Yu C, Zaucha JM, et al. Stable mixed hematopoietic chimerism in dogs given donor antigen, CTLA4Ig, and 100 cGy total body irradiation before and pharmacologic immunosuppression after marrow transplant. Blood. 1999;94: 2523-2529. [PubMed] [Google Scholar]
- 21.Hickstein DD, Locksley RM, Beatty PG, Smith AJ, Stone DM, Root RK. Monoclonal antibodies binding to the neutrophil C3bi receptor have disparate functional effects. Blood. 1986;67: 1054-1062. [PubMed] [Google Scholar]
- 22.Kuramoto K, Follman D, Hematti P, et al. The impact of low-dose busulfan on clonal dynamics in nonhuman primates. Blood. 2004;104: 1273-1280. [DOI] [PubMed] [Google Scholar]
- 23.Hawley RG, Lieu FH, Fong AZ, Hawley TS. Versatile retroviral vectors for potential use in gene therapy. Gene Ther. 1994;1: 136-138. [PubMed] [Google Scholar]
- 24.Trowald-Wigh G, Håkansson L, Johannisson A, Norrgren L, Hård af Segerstad C. Leucocyte adhesion protein deficiency in Irish setter dogs. Vet Immunol Immunopathol. 1992;32: 261-280. [DOI] [PubMed] [Google Scholar]
- 25.Beagles KE, Peterson L, Zhang X, Morris J, Kiem HP. Cyclosporine inhibits the development of green fluorescent protein (GFP)-specific immune responses after transplantation of GFP-expressing hematopoietic repopulating cells in dogs. Hum Gene Ther. 2005;16: 725-733. [DOI] [PubMed] [Google Scholar]
- 26.Wright SD, Weitz JI, Huang AJ, Levin SM, Silverstein SC, Loike JD. Complement receptor type three (CD11b/CD18) of human polymorpho-nuclear leukocytes recognizes fibrinogen. Proc Natl Acad Sci U S A. 1988;85: 7734-7738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ugarova TP, Yakubenko VP. Recognition of fibrinogen by leukocyte integrins. Ann N Y Acad Sci. 2001;936: 368-385. [DOI] [PubMed] [Google Scholar]
- 28.Beatty PG, Ledbetter JA, Martin PJ, Price TH, Hansen JA. Definition of a common leukocyte cell-surface antigen (Lp95–150) associated with diverse cell-mediated immune functions. J Immunol. 1983;131: 2913-2918. [PubMed] [Google Scholar]
- 29.Trowald-Wigh G, Ekman S, Hansson K, Hedhammar Å, Hård af Segerstad C. Clinical, radiological and pathological features of 12 Irish setters with canine leucocyte adhesion deficiency. J Small Anim Pract. 2000;41: 211-217. [DOI] [PubMed] [Google Scholar]
- 30.Schmidt M, Carbonaro DA, Speckmann C, et al. Clonality analysis after retroviral-mediated gene transfer to CD34+ cells from the cord blood of ADA-deficient SCID neonates. Nat Med. 2003;9: 463-468. [DOI] [PubMed] [Google Scholar]
- 31.Storb R, Deeg HJ, Raff R, et al. Prevention of graft-versus-host disease. Studies in a canine model. Ann N Y Acad Sci. 1995;770: 149-164. [DOI] [PubMed] [Google Scholar]
- 32.Storb R, Epstein RB, Ragde H, Bryant J, Thomas ED. Marrow engraftment by allogeneic leukocytes in lethally irradiated dogs. Blood. 1967;30: 805-811. [PubMed] [Google Scholar]
- 33.Goerner M, Horn PA, Peterson L, et al. Sustained multilineage gene persistence and expression in dogs transplanted with CD34+ marrow cells transduced by RD114-pseudotype oncoretro-virus vectors. Blood. 2001;98: 2065-2070. [DOI] [PubMed] [Google Scholar]
- 34.Kiem HP, McSweeney PA, Bruno B, et al. Improved gene transfer into canine hematopoietic repopulating cells using CD34-enriched marrow cells in combination with a gibbon ape leukemia virus-pseudotype retroviral vector. Gene Ther. 1999;6: 966-972. [DOI] [PubMed] [Google Scholar]
- 35.Laukkanen MO, Kuramoto K, Calmels B, et al. Low-dose total body irradiation causes clonal fluctuation of primate hematopoietic stem and progenitor cells. Blood. 2005;105: 1010-1015. [DOI] [PubMed] [Google Scholar]
- 36.Goerner M, Bruno B, McSweeney PA, Buron G, Storb R, Kiem HP. The use of granulocyte colony-stimulating factor during retroviral transduction on fibronectin fragment CH-296 enhances gene transfer into hematopoietic repopulating cells in dogs. Blood. 1999;94: 2287-2292. [PubMed] [Google Scholar]
- 37.Horn PA, Topp MS, Morris JC, Riddell SR, Kiem HP. Highly efficient gene transfer into baboon marrow repopulating cells using GALV-pseudo-type oncoretroviral vectors produced by human packaging cells. Blood. 2002;100: 3960-3967. [DOI] [PubMed] [Google Scholar]
- 38.Hacein-Bey-Abina S, Von Kalle C, Schmidt M, et al. LMO2-associated clonal T cell proliferation in two patients after gene therapy for SCID-X1. Science. 2003;302: 415-419. [DOI] [PubMed] [Google Scholar]
- 39.Dave UP, Jenkins NA, Copeland NG. Gene therapy insertional mutagenesis insights. Science. 2004;303: 333. [DOI] [PubMed] [Google Scholar]